
Abstract
Significance:
Cell homeostasis and redox balance are regulated in part by hydrogen sulfide (H2S), a gaseous signaling molecule known as a gasotransmitter. Given its biological roles, H2S has promising therapeutic potential, but controlled delivery of this reactive and hazardous gas is challenging due to its promiscuity, rapid diffusivity, and toxicity at high doses. Macromolecular and supramolecular drug delivery systems are vital for the effective delivery of many active pharmaceutical ingredients, and H2S stands to benefit greatly from the tunable physical, chemical, and pharmacokinetic properties of polymeric and/or self-assembled drug delivery systems.
Recent Advances:
Several types of H2S-releasing macro- and supramolecular materials have been developed in the past 5 years, and the field is expanding quickly. Slow-releasing polymers, polymer assemblies, polymer nano- and microparticles, and self-assembled hydrogels have enabled triggered, sustained, and/or localized H2S delivery, and many of these materials are more potent in biological assays than analogous small-molecule H2S donors.
Critical Issues:
H2S plays a role in a number of (patho)physiological processes, including redox balance, ion channel regulation, modulation of inducible nitric oxide synthase, angiogenesis, blood pressure regulation, and more. Chemical tools designed to (i) deliver H2S to study these processes, and (ii) exploit H2S signaling pathways for treatment of diseases require control over the timing, rate, duration, and location of release.
Future Directions:
Development of new material approaches for H2S delivery that enable long-term, triggered, localized, and/or targeted delivery of the gas will enable greater understanding of this vital signaling molecule and eventually expedite its clinical application.
Introduction
Next-generation approaches to disease treatment will rely on the precise and controlled delivery of active pharmaceutical ingredients (APIs) such as drugs, peptides, enzymes, and vaccines (11, 43, 84). To this end, drug delivery systems provide numerous benefits, including a wide range of administration techniques, increased efficacy and longer circulation times relative to pure APIs, and the potential for advanced delivery conditions (e.g., targeting, triggered response). Often researchers transport, contain, and/or protect therapeutic agents using synthetic polymers due to their ease of synthesis and wide versatility (25). In fact, many pharmaceuticals on the market use macromolecular/supramolecular delivery approaches, including microparticle depots (e.g., Lupron, Vivitrol), liposomal formulations (e.g., Doxil), nanoparticles (e.g., Abraxane), and implantable systems (e.g., Vitrasert) (5, 23, 29, 37, 78). The development of polymeric drug delivery systems is well studied, with many thorough reviews existing in the literature (3, 19, 47, 64, 70, 90). Despite several decades of work in this field, there remains a clinical need to continue to develop drug delivery systems.
Signaling gases, known as gasotransmitters (65, 98, 102), stand to benefit from drug delivery approaches even more than conventional small-molecule drugs, proteins, and other APIs due to their potency and fast action. Gasotransmitters are biological signaling molecules, which fulfill several well-defined criteria, the most important being enzymatic regulation of their production/metabolism and their independence from specific membrane receptors (99, 100). There exist three known gasotransmitters: nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S). Each is produced and regulated endogenously, and each has specific physiological functions and targets. There exist therapeutics on the market and/or in clinical trials for formulations of each gasotransmitter, either in the form of the gas itself or as small molecules that release or generate the gas in vivo (16, 45, 94).
Unlike other chemical messengers, gasotransmitter signaling is not limited by exocytosis-mediated release, receptor binding, vesicular storage, and/or elaborate removal processes from the site of action (102). Instead, cells quickly turn on or off gasotransmitter production to regulate autocrine or paracrine signaling, and these gases diffuse rapidly through biological membranes. Their short in vivo half-lives match their rapid production, allowing NO, CO, and H2S to work together to regulate cell behavior nearly instantaneously (101). Due to their gaseous nature, fast diffusivity, and high reactivity, gasotransmitters are challenging to administer therapeutically but may offer substantial benefits if delivered at a desired site of action in a controlled manner.
H2S, the most recent addition to the list of gasotransmitters (1, 35, 100), has attracted attention due to its various biological roles, including cardioprotection (6, 15, 76), vasodilation (66, 89, 114), angiogenesis (85, 87, 97), anti-inflammation (105, 112), neurotransmission (50, 51, 104), and others. It is produced endogenously by the enzymes cystathione gamma-lyase, cystathione beta-synthetase, and 3-mercaptopyruvate sulfur transferase, which are present in organs and systems throughout the body. Physiological concentrations of H2S are highly regulated by distinct biosynthetic processes, which contribute toward homeostasis of the organism. The appreciable roles of H2S in physiological and pathophysiological processes have led researchers to develop methods to deliver H2S to study its physiological pathways and therapeutic potential (68, 86, 95). Indeed, small molecules that release H2S in vivo (so-called H2S donors) provide evidence for the beneficial effects of exogenous delivery of H2S (20, 57, 67, 93). However, as is true for other gasotransmitters, H2S is toxic at high concentrations, it has a short half-life, and it has no specific receptor but many targets. Coupled with its propensity to oxidize, these factors make sustained release and precise dosage control vital for H2S delivery, even more so than in delivery of traditional therapeutics (75).
Delivery of H2S can take several forms. Direct inhalation presents the most straightforward method for exogenous H2S administration, delivering the gas without any byproducts or carriers. The inhalation method can deliver the required H2S dosage by modulating gas partial pressure over time. Researchers have successfully employed inhalation to systemically deliver H2S in mice, initiating a state of suspended animation or slower metabolism (10). Despite its advantages, inhaled delivery of H2S is not likely to reach a clinical setting due to the malodorous nature of the gas and its lack of targeting. Stringent safety regulations concerning storage of this flammable and corrosive gas also hinder inhaled delivery.
Inorganic sulfide salts (NaHS, Na2S, and CaS) are common alternatives to gaseous H2S delivery, providing instantaneous access to H2S in its pure form while avoiding the need to handle the gas directly. Sulfide salts are currently the most widely employed delivery strategy in biological and preclinical studies investigating the in vitro and in vivo roles of H2S. Despite their availability and easy administration, sulfide salts have several drawbacks. In particular, the burst H2S release profile from aqueous sulfide salt solutions stands in stark contrast to sustained, tightly regulated enzymatic H2S production.
To address these drawbacks, chemists and pharmacologists have designed synthetic H2S donors over the past decade with control over the release kinetics, dosage, and location of release. Recent reviews cover the many types of existing small-molecule H2S donors (53, 71, 109, 115). However, designing a synthetic H2S donor is not trivial and requires careful consideration of stability, water solubility, H2S release rate, trigger specificity, and toxicity of the donor itself as well as any byproducts generated after H2S release. Thus, despite great progress in the development of H2S donors, advanced delivery methods are still needed.
Due to the benefits of drug delivery systems and the inherent challenges in effectively delivering H2S, there has been a strong interest in macromolecular and supramolecular H2S donors (21, 75, 91). Here, we provide an overview of the development and current state of macromolecular/supramolecular H2S donor systems. In particular, we focus on the distinct advantages that polymeric and self-assembled delivery systems provide over small-molecule H2S donors, and we illustrate how these advantages may enable a greater understanding of H2S (patho)physiology and potentially translate to clinical applications of H2S therapy.
Advantages of Macromolecular Systems
Macromolecular/supramolecular drug delivery systems can modulate the chemical, physical, and pharmacokinetic properties of an active pharmaceutical ingredient (API) without drastically changing the chemical nature of the payload. Through this control over these key properties, many of the challenges associated with delivering therapeutics can be addressed. For example, a large number of potential therapeutic agents are inherently hydrophobic. This presents a problem with solubility in biological systems, ultimately leading to low bioavailability and drug efficacy. Incorporating the hydrophobic drug into a water-soluble polymeric drug delivery system facilitates delivery in aqueous environments (96, 106). Furthermore, the incorporated hydrophobic drug adopts the pharmacokinetic properties of the polymer/supramolecular structure; therefore, changes that alter these properties (i.e., changes to polymer composition, morphology, or functionality) may enable sustained or timed release of the payload (44, 64). In addition, the polymer/supramolecular scaffold shields the entrapped API from the surrounding environment, leading to reduced cytotoxicity in situations where a burst release of the drug might be hazardous or toxic (34, 88). The shielding structure also provides a large area for attachment of targeting functionalities, which may enable triggered or localized release of the payload (60, 110). Altogether, it is clear that integrating small-molecule therapeutics into macromolecular/supramolecular systems provides advantages for delivery.
