
Abstract
Significance:
Sulfides are endogenous and ubiquitous signaling species that share the hemeproteins as biochemical targets with O2, nitric oxide, and carbon monoxide. The description of the binding mechanisms is mandatory to anticipate the biochemical relevance of the interaction.
Recent Advances:
The binding of sulfide to ferric hemeproteins has been described in more than 40 systems, including native proteins, mutants, and model systems. Mechanisms of sulfide binding to ferric hemeproteins have been examined by a combination of kinetic and computational experiments. The distal control of the association process, dissected into the migration of the ligand to the active site and the binding event, reveals that neutral hydrogen sulfide (H2S) reaches the active site and is the predominant binding ligand, while the HS− is excluded by the protein matrix. Experiments with model compounds, devoid of a protein scaffold, reveal that both H2S and HS− can bind the ferric heme if accessing the site. A critical role of the proximal ligand in the prevention of the metal-centered reduction has been experimentally assessed. For metmyoglobin and methemoglobin, the coordination of sulfide leads to noncanonical functions: sulfide storage and its oxidative detoxification have been evidenced under physiological and excess sulfide concentrations, respectively.
Critical Issues:
The bound species is suggested to predominate in the monoprotonated form, although spectroscopic evidence is pending.
Future Directions:
A description of the role of hemeproteins as biochemical targets for inorganic sulfide requires understanding the reactivity of bound sulfide, for example: the metal-centered reduction, the reaction with excess sulfide, oxidants, or other gasotransmitters, among other biomolecules.
Introduction
Hydrogen sulfide (H2
The physiological concentration of H2S in mammalian tissues is a subject of active debate, but is generally accepted to be in the low micromolar region (31, 60). Both the excess and the deficiency of H2S have been related to pathologic events (35, 60). Low-molecular-weight thiols, protein thiols, and metalloproteins—with the ferric center of hemeproteins as constituents of proteins, enzymes, transcription factors, membrane ion channels (55) —have been recognized as biochemical targets for inorganic sulfide (42), and their interactions should be considered in the molecular description of potential therapeutic objectives for sulfide (55). While the participation of the interaction of sulfide species and thiol compounds in biologically relevant processes has been and is still being extensively studied (persulfidation or S-sulfhydration processes) (22), the chemistry and mechanisms of sulfide binding to hemeproteins forming coordination complexes have been less appraised. The subject has actively grown in the last decade to provide tools for the inspection of the outcomes of the interaction, and eventually to focus on the use of hemeproteins as pharmacological targets for sulfide donors.
Among heme-containing proteins, the reactivity of sulfide toward their ferric forms is remarkable because it is known to rule the inhibition of the cytochrome c oxidase, by binding and reducing the ferric heme iron and coordinating with the ferrous form assisted by the copper center, thus hampering O2 respiration (16, 47). Less dramatic, but very interesting from a structural and functional standpoint, is the binding of sulfide to the ferric form of hemoglobin I from Lucina pectinata, a clam inhabiting sulfide-rich environments, with a very high-affinity constant (33, 34). This case was the first evidence of sulfide binding to a methemoglobin, and significantly triggered the interest in the coordination chemistry of sulfide to hemeproteins.
The attempts to understand the different chemical behaviors require the dissected analyses of the role of neighboring amino acids or other functional structures, defining a distal and proximal site (12), for the formation and stabilization of the coordination complex. The distal mechanisms encompass the possibilities of migration of the potential ligand from the bulk to the active site, and the final binding to the ferric heme. As the state-of-the-art covers mostly the interaction of sulfide with hemoglobins, the proximal mechanisms for the stabilization of ligands to heme compounds encompass the interactions of the hydrogen on the Nδ of the conserved histidyl residue acting as fifth ligand, with acceptor surrounding amino acids.
To describe the reaction mechanisms where sulfides participate, it is also necessary to take into consideration the intrinsic complexity of the ligand. From the acid-base perspective, the term inorganic sulfide encompasses the neutral H2S, and the two anionic conjugated bases hydrosulfide and sulfide, HS− and S2−, with pKa1 = 6.98 and 6.76, at 25°C and 37°C,22 respectively, and an accepted value of pKa2 = 19 ± 2, with reported values covering a range of eight orders of magnitude (28, 59). This acid-base behavior is a distinctive feature of H2S among gasotransmitters, as NO and CO lack this chemical reactivity. This anticipates lipid or water solubility favoring the neutral H2S or the anionic HS− species, respectively. Significantly, the solubility of H2S in water, explained in terms of a weak hydrophobic solvation, promoted studies of the partition coefficient of H2S in water and liposome membranes, to address the potential biological distribution of sulfide species (19, 56).
The reactive sulfide species scenario is still more complicated, as it has been recently reported that the 3-MST can also form hydropolysulfides, H2Sn (n > 1), and that some of the roles attributed to H2S are a consequence of the action of H2Sn and not of H2S (31). The reaction between H2S and NO is also capable of forming H2Sn, along with nitroxyl (HNO/NO−) and nitroso persulfide (SSNO−) (5, 23, 24). These cross talk derived species are also part of the complex network capable of mediating roles for H2S (and NO) from hemeprotein platforms (17, 18). Regarding the redox potential, as sulfide species are in the lowest oxidation state that sulfur can achieve, they are reducing agents, and the reduction potential for the two-electron process at pH 7 is E°′ (S°/HS−) = −0.27 V, comparable with the formation of the biologically relevant glutathione disulfide and cystine [E°′ (GSSG/GSH) = −0.24 V at 40°C, and E°′ (CysSSCys/CysSH) = −0.34 V] (37). At neutral pH, the one-electron reduction potential is E°′ (S•−, H+/HS−) = 0.91 V, and as the first pKa of H2S is approximately (ca.) 7, it can be derived that the E°′ (S•−, 2H+/H2S) has a similar value (32). Taking the highest occupied molecular orbital (HOMO) energy as an indicator of nucleophilicity for H2S and HS−, the corresponding negative values of −9.4 and −7.1 eV determined at the density functional theory level indicate that both species will be prone to react with positively charged metal centers as target electrophiles, but favor the anionic form as a reactive species as the calculations retrieve a higher HOMO energy (8, 13).
