
Abstract
Significance:
Along with other gasotransmitters nitric oxide (NO) and carbon monoxide, hydrogen sulfide (H2S) has recently emerged as an important signaling molecule with a particularly complex metabolism. Endogenous H2S reacts with multiple cellular targets, including protein ferric heme groups, to elicit physiological responses, such as regulation of local blood flow.
Recent Advances:
Recent in vitro evidence suggests that H2S at low physiological concentrations is carried in the blood as bound to the small fraction of oxidized ferric hemoglobin (metHb). A relatively stable metHb–sulfide complex forms when H2S and purified metHb react in vitro, with an affinity within the in vivo physiological range of sulfide in the blood. Formation and subsequent redox metabolism of metHb–sulfide complex have also been confirmed in isolated intact red blood cells (RBCs) containing enhanced metHb levels. Thus, H2S may function as an endocrine signaling molecule and elicit responses at sites away from the site of production. In addition, metHb, considered as an inert or pathological hemoglobin derivative, may have a novel potential physiological role in the transport of H2S in the blood.
Critical Issues:
The transport of H2S in the blood mediated by metHb would represent an O2-independent pH-dependent mechanism for the blood-mediated control of blood flow and as such it is critical to understand the in vivo significance of this transport.
Future Directions:
Major challenges must be resolved to understand how metHb may carry H2S in the RBCs, in particular determination of metHb–sulfide levels in the blood and identification of targets in the vasculature.
Introduction
Hydrogen sulfide (H2
H2S is a weak acid in solution (with pKa1 ∼6.8, 37°C) (27), and at the physiological pH of 7.4, ∼80% of total sulfide is present as hydrosulfide anion. H2S is continuously produced by several enzymes including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST), differing in substrate preference, tissue, and cellular localization (22, 56) (Fig. 1). As H2S is readily degraded in the mitochondria via oxidation to numerous products (24), the lifetime of free H2S is limited (seconds to minutes) (57) and major targets for signaling include ferric heme and thiol groups of nearby proteins, with the formation of persulfide (R-SSH) and polysulfide (R-SnH) derivatives (Fig. 1). These may act on protein activity (34) but also function as storage forms of bioactive sulfide, collectively known as bound sulfane sulfur (BSS) (48).
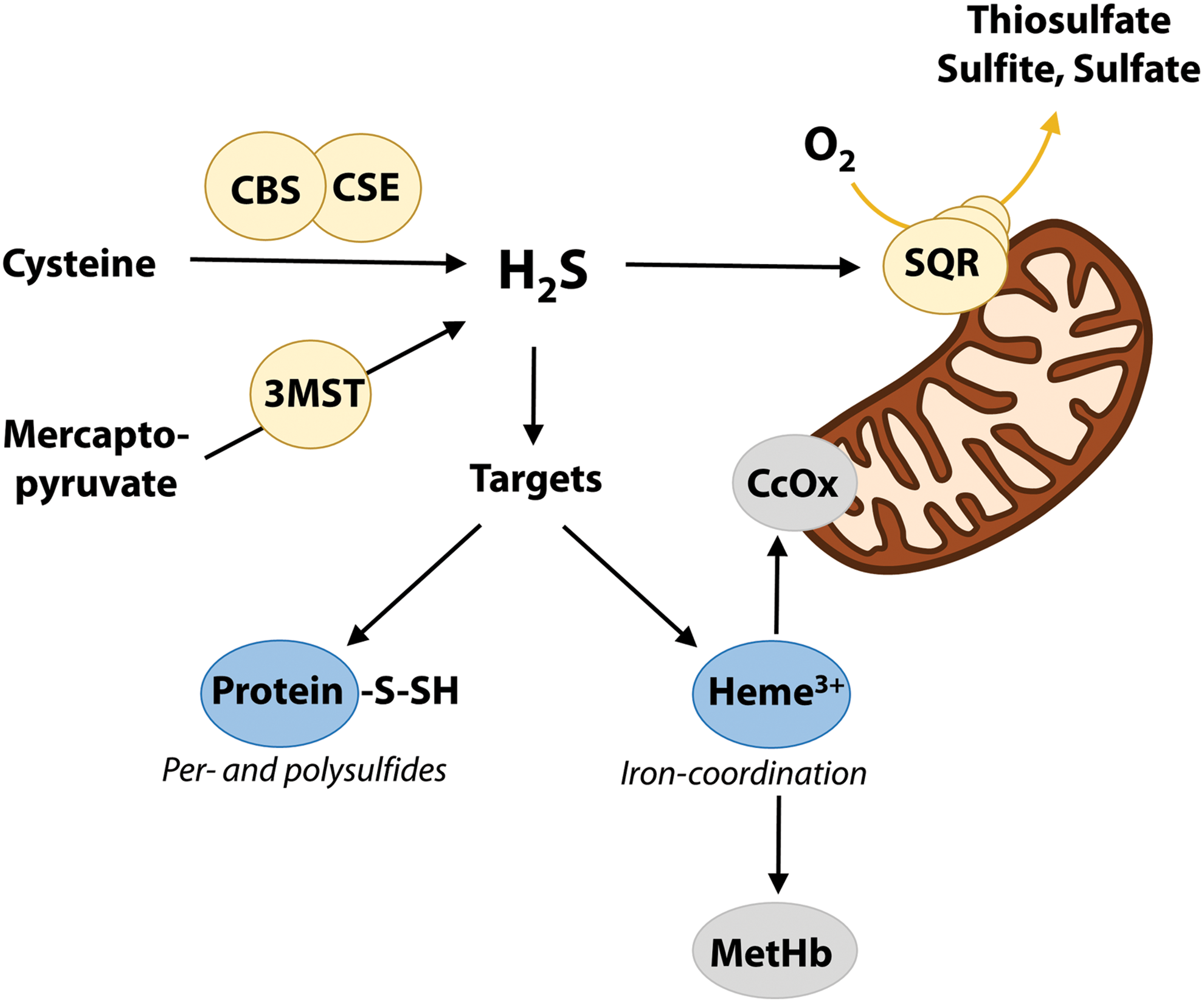
