
Abstract
Significance:
Electrophile signaling is coming into focus as a bona fide cell signaling mechanism. The electrophilic regulation occurs typically through a sensing event (i.e., labeling of a protein) and a signaling event (the labeling event having an effect of the proteins activity, association, etc.).
Recent Advances:
Herein, we focus on the first step of this process, electrophile sensing. Electrophile sensing is typically a deceptively simple reaction between the thiol of a protein cysteine, of which there are around 200,000 in the human proteome, and a Michael acceptor, of which there are numerous flavors, including enals and enones. Recent data overall paint a picture that despite being a simple chemical reaction, electrophile sensing is a discerning process, showing labeling preferences that are often not in line with reactivity of the electrophile.
Critical Issues:
With a view to trying to decide what brings about highly electrophile-reactive protein cysteines, and how reactive these sensors may be, we discuss aspects of the thermodynamics and kinetics of covalent/noncovalent binding. Data made available by several laboratories indicate that it is likely that specific proteins exhibit highly stereo- and chemoselective electrophile sensing, which we take as good evidence for recognition between the electrophile and the protein before forming a covalent bond.
Future Directions:
We propose experiments that could help us gain a better and more quantitative understanding of the mechanisms through which sensing comes about. We further extoll the importance of performing more detailed experiments on labeling and trying to standardize the way we assess protein-specific electrophile sensing.
Introduction
Human's appreciation for molecular shape, configuration, and topology in biology has had a checkered history. These vacillations are perhaps best exemplified by our understanding of enantiomers, although similar arguments hold for other minor structural changes. By the 1800s, we had started to understand that many naturally occurring compounds had the ability to change the orientation of plane-polarized light. The examples included several naturally derived inorganic substances, such as quartz crystals (16), as well as bioactive natural products, such as tartrate (15). In the 1840s, Louis Pasteur performed his famous experiment separating racemic tartaric acid into different crystalline forms, which transpired to contain the resolved enantiomers of the compound (15).
Later, it was noted that the enantiomers of borneol had distinctly different odors, hinting that molecular recognition was linked to smell, and that our bodies can distinguish effectively between enantiomers (6). By the 1930s, the concept of enantiomorphism in biology had permeated the general conscience. A contemporary detective series writer, Dorothy L. Sayers, even used the concept that naturally derived poisons are homochiral and synthetic versions (like would be found in a synthetic poison) are racemic as the denouement to her novel “The Documents in the Case” (48). In the book, the “poison” was muscarine, a sugar analog that is an agonist of the muscarinic acetylcholine receptor (23).
Yet this appreciation for the differential biological properties of enantiomers did little to prepare scientists for the thalidomide crisis in the 1960s, which has left medicinal chemists with the critical knowledge that enantiomers can be divergently bioactive. Ironically, this debacle occurred some 10 years after the observations on the potency of different enantiomers by Pfeiffer (5).
A silent assumption in the examples of molecules interacting with biological receptors/enzymes/proteins just cited is that noncovalent binding is the key driving force of the interaction. Indeed, it is the concept of noncovalent binding and the required spatial recognition between enzyme and substrate (or protein and ligand) that has preoccupied biochemists and biologists arguably since the work of Leonor Michaelis and Maud Menten in 1913, and the development of their eponymous equation (or its precursor, the Henri equation) (11).
With our current understanding that ligand recognition is a key determinant of biological processes, it is clear why the different enantiomers of different molecules can exhibit divergent biological properties, such as having different odors. Because high affinity translates to large binding energy, the stabilization of the system incurred on ligand binding can also be leveraged to induce changes in stabilization, conformation, association, etc., that can be transmuted into meaningful changes in cellular decision making. The binding requirements can be so stringent, that even very similar molecules that are present together in the cellular milieu can be suitably differentiated by enzyme active sites, and hence this “chemical noise” is efficiently filtered out.
The Michaelis–Menten equation also extends to situations where the enzyme forms a covalent bond to the substrate. The covalent intermediate can be harnessed for numerous possible ends, such as: to promote fission, for example, in serine proteases; or to promote reaction with an electrophile, for example, in thymidylate synthetase. Nevertheless, it remains generally true that it is the ability of the enzyme to accommodate the substrate(s) in the active site, and to orientate reactive positions to promote reactivity that ultimately synergize to allow an overall selective process to occur. For instance, deubiquitinating enzymes mostly hydrolyze essentially “the same” substrate, that is, an isopeptide bond formed between ubiquitin's C-terminus and a protein-lysine. Selectivity is engendered by a combination of recognition of ubiquitin chain linkage, and the protein to which the ubiquitin(s) is(are) bound.