The two major strategies for preparing macromolecular drug delivery systems include covalent linkage and physical entrapment. In the covalent linkage approach, APIs are chemically linked to polymer scaffolds. The linkage method often depends on the specific chemical nature of the API, making this approach specific to a given drug. In the physical entrapment approach, APIs are sequestered in polymeric nanostructures without the formation of covalent bonds. While this approach often leads to lower loading rates than covalent linking, the lack of chemical modification enables its use with a wider range of APIs. For example, poly(lactide-co-glycolide) has been used to deliver a wide range of APIs by physical entrapment (38, 46, 77, 80). While these preparation methods differ in their implementation, they both succeed in changing the chemical, physical, and pharmacokinetic properties of small-molecule drugs, proteins, and other APIs.
Macromolecular Systems with Covalently Bound H2S Donors
Chemically linking APIs to a polymer scaffold, despite the need for linker chemistry specific to each API, provides several advantages over physical entrapment. For example, a physically entrapped API is prone to leaching, a process in which the drug escapes from the polymer scaffold in an undesirable manner (i.e., too quickly or before reaching the desired site of release). Covalent linkage of drug to the polymer scaffold minimizes leaching and maintains the desired release profile and/or pharmacokinetics. In addition, chemical incorporation of the API can be used to influence the physical properties of the polymer itself. For example, conjugation of a hydrophobic drug to a hydrophilic polymer can result in amphiphilic behavior, which provides a method for modifying the morphology of the system that is unique to covalently linked donors. Finally, covalent linkers can be designed to release the drugs under specific conditions, such as a change in pH or the presence of a particular enzyme. As a result of these advantages of covalently linked drug delivery systems over physically entrapped systems, a variety of chemically linked H2S-releasing polymers have been developed.
H2S-releasing linear polymers
Covalent attachment of an H2S-donating motif to a linear polymer provides a simple strategy for design of a macromolecular donor. To this end, a variety of systems utilizing small-molecule H2S donors attached to linear polymers have been developed.
A popular potential H2S donor that can be easily attached to polymers is 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH). ADT-OH is the active metabolite in anethole trithione (a bile-secretion stimulating drug used in the treatment of dry mouth) and may have bioactivity aside from releasing H2S (39); beyond this, its mechanism of release is not clear, nor are the factors that affect its rate of release. Despite these issues, it has been conjugated onto several polymer systems, the first of which was reported by Hasegawa et al. in 2014 (Fig. 1A) (40). In this work, ADT-OH was conjugated to the chain end of poly(ethylene glycol) (PEG), producing PEG-ADT. Conjugation to the polymer provided improved solubility of the drug, significantly reducing toxicity in vitro in RAW-Blue macrophages. Interestingly, the authors demonstrated that PEG-ADT conjugates became less cytotoxic with more hydrolytically stable linkages, underlining the importance of covalently attaching ADT-OH to the polymer. The PEG-ADT conjugates entered cells through endocytosis, while small-molecule ADT-OH diffused across the cell membrane into the cytoplasm. The authors attribute the difference in cytotoxicity between small-molecule and polymeric donors to a difference in intracellular distribution due to these unique pathways of cellular entry. Furthermore, the slower release of H2S from PEG-ADT relative to ADT-OH resulted in a significantly enhanced potentiating effect on lipopolysaccharide-induced inflammation.

Also in 2014, our group developed macromolecular H2S-donating polymers with each repeating unit functionalized with S-aroylthiooximes (SATOs) through a postpolymerization modification of pendant aldehyde groups (Fig. 1B) (31). SATOs, reported by our group earlier in 2014, release H2S upon addition of a thiol. H2S release rates from SATOs could be tuned by changing substituents on the aroyl ring (32). The S-aroylthiooxime (SATO)-conjugated polymers retained control of H2S release through electronics of the aroyl structure, allowing for the preparation of several polymers with varying H2S release kinetics. A statistical copolymer, in which polymer repeat units are randomly distributed, containing SATO pendant groups as well as oligo(ethylene glycol) (OEG) pendant groups enhanced the water solubility of these hydrophobic donors.
In 2016, we developed a different H2S-donating polymer system using N-thiocarboxyanhydrides (NTAs) as the H2S-releasing groups (Fig. 1C) (72). Nucleophilic addition of an amine to the carbonyl neighboring the unsubstituted CH2 unit caused ring opening of the NTA and released carbonyl sulfide (COS), which is rapidly converted into H2S enzymatically by carbonic anhydrase (83). We synthesized NTA-functionalized and OEG-functionalized norbornene derivatives for use in ring-opening metathesis polymerization (ROMP). ROMP provided a polymerization method devoid of nucleophiles potentially capable of opening the NTA ring, and copolymerization with the OEG-functionalized norbornene derivative imparted good water solubility to the copolymer. Conjugation of NTA motifs to the polymer resulted in prolonged H2S release, with a threefold increase in release half-life relative to the small-molecule NTA. This change in release profile may be a result of steric crowding around the NTA, retarding nucleophilic addition, which again showcases the protective effect of polymer systems on sensitive prodrugs.
Recently, Li et al. reported an H2S-donating polymer system based on conjugation of 2-nitrobenzenemethanethiol to pendant ketones on a polymethacrylate backbone, creating a photodegradable thioketal linkage (Fig. 1D) (56). Upon UV irradiation, the thioketal linkage degraded to release H2S and 1,2-nitrobenzaldehyde, while also regenerating the parent polymer. Copolymerization of a ketone-functionalized methacrylate with an OEG-functionalized methacrylate produced a highly water-soluble graft copolymer with a molecular weight close to the renal excretion threshold (40 kg/mol), the size below which clearance by the kidneys is rapid (81). The rate of H2S release from the copolymer correlated positively with the intensity of UV irradiation, with no release observed in the absence of irradiation. In addition, the gem-dithiol-functionalized polymer and its byproducts showed no cytotoxicity at concentrations <25 μM in NIH3T3 cells.
Polymeric micelles for H2S delivery
Polymer micelles, a common drug delivery system made from amphiphilic block copolymers, are known for their drug loading capability and long circulation times in the bloodstream (48). In such systems, amphiphilic block copolymers, polymers in which long runs of repeat units are discretely partitioned but chemically linked, self-assemble in water to form micelles due to the differential solubility of the component blocks. Polymer micelles form core-shell architectures in aqueous environments, with hydrophobic blocks forming the core and hydrophilic blocks composing the surrounding shell. As a result, micellar systems can carry various hydrophobic drug molecules within their cores, either through covalent attachment or via physical encapsulation, thereby behaving as efficient drug delivery vehicles. Furthermore, the size range (typically 10–100 nm) of polymer micelles is critical in determining their biodistribution, as larger micelles may exceed the renal excretion threshold and accumulate in sites of interest through the enhanced permeability and retention effect (62). In addition, micellar carriers improve aqueous solubility and prolong the circulation times of encapsulated hydrophobic small-molecule APIs in the bloodstream.
Among the earliest examples of H2S-releasing polymer micelles, Hasegawa et al. expanded upon their previous work with linear H2S-donating polymers to create ADT-containing polymer micelles (Fig. 2A) (41). In this work, ADT was conjugated to poly(acrylic acid) that was polymerized through reversible addition–fragmentation chain-transfer (RAFT) polymerization, a radical polymerization technique capable of producing polymers with controllable molecular weights and end-group fidelity, using a PEGylated chain transfer agent (CTA) to yield an amphiphilic block copolymer. Here, PEG was the hydrophilic block, and ADT-functionalized poly(acrylic acid) was the hydrophobic block (PADT). Analysis by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (55) revealed formation of spherical micelles in water, with an average hydrodynamic radius of 36 nm. Although only small amounts were released, the H2S release profiles observed for these PEG-b-PDAT polymer micelles were similar to those for linear PEG-ADT, indicating that micellization did not hinder H2S release from the encapsulated ADT-OH moieties. As reported for PEG-ADT linear polymers, PEG-b-PADT micelles showed less cytotoxicity than small-molecule ADT-OH in vitro. Interestingly, PEG-b-PADT micelles enhanced the proinflammatory response in gardiquimode-stimulated murine macrophages, whereas ADT-OH slightly decreased the proinflammatory response under similar conditions. The observed difference is likely due to the altered cellular internalization of ADT micelles relative to ADT-OH, similar to that observed for ADT-PEG polymers. This study showed the ability of micellar systems to attenuate certain toxic effects of ADT-OH by shielding it and controlling its entry into cells.