Although much of H2S metabolism remains to be understood, it appears clear that at least some oxidation products such as R-SSH, R-SnH, and thiosulfate may be reduced to regenerate H2S in vivo and contribute to local (paracrine) sulfide signaling by rapid diffusion through the cell membrane (19). In addition, H2S, NO, and CO have overlapping functions by sharing downstream pathways and interact to generate biologically active products (23, 38, 55), thus adding complexity to these already intricate signaling systems.
As is discussed in this review, there is emerging experimental evidence that H2S may be carried in vivo bound to the small fraction of ferric (met) heme of hemoglobin (Hb) present in red blood cells (RBCs). Thus, H2S may function as an endocrine signaling molecule and elicit responses at sites away from the site of production.
The two other gasotransmitters NO and CO also bind to Hb, but only to ferrous heme and not (or weakly for NO) to ferric heme (Table 1). Conversely, H2S binds to ferric heme, but not to ferrous heme (2, 33, 40). Mammalian blood contains detectable levels of NO (29) and CO (25, 54) bound to ferrous Hb. Interestingly, although partial CO saturation (16) or partial heme oxidation (11) may critically impair O2 transport, even high (up to ∼50%) NO saturations of Hb leave the O2 equilibrium curve virtually unaffected due to shifts in NO–heme coordination (14).
Reported Association Rate Constants (k on), Dissociation Rate Constants (k off), and Equilibrium Dissociation Constants (K D) for Various Ligand of Human Hemoglobin in the R State
CO, carbon monoxide; H2S, hydrogen sulfide; Na2S, sodium sulfide; NO, nitric oxide.
The vast majority of Hb in RBCs contains ferrous heme to carry sufficient O2 in the circulation. However, a small amount of ferric hemoglobin (metHb) is present in vivo due to reactions of peroxides or other biological oxidative agents (including NO) with ferrous oxygenated heme or due to continuously spontaneous autoxidation (1, 35, 41). Further oxidation of metHb produces highly reactive protein-centered radicals and oxoferryl heme, capable of oxidizing cellular components (1, 41). For these reasons, a nicotinamide adenine dinucleotide (reduced form) (NADH)-dependent cytochrome b5 metHb reductase system is present in RBCs to counteract the threat of excessive metHb buildup to O2 transport and safeguard cellular integrity (43).
In this review, we highlight recent advances in the study of the interactions between H2S and metHb occurring under physiological conditions. metHb in the blood is normally regarded as physiologically inert due to its inability to participate in O2 transport and is associated with pathological states (1). However, recent experiments converge to a novel potential physiological role of metHb in the transport and metabolism of H2S, with far-reaching consequences in the way we understand the physiology of H2S and metHb.