Up to date, particularly, the sulfide-ferric heme complexes have been ascribed to transport and storage (33), or to activation and detoxification of sulfide (6, 61). Sulfide-mediated potentially functional processes in mammals have been recognized and described in myeloperoxidase (51), methemoglobin (29, 61), metmyoglobin (6, 29), neuroglobin (57). The present Forum article is focused on the coordination chemistry of inorganic sulfides to iron heme platforms. Herein, both the oxidized and reduced forms of heme-containing proteins, protein mutants, and model compounds, both in organic solvents or under biorelevant conditions that have been explored as targets for sulfide species, are presented to provide a comprehensive description of the outcomes of the interaction of heme iron and sulfide.
The Cases of Binding of Inorganic Sulfide to Ferrous Hemeproteins Are Scarce
The interaction of the HS− anion with ferrous porphyrinates (FeII(Por)) in organic media allowed the preparation and characterization of sulfide-bound penta and hexacoordinated, high-spin ferrous compounds with general structures [FeII(Por)(SH)]− and [FeII(Por)(SH)2]2−, respectively (52). As nonpolar solvents were used in the experiments, the crown Kryptofix-222 aided the dissolution of the HS− anion from anhydrous NaHS as source, and is part of the crystal structures. Crystals of the (HS−) derivatives of FeII-octaethylporphyrinate (OEP), FeII(T-p-methoxyphenyl)porphyrinate), and FeII(T-p-mesityl)porphyrinate) were thus obtained and characterized. Alternatively, N(Bu)4SH was used as a source of sulfide in organic solvents, and the reaction with a survey of FeII(5,10,15,20-tetraphenyl porphyrinate [TPP])s yielded the corresponding monosulfide complexes (40). Interestingly, the use of the nonbulky sulfide source allowed the detection of the intermediacy of a bimetallic μ-S complex, for the case of the ferrous octa-fluor-tetraphenylporphyrinate, [Fe2(μ-SH)(F8TPP)2]−. Isolation of a μ-S complex was only attained so far for the case of RuII(OEP), yielding Ru2(μ-S)(OEP)2 (14), and was elusive for the GaIII(TPP) case (41), where metal-centered reduction is inaccessible, suggesting that a reduced form is mandatory for the formation of μ-S-type complexes.
Picket-fence ferrous porphyrinates, sterically impeding the formation of μ-S bridges, were selected to explore the discriminated behavior of the neutral H2S and the anion HS−, hampering the acid-base equilibria with the use of organic solvents of different polarities (26). Gaseous H2S was inert toward ferrous (and also toward the ferric) picket-fence porphyrinates, while the HS− from N(Bu)4SH yielded the coordinated FeII(SH−) form, regardless of the presence of N-methylimidazole as axial ligand. As this speciation behavior was also observed for the FeII(TPP), the results suggest an intrinsic inertness of H2S toward ferrous hemes in organic solvents.
The above results indicate that FeII(SH−) porphyrinates can be prepared and isolated in organic media using HS− but not H2S, starting from the ferrous systems. As described in the following section, FeII(SH−) porphyrinates can also be obtained from the corresponding ferric porphyrinates, after a sulfide-mediated reduction step.
Evidence for the formation of mononuclear ferrous hemeprotein complexes has been reported in the case of myeloperoxidase, where the formation of FeII(SH2) in buffered aqueous solutions was proposed from the reaction of sulfide with the ferric form (in the presence of excess sulfide), from the ferrous form, or from the corresponding compounds I and II (25, 51). A rate constant, rate constant for ligand binding (kon) = 2 × 104 M −1s−1, has been reported for the binding of H2S to the ferrous myeloperoxidase (51).
The binding of sulfide to ferrous myeloperoxidase seems strongly dependent on the nature of the distal amino acids. Based on our observation of the crystal structures of myeloperoxidase, horseradish peroxidase, and lactoperoxidase, the presence of a common distal arginine, absent in hemoglobins, may provide strong stabilization to H2O molecules in the distal cavity, hampering the access of sulfide ligands. Eventually if sulfide ligands access the distal site, the distal arginine may provide stabilization mechanisms.
Microperoxidase 11 (39), which is a minimalistic heme model compound, derived from the proteolysis of cytochrome c that retains the two thioether linkages of the heme to Cys14 and Cys17, the His18 as fifth ligand, and a water molecule as sixth ligand, represents an opposite example. This heme compound is devoid of distal mechanisms for ligand stabilization (4), and the ferrous form is inert toward sulfide species in the ferrous form.
These dispersed data concerning the interaction of sulfide and ferrous heme systems suggest that the sulfide species are not competing ligands toward O2, NO, or CO in hemoglobins, while are likely to inhibit the canonical function of myeloperoxidase (51).
The Binding of Inorganic Sulfide to Ferric Heme Compounds Is Observed Mainly in Aqueous Solutions
Compared with the binding of sulfides to ferrous hemes, there is a list of diverse examples of sulfides bound to ferric heme systems, which keeps growing since the pioneer reports on the sulfide-bound ferric hemoglobins from L. pectinata (33, 34).
Indeed, up to date, there have been reported more than 40 examples, including hemeproteins, mutants, and heme model compounds, where the binding of inorganic sulfide has been detected and kinetically characterized in aqueous buffered solutions (8). A selection of the collected data is presented in Table 1. In organic solvents, conversely, it has only been reported the formation of a low-spin pentacoordinated ferric heme complex in 1,1,2-trichloroethane (21), later disputed (3). More recently, the intermediacy of an FeIII(SH−) low-spin form has been detected after the addition of HS− to two ferric picket-fence model systems only at −80°C in methanol; at room temperature, the fast formation of the metal-centered reduction product was observed (43).