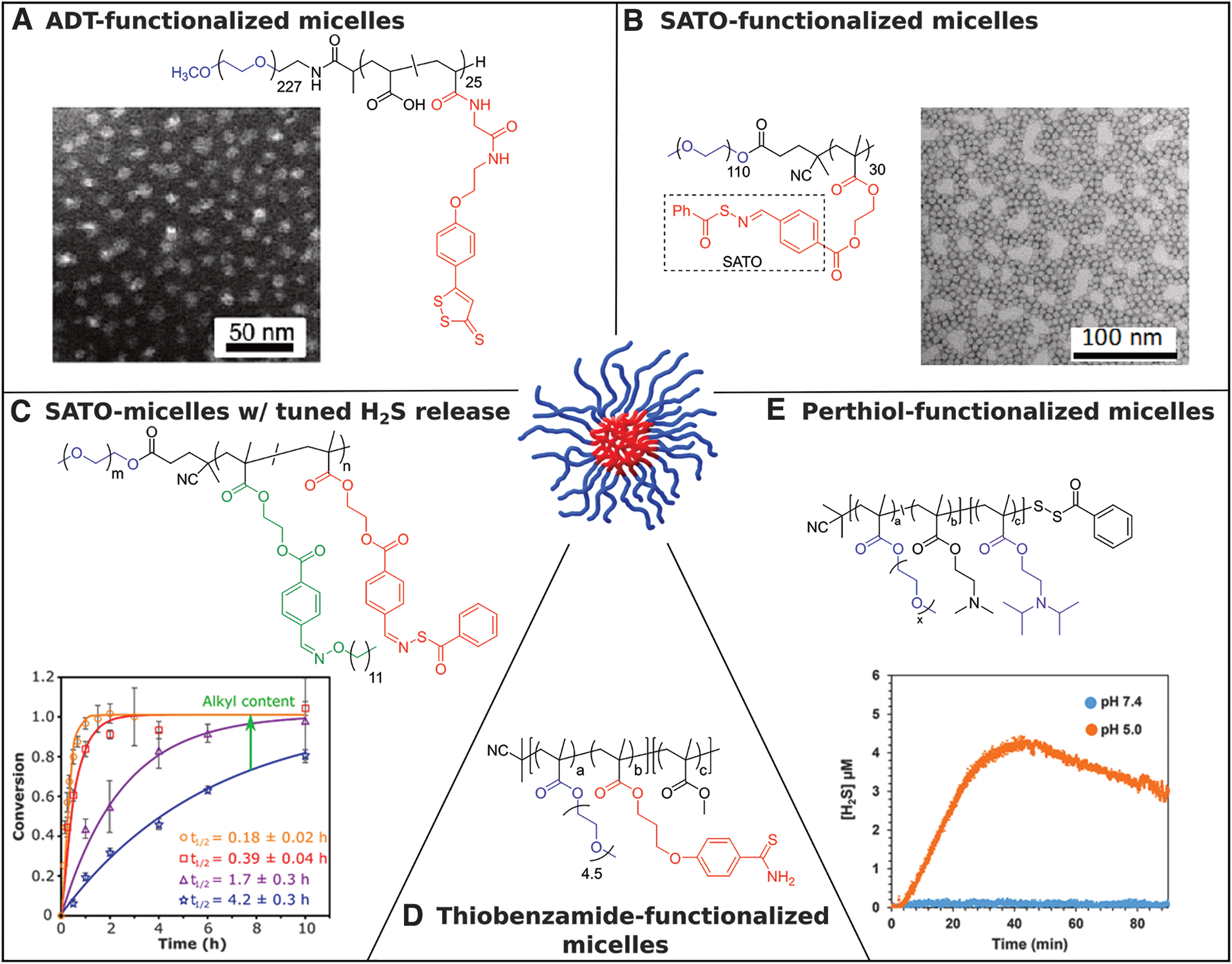
In 2017, we reported an H2S-releasing polymer micelle system to address the rapid clearance rate associated with linear polymers and access better pharmacokinetics (Fig. 2B) (33). In this work, 2-(4-formylbenzoyloxy)ethyl methacrylate (FBEMA) monomer was polymerized via RAFT using a PEGylated CTA to give an amphiphilic block copolymer bearing pendant SATOs in the hydrophobic block. Uniform spherical micelles were then prepared with an average core diameter of 21 ± 2 nm as measured via TEM and a hydrodynamic diameter of 38 ± 4 nm as measured by DLS. Confinement of SATO moieties in the hydrophobic core of micelles resulted in a significant increase in the H2S release half-life, with micelles releasing ninefold more slowly than a small-molecule analog. We attributed this drastic difference in release rate to restricted diffusion of the hydrophilic Cys trigger into the hydrophobic micelle cores, retarding H2S release compared with molecularly dissolved small molecules and linear polymers. This finding highlights the potential for tuned release rate of H2S through control of polymer properties. Furthermore, the polymer micelles were more effective relative to common small-molecule H2S donors (Na2S, a small-molecule SATO, and GYY4137) in decreasing viability of HCT116 colon carcinoma cells. These results reveal the importance of H2S release rate on biological activity and further underline the potential of polymeric H2S donors.
Expanding upon the previous study, in 2019 we developed a method for systematically tuning H2S release rate from SATO-conjugated micelles (Fig. 2C) (30). PEG-b-PFBEMA block copolymers, prepared in a similar manner to the previous work, were conjugated with functionalized SATO derivatives. We hypothesized that controlling the rate of Cys diffusion into the micelle core would control the H2S release rate. H2S release experiments on micelles swollen in water with varying amounts of ethanol provided evidence to support this claim, where the H2S release rate became increasingly faster with greater degrees of micelle swelling. We further hypothesized that H2S release from SATO-conjugated micelles could be systematically tuned by controlling the chain mobility of the micelle core. To this end, we cofunctionalized PEG-b-PFBEMA block copolymers with varying amounts of SATO and an alkyl plasticizing agent, which we expected would increase the mobility of polymer chains. As expected, the resulting series of block copolymers showed a trend in hydrophobic block glass transition temperature (Tg), the temperature above which long-range polymer backbone movement can occur, correlating with the mass ratio of SATO versus alkyl group. In addition, the H2S release rate varied over 20-fold throughout the series, with the lowest Tg hydrophobic block resulting in a release half-life similar to that of small-molecule SATOs. Overall, this work highlighted the structural tunability of polymeric micelles and demonstrated precise control over H2S release from a polymeric system without chemically modifying the donor.
Another thiobenzamide-functionalized macromolecular H2S-releasing system was reported by Davis and co-workers in 2016 (Fig. 2D) (26). Two amphiphilic polymethacrylates were synthesized by copolymerizing 3-(4-cyanophenoxy)propyl methacrylate (CPPMA), oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA), and methyl methacrylate using RAFT polymerization. Pendant thioamides were generated in situ via thionation of benzonitrile pendant groups present on the CPPMA block in a postpolymerization modification. The final polymers, [POEGMA-co-PTHA]-b-poly(methyl methacrylate) (PMMA) and POEGMA-b-[PTHA-co-PMMA], both formed micelles in aqueous buffered solutions. Faster H2S release from [POEGMA-co-PTHA]-b-PMMA versus POEGMA-b-[PTHA-co-PMMA] micelles was attributed to the placement of the H2S-releasing thioamide units in the hydrophilic corona of the micelle in the former system versus within the hydrophobic core for the latter system. The differences observed in the H2S release profiles of the two micellar systems highlight the shielding effect of micellization in slowing down the hydrolysis of donors encapsulated within the core. Furthermore, the H2S delivered from slow-releasing POEGMA-b-[PTHA-co-PMMA] polymer micelles gradually increased levels of cytosolic extracellular signal–regulated kinases and plasma membrane-localized protein kinase C activity in HEK293 cells.
In 2017, Quinn and coworkers reported on H2S-releasing micelles from perthioester-containing polymer constructs synthesized via RAFT polymerization with facile end-group modification (Fig. 2E) (111). The benzyl dithioester chain end was first functionalized with pyridyl disulfide groups and subsequently reacted with thiobenzoic acid to yield polymers with benzoyl-capped perthiol ω-chain ends. The final perthioester-functionalized polymer P[OEGMA]-S-S-(C = O)Ph underwent nucleophilic addition of cysteine to release a free perthiol, which subsequently liberated H2S. The polymer remained stable in buffered aqueous solutions without thiol. As an extension of this concept, the authors prepared an amphiphilic block copolymer by polymerizing butyl methacrylate as a hydrophobic block to yield P[OEGMA-b-BMA]-S-S-(C = O)Ph. This polymer readily formed micelles when dispersed in PBS (pH = 7.4) and showed significantly slower H2S release relative to the homopolymers. Furthermore, a pH-responsive amphiphilic block copolymer was synthesized by copolymerizing N,N-(dimethylamino) ethyl methacrylate (DMAEMA) and N,N-(diisopropylamino) ethyl methacrylate (DIPMA) to generate P[OEGMA-co-DMAEMA-b-DIPMA]-S-S-(C = O)Ph. Cys-triggered H2S release from these pH-responsive micelles significantly accelerated under acidic conditions. This phenomenon was attributed to the increased hydrophilicity of the DIPMA block under acidic conditions, leading to micelle disassembly and increased exposure to cysteine. This work demonstrated the potential for triggered release of gasotransmitters through careful design of a polymeric drug delivery system.