Kinetics of metHb and H2S and Significance in the Blood
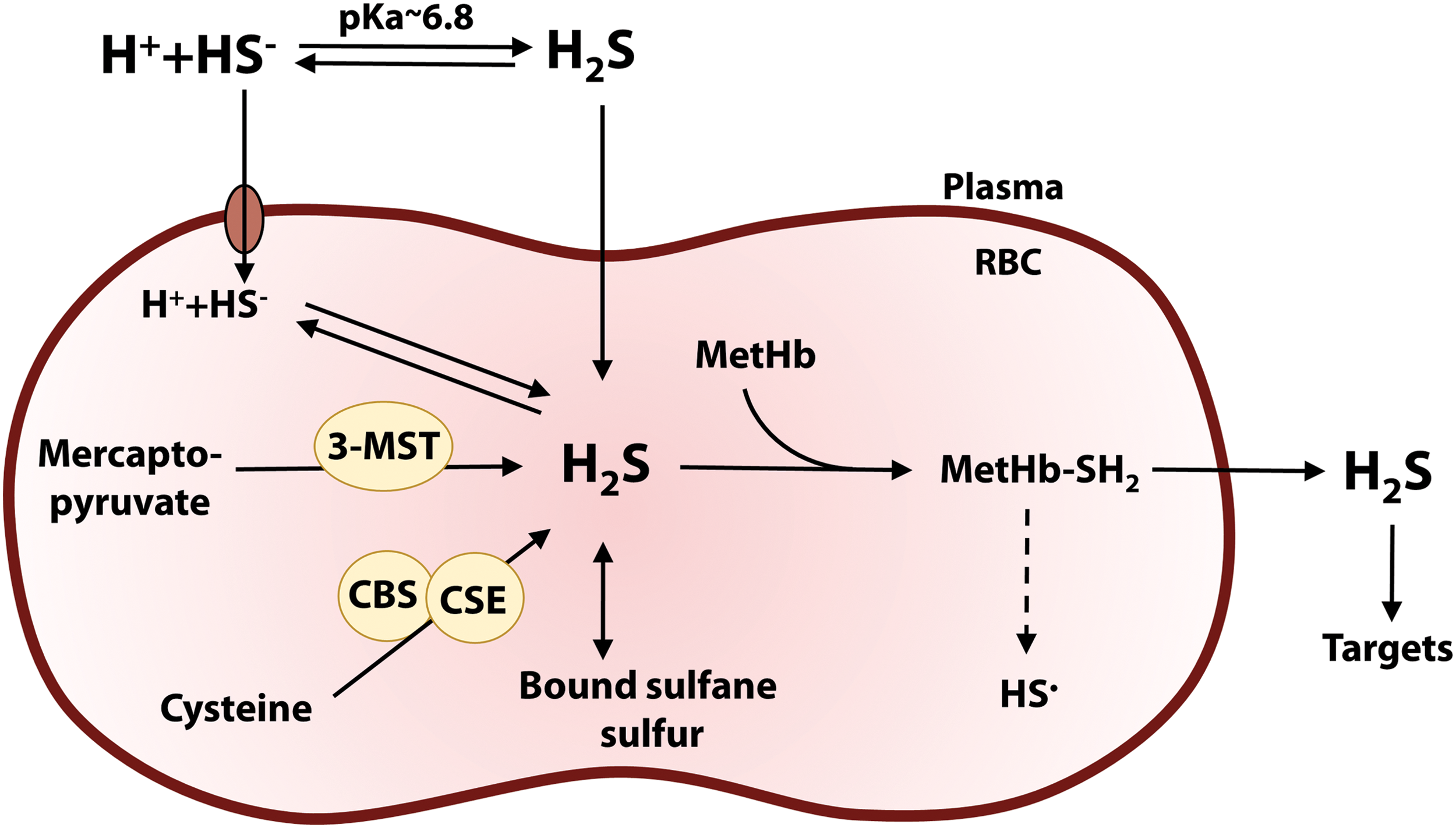
In human blood, ∼1–3% of Hb is in the met form, as recently shown by electron paramagnetic resonance (EPR) measurements (2). This corresponds to a ferric heme concentration in vivo of 100–300 μM, which is well above basal levels of H2S measured in plasma (0.3–1.3 μM) and RBCs (1.2–3.8 μM) (36, 48, 53). H2S can be produced within RBCs, as these contain the H2S-synthesizing enzyme 3-MST (59), and possibly also CBS and CSE, utilizing cysteine as the main substrate (42). There is ∼8 μM sulfide present as BSS in human RBCs and plasma that can be readily converted to H2S by reducing agents (53). In addition, H2S binds rapidly and reversibly (within tens of seconds) to human serum albumin and Hb (53), indicating a rapid exchange between substantial pools of free and bound sulfide in the blood. Furthermore, H2S diffuses out of RBCs via simple diffusion (30) or in the anionic form HS− via the anion exchanger protein AE1 in a very rapid process (19).
