
Abstract
Significance:
Over the past 50 years, the mechanisms for O2 storage and transport have been determined quantitatively on distance scales from millimeters to tenths of nanometers and timescales from seconds to picoseconds.
Recent Advances:
In this review, I have described four key conclusions from work done by my group and our close colleagues. (i) O2 uptake by mammalian red cells is limited by diffusion through unstirred water layers adjacent to the cell surface and across cell-free layers adjacent to vessel walls. (ii) In most vertebrates, hemoglobins (Hbs) and myoglobins (Mbs), the distal histidine at the E7 helical position donates a strong hydrogen bond to bound O2, which selectively enhances O2 affinity, prevents carbon monoxide poisoning, and markedly slows autoxidation. (iii) O2 binding to mammalian Hbs and Mbs occurs by migration of the ligand through a channel created by upward rotation of the His(E7) side chain, capture in the empty space of the distal pocket, and then coordination with the ferroprotoporphyrin IX (heme) iron atom. (iv) The assembly of Mbs and Hbs occurs by formation of molten globule intermediates, in which the N- and C-terminal helices have almost fully formed secondary structures, but the heme pockets are disordered and followed by high-affinity binding of heme.
Critical Issues:
These conclusions indicate that there are often compromises between O2 transport function, holoprotein stability, and the efficiency of assembly.
Future Directions:
However, the biochemical mechanisms underlying these conclusions provide the framework for understanding globin evolution in greater detail and for engineering more efficient and stable globins.
Introduction
This review of 50 years of hemoglobin (Hb) research is based on my opening lecture for the XXth International Conference on Oxygen Binding and Sensing Proteins (O2BIP), held on September 3–6, 2018, at the University of Barcelona in Spain. It is not intended to be a comprehensive literature review of the various subject headings but, instead, provides a historical and personal view of my groups' major contributions to the four conclusions in the abstract. The topics were also chosen to complement and provide background for the other session speakers at the conference and their articles.
In 1968 when I started graduate work with Quentin H. Gibson at Cornell University, Perutz and his colleagues reported the first high-resolution crystal structure of Hb (77). Then, he and Lehmann (75) published an article interpreting the clinical phenotypes of more than 100 different human hemoglobinopathies, setting the stage for structural interpretations of how globins function at the molecular, cellular, and tissue level. In effect, this article represented the first structure/function mutational analysis of a protein and served as a model for site-directed mutagenesis studies that were to follow some 20 years later with the development of recombinant DNA technology. By the early 1970s, work with Hbs containing these naturally occurring point mutations led Perutz (73, 74) and others to develop detailed structural mechanisms for cooperative O2 binding to human Hb, which with modifications have stood the test of time (24, 116), have been presented in most modern biochemistry text books [e.g., Nelson and Cox (60) and Storz (103)], and are summarized briefly in the next two paragraphs.
Native human Hb is a tetramer that comprised 2α (141 amino acids) and 2β (146 amino acids) monomers, which form two distinct sets of subunit interfaces [for structures and helical definitions in myoglobins (Mbs) and Hbs, see Nelson and Cox (60) and Storz (103)]. Monomers first form a heterodimer with strong hydrophobic interactions at what is called the α1β1 interface. This interface remains unchanged when ligands bind either to this dimer in dilute solutions or to the tetramer under physiological conditions. Tetramer formation involves the association of these α1β1 dimers to create two new α1β2 and α2β1 interfaces, which weaken greatly when O2 binds to the native deoxyhemoglobin tetramer. The change in conformation at the α1β2 interfaces is the major cause of cooperative ligand binding (60, 73, 74, 116).
In the low-affinity or T state, the proximal histidine-Fe(II) complex is pulled away from the plane of the porphyrin ring by strong interactions of the F-helix with adjacent helices on the partner subunit at the α1β2 interface. O2 binding requires in-plane movement of the iron atom, which is restricted in the deoxyhemoglobin conformation, and as a result, ligand affinity is low. After one subunit binds O2 pulling its iron atom into the porphyrin plane, the interactions at the α1β2 interface are weakened by movement of the F-helix away from the partner subunit. This ligand-induced change “frees up” the proximal constraints in the adjacent subunit, allowing in-plane movement of the His(F8)-Fe complex and greater reactivity of the iron atom. A key issue of debate has been whether or not these ligand-induced changes at the α1β2 interface occur stepwise or are concerted with a single conformational change of the entire tetramer [see Yuan et al. (116) and Eaton et al. (24) and references therein]. Regardless, it is clear that decreases in proximal stereochemical constraints are the underlying cause for the increase in O2 affinity in Hb as progressive binding occurs.
Later studies with recombinant globins in the 1990s showed that proximal constraints also regulate O2 binding in monomeric globins. In these cases, it is often the orientation of the proximal histidine that regulates iron reactivity [see Olson and Ghosh (65)]. When the edges of the proximal imidazole in the unliganded protein are located in an eclipsed conformation right underneath the pyrrole N atoms, movement of the high-spin iron into the plane of the ring is inhibited, decreasing both association rates and affinities for O2 binding. This eclipsed conformation is seen in mammalian Mbs that have moderate affinities (P50 ≥ 1 μM or 0.6 mm Hg). In contrast, leghemoglobin and many invertebrate globins have staggered imidazole/pyrrole nitrogen geometries and conformations that facilitate in-plane movement of the iron, accounting for their ultrahigh ligand affinities (P50 for O2 ≤ 0.1 μM) (22, 36, 65).
My graduate studies at Cornell focused on trying to measure differences in the ligand binding rate constants of the α and β subunits within tetrameric human Hb. Because site-directed mutagenesis was not available, we chose to explore the accessibility and iron reactivities of the subunits using larger alkyl isocyanide ligands, adopting an approach that was pioneered by St. George and Pauling (102) and then used by Antonini and Brunori's group in Rome (105). One motivation was Perutz's suggestion in his classic 1970 article that ligands could not bind to β subunits in deoxyhemoglobin until ligands bound to α subunits causing a switch to the R or high-affinity conformation (73). He proposed that access to the iron atom in β subunits was sterically restricted. However, our experimental kinetic results suggested the opposite conclusion. Ligands bind to and are released from β subunits more rapidly than α subunits in both the low- and high-affinity quaternary states, but the equilibrium constants for the subunits are identical, a property that is required for cooperative ligand binding (66, 84). The differences in accessibility for entry and exit depended on ligand size, with β subunits showing 2-fold higher rates of association and dissociation for O2 binding and 10- to 20-fold higher rates for the larger ligand, n-butyl isocyanide (84).
After a year of postdoctoral studies on xanthine oxidase and related molybdoflavoproteins with Vincent Massey and Graham Palmer at the University of Michigan, I took an assistant professor position at Rice University in the summer of 1973. Later in that year, Graham Palmer decided to take a senior faculty position at Rice. His decision allowed us to continue working together on xanthine oxidase, but, at the same time, I had to establish an independent program so I went back to studies of O2 transport by globins.
Part of our early work at Rice University involved systematically looking at the effects of ligand size and chemistry on the parameters for ligand binding to native human Hb and related globins. We expanded our studies to include a complete series of alkyl isocyanides [from methyl to hexyl, including various stereoisomers (84)] and more detailed comparisons between O2, nitric oxide (NO), and carbon monoxide (CO) (43, 56). The isocyanides served as “substrate analogs” to map out steric restrictions in the active sites of Mb and the α and β subunits of Hb (56). However, these initial maps were empirical and not very satisfying until crystal structures of the isocyanide complexes were determined (7, 8, 99). Detailed mechanistic comparisons between O2, NO, and CO had to wait until libraries of site-directed mutants and ultrafast-laser photolysis techniques could be used to measure directly individual rates of ligand entry, exit, and bond formation within the various globins.
Unstirred and Cell-Free Layers
In the late 1970s and 1980s, we also examined quantitatively the factors that govern O2 uptake and release by Hb encapsulated in red cells, either in simple rapid mixing experiments or during flow in capillaries. The motivation was threefold: (i) the experiments and analyses were intellectually challenging and had not been done systematically; (ii) the results were crucial to understanding the factors that govern O2 transport in vivo and in developing hemoglobin-based oxygen carriers (HBOCs) for transfusion therapy; and (iii), more selfishly, it was important for me to set up a research program that was distinct from my PhD advisor, Quentin Gibson, who was developing laser photolysis technology to examine the kinetics of cooperative ligand binding and associated conformational transitions in human Hb.
The first question to answer was why, in simple rapid mixing experiments, human red cells take up oxygen so much more slowly than Hb free in solution. An example from our own work using a Gibson-Dionex stopped-flow apparatus is shown in Figure 1 (17). In this experiment, either free Hb or a suspension of red cells was mixed 1:1 with air-equilibrated, isotonic phosphate buffer. Absorbance increases at 576 nm (HbO2 peak) and decreases at 560 nm (deoxyHb peak) were measured simultaneously and then subtracted to increase the signal for O2 binding and to cancel out light scattering effects, which were large and complex due to the change from turbulent to laminar flow followed by no stirring and cell settling.

As shown in Figure 1, the reaction with free Hb is very rapid with a half time on the order of ∼2 ms, and much of the reaction occurs in the dead time of the apparatus (∼2 ms). In contrast, uptake of O2 by red cells containing the same amount of Hb is dramatically slower, with a half time of ∼80 ms. Hartridge and Roughton (42) observed this same difference in 1927 using the first successful rapid mixing device for measuring Hb oxygenation. In his original article and several later publications, Roughton suggested that part of the slowing was due to the need for O2 to diffuse through the dense cytoplasm of the red cell to reach all the Hb molecules (42, 61, 86). When this diffusion resistance is simulated using a packet model of Hb surrounded by well-mixed buffer (Fig. 1, left bottom panel), the rate of O2 uptake does slow down approximately eightfold, yielding a half-time of ∼16 ms. However, the measured rate of uptake by red cells is still approximately fivefold slower (Fig. 1, right panel).
Roughton and colleagues correctly concluded that slow O2 uptake by red cells is due to external diffusion processes, whereas chemical reaction with internal Hb is much faster and not limiting (42, 61, 86). He and others suggested that the limiting process was diffusion through the red cell membrane and proposed that the O2 movement across the lipid bilayer was ∼5 to 10 times slower than the movement through an equivalent volume of buffer or cell cytoplasm. As shown in Figure 2 and Equations 1–3, his membrane model indicates that the rate of O2 uptake can be approximated by a simple interpretation of Fick's first law for diffusion across the red cell membrane. The flux, JO2, in molecules of O2 per cross-sectional area across the membrane is given by the following:
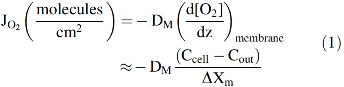
where DM is the diffusion constant for O2 in the cell membrane, ΔXm is the thickness of the membrane, and Cout and Ccell are the concentrations of free O2 outside and inside the cell, respectively. If this diffusion process is rate limiting for uptake, then the rate of increase in internal [O2] will be given by the flux times the surface area divided by the volume of the cell. Assuming a high concentration of internal Hb (∼20,000 μM) and a relatively high affinity (P50 ≈ 10–20 μM or 6 to 12 mm Hg), every molecule of O2 that enters the cell will quickly be bound to deoxyhemoglobin. Under these conditions, the rate of saturation of the internal Hb can be approximated by the following:

As a result, the rate of decay of deoxyHb is diffusion limited and appears zero order, accounting for the curvature (acceleration) in the logarithmic plot shown in the right panel of Figure 1. The idea that O2 uptake by red cells is limited by diffusion through the erythrocyte membrane persisted up until the early 1970s when my group became interested in the problem. However, by then, it was becoming clear that membranes offer little resistance to the diffusion of any of the physiologically relevant diatomic gases (N2, O2, CO, and NO) (51). Measurements of O2 diffusion constants, Dm, in biological membranes suggested a three- to fourfold reduction compared with the values in water. However, the solubility of O2 in membranes is roughly three- to fourfold higher, which compensates for the slower movement. As a result, the net rate of transport of O2 across most biological membranes is the same as that across a similar volume element of solvent if the gradients are the same (111).