Polymeric liposomes
Related to micelles, liposomes represent another nanostructure accessible from surfactants or amphiphilic block copolymers. The structure of liposomes resembles that of a lipid bilayer as found in cell membranes—the amphiphilic constituents of liposomes arrange in parallel to form a bilayer with hydrophilic units forming the lining of the bilayer and hydrophobic tails forming the core. As a result, liposomes can encapsulate hydrophobic drugs in the bilayer core and hydrophilic drugs in the aqueous inner compartment. In addition, manipulation of membrane properties allows for controlled release of bilayer-encapsulated drugs and/or penetration of triggering molecules into the hydrophilic core.
In 2017, Quinn and coworkers reported on a liposomal H2S-donating macro- and supramolecular system utilizing PEG-cholesterol (PEG-Chol) conjugates with trisulfide linkages (27). Organic polysulfides, which are biologically active components in various natural products, release H2S upon thiol exchange reactions (4, 7, 92). The authors hypothesized that incorporation of the trisulfide linker into the PEG-Chol conjugate would allow for thiol-triggered H2S release, and the resultant disruption of the liposomal membrane might induce release of a hydrophilic payload trapped inside the liposome. Thiol-triggered release of H2S from micelles formed by the trisulfide-linked conjugates was confirmed using SF4, an H2S-selective fluorescent probe. In contrast, no H2S release was observed from micelles that included disulfide or amide linkages in place of the trisulfide. The PEG-Chol conjugates were then incorporated into preformed liposomes using the cholesterol group to anchor the conjugates into the bilayer. H2S release from these liposomes was evaluated through amperometry using an H2S-selective microsensor. Trisulfide conjugates incorporated into liposomes showed a reduced peaking concentration of H2S relative to micelles. The authors attributed this decrease in total H2S release to partial inaccessibility of trisulfides in the bilayer membrane, which suggests a potential for tuning release from liposomes through control of membrane properties.
Polymeric micro/nanoparticles
Nanoparticles range in diameter from a few to a few hundred nanometers, making them suitable for injection directly into the bloodstream. This allows for free circulation throughout the vasculature, targeting to specific tissues, and long-term systemic release of APIs for long-circulating nanoparticles. Polymer nanoparticles differ from polymer micelles, in that they are typically made using specific processing methods such as nanoprecipitation, a phase separation process rather than a self-assembly process. Conversely, microparticles range in diameter from 1 to 100 μm and are therefore restricted in migrating from their site of injection. As a result, microparticles are more useful for applications requiring long-term localized release through injection or implantation at a desired site (e.g., muscle tissue, peritoneum). Due to their much smaller surface area to volume ratio compared with nanoparticles, microparticles degrade more slowly and release their cargo more slowly. Combined with their retention in tissue, these features make microparticles suitable for long-term delivery of APIs.
In 2015, Bowden and coworkers reported a polymeric H2S-donating microparticle system consisting of a polylactide backbone decorated with thiobenzamide groups (61). Ring-opening copolymerization of
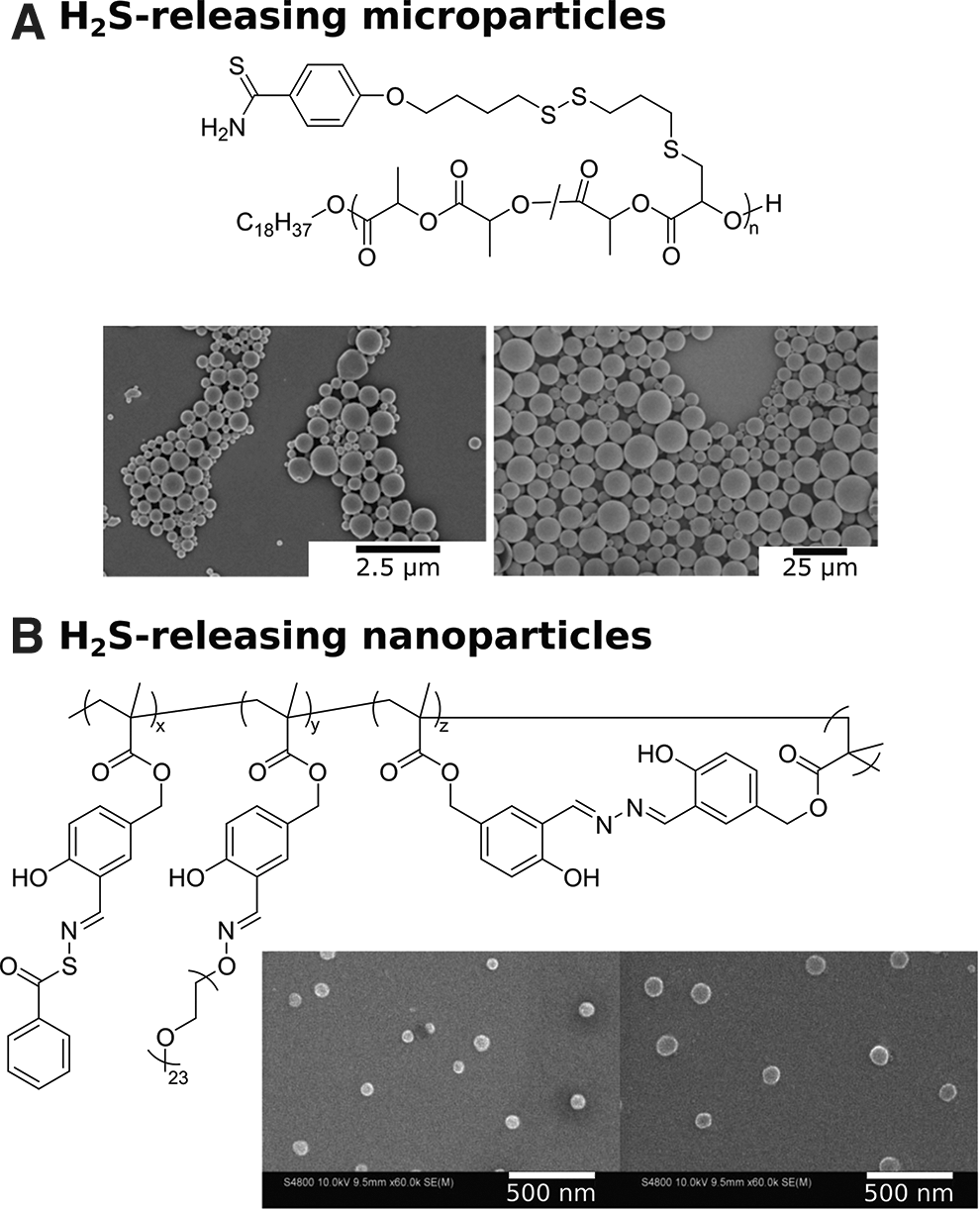
In related work focusing on nanoparticles instead of microparticles, Lin and coworkers recently described self-fluorescing, H2S-releasing nanoparticles containing SATOs and aggregation-induced emission fluorophores (AIEgens) (59). RAFT polymerization of 3-formyl-4-hydroxybenzyl methacrylate (FHMA) yielded poly FHMA (PFHMA) decorated with salicylaldehyde (ortho-hydroxybenzaldehyde) units as reactive handles, which were functionalized in three different ways (Fig. 3B). A small percentage of aldehyde units were functionalized with aminooxy PEG to impart water solubility to the polymer, while others were crosslinked with hydrazine to form salicylalhydrazine as an AIEgen (22). Crosslinking with hydrazine was carried out at a high dilution to promote intrachain crosslinking of aldehyde units, yielding a water-soluble, AIE-active polymer nanoparticle. Finally, the remaining salicylaldehyde units were converted into SATO groups, yielding H2S-releasing, aggregation-induced emission (AIE)-active nanoparticles. The crosslinked polymers were then dispersed in water with vigorous stirring, and nanoparticles were in the range of 100 nm, as confirmed by scanning electron microscopy (SEM) and DLS. Cellular internalization of the polymeric nanoparticles was observed by confocal laser scanning microscopy, highlighting the self-fluorescence characteristics of the nanoparticles. This was the first example of H2S-releasing nanoparticles that could also be traced within the living systems. This work provides insight into the multifunctional capabilities of polymeric drug delivery systems and exhibits the potential for in vivo H2S quantification directly at the targeted sites.