Literature Data on Kinetic Constants for Sulfide Binding to Ferric Hemeproteins and Heme Model Compounds
Data selected around neutral pH, or closest values reported.
Calculated.
pH 7.5.
Maximum value.
Thermodynamic affinity constant.
In most of the examples reported in Table 1, the FeIII-sulfide bond formation has been assessed by resonance Raman (4, 48) and/or EPR techniques (6, 51, 61, 65). The complexes are described as hexacoordinated, low-spin ferric structures (46). It can be observed that while the values of kon are all ca. 104 M −1s−1, the values of rate constant for ligand release (koff) are more dispersed. As a consequence, the derived affinity constant (Kaff) values (Kaff = kon/koff) range from 103 to 109 M −1.
The fact that, analytically, the ferric bound sulfides are found as part of the pool of acid-labile sulfur is in line with the higher lability of the neutral form (55). The protonation state of the preferred bound sulfide species has not been addressed spectroscopically under biorelevant conditions so far.
Either H2S or HS− Can Bind to Ferric Heme Compounds in Aqueous Solutions if Accessing the Binding Site
In different hemeproteins, the participation of either H2S or HS− has been tentatively assumed as reactive (and bound) species, participating in proposed reaction mechanisms. In an attempt to clarify the issue, the rate constant for the binding process has been estimated for different hemeproteins as a function of pH, to envisage the discriminated participation of H2S or HS−. Studies of the kinetic behavior of the interaction between ferric hemeproteins and sulfide as a function of pH have been reported for hemoglobins I, II, and III of L. pectinata (33), human and porcine hemoglobins (29, 44, 61), myoglobin (6, 8, 29, 44), Vitreoscilla hemoglobin (62), and neuroglobin and H64A neuroglobin mutant (57). We included the data in Figure 1, depicting the values of kon of sulfide as a function of pH for the ferric forms of the L. pectinata and Vitreoscilla hemoglobins, human and porcine hemoglobins, and myoglobin.
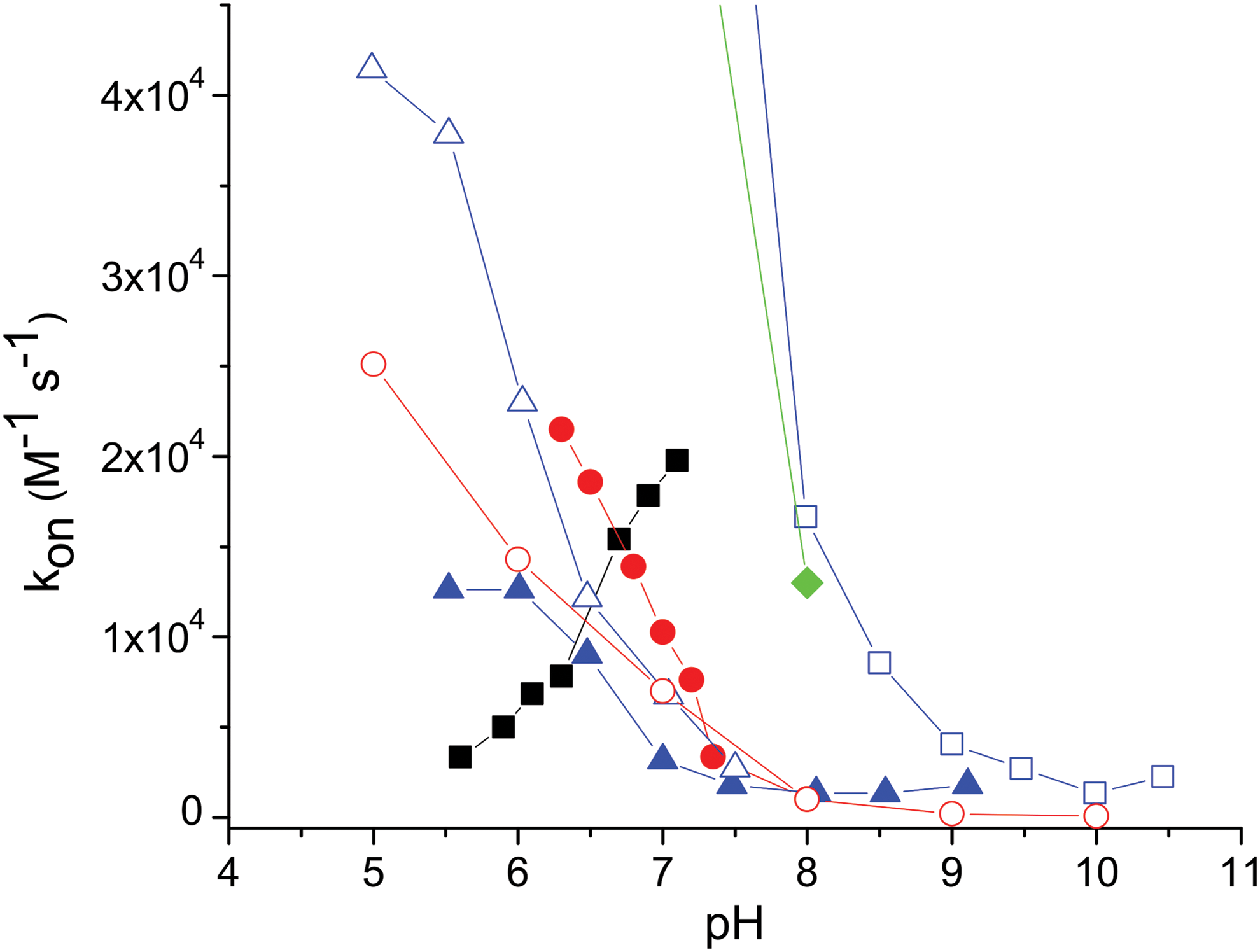
As can be observed in Figure 1, the pH intervals ranged from 5.5–6 to 8–9, and the results are coincident, in that the neutral H2S appears as the fastest reactive species. Furthermore, in the alkaline region, where HS− is the most abundant species, the reaction rate seems negligible for all the cases reported. This has been assessed for the case of metmyoglobin, where the intrinsic rate constants (kint) for H2S and HS− have been estimated retrieving a seemingly null value for the kint (HS−) (8, 29). It is relevant to address that the pH intervals explored are compromised with the exclusion of other pH-dependent events, affecting the binding within the protein matrices.