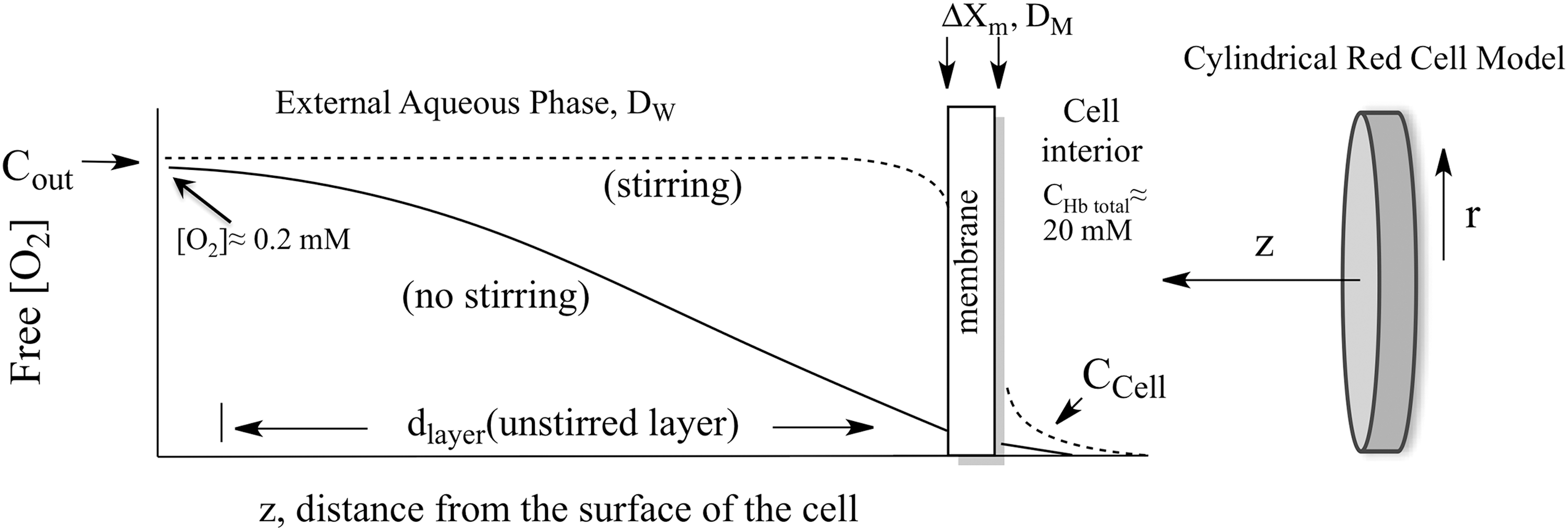
Instead of slow diffusion in the membrane, the major external resistance in red cell uptake experiments is the need for most of the O2 to diffuse through unstirred layers of solvent adjacent to the surface (Fig. 2). This problem arises from the high concentration of Hb present in the red cells, ∼20 mM Hb subunits, compared with the free concentration of O2 in air equilibrated buffer, ∼0.2 mM. If the thickness of a human red cell is ∼1.6 μm, then it would take a slab of buffer ∼160 μm thick to provide enough O2 to saturate the internal Hb. Gad-El-Hak et al. (30), Kutchai (52), and others suggested that these unstirred layers are the major cause for slow uptake of O2 by either stationary or red cells in stopped-flow, rapid mixing experiments.
Very quickly after flow stops in rapid mixing experiments, a “steady-state” gradient forms at the surface of the red cell and extends out to roughly 8–10 μm (17). The [O2] inside the cell (Ccell in Fig. 2) is roughly zero due to combination with deoxyHb and the concentration of external O2 does not reach that in bulk solvent, Cout in Figure 2, until about 10 μm away from the flatter surface of the cell. In our and Roughton's initial models, the red cell was approximated as a thin slab using a single distance parameter in the z direction in Figure 2 (17). We later examined systematically the unstirred layer diffusion problem using spherical and cylindrical models for human erythrocytes and taking into account the increase in the effective unstirred layer as the red cells are exposed to turbulent flow in the mixer, transient laminar flow as the mixture moves through the observation cuvette, and then no convective mixing shortly after flow stops (111). We were able to simulate quantitatively the shapes and dependences of time courses for O2 uptake on ligand concentration, internal Hb concentration, and external viscosity using cylindrical coordinates and the time dependence of the unstirred layer (dlayer, Fig. 2) after mixing (111).
Detailed analyses of O2 uptake and resistance due to unstirred layers are very complex and require solving partial second-order differential equations for both [HbO2] and [O2] inside the red cell and [O2] outside. However, the overall rate of uptake can be approximated by an expression similar to that derived for Roughton's original membrane limitation model (Eq. 2). If the rate of uptake is limited only by the flux across the unstirred, external layer, the net rate of O2 uptake by the cell, d[O2]total/dt ≈ d[HbO2]cell/dt, can be approximated by the following:

Again, the reaction is zero order with respect to [deoxyHb] (i.e., no chemical reaction limitation), but does depend linearly on [O2] in the solvent, Cout, and the diffusion constant of O2 in the solvent phase, Dw. Coin and Olson (17) verified this interpretation by showing that the observed rate of uptake depends inversely on the viscosity of the solvent phase using high concentrations of serum albumin and Stractan, an arabinoglactan polymer, to lower Dw.
Equation 3 also provides a semiquantitative explanation of the dramatic dependence of the rate of O2 uptake on red cell size (Fig. 3, right panel). In 1966, Holland and Forster (44) measured the apparent bimolecular rate of O2 uptake by red cells, k′O2, for a series of animal red cells with significantly different mean corpuscular volumes. We extended the range of sizes using large nucleated red cells (diameter ≈50 μm, thickness ≈6 μm) from the salamander, Amphiuma means, and artificial red cells generated by encapsulating human Hb in liposomes (diameter ≈0.2 μm, spheres), using human red cells as our control (diameter ≈8 μm, thickness ≈1.6 μm) (Fig. 3) (110). The values of k′O2 were taken from the slopes of plots of the observed rates of O2 uptake as function of [O2] or Cout in Equation 3. As shown in the right-hand panel in Figure 3, the apparent bimolecular rate of O2 uptake, k′O2, depends markedly on the surface area to volume (S/A) ratio of the red cell. The rate for artificial cells is off the observed scale and is roughly equal to the bimolecular rate for binding to acellular Hb, indicating little or no diffusion limitation. The latter result also argues strongly against any limitation by diffusion of O2 across a lipid bilayer.
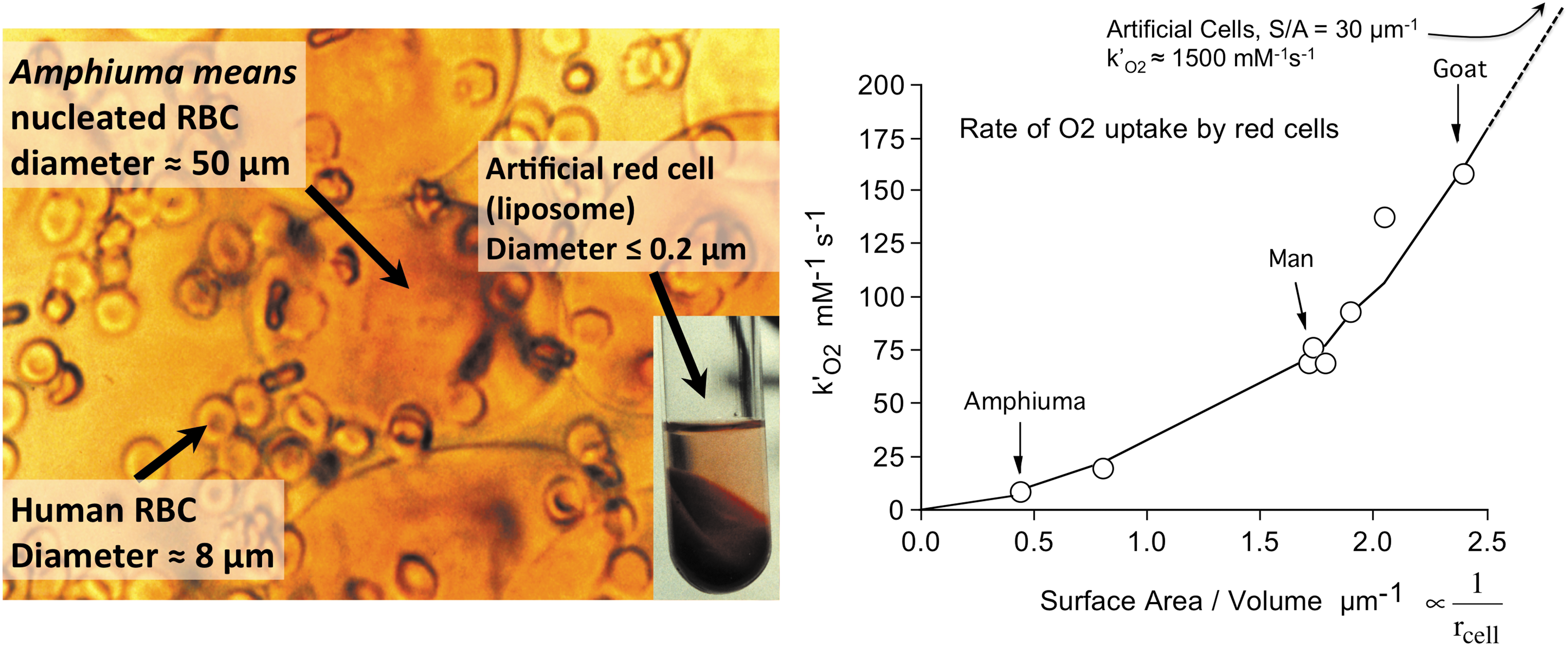
The unstirred layer model described by Equation 3 explains the parabolic dependence of the rate of O2 uptake on the S/A ratios of the different animal erythrocytes shown in Figure 3. If the cells were spherical, the S/A ratio would be given by 3/rcell. The thickness of the unstirred layer in the steady state is roughly equal to the radius of the cell (i.e., dlayer ≈ rcell), particularly for spherical shapes (111). Thus, Equation 3 reduces to the following:

The observed bimolecular rate of O2 uptake is predicted to depend parabolically on the S/A ratio of red cells, which for a sphere would be proportional to the reciprocal of the radius squared. Thus, the large red cells of amphibians are poor at taking up and releasing O2. These animals rely primarily on anaerobic skeletal muscle metabolism, whereas ruminants have the smallest red cells, which allow more efficient O2 transport for aerobic skeletal muscle metabolism (44, 110). The results in Figure 3 also explain why the fastest growing aerobic bacteria are small and nonspherical to enhance their S/A ratios and why unicellular microorganisms have often evolved flagella to stir themselves with random motions to reduce the resistance to diffusion of metabolites up to their surfaces (110).