H2S rapidly combines with metHb present in excess (k on 3.5 × 103 M −1 s−1 at 25°C; Table 1) to generate the metHb–sulfide complex, with H2S bound to the ferric heme. Recent data from our group show that the metHb–sulfide derivative (having a characteristic absorption spectrum) forms within seconds to minutes with purified metHb in excess under conditions mimicking physiological ratios of metHb to H2S (20). In agreement with these observations, recent EPR spectroscopic data on intact isolated RBCs containing enhanced levels of metHb (obtained by prior reaction with NO) confirm that the metHb–sulfide complex forms rapidly in the reaction with stoichiometric H2S (2).
Although relatively stable in the purified protein, the metHb–sulfide complex formed in RBCs undergoes redox reactions with formation of oxyHb, R-SSHs, and thiosulfate (2). Under physiological conditions of low to stoichiometric H2S to heme and using purified metHb (20) and intact isolated RBCs (2), sulfheme does not form appreciably and for this reason will not be dealt with in this article. Detailed overviews of sulfheme formation can be found elsewhere (44, 45).
When plotted against the total sodium sulfide (Na2S) concentration added, the association rates of metHb increase with decreasing pH (20, 33, 59) (Fig. 2A). However, when plotted against the effective H2S concentrations at each pH value (calculated as [H2S] = [Na2S]/(1 + 10pH−pKa1)), the association rate becomes independent of pH (20) (Fig. 2B). This observation indicates that the heme-attacking species is the neutral H2S and not the charged and highly hydrated HS−, as predicted by modeling studies (6). Monophasic kinetics reveal similar reactivity of ferric α and β hemes with H2S (20).

Once bound to the ferric heme of Hb, H2S can dissociate in a reversible process with k off of 3.5 × 10−3 s−1 at pH 7.4 and 25°C (Table 1) (20). Since dissociation rates increase with decreasing pH (20) (Fig. 2C), it appears plausible that protonation of the distal His may widen the heme pocket and promote faster escape of dissociated H2S.
The reversible H2S dissociation from metHb–sulfide complex is a process that is not observed in experiments wherein metHb and H2S react in a sealed cuvette, as usually done to avoid disappearance of gaseous H2S from the solution. However, when the metHb–sulfide complex is transferred into a new sulfide-free buffer, the metHb spectrum gradually reappears, indicating the reversible dissociation of H2S (20). This rapid dissociation of gaseous H2S is expected also to take place during blood sample preparation and may explain why only metHb but not the metHb–sulfide complex is observed in RBCs (2). Thus, the reaction between metHb and H2S can be considered as an equilibrium that can be shifted to either direction, due to changes in the levels of H2S in solution. Simply by virtue of these rapid shifts in equilibrium, the metHb–sulfide complex would be capable of contributing to H2S homeostasis in the blood and in the circulation.
The pH-dependent k off dissociation of H2S from metHb may at first suggest a faster unloading of H2S in regions of local acidosis in the systemic circulation. However, the in vivo rate for the approach to equilibrium is equal to k on × [metHb] + k off, and because k on and in vivo [metHb] are much larger than k off (the only pH-dependent constant; Table 1), the rate by which the equilibrium between metHb and H2S is reached in the capillaries would be only marginally affected by pH changes. Nevertheless, assuming a [metHb] in RBCs of 100 μM, the rate constant for the approach to equilibrium between H2S and metHb would be of 3.5 × 103 M−1 s−1 × 0.0001 M + 0.0035 s−1 ∼ 0.35 s−1, which is compatible with the estimated transit time of blood within capillaries of ∼0.7 s−1 (61). Thus, even when measured at 25°C, the in vitro kinetics of the reaction with H2S support a physiological role of metHb as a physiological H2S carrier in the blood in vivo.
metHb–H2S Equilibrium Curves and pH-Dependent Regulation
We have recently reported for the first time a metHb–H2S equilibrium curve (20) measured in vitro, showing that H2S binds to metHb with an affinity expressed as an apparent KD of 0.68–1.02 μM at 25°C and physiological pH of 7.4 (Table 1 and Fig. 2D). This affinity value is in the range of measured H2S levels in plasma (0.3–1.3 μM) and RBCs (1.2–3.8 μM) (36, 48, 53), whereby metHb would be able to bind and release H2S within its physiological range of blood H2S as required for a sulfide carrier.