Complex self-assembled and crosslinked materials
Hydrogels are three-dimensional networks of entangled covalent or supramolecular polymers that trap a substantial amount of water. In the field of drug delivery, hydrogels are studied widely due to their resemblance to soft tissue in terms of morphology and mechanical properties combined with their ability to sequester and release APIs. Hydrogels can be broadly categorized as physically or chemically crosslinked gels depending on the type of network crosslinking. Physically crosslinked hydrogels retain their 3D structures through reversible, noncovalent supramolecular interactions, whereas covalent bonds link individual chains in chemically crosslinked hydrogels. Hydrogels can be particularly advantageous for delivering H2S in a localized manner. While macromolecular assemblies such as micelles and nanoparticles may deliver H2S to the targeted tissues through systemic administration, hydrogels can be injected or implanted at the site of interest and deliver H2S locally.
In 2015, our group reported a supramolecular, peptide-based H2S-releasing hydrogel system containing a covalently bound SATO donor (17). The 6-mer peptide sequence included a β-sheet-rich region (where peptide chains are arranged parallel/antiparallel to each other and are stabilized by a network of intermolecular H-bonds), a hydrophilic region containing three glutamic acid residues, and the N-terminus functionalized with a SATO group. Driven by hydrophobic interactions and stabilized by hydrogen bonding in the β-sheet-rich region, the SATO-functionalized peptides self-assembled into nanofibers in aqueous solutions. At concentrations as low as 1 wt%, a robust hydrogel formed upon charge screening through the addition of CaCl2. Due to shielding of the SATO groups in the nanofiber core, extended release was observed, with measurable H2S levels >15 h.
More recently, we conducted deeper investigations into these peptide-based materials by varying substituents on and around the SATO group (Fig. 4A) (49, 74). Specifically, we found that R group substitution on the aroyl ring of the SATO unit affected the internal structure of the self-assembled nanofibers, which in turn regulated gel stiffness. H2S release rates from the hydrogels depended on a complex balance between substituent electronic effects and internal nanofiber morphology, but extended release profiles were observed for all hydrogels. These H2S-releasing, peptide-based hydrogels may be ideal for localized H2S delivery because the peptide components of the gels can be degraded into benign metabolites.
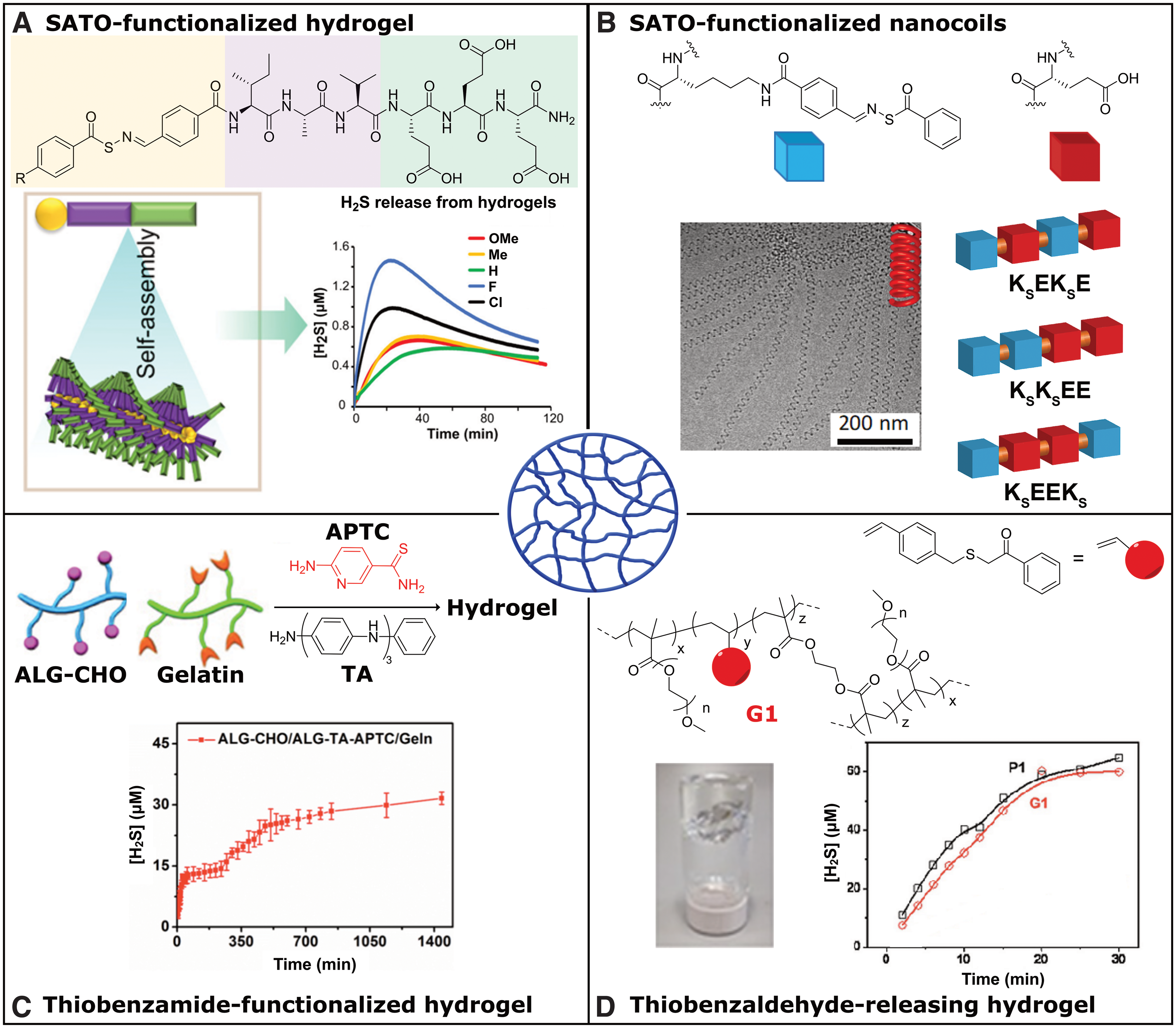
Recently, we reported a self-assembled H2S-releasing system based on constitutionally isomeric peptides (103). A series of three tetrapeptides was designed with two lysine (K) and two glutamic (E) acid units varying in their placement along the peptide backbone (Fig. 4B). The lysine residues were modified with SATO groups, yielding three peptides, KsEEKs, KsEKsE, and KsKsEE (Ks = SATO-modified lysine). Peptide KsEEKs self-assembled into an unusual nanocoil morphology, whereas the other two peptides formed conventional twisted nanoribbons. Interestingly, H2S release from the nanocoils was slower than release from the nanoribbons, which we attributed to slower diffusion of cysteine into nanocoils relative to nanoribbons. Furthermore, nanocoils were more effective compared with small-molecule H2S donors (GYY4137 and Na2S) as well as nanoribbons in rescuing H9C2 cardiomyocytes from doxorubicin toxicity, likely due to the significant differences in H2S release rates among the donors.
This work demonstrated the influence of morphology on donor release profiles and provided evidence for the increased efficacy of prolonged-release donors relative to burst release. Comparisons in biological studies between comparable H2S donors with different release rates will be vital for determining the optimal rate in various settings. Tuning nanostructures through supramolecular approaches may offer a method to modify release rate without changing H2S donor chemistry or released byproducts, allowing for clear comparisons between H2S donors with variable release half-lives.
Recently, Liang and coworkers reported a smart, H2S-releasing hydrogel for potential treatment of myocardial infarction (58). The researchers synthesized a macromolecular H2S donor by grafting 2-aminopyridine-5-thiocarboxamide (APTC), a thiol-triggered H2S donor, onto partially oxidized alginate (Alg-CHO). The macromolecular donor was further functionalized by tetraaniline (TA) to impart conductivity to the final Alg-APTC-TA construct, which was crosslinked with gelatin to form an H2S-releasing conductive hydrogel (Fig. 4C). The hydrogel showed strong adhesion to tissue due to the presence of aldehyde groups on the hydrogel surface, which can react with nucleophilic amines in tissue proteins. The authors observed a significant increase in H2S release half-life of small-molecule APTC from 3 to 270 min by encapsulating it within the hydrogel, and the hydrogel showed extended H2S release exceeding 24 h. The hydrogel was evaluated in mice hearts, exhibiting positive effects on function and remodeling of the left ventricle by significantly reducing cardiac fibrosis and ventricle wall thickening. This multifunctional hydrogel system underscores the importance of localized H2S delivery for conditions like myocardial infarction and further demonstrates the potential for self-assembled systems to achieve these advanced drug delivery goals.