In larger complex animals, circulatory systems evolved to push red cells through small capillaries under conditions of laminar or turbulent flow to reduce the unstirred layers to roughly 1–3 μm compared with ∼10 μm when no stirring is occurring. This convective mixing greatly enhances the rate of O2 uptake and release, using the mechanical energy created by heart muscle contractions. However, an additional diffusive resistance occurs as cells flow through blood vessels due to the Fåhraeus effect, which describes how large particles tend to accumulate in the center of small tubes where the fluid velocity is maximal. As a result, cell-free layers are created adjacent to the vessel walls, and free O2 must diffuse through these layers before reaching the endothelium and the tissues on the other side of the vessel walls.
In collaboration with Dr. J. David Hellums' group in the Chemical Engineering Department at Rice University, we constructed artificial capillary, microspectrophotometer systems to investigate the factors that govern O2 transport under conditions simulating red cell flow in human blood vessels. Detailed descriptions of the apparatus and analyses of the observed transport curves are given in a series of articles between 1987 and 1997 (10, 53, 70 –72). A color-enhanced view of the cell-free layer during red cell flow is shown in Figure 4 under conditions resembling the situation in human arterioles (69).
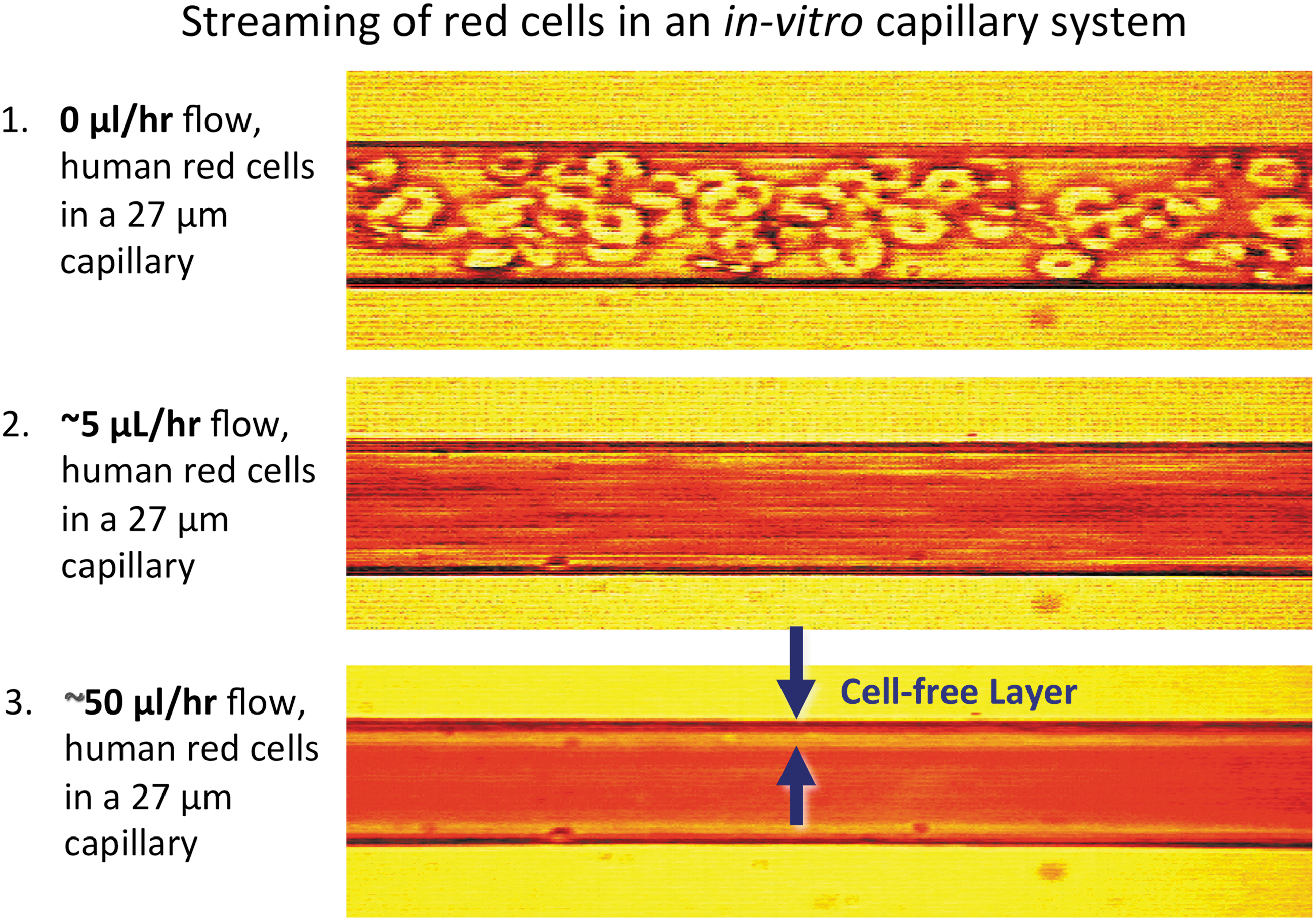
Although the thickness of this layer is small, Hellums' and our groups showed that the diffusion resistance due to this layer and that due to the small unstirred layer surrounding the red cells causes a two- to threefold decrease in the efficiency of O2 uptake and release by red cells compared with solutions of acellular Hb with the same total Hb concentration. Hellums and his students showed that this same twofold decrease in efficiency also occurs in small-bore capillaries due to “slug” flow of the cells (3, 71). In this situation, the vessel wall is in direct contact with a red cell with high rates of O2 transport for a short period of time followed by a similar period of time in which only plasma contacts the wall and little transport occurs. In contrast, high rates of transfer are continuous if an equivalent amount of acellular Hb is present.
There are three key take-home lessons from our work with red cells. First, small cells, small vessels, and laminar or turbulent flow are required for efficient O2 transport to reduce both the unstirred layers adjacent to the red cell surface and the cell-free layers adjacent to the blood vessel wall. Even when these factors are optimized, cell-free Hb is always ≥2 times more efficient at transporting O2 in capillary systems than red cells. This conclusion raises the question why animals retained red cells for O2 transport. The two main reasons are as follows: (i) to decrease oncotic pressure by reducing the number of discrete solutes and particles in blood, and (ii) to provide metabolic pathways for preventing Hb oxidation and degradation, which occur relatively rapidly (1–2 days) for purified Hb at 37°C. Degradation of acellular Hb leads to vascular damage if red cell lysis occurs in vivo. In addition, acellular Hb can penetrate blood vessel walls, scavenge NO, interfere with smooth muscle relaxation, and cause hypertension. Despite degradation problems, however, acellular Hb is more efficient at transporting O2, which explains why acellular HBOCs can sustain effective tissue oxygenation at Hb concentrations two- to threefold less (−4 to 5 g/dL) than that of whole blood (∼10–15 g/dL) (18, 20, 21, 28, 72).
The second key lesson from our red cell studies is that efficient release of O2 in muscle and neuronal tissue capillaries requires a high P50 because the rate of transfer is determined by the flux across the capillary wall. This flux is proportional to the O2 concentration gradient. At 50% unloading, this gradient is roughly the free [O2] in the blood, which is given by the P50 of the red cell Hb, minus the free [O2] in the muscle or neuronal tissue, which is ∼0 due to high rates of mitochondrial respiration. Thus, the rate of O2 efflux through capillaries into actively respiring tissues is proportional to the P50 of the red cell Hb. Lower oxygen affinity (higher P50) normally leads to more rapid unloading and greater extents of transport because of the higher rates of diffusion across the vessel walls.
In alveolar capillaries, the gradient at 50% uptake is given by the free [O2] in the liquid part of the alveolar sacs minus the free [O2] in the blood, which again is roughly equal to the P50 of the red cell Hb. In the alveolar fluid equilibrated with air, [O2]free ≈ 250 μM or ∼150 mm Hg, whereas P50 for the red cell Hb is ∼15–30 μM or 9–18 mm Hg, depending on pH and intracellular 2,3 bisphosphoglycerate concentration. Thus, the flux or rate of O2 uptake is roughly independent of the P50 of the Hb sample, unless it becomes abnormally high, that is, P50 ≥ 100 μM or 60 mm Hg. The large oxygen concentration gradient in the lung vessels also facilitates almost complete saturation of red cell Hb during the brief time spent in the alveolar capillaries (0.5 to 1 s), whereas the 10-fold smaller gradients across muscle and neuronal capillaries prevent complete unloading and limit the overall extent of O2 transport from the lungs to respiring tissue. Animals that need high rates of transport have large numbers of capillary beds, small red cells, highly cooperative intracellular Hb, and mechanisms for increasing P50 markedly by the binding of organic phosphates, protons (alkaline Bohr effect), and carbon dioxide.
The third key conclusion is that under most physiological situations, external diffusion processes limit O2 uptake and release across the red cell membrane and the blood vessel wall and not chemical reaction with the ferroprotoporphyrin IX (heme) iron atoms in the protein. We and others have shown both theoretically and experimentally that if the ligand association rate constant for reaction with free Hb, k′O2, is ≥ ∼1 × 106 M −1s−1 and the dissociation rate constant, kO2, is ≥ ∼10 s−1, there is no dependence of the time course of uptake and release of O2 by red cells on the absolute values of these parameters in either simple rapid mixing or capillary experiments (53, 111). However, as described above, the ratio of these constants, which defines the overall oxygen affinity of the Hb, does matter because the P50 value defines the gradient across capillary walls during release in actively respiring muscle and neuronal capillaries. Thus, red cell Hbs in almost all vertebrates have evolved to have very high rates of O2 association and dissociation, and almost all have relatively high P50 values ≥ ∼20 μM or 12 mm Hg under physiological conditions.
A similar situation occurs for mammalian Mbs, which act as O2 storage and release proteins in striated muscle. The rates of uptake are rapid (k′O2 ≥ ∼15 × 106 M −1s−1) when O2 is delivered to myocytes during blood flow, and release to mitochondria cytochrome oxidase is also very rapid (kO2 ≥ ∼15 s−1) during contraction when no blood flow can occur. The ratio of these rate constants (P50 ≈ 1 μM or 0.6 mm Hg) is crucial in order for the Mb to pick up O2 from lower affinity Hb in blood (P50 ≈ 10–20 μM), but still small enough to release the O2 to mitochondrial cytochrome c oxidase, which has a Km ≤ 0.1 μM.
Electrostatic Stabilization of Bound O2
As described above, the absolute O2 affinities of Hb and Mb are crucial to their physiological function both in terms of fluxes of O2 across blood vessel walls and in terms of storing O2 for release to mitochondrial cytochrome c oxidase during respiration. The protein portions of these globins play a crucial role in regulating the absolute affinities of the heme iron atom for the diatomic gases NO, CO, and O2 and for discrimination between them. For simple pentacoordinate ferrous model heme compounds in organic solvents, the overall association equilibrium constants for NO, CO, and O2 are on the order of ∼1012, 108, and 104 M −1 (65, 67, 81). The resultant P50 or Kdissociation for O2 is too high (≥100 μM or 60 mm Hg) to allow efficient uptake from air, and autoxidation to the ferric forms is too rapid to allow formation of a stable oxygenated complex. In addition, the affinity of these model heme compounds for CO is ∼10,000 times greater than for O2. As a result, CO produced internally by heme oxidation or present in the environment would effectively displace any O2 that did bind, making CO highly toxic even at very low levels. Thus, there is strong evolutionary pressure for globins to evolve mechanisms for increasing O2 affinity, discriminating in favor of O2 over CO, and dramatically reducing autoxidation rates to allow efficient O2 transport and storage. Over the past 30 years, large libraries of recombinant Mb and Hb mutants were constructed to examine these processes.