Consistent with the pH dependency of H2S dissociation but not of association rates, the H2S equilibrium curves of metHb are right-shifted when pH decreases (20) (Fig. 2D), thereby increasing the fraction of H2S unloaded to metabolizing tissues, a functional regulation equivalent to the Bohr effect of the O2 equilibrium curve of the ferrous Hb. The magnitude of the pH dependency of H2S affinity of metHb (ΔlogKD/ΔpH) is almost identical to the Bohr effect of ferrous Hb of ∼−0.5 (20). This suggests a parallel unloading in vivo of O2 from ferrous Hb and H2S from ferric Hb for a given decrease in blood pH, thereby enhancing local O2 supply and vasodilation in response to an increase in metabolic activity.
Besides from being sensitive to pH changes, metHb–H2S equilibrium curves are slightly sigmoidal and show a small degree of apparent cooperativity (Hill coefficient of 1.6 ± 0.1) (20) (Fig. 2D). Given that metHb fully ligated with H2S is in the R quaternary arrangement (58), this degree of heme–heme interaction reasonably originates from tertiary effects upon H2S ligation to ferric heme occurring within the quaternary R state, sequentially enhancing H2S affinity of the remaining accessible ferric hemes. An alternative explanation is that this positive cooperativity arises from shifts between R state like quaternary conformations of metHb having slightly different affinity for H2S. Either of these possible mechanisms would differ from the structurally extensive T-R quaternary shift known from Hb oxygenation, generating a substantial degree of cooperativity in O2 binding (Hill coefficients of ∼2.5) and markedly sigmoidal O2 binding curves.
As for O2 transport, the physiological role of a positive cooperativity in H2S binding is to enhance the amount of H2S bound and released in the circulation for a given H2S concentration gradient.
Redox Reactions Between metHb and H2S
Once formed, the metHb–sulfide adduct is stable for ∼10–30 min in intact RBCs (2) and up to 1 h in cell-free solutions (20), but over time, metHb is slowly reduced to the ferrous form (2, 20), and thiosulfate and R-SnHs are produced and released from RBCs when H2S is added in excess of metHb (2, 59).
The proposed sequence of reactions (Fig. 3A) proceeds through initial deprotonation of the heme-bound H2S to the HS− form Fe(III)–SH−, likely assisted by the distal His, formation of a Fe(II)–SH• adduct, which readily decomposes into ferrous heme and a HS•, consistent with the observation that ferrous heme does not bind sulfide. In analogy with the reductive nitrosylation of NO and metHb also generating ferrous heme (Fig. 3B), this process has been termed reductive sulfhydration (2) and involves the hemolytic cleavage of the Fe(II)–HS• bond (2), which follows first-order kinetics (32).