According to the above results, it might be tempting to assume that only the neutral H2S is active for the binding interaction, while the anion HS− appears as almost inert. To address the issue, a similar experiment using the heme model microperoxidase 11, where the active site is solvent exposed, reveals instead that the anion HS− is the fastest to bind the ferric heme (Fig. 1), and that the binding of H2S is also significant, although with a lower intrinsic binding constant. An interpretation at the molecular level of the events describing these results has been attained computationally and is included in the next section (7, 8).
The results suggest that the discriminated behavior of H2S and HS− toward ferric heme iron in aqueous solutions cannot be extrapolated either from the observations in organic media or from the analysis of kon versus pH for hemeproteins. In contrast with the case of HCN and CN−, where only the anion is active as binding species regardless of the structural features of the active site and the reaction media (2, 20), for the sulfide species it is mandatory to consider a detailed analysis of the binding mechanism for each species.
Distal Effects Control the Association and Dissociation of Inorganic Sulfide to Ferric Heme Compounds
The association process can be divided into two elementary steps, namely, the ligand migration from the solvent to the distal site, and the formation of the FeIII-sulfide bond. The measurement of the rate-binding constant, kon, encompasses both processes. An observation of the reported values of kon for inorganic sulfide binding (Table 1) suggests a similar binding mechanism operative for the heme compounds listed, as most measurements retrieve a value ca. 104 M −1s−1.
Using classical molecular dynamics (MD) simulations, it has been suggested how the distal effect interactions modulate the association rate of sulfide species to hemeproteins. Specifically, steered MD was used to evaluate the ligand migration free energy profile for H2S and HS− for the cases of met-hemoglobin (Hb)I from L. pectinata (7), and horse heart metmyoglobin(8) (Fig. 2). Altogether, these results were interpreted in terms of the high desolvation requirement of the charged HS− during the migration process from the bulk to the distal cavity, responsible for the specific selectivity of these hemeproteins for the neutral H2S. The computational evaluation for the migration step of the binding process is in agreement with the tendency of kon as a function of pH reported for both cases (6, 8, 29, 33). Thus, MD assists the interpretation of the role of pH in the binding of sulfide species to hemeproteins, suggesting that the binding is seemingly null for the anionic form only due to its impossibility to access the binding site, and not as a consequence of a weaker reactivity. Significantly, for the case of microperoxidase 11, the absence of a protein matrix accounts for the significant reactivity of HS− and H2S, due to lack of restrictions for access to the binding site (8). The binding mechanism of the anion HS− to the microperoxidase model compound, intuitively favored by its higher nucleophilicity (8), deserves further research for a proper description of the reaction mechanism.
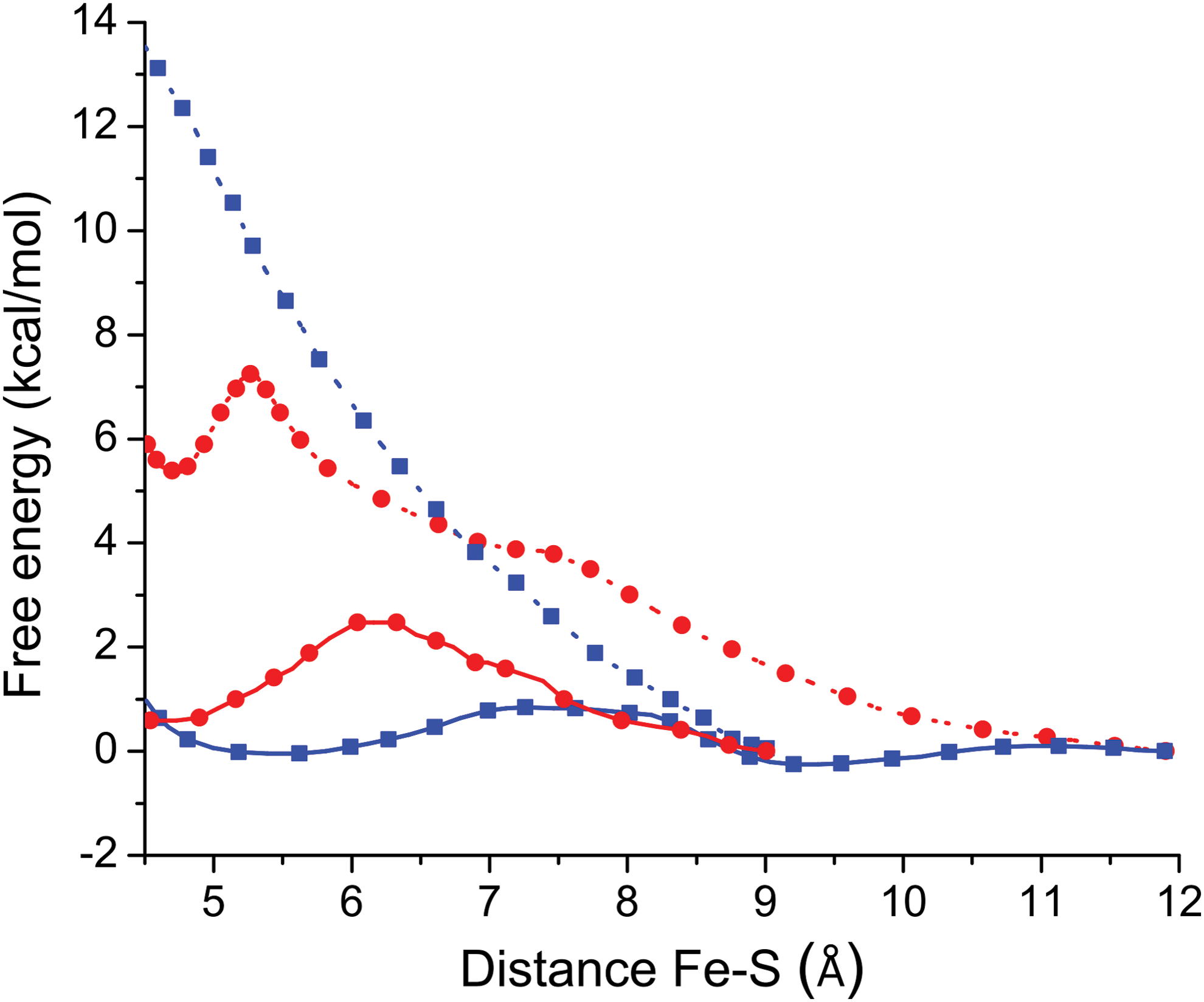
The binding of small ligands to ferric hemeproteins is hampered by a coordinated water molecule. The value of kon ca. 104 M −1s−1 (Table 1) systematically observed for ferric hemeproteins reports the sole binding of H2S, and can be tentatively explained in terms of a dissociative mechanism involving the water molecule coordinated to the distal position. In fact, a simple Eyring analysis shows that the release of the coordinated water molecule (koff(H2O) ≈104 s−1) retrieves a value of ΔG≠ of ca. 12 kcal/mol, which is consistent with the interaction free energy of a water molecule coordinating with a ferric porphyrin system (11, 50).