A light-responsive hydrogel was reported by Connal and coworkers in 2017 (108). They synthesized light-sensitive thiobenzaldehyde precursors that released thioaldehydes upon irradiation. The released thioaldehydes further reacted with amines to form imines, releasing H2S as a byproduct. The small-molecule thiobenzaldehyde precursors released a small amount of H2S upon irradiation followed by enhancement in H2S release rate upon addition of amines. The authors then synthesized a vinyl monomer incorporating the thiobenzaldehyde precursor group, which was copolymerized with polyethylene glycol methacrylate to form water-soluble, light-sensitive, H2S-releasing polymers, and further crosslinked with ethylene glycol dimethacrylate to provide light-sensitive H2S-releasing hydrogels (Fig. 4D). Slow H2S release rates were observed from the hydrogels and linear polymers relative to the small-molecule donor. To that end, the hydrogel was tested as a surface for cell culture to study H2S delivery in vitro, showing successful growth of cells on the gel surface and subsequent H2S delivery.
De-polymerizable H2S-donating systems
An area of macromolecular drug delivery strategies that holds great potential but is in its infancy is depolymerizable systems. In such systems, therapeutic agents are covalently attached to or physically entrapped in polymers, which can systematically depolymerize (also called self-immolation) upon application of a specific stimulus (69, 79). These strategies are appealing because they enable methods for triggered release of payloads and avoid the potentially long biological clearance times of polymer byproducts associated with conventional macromolecular drug delivery systems.
In 2019, we developed a depolymerizable COS/H2S donor based on oligo(thiourethanes) (73). A class of benzyl thiocarbamates developed by Pluth are COS/H2S donors that can be triggered by a variety of stimuli (54, 82). Operating through a 1,6-elimination mechanism, cleavage of a protecting group initiates the reaction, generating the COS product that can be converted into H2S by carbonic anhydrase. Inspired by this work, our group synthesized an aryl isothiocyanate monomer for use in a polyaddition reaction to form an oligothiourethane. This monomer was polymerized and end-capped with 4-azidobenzylalcohol to achieve an aryl azide end-capped oligothiourethane termed self-amplified depolymerizable polymer 1 (SADP1, Fig. 5A).
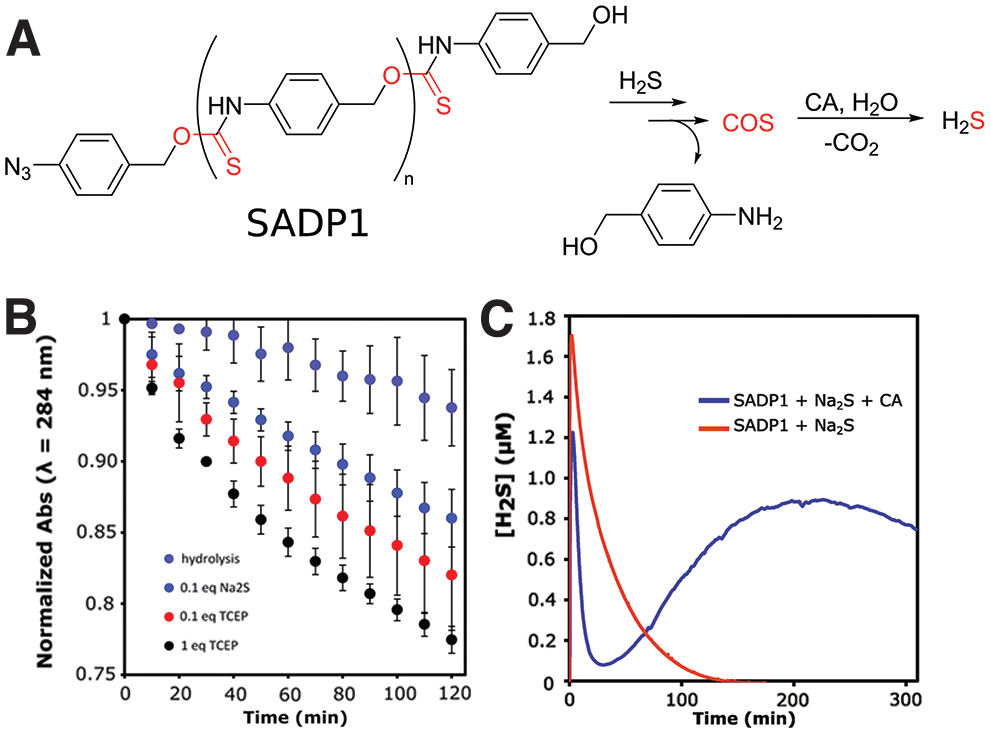
Previous work suggested the potential for reduction of the aryl azide to the corresponding aryl amine upon reaction with two equivalents of H2S, which provided a pathway for self-amplified depolymerization (42). The molecular weight was limited to 1600 g/mol due to the low thermodynamic driving force for the polymerization, but depolymerization triggered by reducing agents revealed a decrease in polymer peaks and the appearance of peaks representative of 4-aminobenzylalcohol by UV-Vis spectroscopy (Fig. 5B). The H2S release profile of the oligothiourethane was observed by an H2S-selective electrochemical probe (Fig. 5C). In the absence of carbonic anhydrase, addition of Na2S to a SADP1 solution showed only a spike in H2S concentration from the initial Na2S addition. However, the experiment performed in the presence of carbonic anhydrase produced a sudden spike followed by a gradual increase in H2S concentration over several hours. The gradual increase after the initial spike was a result of slow generation of COS through the depolymerization mechanism, which was then converted into H2S by carbonic anhydrase. In this way, the system self-amplified H2S production, where two equivalents of H2S reduced the azide end-group to trigger the depolymerization, generating several more equivalents of H2S, which then went on to trigger more depolymerization, producing more H2S. This work hints at the possibilities of depolymerizable polymers as carriers for delivery of “caged” small molecules such as H2S, with potential to tune the triggering stimulus, the rate of H2S production, and the released byproducts.
Macromolecular Systems with Physically Entrapped H2S Donors
Physical entrapment of APIs within macromolecular or supramolecular materials is experimentally simpler than covalent binding of APIs to the scaffold. Consisting at a minimum of only a polymer and the drug to be delivered, no linker chemistry needs to be developed, so chemical designs are less elaborate. In addition, a wide range of APIs can be sequestered in the same polymer matrix, and multiple APIs can be entrapped if desired. On the macroscale, physical entrapment tends to affect the physical properties of the polymer matrix less than covalent attachment, so physical, morphological, and biological properties of the matrix can be determined and optimized independent of the API. A number of delivery systems have been developed based on physically entrapping H2S donors.
Hydrogels
In 2019, Xiao and coworkers reported a dual pH- and enzyme-responsive H2S-releasing hydrogel based on collagen for the potential treatment of indivertible disk degeneration (IDD) (116). A pH-sensitive small-molecule H2S donor, JK1, was encapsulated within the porous network of the collagen hydrogel to generate Col-JK1 gel (Fig. 6A). Col-JK1 could be slowly degraded by matrix metallopeptidase 9 (MMP9), an enzyme that is overexpressed under IDD conditions. Degradation of the collagen gel led to release of JK1 into the low pH environment of the inflamed tissue, where it underwent intramolecular cyclization to release H2S. H2S release from Col-JK1 was slower compared with the small-molecule donor alone and was accelerated in the presence of MMP9. Furthermore, Col-JK1 successfully inhibited apoptosis in nucleus pulposus cells and prevented degradation of extracellular matrix (ECM), indicating its potential for IDD treatment. Encapsulating the donor within the collagen hydrogel not only increased its retention time in the IDD lesion and prolonged the H2S release time, but it also created an enzyme-responsive, on-demand release system. However, H2S release from Col-JK1 gel in the absence of MMP9 was still substantial, highlighting the need for continued development of enzyme-responsive systems that release H2S only in the presence of the desired enzyme.