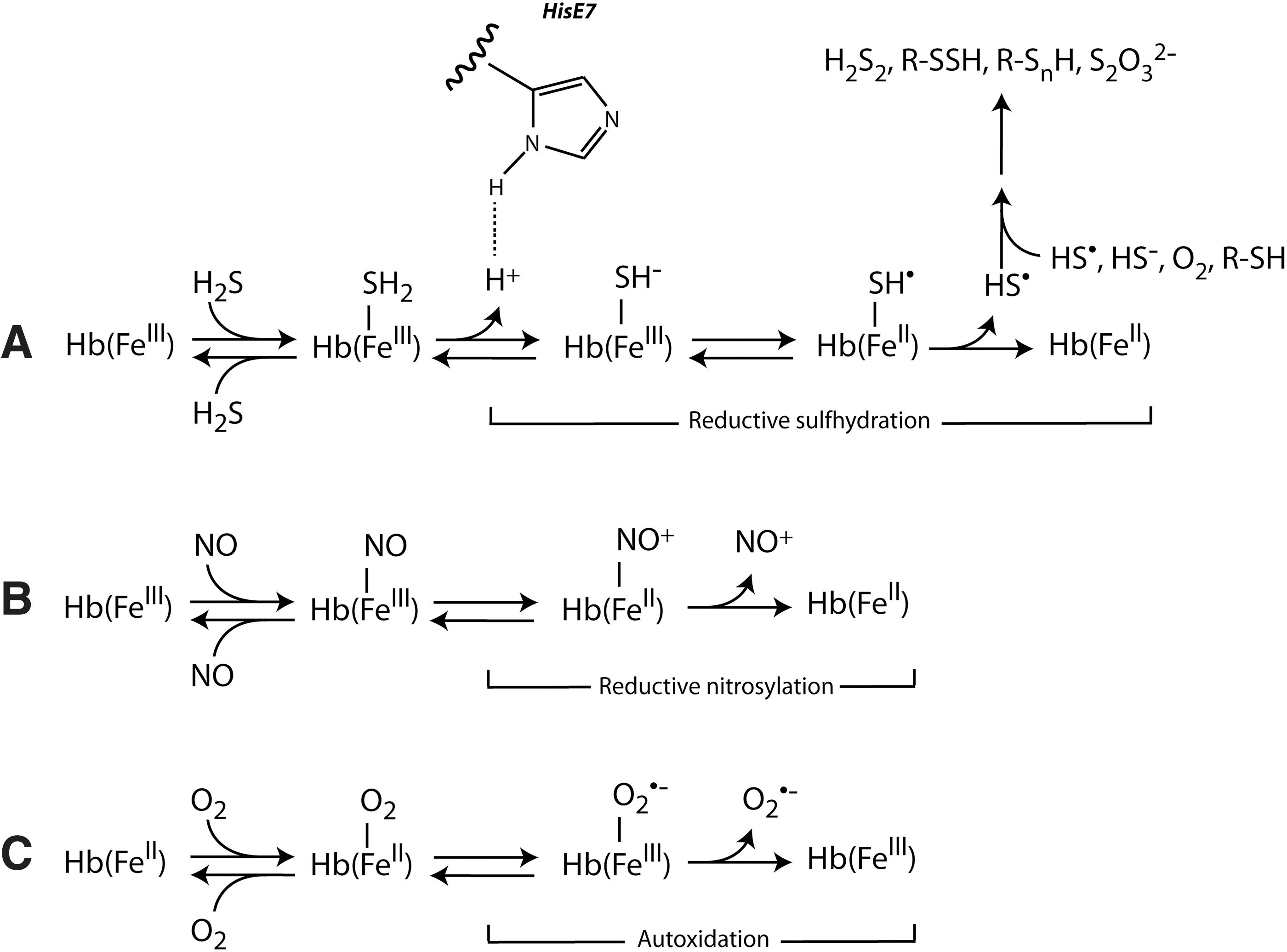
Although reversible ligand binding and release are the dominant reactions of Hb, a small part of Hb seems invariable to take part in slow redox side reactions with gaseous ligands. These occur when ferrous heme reacts with O2 (autoxidation, generating ferric heme and superoxide) and when ferric heme reacts with either NO (reductive nitrosylation) (17) or H2S (reductive sulfhydration) (2) to generate ferrous heme (Fig. 3). CO-mediated reduction of ferric Hb has also been reported (3). The interplay of these reactions, connecting O2 transport with H2S, NO, and CO transport and metabolism in the blood, and their in vivo implications remain to be understood.
Although occurring at much lower rates than the formation of the metHb–sulfide complex, the reductive sulfhydration process takes place at low (<0.5) substoichiometric ratios of H2S to ferric heme, and becomes slightly faster the lower the H2S (20), indicating that this slow redox reaction may be of increasing importance at lower in vivo H2S concentrations. In particular, the generation of ferrous heme by reductive sulfhydration mediated by H2S would contribute to the reduction of metHb within RBCs as a NADH-independent supplement to the cytb5 system. The decay of the metHb–sulfide adduct does not seem to be affected by Na2S, cysteine, or glutathione (2). At low H2S levels, heme-dissociated free HS• could then rapidly react with another HS• to form HSSH, detected spectroscopically (20).
At high H2S levels, HS• may react with other sulfide and thiol species to generate per- and R-SnHs, and thiosulfate in the presence of O2 (2, 12) (Fig. 3A), as recently shown in intact RBCs (2). These products prevail when metHb acts as a scavenger of toxic free H2S present in excess (58, 59), and due to their inherent chemistry their levels increase the higher the H2S present.