But we now understand that there are numerous very simple biological signaling molecules that are proposed to regulate numerous important pathways. These molecules, however, appear to be assumed to have little ability to interact noncovalently with their targets. So how can, and indeed, do these molecules interact selectively? In this opinion piece, we compare and contrast the thermodynamics and kinetics of covalent and noncovalent binding, to try to better understand how reactive electrophiles work, and to try to derive some insight into how selectivity and concepts of biochemistry can be used to begin to interrogate electrophile signaling in more detail.
Our ultimate hope is that this work will serve as a reminder of the importance of noncovalent interactions in marshaling even overwhelmingly strong covalent bond-forming events, and further to underline the insight that can be brought to the table by examining stereo- and chemoselectivity in biological systems.
Thermodynamics of Covalent Binding Compared with Noncovalent Bonding
Covalent bond formation and noncovalent bonding are critically different. The bond energy of an sp3-hybridized C–H bond is around 420 kJ mol−1. Even a relatively weak sp3-hybridized bond, where orbital overlap is considered poor, such as C–S, has a bond energy of around 290 kJ mol−1. Alternatively, the π-bond in C = C has an energy of 266 kJ mol−1. By contrast, “only” 11.5 kJ mol−1 represents the transition-state stabilization energy required for a reaction to proceed with 99% enantiomeric excess at room temperature. This energy difference is less than five times the corresponding available thermal energy. Less stabilization is required as the temperature decreases (8 kJ mol−1 at −78°C).
Even what are considered high-affinity, noncovalent binders (typically K d = 0.1–10 nM, equivalent to ΔGbinding = ∼60 to 46 kJ mol−1) have binding energies that are significantly lower than those of particularly weak covalent bonds. Further, to achieve even “nanomolar” dissociation constants, we know that arrangement of atoms in space relative to the binding pocket must be “close to perfect.” For instance, even epimerization of the hydroxyl group in hydroxyl proline renders this ligand essentially unable to interact with its cognate partner, the von Hippel-Lindau E3 ligase (7). Loss of the hydroxyl group altogether decreases the affinity by three orders of magnitude.
To illustrate this point further, we contrast one of the strongest noncovalent interactions known (the biotin–streptavidin association) with a representative covalent bond-forming event (the Michael addition of a thiol to an α,β-unsaturated carbonyl or nitrile to generate a β-thio-carbonyl or -nitrile, respectively). The biotin–streptavidin interaction (K d ∼ 10−15 M; ΔGbinding = ∼ −85 kJ mol−1) (17, 36, 41) is certainly stronger than the interaction of even the strongest manmade noncovalent inhibitors with their target proteins (Fig. 1). For instance, Immucillin H, a drug approved in Japan for treating peripheral T cell lymphoma, is an inhibitor of purine nucleoside phosphorylase with K d = 6 × 10−11 M; ΔGbinding = −60 kJ mol−1 (26). Indeed, aside from some exceptions of which biotin–streptavidin is the most well known, pM affinity is believed to be near the limit of what is accessible for solely noncovalent-binding drugs and ligands (50).

By comparison, thiol Michael adductions are particularly exergonic. The enthalpy change in going from an α,β-unsaturated amide or nitrile to a β-thio amide is around −100 kJ mol−1. Calculated values for ΔG of simple thiol Michael additions vary, in some instances by around twofold, in the literature; are, indeed, difficult to calculate; and require specialized methods. Nevertheless, for instance, for methyl thiol adduction to but-3-en-2-one, an average ΔG of −90 kJ mol−1, with a standard deviation of 17 kJ mol−1, has been reported. Similar exergonicity with similar variances were quoted for thiol adduction to nitroethene and acrylonitrile. Relatively similar energy changes have been reported for numerous other Michael-type adductions: enolate addition to α,β-unsaturated esters (ΔH ∼ −88 kJ mol−1) (25); nitromethane coupling with cinnamaldehyde (ΔG ∼ −70 kJ mol−1) (52), with similar values being reported for acetylacetone adding to the same electrophile (46); and various amines adding to parthenolides (ΔG ∼ −80 kJ mol−1) (62), although values quoted for the aza-Michael adduction across a large range of substrates are quite variable (13) (Fig. 1).
There are two other points that are relevant to this comparison. The first point deals with the unusually high atom efficiency of binding of most Michael acceptors. One way of estimating the efficiency of binding of a ligand to a target in terms of atom economy is ligand efficiency, defined as the negative of the Gibbs free energy of binding divided by the number of nonhydrogen atoms. For biotin, ligand efficiency is 5 kJ mol−1/nonhydrogen atom (the maximum efficacy for noncovalent binding is ∼6 kJ mol−1) (24); the efficiency for Immucillin H is around 3 kJ mol−1/nonhydrogen atom. On the other hand, Michael acceptors, for example, but-3-en-2-one and ethene-nitrile, have ligand efficiencies of 18 and 23 kJ mol−1/nonhydrogen atom, respectively. From this perspective, the variation in ΔG values for Michael adduction in the literature is minimal. This is because the values of ligand efficiency for ethene-nitrile is close to the highest values for interaction per nonhydrogen atom known in biology (24), which typically corresponds to metal-ion interactions with proteins (Fig. 2A).