Electrospun fibers
A simple method to prepare drug-releasing polymers with control over release rates, mechanical properties, and matrix degradability is electrospinning. Electrospinning is a process of generating nano/microfibers by spraying a continuous stream of polymer solution and stretching it under high voltage. Electrospun fibers, similar to polymeric and self-assembled hydrogels, resemble the ECM in morphology and mechanical properties, and therefore find use as materials for tissue engineering and regenerative medicine. Electrospun fibers, however, are much larger in diameter than peptide-based nanofibers or polymer chains (in the range of ∼1 μm vs. 1–10 nm). The large diameter allows the fibrous network to physically entrap and stabilize drugs and other bioactive molecules with high drug loading capacity. Chemical structure and morphology affect polymer degradation kinetics, as does fiber diameter, giving researchers several methods to alter drug release profiles. With its tunability and versatility, the inexpensive technique of electrospinning is a natural fit for H2S-releasing materials, where control of release rate is vital.
Wang and coworkers reported the first electrospun H2S-releasing microfibers in 2015 based on a biodegradable polycaprolactone (PCL) polymer matrix (28). A solution of PCL at different concentrations (6%, 8%, and 12%) and a thiol-activated H2S donor (NSHD1) were subjected to electrospinning to yield microfibers with diameters ranging from 0.5 to 1.5 μm (Fig. 6B). An increase in microfiber diameter was observed with increasing PCL concentration. SEM revealed smooth microfiber surfaces indicative of uniform distribution of NSHD-1 within the fibers, and FTIR spectroscopy confirmed NSHD-1 encapsulation. H2S release half-lives for the microfibers were longer than NSHD-1 alone, with measurable H2S levels extending past 24 h. Release rate depended on fiber thickness, with thicker fibers releasing more slowly than thinner ones. In addition, these electrospun, H2S-releasing microfibers protected H9C2 cardiomyocytes subjected to oxidative stress by addition of H2O2. They also enhanced proliferation of 3T3 fibroblasts, which is potentially useful for wound healing.
A few years later, the same group applied H2S-releasing PCL nanofibers, with diameters ∼300 nm, in the development of a nanofibrous coating for wound healing (107). The fabrication and characterization of the nanofibers were similar to PCL-NSHD-1 nanofibers, but the pH-responsive H2S donor JK1 was included instead. H2S release from PCL-JK1 fibers was higher at pH 6 relative to physiological pH and was significantly slower than small-molecule JK1 donor alone, with measurable H2S levels past 2 h (Fig. 6C). One-time application of PCL-JK1 nanofibrous scaffolds to full-thickness cutaneous wounds in mice showed successful wound regeneration over 20 days with healing rates significantly higher than PCL fibers alone. It is not clear how the release profile changed in vivo compared with the release over a few hours in vitro, but it likely released H2S for a much longer period based on the results from the in vivo studies.
In 2018, Melino and coworkers reported electrospun microfibrous polylactic acid (PLA) membranes containing garlic-derived organosulfur compounds fabricated using two different approaches (13). In the first approach, garlic oil-soluble extract (GaOS) was directly coated onto the electrospun microfibers (∼700 nm in diameter) to yield GaOS-coated PLA fiber membranes (PFM+GaOS). In the second approach, GaOS or diallyl disulfide (DADS) was encapsulated within the PFMs during the electrospinning process to generate GaOSPFM or DADSPFM fibers, respectively. Both doped fibers released substantially less H2S than the coated fibers. H2S release experiments were conducted in pH 8 buffer at 37°C, and although the authors did not conduct a typical release experiment to measure cumulative or real-time H2S concentrations over time, the electrospun mats clearly protected the extracts in the dry state for several days. Studies on cardiac mesenchymal stem cells showed that the fiber mats protected the cells from oxidative stress.
Nanoemulsions and liposomes
Nanoemulsions and liposomes physically loaded with H2S donors have similar therapeutic potential to polymer micelles described above. They can protect encapsulated APIs to improve stability, increase circulation times, and accumulate at sites of interest (e.g., tumors). These types of formulations are generally prepared through simple and scalable processes such as nanoprecipitation. Careful process control allows for tunable and uniform droplet sizes. The facile preparation and wide range of formulation possibilities are ideal for the development of various macromolecular H2S donor systems.
In 2016, Melino and coworkers reported on an H2S-releasing protein nanoemulsion formulation utilizing DADS, α-linoleic acid (ALA), and bovine serum albumin (BSA) (Fig. 7A) (63). DADS is found in various natural extracts and releases small amounts of H2S upon reaction with a thiol (14). Similarly, ALA promotes cardiovascular health, reduces the risk of cardiovascular attacks, and exhibits chemoprotective effects (8, 113). The therapeutic applications of both DADS and ALA are limited by low aqueous solubility and stability. The authors suggested that a nanoemulsion formulation of both compounds may improve their bioavailability, thus increasing their therapeutic potential. Protein nanoemulsions of BSA-ALA-DADS-nanoemulsions (BAD-NEs) and BSA-DADS-nanoemulsions (BD-NEs) were prepared by sonication, with average diameters in the range of a few hundred nanometers. Interestingly, in thiol-containing buffer, BAD-NEs, which contained all three components, released more total H2S per unit of DADS relative to both BD-NEs, which lacked the ALA lipid component, and DADS alone. The authors attributed this result to both increased stability of DADS in the nanoemulsion and the antioxidant properties of ALA. Incubation of BAD-NE nanoemulsions with supercoiled plasmid DNA in an oxidative environment revealed superior antioxidant properties relative to ALA and DADS alone, as well as their binary mixture. Furthermore, BAD-NEs significantly decreased cell viability in MCF-7 mammary adenocarcinoma and HuT 78 T-cell lymphoma cells, suggesting their potential as an anticancer therapeutic.
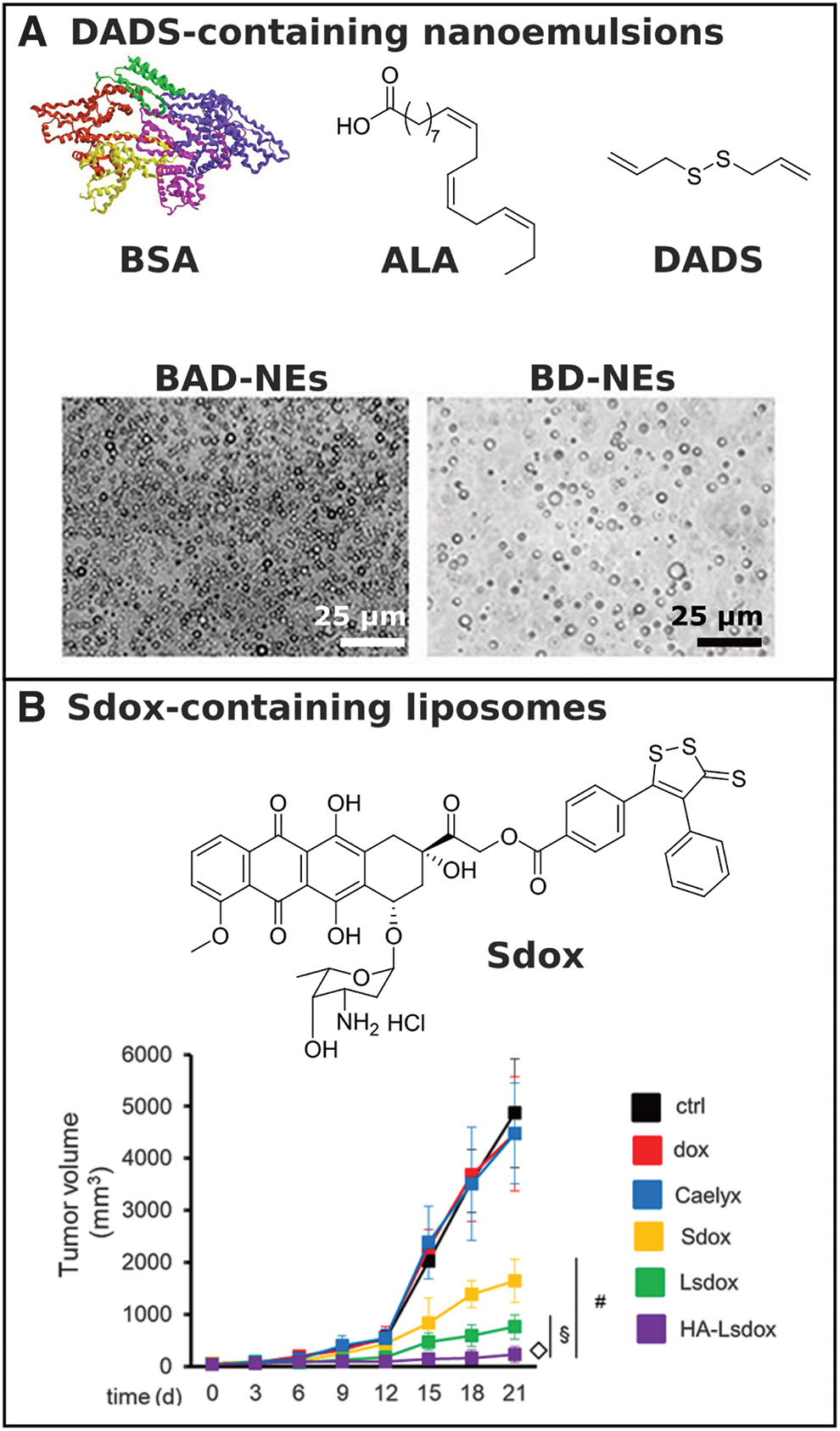
In 2019, Arpicco, Riganti, and coworkers reported on liposomal formulations of doxorubicin (Dox) conjugated to an H2S donor (Sdox) (36). The Sdox compounds, reported by the authors in previous work (9, 12, 18), showed reduced cardiotoxic effects compared with free Dox while retaining their efficacy against human bone osteosarcoma cells (characterized for their resistance to Dox). The authors suggested that encapsulation of Sdox in liposomes conjugated with hyaluronic acid (HA) might promote targeting to osteosarcoma cells due to their expression of the HA-receptor CD44. Liposomal Sdox (Lsdox) and hyaluronated liposomal Sdox (HA-Lsdox) were prepared through hydration of drug–lipid films, and successful insertion of Sdox into the liposomal membrane was confirmed by differential scanning calorimetry. HA-Lsdox showed a higher accumulation in CD44-expressing cells in vitro relative to Dox, Sdox, Lsdox, and Caelyx®, a commercial liposomal Dox formulation (Fig. 7B). In addition, HA-Lsdox induced higher cell damage, apoptosis, and lower cell viability relative to the other formulations in vitro. In vivo studies revealed that HA-Lsdox greatly outperformed Sdox and the other liposomal Dox formulations with respect to inhibition of K7M2 tumor growth.