Ligand binding to the ferrous form of Mb involves at least four distinct processes, which are shown in Figure 5, and this basic mechanism also applies to the reduced subunits of all animal and plant Hbs. The first step requires the displacement of water molecules that are held in the distal pocket by hydrogen bonding to polar residues. In the case of sperm whale Mb (SwMb), a water molecule is found 2.7 Å away from His64(E7) in deoxygenated crystals and represented by the blue sphere in the upper left-hand panel in Figure 5 (83). The electron density observed at room temperature suggests an occupancy of roughly 80%. Diatomic ligands cannot enter the active site if this water molecule is present. The second step involves movement of a diatomic ligand into the empty volume of the distal pocket to form a “caged” complex in which the diatomic gas is sequestered in the protein close to the sixth coordination position of the heme iron atom in the center of the porphyrin ring. This transient intermediate is represented by the Mb••••X state in Figure 5. The third step involves formation of the covalent bond with the iron atom to generate the bound state, MbX, and is the major driving force for the overall binding process. The last step involves minor rearrangements that lead to electrostatic stabilization of the bound ligand, which in the case of mammalian Mbs and Hb subunits involves formation of a hydrogen bond donated by H-Nɛ of His(E7), as shown in the right-hand panel in Figure 5.
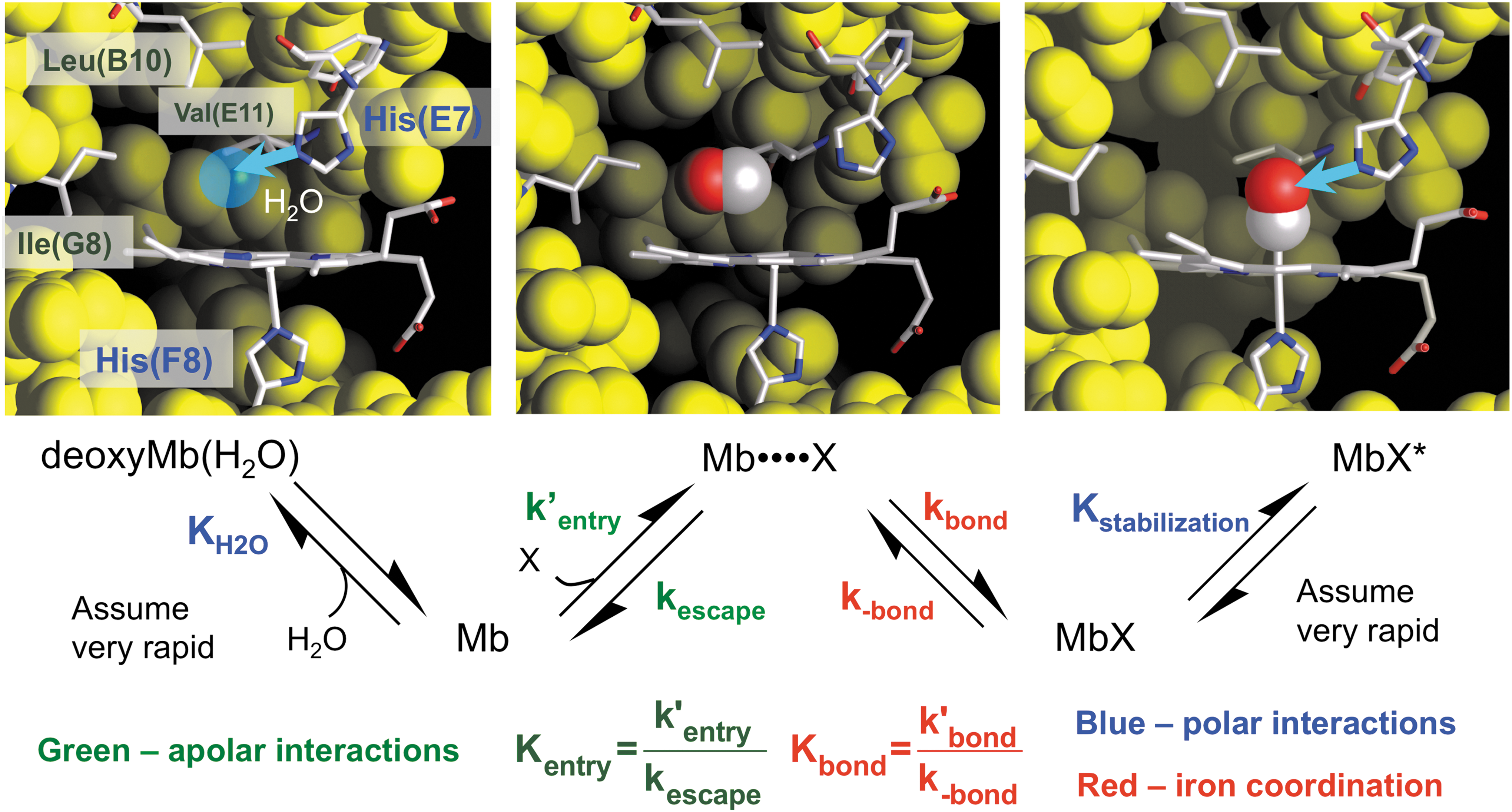
Using this mechanism, we derived an expression for the overall association equilibrium constant, which is given below, using the equilibrium constants defined in Figure 5 (67, 68):

The four terms in Equation 5 provide explanations for what determines overall ligand affinity. The 1/(1 + KH2O[H2O]) term represents the fraction of empty distal pockets in deoxyMb, which ≈0.2 based on the observed fractional occupancies (∼0.8) of distal pocket water in crystal structures of sperm whale deoxyMb. This factor is independent of the size and chemistry of the ligand molecule. Kentry represents the bimolecular association equilibrium constant for nonspecific binding in the distal cavity, which is driven by a hydrophobic effect because the diatomic gases are apolar. The value of Kentry is on the order of 1 M −1 and the same for all three diatomic gases, which have approximately the same size, polarity, and solubility in water (1–2 mM at 1 atm). Thus, there is no discrimination between the ligands for the first two steps.
The overall binding reaction is driven by iron-ligand bond formation, where Kbond is on the order of 1012, 108, and 104 for internal NO, CO, and O2 coordination to the heme iron atom and reflects the intrinsic chemical differences between the ligands. The absolute value of Kbond can be altered markedly by steric hindrance from large amino acids on the distal side of the heme ring and by steric restraints on the proximal side that inhibit in-plane movement of the iron-His(F8) complex. These proximal constraints can be due to conformations that restrict movement of the F-helix as in human Hb or an eclipsed orientation of the proximal imidazole ring with respect to pyrrole N atoms, both of which lower ligand affinity.
Distal steric hindrance and proximal constraints cause roughly equal decreases in the affinities of all three diatomic gases. There is virtually no discrimination between the linear versus bent geometries of the Fe-O2 versus Fe-CO complexes even when larger amino acid side chains are inserted at the four key positions (CD1, B10, E7, and E11) adjacent to the bound ligand. Larger distal side chains do decrease the affinities for NO, CO, and O2 markedly, but by the same amount (67, 68). However, as expected, steric hindrance in the distal portion of the heme pocket does dramatically discriminate between isocyanide ligands with differing sizes and shapes (7, 8, 99).
The last term in Equation 5, (1 + Kstabilization), represents the extent of electrostatic stabilization of the bound ligand and does differ dramatically between the bound iron-diatomic ligand complexes. The Fe(II)-O2 complex is highly polar due to partial oxidation of the iron atom leading to a significant partial negative charge on the second bound O atom (65). This partial charge allows bound O2 to accept a strong hydrogen bond from H-Nɛ of His(E7) or Gln(E7). In contrast, the Fe(II)-CO and Fe(II)-NO complexes are apolar and only weakly stabilized by hydrogen bonding interactions. The extent of this stabilization can be measured quantitatively by examining the effects of mutations in the distal pocket on ligand dissociation rate constants.
If water binding in the first step and hydrogen bonding in the last step of the mechanism shown in Figure 5 are assumed to be very fast, expressions for the overall bimolecular association and unimolecular dissociation rate constants (k′association, kdissociation) can be derived by assuming a steady-state approximation for the transient Mb••••X intermediate (68, 88).

The expressions in Equation 6 show that the overall association rate constant is directly proportional to the fraction of water-free or empty active sites in deoxyMb, whereas the dissociation rate constant is directly proportional to the fraction of bound ligand molecules that are not electrostatically stabilized by the polar residues in the active site [i.e., 1/(1 + Kstablization)]. Thus, the most direct way to measure the extent of preferential electrostatic stabilization of bound O2 is to compare the oxygen dissociation rate constant of the native protein with those for variants in which the distal His(E7) has been replaced by an apolar amino acid of comparable size.
In 1999, George Phillips' and my group correlated the overall rates of ligand dissociation from a library of 29 different mammalian Mb variants with electrostatic fields near the bound ligand atoms that were calculated from their corresponding high-resolution crystal structures (81). The results are shown in Figure 6 and define the extent of preferential electrostatic stabilization of bound O2 quantitatively. In principle, we could use the expression for kdissociation in Equation 6 and then attempt to determine the values of kescape, kbond, and k-bond from global analyses of rapid mixing and ultrafast laser photolysis experiments for each variant to try to estimate 1 + Kstabilization. However, if the primary goal is to examine stabilization in the native protein, then replacement of the distal histidine with an apolar amino acid of similar size allows a simpler, experimental determination of the extent of stabilization. The observed rate of ligand dissociation without polar interactions should be given by just the (kescape/(kescape + kbond))k-bond term in Equation 6, which is the internal rate of Fe-ligand bond dissociation times the fraction of internal dissociated ligands that escape the distal pocket. Thus, the kdissociation for wild-type Mb should equal the kdissociation for the apolar variant reduced by the extent of electrostatic stabilization due to His(E7).