A more detailed analysis should take into consideration the interactions of the coordinated water molecule with the surrounding amino acids, expected to affect the value of kon. Indeed, those interactions probably explain the special cases among the reported data (Table 1), where specific mutations on the distal side affect the value of kon within one order of magnitude, at the most. For example, the GlnE7Val and PheB10Tyr mutants of met-HbI of L. pectinata show an increase and a decrease of one order of magnitude of kon, respectively. With the E7 mutation, the water molecule is destabilized, but the inclusion of a TyrB10 as a distal amino acid may provide further stabilization to the coordinated water molecule (53). Other E7 mutations that still provide stabilization through hydrogen bonding to the coordinated water molecule (GlnE7Asn or GlnE7His) do not modify the kon value significantly, when compared with the wild type. The kon results for these mutations suggest that the E7 position might not be acting as a gate, and highlight the role of stabilization of the coordinated water molecule in ferric hemeproteins, contrasting with the analysis of ferrous systems.
Additional evidence of the variation of kon as a function of distal mechanisms stabilizing the coordinated water molecule has been provided by mutations on the truncated hemoglobin O of Thermobifida fusca (trHbO). Similar to the case of HbI mutants, the mutations of H bond donor residues by phenylalanine residues reflect an increased value of kon from one to three orders of magnitude of kon, depending on the number of mutated residues (Table 1) (48, 49).
In this context, it can be speculated that the anomalous case of ferric horseradish peroxidase, which is inert toward sulfide species at neutral pH, can be explained in terms of a strong stabilization of the coordinated water molecule by a distal positively charged arginine residue.
As a general conclusion, it can be derived that the dispersed kon values for the binding of O2, NO, or CO to ferrous hemeproteins, devoid of a sixth ligand, are a consequence of a combination between the distinct interactions of the ligands while migrating from the bulk to the distal site, and eventually the requirement of the displacement of water molecules stabilized by the distal amino acids near the metal center (10). Conversely, for the case of the ferric hemeprotein, the rate-limiting step is the release of a coordinated water molecule, rendering a low fluctuation in the kon values for the binding of sulfide species.
The stabilization of bound sulfide species was first addressed in the paradigmatic HbI of L. pectinata, where the elevated content of aromatic amino acids in the distal site (3 × phenylalanine) was presumed to impose a hydrophobic environment, favoring the stabilization of neutral H2S as a coordinated ligand (53).
The pKa shift between free and coordinated H2S can be roughly compared with the pKa shifts observed for H2O in ferric hemeproteins. Taking into consideration that the pKa of the bulk water is about 14, experimental and computational results show that the pKa shift between the bulk H2O and the coordinated H2O is ≈4 pKa units (9, 33, 38, 58). Using the same assumptions, and considering that the pKa of H2S in aqueous solutions is ≈7, this analysis suggests that the coordinated H2S is deprotonated at physiological pH. This is in agreement with the reported quantum mechanics/molecular mechanics (QM/MM) energy profile evaluating the energy cost for the deprotonation step, suggesting that the coordinated H2S spontaneously deprotonates to give HS−, with a ΔE value of ca. −12 kcal/mol for HbI of L. pectinata, assisted either by GlnE7 or by the entrance of water molecules to the active site (Fig. 3) (7).

As detailed for the stabilization of the coordinated water molecule, the stabilization of the HS− ligand can also be explained in terms of distal hydrogen bonding interactions, and is reported by the values of koff (Table 1). Evidence of this interaction can be also obtained from MD simulations. Figure 4 shows the snapshots taken from the reported MD simulations for the HS− complexes of HbI of L. pectinata (left panel) (7), and the truncated hemoglobin from trHbO of T. fusca (right panel) (48).

Following a similar analysis for the case of neuroglobin, the 700-fold higher koff value observed for sulfide for the His64Ala mutant compared with wild-type neuroglobin is consistent with the role of distal histidine residue in stabilizing sulfide (57). In addition, single mutations on trHbO of T. fusca dramatically affect the stabilization of the coordinated HS− ligand, with an ∼300-fold increase in the koff for the TrpG8Phe mutant (48). The value of koff for microperoxidase 11 (Table 1), the highest reported so far, is explained in this context by the absence of distal effects.