This work highlights the potential for supramolecular donor systems in clinical applications of H2S therapy. Sdox is an advanced form of Dox that may reduce side effects of this common chemotherapy drug, but this work shows that drug delivery vehicles are still needed for Sdox to maximize its anticancer effects while limiting cardiotoxicity.
Key Challenges and Future Perspectives
In recent years, it has become clear that H2S, along with the other known gasotransmitters NO and CO, are molecules of supreme importance in mammalian physiology. Our understanding of the biological significance and therapeutic potential of H2S continues to expand, and there exists a need for a wide variety of strategies to deliver the gas at the desired location in the body, at the intended time, for the appropriate duration, and with the chosen rate of release. Due to its reactivity and short half-life, controlled delivery becomes even more important for H2S than conventional small-molecule drugs, which are typically selected from a variety of possible structures with considerations toward their pharmacokinetics and target specificity. Small-molecule H2S donors remain vital in this endeavor, but H2S-releasing macro- and supramolecular materials offer additional possibilities for regulating these key drug delivery parameters.
Polymers functionalized with H2S donors were the first macromolecular materials developed for H2S delivery, with initial reports in 2014. This strategy provides a straightforward method for extending the duration of H2S release and potentially altering its biodistribution and pharmacokinetics compared with small-molecule donors. However, the polymer itself in this strategy typically brings limited if any additional control over release compared with small-molecule analogs. In addition, the fate of the polymeric byproduct(s) must be considered in these delivery systems. If the polymer is small enough, it may be renally excreted; otherwise it may accumulate if it is not biodegradable. Depolymerizable H2S-releasing polymers, first reported in 2019, may hold advantages over functionalized nondegradable or slowly degradable polymers, including the potential for a specific trigger to initiate depolymerization.
Compared with soluble homopolymers and copolymers, H2S-releasing block copolymers that form assemblies, such as micelles and liposomes, enhance release control. The hydrophilic block can protect H2S donors linked to or sequestered in the hydrophobic domain of a micelle or liposome, and micelle core mobility can regulate release rate. Beyond these advantages, micelles, liposomes, and other self-assembled structures have been studied extensively as drug delivery vehicles, so H2S-releasing components can be included in existing platforms. H2S-releasing micelles with targeting groups on their peripheries hold potential for site-specific H2S delivery through systemic injection. Furthermore, polymer assemblies enable H2S delivery across biological membranes such as the blood–brain barrier, which may provide a unique method for studying the biological effects of H2S on neuromodulation (52).
Localized H2S delivery may prove invaluable in studying the effects of H2S on specific organs because systemic drug delivery vehicles never have perfect targeting capacity. Hydrogels, either with covalently attached H2S donor motifs or with sequestered donors, lead the way in localized H2S delivery. Hydrogels can be made from crosslinked water-soluble polymers (chemically crosslinked hydrogels) or from self-assembling small molecules such as peptides (physically crosslinked hydrogels). Both can be implanted (or injected from a needle in the case of shear-thinning hydrogels, which disassemble in response to shear stress during injection and reassemble in vivo after injection) at a site of interest to deliver H2S locally.
Increased capacity to control the H2S release rate, hydrogel stiffness, and degradability of the hydrogel carrier material will further enhance the utility of H2S-releasing hydrogels. The potential to functionalize hydrogels with additional bioactive units offers exciting possibilities in influencing cellular behavior and promoting healing. H2S-releasing microspheres and electrospun scaffolds, which may offer longer release periods than hydrogels, may also enable a number of biological studies and applications where long-term delivery (i.e., weeks to months) is desired. All of these platforms, several types of which have thoroughly characterized degradation rates and mechanisms, are also well suited to pairing H2S with other small-molecule drugs where synergistic effects could be observed.
Many challenges remain in this field. While triggerable, small-molecule H2S donors have many advantages over sulfide salts, they are inherently unstable compounds that must be handled with care to avoid triggering release prematurely. This need for careful handling and storage extends to H2S-releasing materials, and it can limit storage times and shipment methods. The instability of H2S donors also limits delivery of H2S to active delivery, where exogenous H2S is administered in response to a specific condition. In contrast, proactive delivery strategies involve materials containing latent APIs that respond only to a biological trigger to release their payload. Development of proactive H2S-releasing materials with precisely triggered release mechanisms is needed to continue moving the field forward.
Beyond stimuli-responsive delivery strategies, materials that release H2S over the course of weeks to months are needed. Most extended biological studies on the effects of H2S treatment in slow processes, for example chronic wound healing, are conducted using daily dosing of donor compounds. This dosing schedule results in fluctuations in H2S levels in circulation and/or at the treatment site. Delivery from materials, such as hydrogels, microparticles, electrospun scaffolds, or polymer films, has the potential to stabilize H2S levels through sustained release. Furthermore, long-releasing H2S donors may enable treatment of chronic disease, such as high blood pressure, through H2S therapy. Indeed, delivery from various materials is used widely to decrease dosing frequency and extend release times in many common pharmaceuticals (2, 24).
Sustained delivery of H2S from materials has great therapeutic potential, but analytical challenges must be addressed to properly characterize H2S release profiles from these materials, both in vitro and in vivo. Methods for determining H2S release profiles, such as the methylene blue method, fluorescent sensing methods, and electrochemical probes, are well suited for measuring release times on the order of minutes to hours, but each has limitations beyond this time period. Reliable and sensitive methods for long-term measurement of H2S concentrations are needed.
Addressing these challenges in H2S-releasing materials may provide insight into H2S biology and physiology, and answer questions regarding potential H2S therapies. For example, will targeted and/or localized delivery reduce the dose needed compared with systemic injection? Another question is how release rates affect therapeutic outcomes. Ongoing work in our laboratory and other laboratories is beginning to show that sustained H2S concentrations at low levels are more effective than instantaneous H2S donors or very long-acting donors. These results are consistent with a bell-shaped exposure–response curve, in which H2S exerts its maximum beneficial biological effect at intermediate concentrations. More efforts to compare similar donors with varying rates of H2S release are needed to verify these early results, and the outcomes of these experiments will inform clinical possibilities for H2S therapy.
Compared with other drugs, the field of H2S-releasing materials is in its infancy; drug delivery methods have been used clinically for decades and continue to be developed for other APIs. In the coming years, we expect to see materials capable of controlling many aspects of H2S delivery, revealing (patho)physiological insights and moving toward clinical applications.
Footnotes
Funding Information
This study was supported by the National Science Foundation (DMR-1454754) and the National Institutes of Health (R01GM123508).