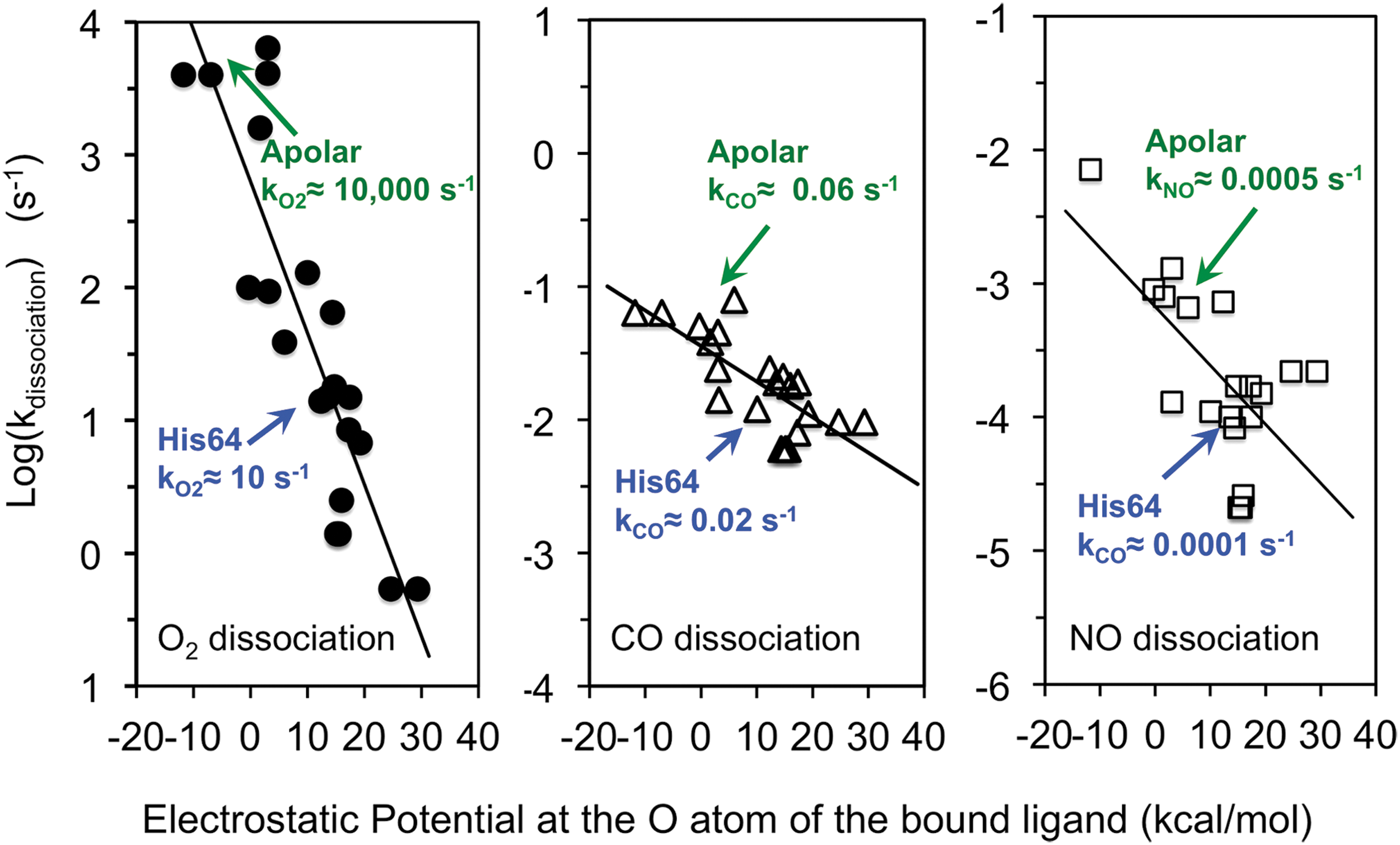

Based on Equation 7, the extent of electrostatic stabilization is given by the ratio kdissociation(apolar)/kdissociation(polar or wild-type).
As shown in Figure 6, there is a strong linear correlation between the log(kdissociation) and the computed electrostatic field at the second bound ligand atom. The details of these calculations are given in Phillips et al. (81). Positive fields are due to single or multiple H atom donors (i.e., His, Gln, Asn) and positive multipoles (Phe) in the active site. Neutral fields indicate an apolar pocket, and negative fields reflect nonbonded electrons of O atoms pointing toward the bound ligand (i.e., Thr, Tyr). These data show unambiguously that bound O2 is markedly affected by electrostatic fields, with kO2 values ranging from roughly 10,000 s−1 to ≤0.5 s−1, where multiple H-bond donors [His(E7), Asn(E11)] are present. The value of (1 + Kstabilization) in wild-type MbO2 with His64(E7) is roughly 1000 based on the values shown in Figure 6, left panel. Significantly less dependence is seen for both CO and NO dissociation. In the case of MbCO, the rate of dissociation is on the order of 0.06 s−1 for apolar variants and 0.02 s−1 for wild-type Mb, indicating that H-bonding from His64(E7) only stabilizes bound CO by a factor of ∼3. In the case of NO, the extent of stabilization is roughly fivefold (i.e., kNO(apolar)/kNO(wild-type) = 0.0005s−1/0.0001s−1).
The data in Figure 6 show that positive electrostatic fields stabilize bound ligands, with the greatest decrease in kdissociation occurring for the polar Feδ(+)-O2 δ(−) complex. The net effect on overall ligand affinity, Kassociation, is smaller because as shown in Figure 6 and Equation 5, there is also a nonspecific inhibitory effect of having a polar amino acid in the active site due to stabilization of a distal pocket water molecule in the deoxyMb state. The net effect of introducing a polar amino acid into the distal pocket is determined by the ratio (1 + Kstabilization)/(1 + KH2O[H2O]).
The inhibitory factor, 1 + KH2O[H2O], can be determined from either (i) the water occupancy observed in room temperature crystal structures of deoxyMb or (ii) the effects of replacement of His64(E7) with apolar amino acids on the association rate constants, which are proportional to the fraction of empty distal pockets (Eq. 6). For mammalian Mbs, the value of 1 + KH2O[H2O] is roughly 5 [i.e., 80% water occupancy in the deoxyMb structures and 5–10-fold increases in k′association when His(E7) is replaced with Leu, Ile, and Phe (67, 81)]. This value for 1 + KH2O[H2O] allows a quantitative evaluation of the role of the distal histidine in discriminating in favor of O2 binding compared with either CO or NO binding. The net change in O2 affinity due to electrostatic interactions with His(E7) is (1 + Kstabilization)/(1 + KH2O[H2O]) ≈ 1000/5, a 200-fold increase in Kassociation. In the case of CO, the distal His(E7) causes a small net decrease in affinity of ∼0.5 because the inhibitory effect of the distal pocket water in deoxyMb is greater than stabilization of the bound ligand. In the case of NO, the stabilization effect is roughly equal to the inhibitory effect, and there is no net change in overall affinity between variants with and without a distal histidine.
Favorable electrostatic interactions with polar amino acids in the distal pocket also play a crucial role in inhibiting autoxidation of MbO2 and HbO2 complexes. In the early 1990s, we used our large library of mammalian Mb variants to show that autoxidation occurs by two mechanisms (13). At high [O2], autoxidation occurs by protonation of bound O2 and direct dissociation of HO2•. In the absence of the distal histidine, this process is fast (kox ≈ 10 h−1 or t1/2 ≈ 4 min at 37°C, pH 7 for apolar E7 mutants of SwMb) because the extent of protonation of the Feδ(+)-O2 δ(−) is fairly high (pKa ≈ 6). When a strong hydrogen bond is donated to the bound O2 by His(E7), protonation is dramatically inhibited (pKa ≤ 4) and the rate of autoxidation decreases to 0.05 h−1 with a t1/2 ≈ 14 h or 840 min (13). Thus, electrostatic stabilization of bound O2 reduces autoxidation roughly 200-fold at high [O2], which is roughly the same extent that it increases O2 affinity.
At low [O2], near or below the Kd or P50 of the globin sample, the rate of autoxidation occurs by an outer sphere mechanism. In this case, solvent O2 reacts indirectly with a deoxygenated ferrous heme group that contains a weakly bound sixth ligand (water, azide, and cyanide) that favors oxidation to the ferric state (13). The rate of this autoxidation process shows a complex bell-shaped dependence on [O2] with a maximum rate at the P50 of the MbO2 or HbO2 variant. The rate of this outer-sphere mechanism decreases to 0 at both low and very high [O2] because it requires the reaction of the deoxygenated protein with free dioxygen. Under hypoxic conditions, there is no O2 available, and at high O2, the Hb or Mb is fully saturated and no deoxyMb or deoxyHb is available. In this case, stabilization of bound O2 by favorable electrostatic interactions with the distal histidine lowers the P50 of the sample, which decreases the rate of autoxidation under normoxic conditions by decreasing the amount of unliganded protein available for the outer sphere oxidation process.
Thus, it is clear that most globins evolved to have hydrogen bond donors in their active sites to discriminate in favor of O2 binding over CO (and NO) and to greatly inhibit autoxidation. In the case of mammalian Mbs and Hbs, the key residue is at the E7 helical position, which is almost always a His (or in rare cases a Gln) with hydrogen bond donation from the H-Nɛ atoms. In other animal and bacterial Hbs, multiple hydrogen bond donors can be present to increase O2 affinity to values equal to or greater than those for CO (P50 ≤ 0.01 μM or ≤0.006 mm Hg) and effectively prevent any oxidation without the addition of oxidizing agents (22, 65). Some of these proteins appear to function as O2 scavengers, and, in most cases, the observed properties can be explained by computing electrostatic fields adjacent to the bound ligand atoms as was done for the mammalian Mbs in Figure 6.
Baseball Glove Model for O2 Entry and Capture
As described in the first section of this review, O2 uptake and release by red cells flowing through capillaries are limited primarily by external diffusion processes, which occur on μm distance scales adjacent to the erythrocyte membrane and across the vessel walls. The rates of chemical reaction with the intracellular Hb and muscle Mb evolved to be much faster in order not to limit transport and storage under physiological conditions. With the development of recombinant Mb and Hb expression systems and ultrafast laser photolysis methods in the late 1980s, we began to examine the pathways for ligand movement in these proteins on much shorter distance and timescales and to assign specific values to the rate constants for entry, escape, and bond formation shown in the scheme in Figure 5.
These assignments require combined measurements and analyses of overall association rate constants on ms to μs timescales and of the ultrafast internal rate parameters that are measured on picosecond to nanosecond timescales (68, 88, 95). Immediately after a short laser pulse (≤5 ns), the bound ligand is photodissociated into an initial geminate state in which the Fe coordination bond is broken, but the ligand is still “caged” or sequestered near the heme group by amino acid side chains in the distal pocket (i.e., the Mb••••X intermediate in Fig. 5). From this position, the ligand can either escape from the protein at a rate, kescape, or rebind geminately at a rate, kbond, forming a bond between the same ligand and Fe (i.e., a geminate pair).
The value of kbond depends on the reactivity of the ligand molecule (NO > >O2> > CO), the ease iron movement back into the plane of the porphyrin ring, and the steric accessibility of the iron atom on the distal side of the porphyrin ring. However, kbond is little affected by electrostatic fields. The rate of ligand escape is roughly independent of the nature of the diatomic ligand because NO, CO, and O2 all have roughly the same size, but is affected by the location and shape of the exit pathway. Similarly, the bimolecular rate constant for ligand capture in the distal pocket to form the transient Mb••••X transition state, k′entry, is also independent of the chemical nature of the diatomic ligand and depends only on the size and shape of the entry channel.
Two major ligand rebinding phases are seen in all nano- or picosecond laser photolysis experiments at room temperature. The first phase is very fast, unimolecular, and does not depend on the external ligand concentration. As shown in Figure 7, it represents internal ligand rebinding and escape from the Mb••••X transition state, shown in the upper middle panel in Figure 7. The observed geminate rate for this ultrafast phase is kgem ≈ kbond + kescape, and its amplitude is given by the fraction of photodissociated ligands that rebind internally, Fgem ≈ kbond/(kbond + kescape). The second slower phase represents rebinding by ligands present in the solvent. In this case, the observed rate does depend on the external concentration of ligand and is determined by the overall bimolecular association rate constant k′association, which is defined by the upper expression in Equation 6. In this case, the photodissociated ligand does not rebind to the same iron atom because it becomes mixed with the large number of ligands in the buffer solution. As shown in Figure 7, measured values of kgem, Fgem, and k′association can be used to compute the entry, escape, and bond formation rate parameters. The values of kbond and kescape are given by kgemFgem and kgem(1 − Fgem), respectively, and the value of k′entry is given by k′association/Fgem.