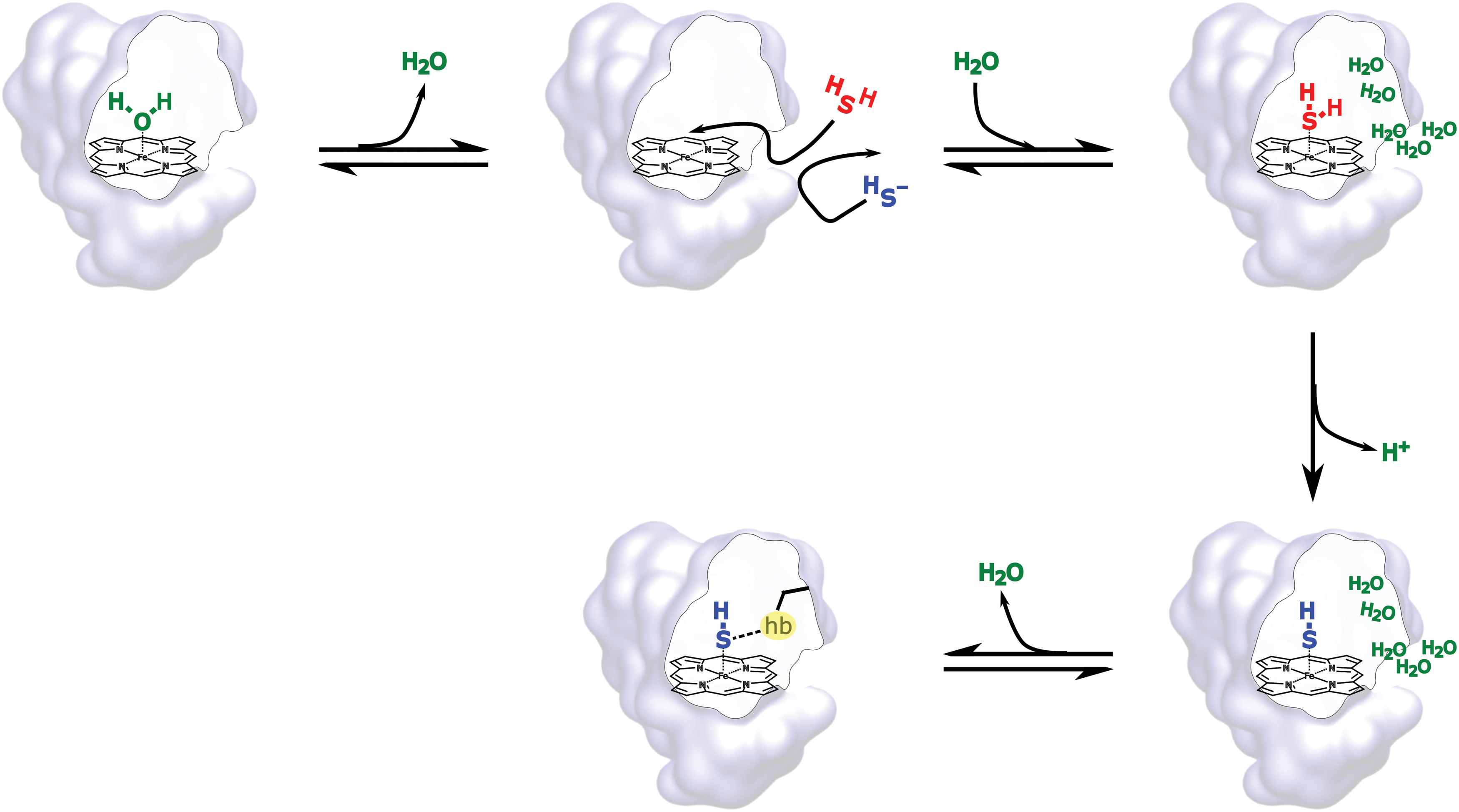
A global reaction mechanism derived from the experimental and theoretical analysis is presented in Figure 5. The first step encompasses the release of the coordinated water molecule, interpreted as a rate-limiting step after the analysis of the kon values included in Table 1. As HS− is excluded by the protein matrix as a consequence of its high desolvation cost, only the neutral H2S is available for the binding step. After coordination, a barrierless deprotonation step, and surrounding water molecules or distal amino acids act as proton acceptors. The HS− bound ligand is finally stabilized by hydrogen bonding interactions with distal amino acids.
The Role of the Proximal Ligand in the Stabilization of Coordinated Sulfide to Ferric Hemeproteins is Evidenced in Models Devoid of Distal Interactions
The absence of a nitrogen containing ligand in the fifth coordination position of ferric porphyrinates drives the addition of sulfide to the fast formation of the metal-centered reduction product (43). Conversely, the presence of nitrogen containing bases as proximal ligands reveals that complexes with varied stability can be prepaired. The release constant, koff, has been determined in many cases, and values of koff spanned within six orders of magnitude (from 10−5 to 10 s−1, Table 1), with an impact in the kinetically derived Kaff = kon/koff.
To discriminate the proximal contribution to sulfide affinity of ferric heme compounds, the binding has been reported, at least, in three diverse model compounds sharing as common features a nitrogen containing fifth ligand, and structurally hampered, or even unavailable, distal mechanisms for ligand stabilization.