A schematic overview of the main reactions, transport mechanisms, and potential effects of H2S carried in RBCs is shown in Figure 4.
Future Challenges and Perspectives
To understand to which extent metHb may act as a physiological H2S carrier at the levels existing in RBCs, it is critical to determine metHb–sulfide concentrations in vivo. Despite recent methodological advances in obtaining reliable measurements of free H2S and related metabolites in tissues, it has proven difficult to quantify levels of metHb–sulfide complex in RBCs, probably because of perturbations of the existing equilibrium, resulting in heme dissociation of H2S.
The monobromobimane derivatization technique allows to identify dissolved free sulfide (as H2S gas) along with its bound forms (47, 48, 53). These include (i) the BSS pool (comprising R-SSHs, R-SnHs, thiosulfate, thiosulfonates, polythionates, and elementary sulfur), which liberates H2S gas when samples are incubated with reducing agents, and (ii) the acid-labile pool (comprising mainly iron–sulfur clusters), which liberates H2S gas when samples are incubated with strong acids (48). With this technique, any metHb–sulfide complex present in RBCs would be detected as free sulfide due to shift of the equilibrium shown in Figure 3A along with any H2S generated enzymatically, but it would also appear in the acid labile fraction (as low pH promotes heme dissociation), and possibly in the BSS fraction due to heme reduction.
A better technique would be the visualization of the metHb–sulfide complex by EPR, although the resolution limit may be too high (2). Furthermore, the role of the reactive Cysβ93 of human Hb as a possible site of S-sulfhydration has not been investigated yet. In analogy, Cysβ93 of human Hb can be S-nitrosated and contribute to (S)NO-dependent hypoxic vasodilation of the systemic circulation (21, 51, 52, 63), although some controversy exists (15, 18).
Another major challenge relies in the identification of biological targets of metHb–sulfide in the vasculature and tissues. If the reversible dissociation of H2S is the dominant pathway (Fig. 3A), H2S would be able to react directly with a ferric heme containing target, such as cytochrome c oxidase in the mitochondria. In such case, the possible effect of a low H2S level would be to increase or decrease respiratory rate (7 –9) depending on the relative levels of H2S to heme. In addition, H2S may initiate cardioprotective redox signaling reactions (49) via R-SSH and thiosulfate, as met myoglobin appears more prone to reductive sulfhydration than metHb (5, 20, 59).
Under conditions wherein redox interactions of H2S with metHb are predominant (Fig. 3A), these would contribute to circulating R-SSH (and thiosulfate). Both H2S and R-SSHs are able to activate protein thiols, as for KATP channels in vascular muscle cells and for endothelial NO synthase, thus acting in the modulation of vasodilation and vasoconstriction, respectively, of systemic and pulmonary circulation (50). Most likely, a balance between redox and nonredox reactions depending on H2S levels, pH, and oxygen availability would be determinant for the type of chemistry operating in vivo under varying conditions.
Finally, transport of H2S in the blood mediated by metHb would then represent an O2-independent, but pH-dependent, mechanism for blood-mediated control of local blood flow, regulated by own allosteric cooperative interactions distinct from those regulating O2 transport. In contrast, generation of NO from blood occurs during conditions of local tissue hypoxia via generation of NO from deoxy Hb and circulating nitrite (10, 28) or via liberation of NO or vasoactive S-nitrosothiol from the small amount of circulating S-nitrosated Hb (51, 52), both mechanisms requiring quaternary R to T shifts of Hb in hypoxia. In either case, blood-mediated NO generation requires retrograde action of NO from capillaries to resistance arterioles to mediate vasodilation, whereas H2S-mediated vasodilation or vasoconstriction does not require concomitant gas exchange in capillaries and would act upstream of NO in the circulation, directly on arterioles' vasculature.
In conclusion, emerging experimental evidence points toward a novel physiological role of metHb in the transport and metabolism of H2S. This potential mechanism of blood-mediated control of local blood flow should be explored in future studies to fully understand its physiological relevance.
Footnotes
Acknowledgment
We thank an anonymous reviewer for excellent feedback on the article.
Funding Information
This study was supported by the Aarhus University Research Foundation (NOVA grant AUFF-E-2016-9–37 to A.F.) and the Independent Research Fund, Denmark, Natural Sciences (grant 4181-00094 to A.F.).