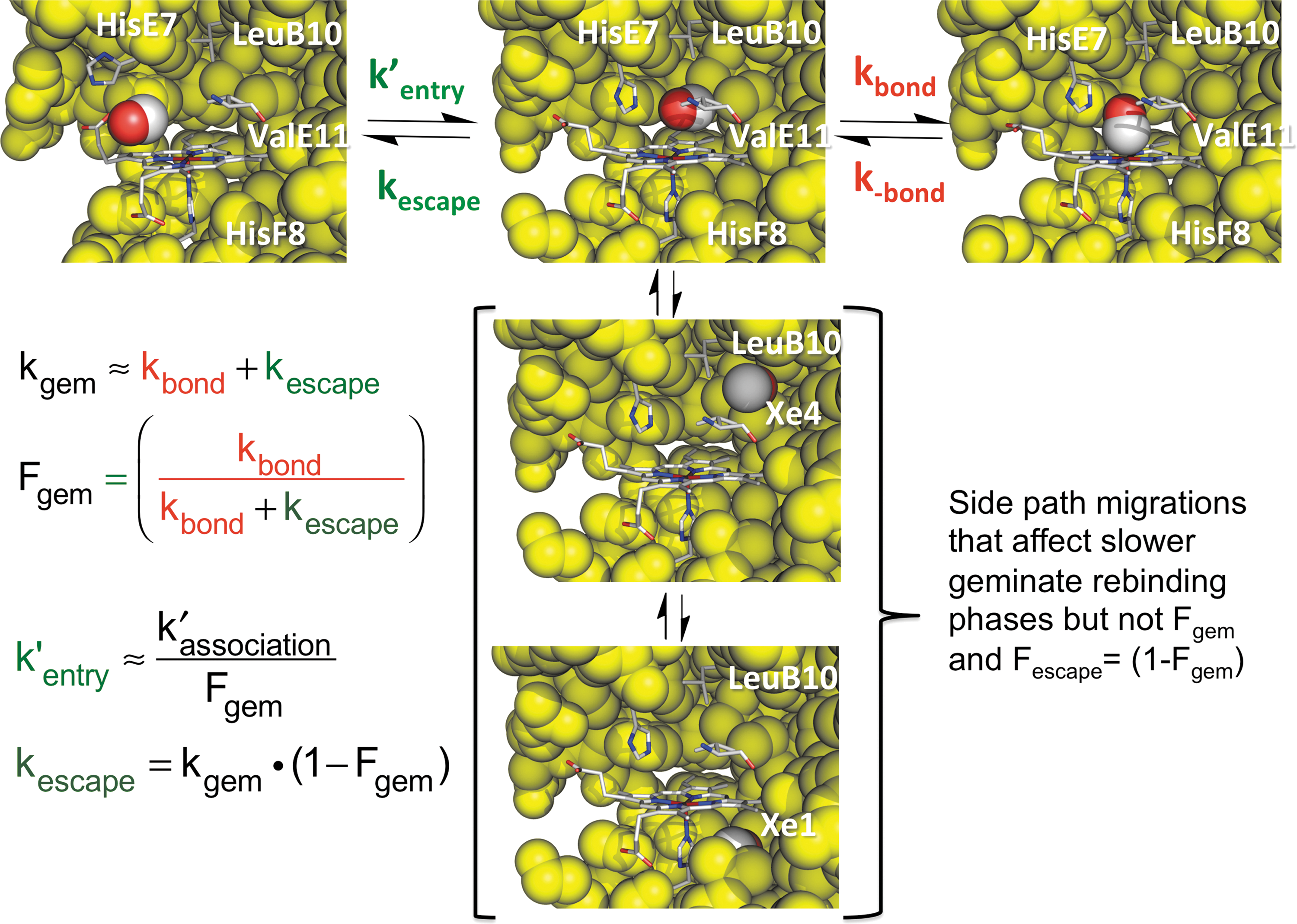
These relationships allowed us and others to show that the values of kbond for internal NO, O2, and CO binding to wild-type mammalian Mbs are on the order 1011, 107, and 105 s−1, respectively, at room temperature and that the bimolecular and unimolecular rates of ligand entry, k′entry, and escape, kescape, are on the order of 2–3 × 107 M −1s−1 and 107 s−1, respectively, for all three diatomic ligands (95). These values explain why the fractional amount of geminate rebinding of NO after photolysis is effectively 1 because for this ligand the ratio of kbond/kescape is 104 and all of the photodissociated NO molecules rebind before they have time to escape. The barrier to internal NO rebinding to Fe(II) is very small because both the pentacoordinate iron atom and the ligand have unpaired electrons and are high-spin states (29). As a result, the overall association rate for NO binding can be used as an independent measure of the bimolecular rate for ligand entry because k′NO = k′entryFgem ≈ k′entry, which for Mb is 2–3 × 107 M −1s−1(88, 95). Photolysis of MbCO shows the opposite result, and there is almost complete escape of the CO into solvent after photodissociation because the ratio kbond/kescape ≈ 0.01. As a result, the Fgem values for most native MbCOs are ≤0.05 at room temperature, making it difficult to determine the individual geminate parameters for this ligand. In the case of MbO2, the values of kbond and kescape are roughly equal and Fgem ≈ 0.5. As a result, the individual geminate rate parameters and k′entry for O2 binding are readily measured under room temperature and physiological conditions.
In 1996, Quentin Gibson decided to retire from Cornell University and moved his laboratory and ns and ps laser photolysis instruments to Rice University to work directly with my group for the next seven winters. In collaboration with Steve Sligar's, Anthony Wilkinson's, and Masao Ikeda Saito's groups, we had already generated a library of well over a 100 different Mb variants and wondered if these mutants could be used to define the pathways for ligand movements in Mb after laser photolysis. At the time, many workers in the field suggested that ligands escape from the distal pocket by initial movements into the interior of the globin and then leave by multiple pathways out to solvent based on molecular dynamics simulations [for a review, see Elber (26)].
Tilton et al. (107) suggested that ligands move into the apolar cavities that are occupied by Xe atoms in crystal structures of Mb, which were determined under high pressures (≥10 atm) of Xe gas. The highest occupancy based on electron density is at the Xe1 site, which is a large cavity underneath the heme group (bottom middle panel, Fig. 7), and the next highest occupancy is at the Xe4 site, which is located on the distal side of the heme pocket, toward the interior of the globin but connected to the active site (second middle panel, Fig. 7). Initial time-resolved crystallography results suggested that a portion of photodissociated CO molecules move first to the Xe4 site and then to the Xe1 site (106). My graduate student Scott and Gibson (94) tested these ideas by examining geminate recombination in MbO2 variants with mutations that blocked access to or filled the Xe4 and Xe1 cavities and in samples exposed to high pressures of Xe gas.
Time courses for geminate O2 rebinding in Mb often show two phases. The major, more rapid phase represents rebinding to and escape from the initial Mb••••X intermediate shown in Figures 6 and 7. The smaller and slower geminate phase (≤30%) was thought to represent rebinding of O2 molecules that had moved further into the protein interior, primarily into the larger Xe1 cavity. When Scott and Gibson (94) looked at the effects of mutations that either blocked movement into or filled the Xe4 and Xe1 cavities, the slower geminate recombination phase disappeared and only a single rapid phase was observed. The same result was obtained by looking at O2 geminate rebinding in the samples exposed to high enough pressures of Xe gas to fully occupy the Xe1 cavity.
These results showed that photodissociated ligands can access the interior Xe cavities, which was later confirmed by high-resolution time-resolved crystallographic studies of photodissociated MbCO crystals (92, 93, 101). However, to me, the most remarkable result of these first studies was that mutations that filled the Xe1 cavity or the presence of bound Xe did not change either the overall fraction of geminate recombination (Fgem ≈ 0.5) or the overall bimolecular association rate constant for O2 binding (k′O2 ≈ 15 × 106 M −1s−1) (95). The latter observation suggested the “side path” scheme for ligand recombination shown in Figure 7, which had first been proposed by Magde and colleagues (114). In this mechanism, ligands that move into the interior cavities have to return to the distal pocket to escape through a pathway created by the outward movement of the imidazole side chain of His(E7). This distal histidine or E7 gate pathway is the shortest route out of the active site and had been proposed initially by Perutz in 1969 (76) and then later by workers who examined the crystal structures of Mb derivatives containing large bound ligand molecules (11, 48, 85, 99), which move His(E7) outward toward the solvent (upper left panel, Fig. 7).
In the late 1990s, we developed a mutagenesis mapping approach to try to define experimentally and unambiguously the pathway for ligand entry and exit in Mb. Our approach expanded on the work of Huang and Boxer (45) who created a library of random point mutations in human Mb and then screened robotically bacterial expression lysates for changes in bimolecular and geminate CO rebinding. They then mapped the results on the structure of Mb by highlighting the amino acids where mutations had the largest effects on CO rebinding parameters. Although most of the effects mapped to regions near the distal His(E7) gate or in the distal pocket, a significant number of changes were found far removed from the active site, and, as a result, Huang and Boxer (45) suggested that their results supported the multiple pathways and motions out of the Xe cavities that had been seen in molecular dynamics simulations.
We adopted a more labor-intensive approach and constructed large to small amino acid mutations at positions that line the pathways proposed by simulations for ligand entry and escape. The assumption is that if the amino acid side chain is on the pathway, a change from Trp or Phe to Ala or Gly should result in large increases in the rates of both entry and escape (k′entry and kescape in Figs. 6 and 7). Our approach required the following: (i) producing more than 100 different Mb variants in large amounts; (ii) measuring overall and geminate rates of O2, CO, and NO binding in laser photolysis experiments to compute the k′entry and kescape values; and (iii) determining the crystal structures of the key mutants to ensure that no large conformational changes occurred. The initial work for Mb was published in 2001 by Scott et al. (95) and then later expanded to include more positions in 2012 by Salter et al. (88).
To map the results on the Mb structure, an entry/escape rate enhancement factor, Renhance, was defined as follows:

Large to small mutations should increase both the entry and escape rates if they are located on the main pathway for ligand binding and give rise to large positive values for both logarithmic terms in Equation 8. Replacements at positions not on the pathway should have no effect on k′entry and kescape with Renhance values close to zero. Some mutations in the heme pocket can cause significant changes in the overall association and dissociation rate constants due to changes in the internal rate of iron-ligand bond formation, water occupancy, and electrostatic stabilization but have no effect on rates of entry and escape (see Eq. 6).
The Renhance values for the various amino acid positions were mapped on the Mb structure in the left-hand panels of Figure 8, using the following coding pattern: red spheres, Renhance ≥ 1.5 (>10-fold increases in entry/escape rates); orange spheres, 1.5 < Renhance ≥ 1.0 (between 5- and 10-fold increases); yellow spheres, 1.0 < Renhance ≥ 0.5 (between 3- and 5-fold increases); and gray sticks, Renhance < 0.5 (less than 2-fold changes). In our view, the results are unambiguous; the only amino acid positions that “light up” are at the His(E7) gate pathway and in the distal pocket. No large to small replacements at positions at or near the Xe1 and Xe4 sites or along other proposed interior pathways affect the rates of entry and exit in Mb. Similarly, the presence or absence of bound Xe has little or no effect on the rates of ligand entry and exit.