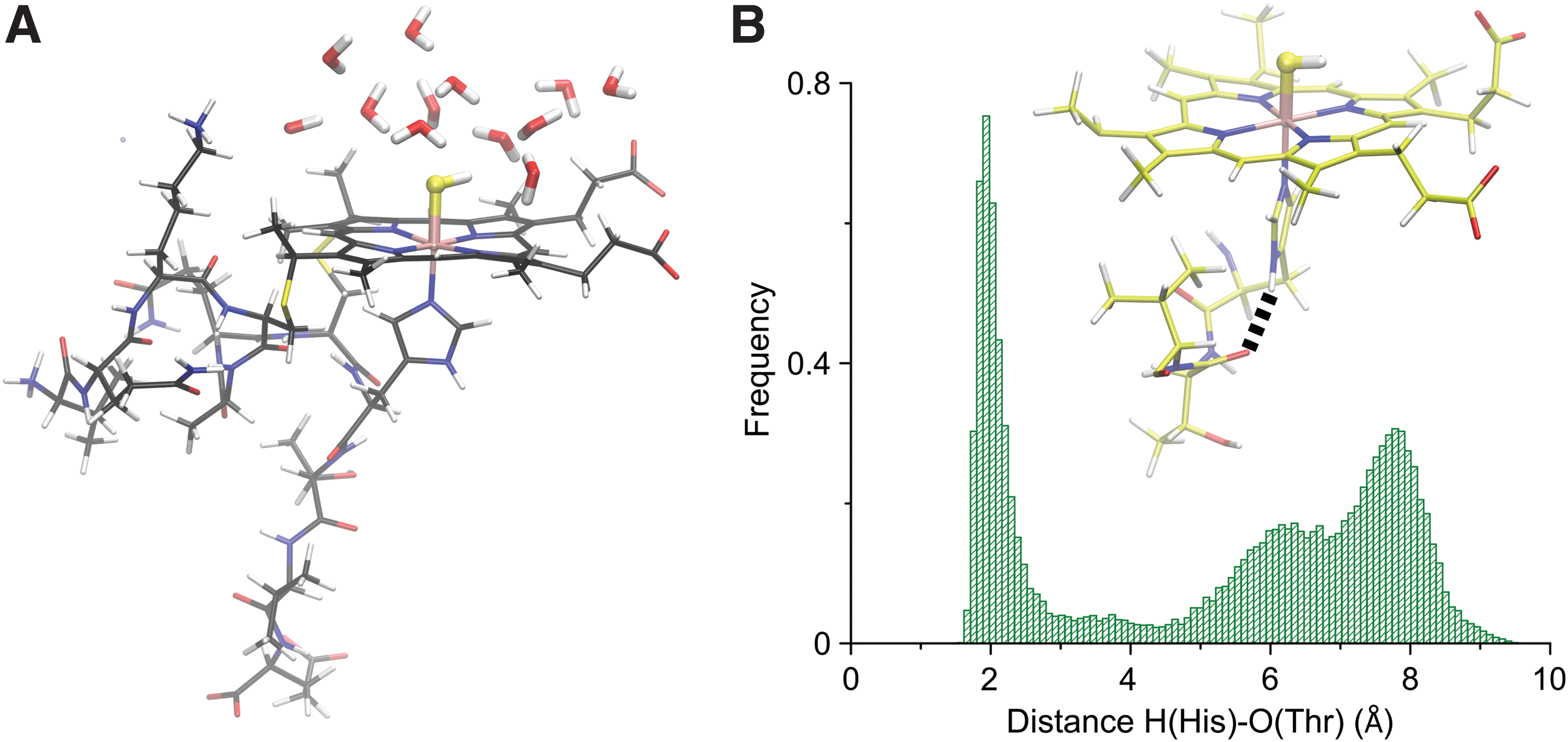
Using microperoxidase 11, the moderately stable binding of inorganic sulfide was observed, and the complex was characterized by means of resonance Raman and UV-Vis spectroscopies (4). To assess the opportunities of the amino acidic residues for the stabilization of sulfide species as ligands, the system was studied by means of MD simulations, which suggested that none of the amino acidic residues interacted with coordinated HS−, and thus, the FeIII(SH−) complex is solvent exposed (Fig. 6A). Furthermore, the results suggested that a conformation involving the interaction of the H atom of the Nδ of the proximal histidine with the carbonyl group of the Thr19 was significantly populated (Fig. 6B), with the consequent change in charge distribution around the heme center (4). The maximum population is centered around 2 Å, suggesting a H bonded interaction. The kinetic study of the binding of inorganic sulfide at neutral pH revealed that the kon value was similar to that of most of the native hemeproteins studied (Table 1), while the koff was increased, thus retrieving a value of Kaff in the lower limit of those reported so far (8). Significantly, the case highlighted the isolated role of the proximal amino acid in the stabilization of the FeIII-sulfide complex.
This computational description of the proximal side has been experimentally verified in a series of heme b type model compounds, namely, a series of deuterohemin-His-peptides, where the binding of sulfide was evaluated in the presence of an increasing number of proximal interactions with the Nδ of the coordinated histidine (66). The kon value showed an increase of half an order of magnitude when allowing one H bonding interaction with the peptide backbone (Nδ-Thr), and an additional increase in one order of magnitude when including a second interaction (Nδ-Thr and Nδ-Glu). In parallel, the koff values for the binding of sulfide to these deuterohemin derivatives were unchanged after the inclusion of the Nδ-Thr interaction, but decreased in the addition of the Nδ-Glu interaction. As a consequence, the kinetically derived Kaff showed an increase only as a result of the participation of one or two hydrogen bonding interactions of the amino acid backbone with the Nδ of the coordinated histidine to the FeIII, in the absence of distal stabilization mechanisms.
Additional evidence for the discriminated role of the proximal ligand was provided by the use of a 1:1 inclusion complex of a water soluble iron porphyrinate, namely FeIII(5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinate), in a methylated β-cyclodextrin (CD) dimer, linked via a pyridine ligand (65). These systems, called methemo CDs or met-hemoCDs, have been characterized and used to mimic hemeproteins in aqueous solutions, and provide a hydrophobic environment in the surroundings of the binding site. The thermodynamic Kaff was reported and is slightly favored compared with the water-exposed active sites of the two models presented above.