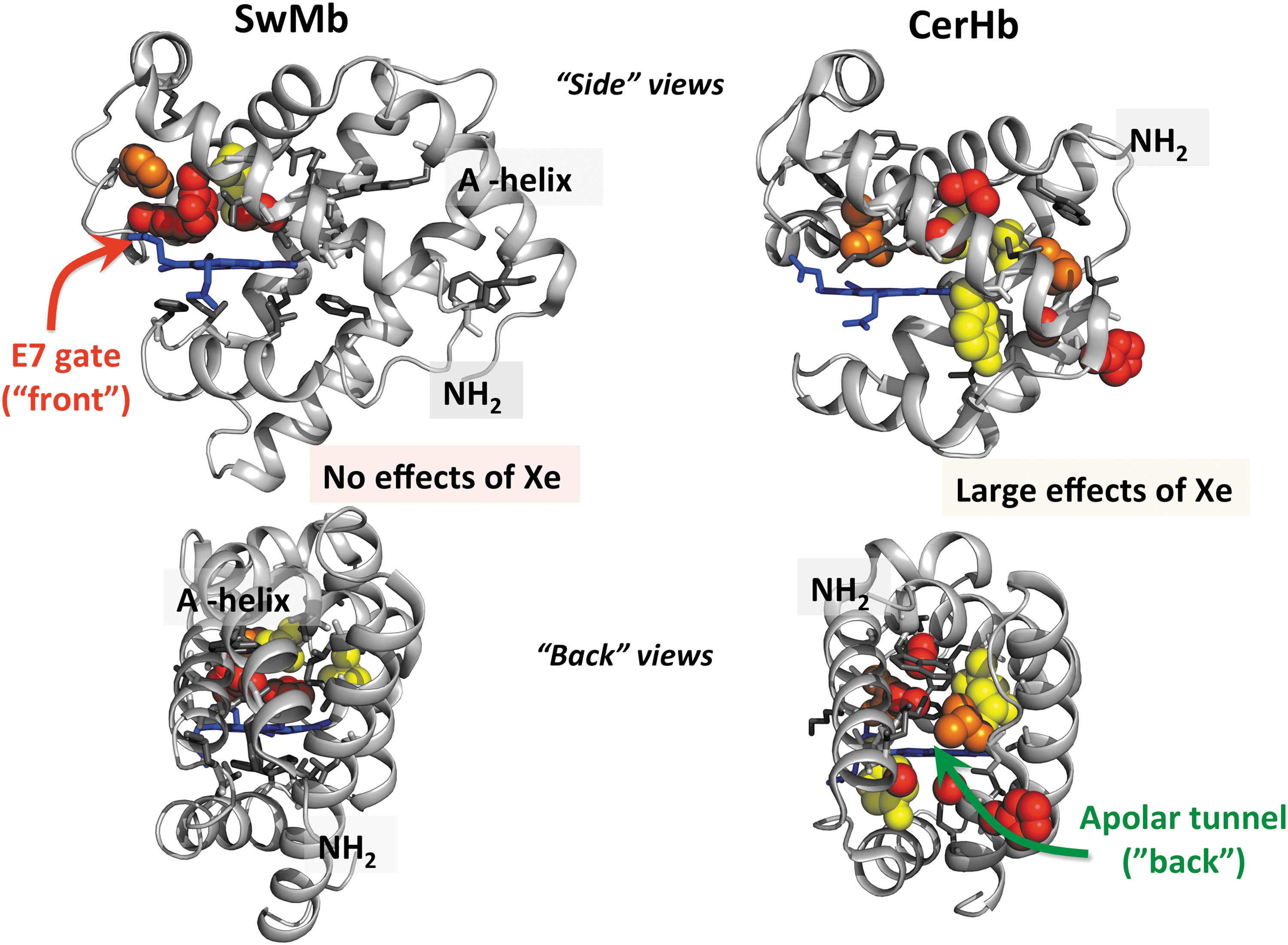
Although our initial mutagenesis map was published in 2001 (95), a large number of molecular dynamics simulation articles continued to argue strongly that (i) ligands escape through multiple pathways in the interior of the Mb as well as through the E7 channel and (ii) that some fraction of dissociated ligands move from the Xe pockets to the solvent phase without returning to the distal pocket (12, 26, 63, 87). We began to wonder if there was a flaw in our mutagenesis mapping strategy and looked for an Hb system that had a pathway for ligand entry through the interior or “back” of the globin structure. Austen Riggs at the University of Texas was kind enough to suggest that we contact Luc Moens and Sylvia Dewilde at the University of Antwerp and Martino Bolognesi at the University of Milan and help them study the kinetics of ligand binding to the neuronal mini-Hb from the Nemertean sea worm Cerebratulus lacteus (CerHb) (78). Bolognesi's group (79) had reported a crystal structure for this globin that showed the presence of an apolar channel leading from the C-terminal ends of the E and H helices through the “back” of the protein to the distal portion of the heme pocket (Fig. 8).
Together with Dewilde's group, we constructed a library of ∼120 different CerHb mutants, measured both the overall association and dissociation rate constants and the geminate rebinding parameters for all these variants, and together with Bolognesi's group determined the crystal structures of a large number of these proteins (78, 80, 88). We then used Equation 8 to map the effects of large to small replacements on k′entry and k′escape on the structure of CerHb and compared the results with those for SwMb, as shown in Figure 8 (88).
Again, the results are clear; ligands enter and exit CerHb by the interior apolar tunnel and not the E7 gate. We were able to partially close the entrance to the tunnel by Trp insertions at the C-terminal ends of the E and G helices and to inhibit entry and exit by the binding of Xe atoms in the tunnel using high pressures of Xe gas (80). Interestingly, the overall rates of O2 uptake and release are almost 10-fold larger for CerHb even though the apolar channel is longer than the E7 gate channel in Mb. CerHb acts as a neuronal O2 storage protein, surrounding the brain and axons of the Nemertean sea worm (112). Thus, rapid release of O2 allows high neuronal activity under hypoxic conditions on the seafloor when the worm hunts for prey (88, 112).
In addition to being intrinsically interesting and relevant to its physiological function, the mapping results for CerHb demonstrate that our mutagenesis mapping strategy can “find” an interior apolar tunnel and add strength to our conclusion that there are no such pathways in Mb. In Mb, the A-helix blocks any ligand movement out through the interior of the Mb structure, whereas it is missing in the smaller CerHb structure. Since the publication of our original Mb mapping study in 2001, a significant number of articles, including time-resolved crystallography experiments (92), work with larger isocyanide ligands (7, 8, 99), molecular dynamics simulations (9, 14, 15, 54), and transition path theory (115), have been published supporting the E7 gate pathway in mammalian Mbs.
We have also carried out laser photolysis studies of libraries of human α and β subunit mutants, all of which argue strongly that the E7 gate is the major pathway for ligand entry and exit in human Hb (4 –6). Royer's group used a similar mutagenesis strategy to show that ligands enter and exit the active sites of the Scapharca inaequivalvis Hb dimer by a similar E7 gate even though this amino acid is very close to the dimer interface (50). Thus, the E7 channel appears to be the dominate pathway for ligand entry in most Hbs, with the only exceptions so far being apolar tunnels in CerHb and some truncated bacterial and plant globins [see Bustamante et al. (14, 15) and references therein]).
The E7 gate mechanism shown in Figure 8 indicates that Mb binds O2 in a manner analogous to catching a ball with a baseball glove. The distal histidine, like the thumb of the glove, has to move outward to create an opening to allow entry of an incoming ligand into an interior pocket that is big enough to keep O2 there until the imidazole side chain can rotate back to close the channel and sequester the ligand adjacent to the heme iron atom. Thus, the ease of movement of the distal histidine and sizes of both the E7 channel and the distal pocket play key roles in ligand capture.
The “baseball glove” model also provides a quantitative explanation for how mammalian Mbs and Hbs efficiently detoxify NO by deoxygenation, a property that is highly conserved among animal and some bacterial, fungal, and plant Hbs (25, 32, 64, 97). Even when O2 is bound, there is still enough room in the interior portion of the distal pocket of Mbs and Hbs to transiently capture NO (21, 25). Once inside the active site, the NO radical immediately reacts with bound O2, which has superoxide character due to partial oxidation of the iron atom. In collaboration with us, Paul Gardner's group (31) showed that both atoms of bound O2 are incorporated into the final NO3 − product, verifying the process as NO dioxygenation (NOD). Remarkably, the bimolecular rate of NOD, k′NOD, equals the observed rate of simple NO binding, k′NO, and the calculated values of k′entry for all the ferrous Mb variants we have examined. Thus, even when O2 is bound, NO enters the protein through the E7 channel shown in Figures 7 and 8. We used this mechanism to successfully engineer recombinant hemoglobin (rhB)-based oxygen carriers (rHBOCs) with reduced rates of NOD by filling the interior of the distal pocket with large aromatic amino acids at the B10, E11, and G8 positions, and these crosslinked rHBOCs showed little or no hypertensive side effects due to endothelial NO scavenging when administered to animals in transfusion experiments (20, 21, 64).
Hb Assembly and Molten Globules
During the construction and expression of libraries of recombinant Hbs and Mbs, we became very interested in determining the structural factors that govern (i) resistance to denaturation to engineer more stable globins, both for our experiments and for potential HBOCs; and (ii) holoprotein expression in bacteria to increase our production yields of recombinant Hbs and Mbs. The latter process is also relevant for understanding Hb assembly during erythropoiesis and evaluating hemoglobinopathies associated with differential expression of α and β subunits, inefficient assembly, and globin instability.
A comparison of the mechanisms for Mb and Hb assembly is shown in Figure 9. The two-step pathway for apoMb folding was established by Baldwin, Barrick, Hughson, and Wright (1, 2, 46, 47) in the early 1990s and then verified in extensive mutagenesis studies over the next 25 years (23, 27, 37, 49, 55, 58, 62, 98). During the same time period, we looked at the first steps in holoMb unfolding and denaturation, which involve autoxidation and ferriprotoporphyrin IX (hemin) dissociation. We engineered an apoMb hemin scavenging reagent by replacing the distal His(E7) with a Tyr to allow hexacoordination with the phenolic O atom when hemin is bound, producing a “green” ferric holoprotein with a strong absorbance peak at 610 nm. Val(E11) was replaced with Phe to increase significantly the stability of the corresponding apoglobin double mutant (39). This reagent, H64Y/V68F apoMb, was then used to measure the rates of hemin dissociation from our libraries of ferric oxidation state (met) Mb variants (35, 41). We also examined reversible holo-metMb unfolding (38) and, in 2010 (19), developed a combined analysis of apoMb and holo-metMb unfolding curves to obtain both unfolding parameters and equilibrium association constants for hemin binding to all three apoglobin unfolding states. Results from all these studies are summarized in the upper scheme in Figure 9.