Interestingly, the reactivity of the ferric sulfide complex of met-hemoCD was evaluated toward CO and O2, and showed the formation of the corresponding ferrous hexacoordinated complexes, with the release of thiyl radicals, suggesting that the FeII(SH•) resonant structure is crucial to interpret the reactivity of the complex (65). Particularly, it is interesting to address that the interaction with O2 mimics the initial step of the catalytic oxidative decomposition of sulfides, described for methemoglobin and metmyoglobin (6, 29, 61).
Sulfide-Bound Species Initiate the Metal-Centered Reduction of Hemeproteins
The experiments of binding of inorganic sulfide to ferric hemeproteins under anaerobic conditions reveal the final formation of the ferrous form and sulfur; the kinetics of the process varies significantly in the different systems (42, 51, 53, 61).
Using methanol as solvent of hemeprotein models devoid of a proximal ligand, a fast reduction has been reported, and by lowering the temperature at −80°C the intermediacy of coordinated sulfide was detected. The reduction product is formed after the cleavage of an FeIII(SH−) form forming thiyl radical (HS•), as demonstrated by trapping experiments using a radical trap as 5,5-dimethyl-1-pyrroline N-oxide (43). The experimental result reveals that the resonance form FeII(SH•) represents a significant contribution to the reactant moiety. Experimental evidence of the relevance of the resonance structure FeII(SH•) was also provided by experiments on the met-hemoCD complex with CO and O2 (65).
Under biorelevant conditions, up to date, the reduction mechanisms on hemeproteins have only been hypothesized. A comparative reduction tendency for a series of mutants of the HbI of L. pectinata was suggested to be governed by a strong dependence on the proton acceptor amino acids in the distal side (53). This analysis considers that hemeproteins with low-polarity distal amino acids form stable ferric-SH2 heme species, and sulfide release is dictated by low H2S dissociation without inducing a significant metal-centered reduction. The presence of strong proton acceptor groups, in contrast, was suggested to explain the metal-centered reduction. Mechanistically, the FeIII/FeII reduction was suggested to be favored after the rapid deprotonation of the bound H2S species, forming the reactive FeII-SH• species, in turn reacting with a second H2S molecule (53, 54).
However, if this were the case, the ferric heme model compounds devoid of a distal cavity that carry a solvent-exposed ligated sulfide species would be prone to fast reduction (4, 65, 66). Indeed, the complexes have a lifetime of hours, regardless of the potential role of surrounding water molecules acting as proton acceptors. In addition, our computational results on the wild-type HbI of L. pectinata suggest that the first deprotonation of the preferred bound species is quickly assisted by water molecules or distal amino acid rearrangements induced by the concomitant formation of FeIII(SH2). Further QM/MM calculations showed that the deprotonation step is barrierless, yielding FeIII(SH−) (7). Thus, the deprotonation of FeIII(SH2) does not seem to participate as a rate-limiting step; the analysis for the formation of the metal-centered reduction product should be commenced after the formation of the bound species HS−. Rationalized in terms of the resonance forms FeIII(SH−)/FeII(SH•). In accordance with this analysis, a mechanistic proposal for the metal-centered reduction mediated by sulfide has been presented for the case of myeloperoxidase, and suggests the release of the thiyl radical (51). The Fe-S bond cleavage appears as a plausible rate-limiting step for the process in agreement with the timescales experimentally observed. Interestingly, Jensen and Fago recently reported on the role of stoichiometry on the timescale of the reduction process for metmyoglobin and methemoglobin (29). This contribution sums up to the above-reported analysis for the description of the reduction mechanism of hemeproteins mediated by sulfide. Still, the molecular determinants of the metal-centered reduction of hemeproteins mediated by inorganic sulfide are not completely elucidated and deserve further inspection.
Unresolved Problems
Recent research elucidating the biochemical relevance of the interaction of sulfide and hemeproteins highlights the role of this chemical process. Intrinsic features of the H2S molecule—acid-base equilibria, redox equilibria, and formation of polysulfides—disclose different levels of complexity when attempting to describe the molecular determinants of the interaction. The analysis of the protonation state of the sulfide species bound to ferric hemes has not been assessed experimentally so far, probably limited by both difficulties in sulfur atom enrichment and by spectroscopic constraints of the Fe-S bond and sulfide-bound exchangeable protons.
Most of the cases in the literature describe the interaction of sulfide with ferric hemoglobins, with a deep insight on the hemoglobins of the clam L. pectinata, different metmyoglobins and methemoglobins, and the description of the interaction has been enriched by a combination of spectroscopic methods, experiments with mutants or model compounds, and computational experiments. The interaction of sulfide with peroxidases has been reported for the mammalian peroxidases, especially for the myeloperoxidase, but the molecular details of the interaction remain to be elucidated.
Concluding Remarks
The coordination chemistry of sulfide and hemeproteins, and the activation of sulfide by hemeproteins, is expected to be involved in biorelevant processes as the reaction with O2 forming sulfur oxygen reactive species, formation of sulfheme compounds, the cross talk with NO or CO, and formation of disulfide or polysulfides. Herein, we attempted to present the state of the art in the interaction of sulfide species with hemeproteins in the ferrous and ferric oxidation states, providing a picture of the occurrence and an analysis of the kinetic descriptors of the binding. The interaction of sulfides and the heme iron should be taken into consideration when interpreting sulfide biochemistry, signaling, and pharmacology, along with the reactions with proteins or low-molecular-weight thiols.
Footnotes
Funding Information
This work was supported by grants from Universidad de Buenos Aires (UBACYT 20020170100043BA), Agencia Nacional de Promoción Científica y Tecnológica (PICT 2014-1022, PICT 2015-2761), and Consejo Nacional de Promoción Científica y Técnica (11220150100303CO, 11220150100394CO).