ApoMb folding occurs in two major steps (23). The first involves nucleation of the A, H, G helical secondary structures in the unfolded polypeptide, U, to form a molten globule intermediate with a native-like, interior core and a disordered or “melted” heme pocket. The second step involves folding of most of the heme pocket to generate the final native apoMb state, N, in which only the F-helix and parts of the C-helix and CD corner are disordered. This mechanism is strongly supported by unfolding studies in which mutations that fill the heme pocket with large apolar amino acids (i.e., His(E7) to Leu or Phe, Val(E11) to Phe, Leu(B10) to Phe) greatly stabilize the N state, whereas mutations that introduce more polar residues (i.e., Val(E11) to Thr or Asn, Leu(B10) to Asn) destabilize the N state and facilitate formation of the molten globule intermediate (37, 91).
Hemin binding to native apoMb results in the final, completely folded holoprotein. Hemin binding to the molten intermediate results in a hemichrome species, which has distinct low-spin iron peaks around ∼540 and ∼570 nm, indicating hexacoordination with two His residues in the heme pocket (19). This molten, hemin-containing intermediate is on the pathway for folding to native aquomet-holoMb and immediately forms a fully folded pentacoordinate deoxyMb species when reduced.
The resistance of holoMb to unfolding and denaturation is governed by heme affinity and not the intrinsic stability of the apoglobin structure. The value of the heme dissociation equilibrium constant (K-H) is on the order of 10−14 M for native metMb and at least 100 times smaller for the reduced forms (i.e., deoxyMb, MbO2, MbCO where K-H ≤ 10−16 M) (38). As a result, much larger amounts of denaturant or lower pH are required to unfold the reduced forms of Mb and Hb, and the limiting step for their denaturation is autoxidation.
In 1996, we showed that holo-metMb variants with poor hemin affinity denature at much lower [GdmHCl] than variants with higher affinity, even when the apoMb forms of latter variants are significantly less stable (38). In most cells, metMb and metHb can be quickly reduced back to the stable ferrous forms. However, if hemin dissociates before reduction, complete denaturation occurs because apoMb is unstable at 37°C. Thus, in practice, it is the resistance of the ferric forms of the hologlobins to hemin dissociation that determines their overall stabilities in vivo and during storage as cell-free proteins in the presence of reducing agents.
In contrast, the expression yields of holoMb correlate directly with the measured stability of the apoprotein and not at all with hemin affinity (91). In 2000, we discovered that apoMbs from deep diving mammals (whales and seals) are 100 to 600 times more stable than the apoMbs from terrestrial mammals (human, pig, etc.) (96). These results suggested an explanation for why it is easy to produce large amounts of recombinant sperm whale holoMb in Escherichia coli. In contrast, neither pig nor human Mb can be expressed as holoproteins in E. coli and instead have to be produced as unfolded apoproteins in inclusion bodies. Roughly 15 years later, we verified this idea by examining quantitatively the dependence of expression yield of holoMb on overall apoMb stability.
Expression of purified holoMb was measured using a wheat germ-based, in vitro translation system and a series of mutant and naturally occurring mammalian Mb genes (91). There was a direct correlation between the expression yield of the isolated holoprotein and the logarithm of the overall stability of the apoprotein. There was no correlation with the measured or estimated hemin affinity. The observed expression yield increased over 10-fold in going from pig Mb to a distal pocket double mutant of dwarf sperm whale Mb, which had an ∼600-fold higher apoglobin stability constant. Similar correlations with apoglobin stability constants were observed for expression yields measured from MbCO spectra in E. coli cell suspensions (98) and for measured values of holoMb levels in the tissues of mammals (57).
To explain these correlations, we argued that during translation there is a competition between nonspecific, irreversible aggregation of newly synthesized unfolded apoMb polypeptides and hemin binding to the folded native apoprotein (91). The latter process is effectively first order, with a rate determined by the fraction of folded apoMb times the rate of hemin dissociation from membranes and protein carriers. The irreversible aggregation and precipitation process is second order, with a bimolecular rate that depends on the square of the absolute concentration of newly translated apoprotein times the fraction that is unfolded. At high rates of translation, aggregation of unfolded chains will increase markedly in rate due to the dependence on [total protein × fraction unfolded]2 and make the expression process highly inefficient unless the overall stability of the monomer is so high that there are virtually no unfolded states present to react with each other. This mechanism explains the selective pressure to increase dramatically the stability of whale apoMbs, because, as described by Berenbrink and coworkers (57), these globins have to be translated in large amounts to act as O2 storage proteins in the skeletal muscle of deep diving animals.
Up until 2 years ago, no one had succeeded in examining quantitatively reversible apohemoglobin unfolding. Most investigators avoided studying adult human hemoglobin A (HbA) unfolding because of the following: (i) the instability of apoHb and its isolated chains, both of which precipitate in conventional buffers at temperatures above 10°C; (ii) the dependence of the unfolding parameters on protein concentration due to oligomerization of Hb subunits; and (iii) disulfide formation between exposed cysteine thiols in both folded and unfolded states. Samuel et al. (90) solved these problems by examining GdmHCl-induced unfolding of apoHbA as function of protein concentration in 200 mM phosphate buffer, pH 7 at temperatures ≤10°C in the presence of 1 mM dithiothreitol (DTT).
The first key observation was that removal of hemin in the presence of DTT led to 100% formation of α1β1 apodimers, with no evidence of tetramers or monomers. As in the case of apoMb unfolding, two phases were observed for the unfolding of these apoHb dimers. The first phase was independent of protein concentration and shifted to higher [GdmHCl] when the distal His(E7) and Val(E11) were replaced with Leu and Phe, respectively (90). These results indicated that, as in apoMb, the first phase represents unfolding of the heme pocket to create a molten globule intermediate, ID, which, in the case of apoHb, is dimer with the α1β1 interface still intact and stabilizing the A, H, and G helices. The second phase shifts to higher [GdmHCl] at higher total apoHb concentration, which is expected for a process involving dissociation of the apodimer intermediate into two unfolded monomers, Um.
The second phase also shifts to higher denaturant concentration for human fetal Hb (HbF), which has a more stable α1γ1 interface and for a recombinant Hb that is genetically crosslinked (90). In the latter case, removal of hemin leads to an “open” tetramer with two α1β1 dimers covalently linked by a glycine connecting the C-terminus of the first α chain to the N-terminus of the second one. Again the first step involves unimolecular unfolding of the heme pockets of all four subunits and is followed by dissociation into two unfolded monomeric β chains and an unfolded di-α chain. The second phase is shifted to high [GdmHCl] because two interfaces have to be disrupted for complete dissociation of the molten intermediate tetramer. As in the case of Mb, hemin can bind to the molten intermediate states to form hemichrome species, which can fold into native holoHb dimers and then associate to make the final Hb tetramer (89).
Although no systematic and quantitative study has been carried out, holoHb expression yields in vivo and in E. coli do not seem as highly dependent on apoglobin stability as was observed for Mb. We observed little difference in the production yields of recombinant HbA versus HbF or crosslinked rHbA or for mutations in which the distal His(E7) and Val(E11) were replaced with large apolar amino acids. The simplest explanation for this result is that bimolecular association of α and β subunits to form the molten dimer intermediate competes effectively against nonspecific bimolecular aggregation and precipitation of unfolded subunits. As a result, increasing the rate of translation increases equally the rates of both bimolecular processes and does not affect the efficiency of holoHbA formation. However, the individual α and β genes cannot be expressed alone as holoproteins. Instead, these genes have to be transcribed and translated at equal rates to ensure rapid formation of the apo-heterodimer before the unfolded monomers begin to aggregate.
As in the case of holoMb, the resistance of HbA to denaturation is still governed by its rates of autoxidation and hemin dissociation. The latter two processes occur 10 to 50 times more rapidly for dimers and monomers, respectively, than for tetramers, and, as a result, the rate of denaturation of Hb is strongly dependent on the protein concentration (40, 59). Thus, inside a red cell where the Hb concentration is very high (∼0.02 M), autoxidation is slow, and the red cell reduction system prevents denaturation by rereduction before any hemin can be lost. In contrast, after red cell lysis, acellular Hb auto-oxidizes, loses hemin, and denatures much more rapidly when it is diluted into plasma because of dimerization.
Another important conclusion from the assembly and denaturation mechanisms shown in Figure 9 is that globin function often requires a compromise in intrinsic apoglobin stability. In the cases of Mb and Hb, the polar distal histidine is required to enhance selectively O2 affinity (100), inhibit autoxidation by preventing protonation of bound O2 (13), and inhibit hemin loss by hydrogen bonding to the coordinated water in the aquomet forms (41). However, when His(E7) is replaced by large apolar amino acids, the stabilities of both apoMb and apoHb are increased significantly by facilitating folding of the heme pocket and stabilization of the native apoglobin state (19, 37, 90, 98). In the case of Mb, these mutations significantly enhance expression yields in E. coli and in vitro translation systems (98). The dilemma is that these mutants auto-oxidize and lose hemin rapidly, making the holoproteins highly unstable, not functional, and selected against during evolution.
Conclusions
Over the past 50 years, detailed mathematical descriptions of O2 uptake and release by red cells have been developed. These analyses provide quantitative explanations for why small erythrocytes and rapid flow are needed to reduce the size of unstirred layers adjacent to the cell surface and why small capillaries are needed to reduce the size of cell-free layers adjacent to the vessel walls.
At the molecular level, studies with libraries of mammalian Mb and Hb mutants have shown that electrostatic interactions, primarily with the distal histidine, selectively enhance O2 affinity, inhibit autoxidation, and lower rates of hemin loss. A detailed multiple-step kinetic mechanism for O2 binding to these globins has been established and is analogous to how a baseball is caught by a fielder, including opening of the glove by outward movement of the distal histidine and capture in a large, flexible interior pocket on the distal side of the porphyrin ring.
The assemblies of mammalian Mbs and Hbs show mechanisms involving first the formation of a molten intermediate with disordered or “melted” heme pockets, but well-formed, native-like secondary structures for the A, H, and G helices in the interior of the protein. The second step involves folding of the heme pocket and then binding of hemin to generate the native holoprotein. In the case of adult human Hb, the initial folding intermediate is a molten globule dimer where the interior helices are stabilized by formation of a very strong α1β1 interface. Heme binding to the fully folded HbA dimer then promotes formation of the α1β2 interface generating the native tetramer, which is capable of cooperative O2 binding.
I have been fortunate to be part of the development of these biochemical mechanisms for how Mbs and Hbs are assembled and how they function in O2 storage and transport. These mechanisms are now being used to interpret how globins evolved specific properties to adapt to various environments, including high altitudes, cold temperatures, anoxia during diving, and embryonic development (16, 57, 103, 104). In all cases, the pathways for amino acid changes required consideration of effects on O2 transport function, holoprotein stability, and the efficiency of folding and assembly. These mechanisms also provide the framework for engineering globins to produce cell-free O2 carriers and storage proteins for medical and industrial purposes. Again, multiple properties have to be considered and optimized, including compromises between the desired function, resistance to degradation during storage or use, and production yields in E. coli or during chemical modifications (21, 34, 113). The mechanisms for ligand binding to globins have also been used to understand the evolution of highly selective, heme protein-based NO, CO, and O2 sensors, which are found in all kingdoms of life (33, 82, 108, 109).
Footnotes
Acknowledgment
I would like to thank all of my former students, fellows, technicians, staff scientists, and colleagues, who made all of this work possible.
Funding Information
The work described in this review was supported for more than 40 years by NIH grants GM035649 and HL047020, and Grant C-0612 from the Robert A. Welch Foundation, and for the last 5 years by NIH grant P01 HL110900.