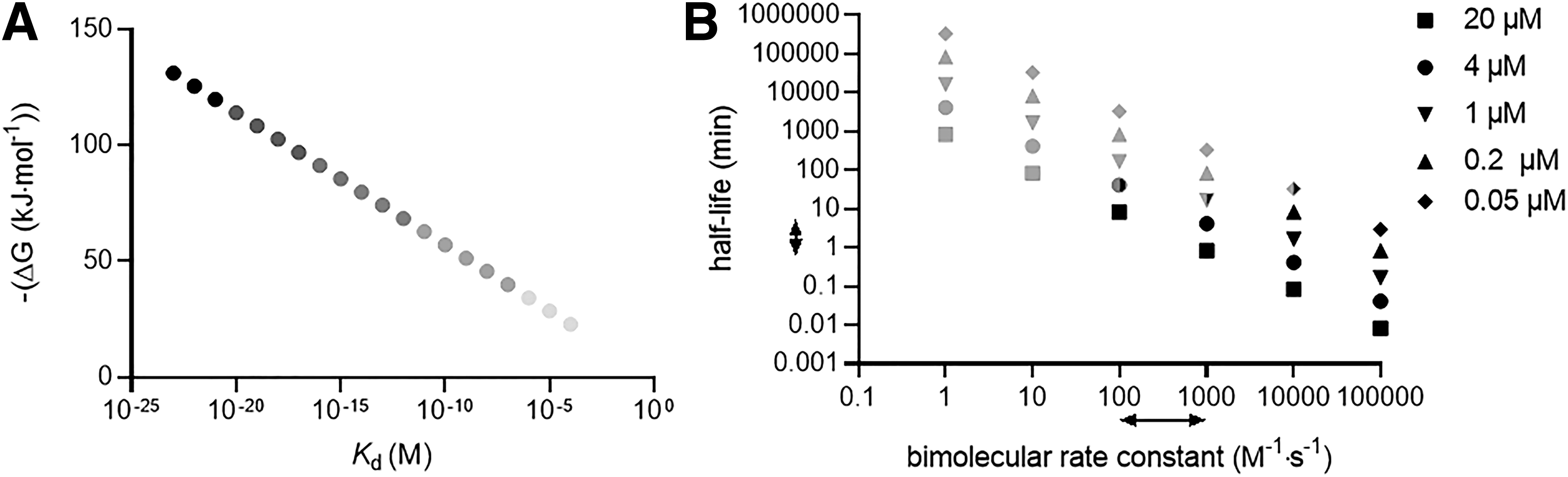
The second standout point of our literature analysis of Michael adduction is that energies of binding are relatively independent of substitutions, although clearly there are some variations. Similar conclusions have also been made in the case of reversible Michael adductions (21). By contrast, the biotin–streptavidin interaction is sensitive to relatively small changes in structure, such as loss of the thioether motif (K d ∼ 10−10 M; ΔG = −57 kJ mol−1) (34) (Fig. 1), whereas changes to the urea moiety can affect binding constants by 2.5 orders of magnitude for O to S substitution, and around 4.5 orders of magnitude for O to NH substitution (2, 17), although on protonation, the imino functionalized analog binds streptavidin quite weakly (7–10 orders of magnitude change) (43, 47).
Covalent Bond-Forming Events to Simple Biological Electrophiles are Overall Slow and Show Little Selectivity
From the preceding discussion, there is overwhelming thermodynamic driving force for bond formation that should render Michael adduction more favored than even the most potent noncovalent binding interactions. One could therefore naively ask what need there is for molecular recognition during Michael adduction. This question highlights a darker side of the large thermodynamic driving force and substituent independence of covalent bond-forming events: Their thermodynamic favorability is also quite independent of the nucleophile. So, what could such relatively weak interactions benefit in the face of such large driving force and substituent independence?
This question clearly ignores the amount of time required for biological processes to occur. Specifically, when we discuss electrophile modification, by either an electrophilic drug or an electrophilic signaling molecule, we typically consider biological signaling processes, similar to phosphorylation or ubiquitination. Most drugs act on enzymes, typically inhibiting their function, which is often linked to driving a specific signaling pathway required by the target disease. For a covalent drug that is intended to react with its target selectively, typically rapid second-order kinetics of binding are needed. The parameter most commonly considered is the effective second-order rate constant for forming the covalent enzyme inhibitor complex, E–I. This rate constant is described by k inact/K i, a parameter that reflects both the speed of formation of E–I from the E•I noncovalent complex (k inact) and the binding affinity of the enzyme for the inhibitor (K i).
Typically, for an effective covalent drug, rate of inhibition must compete against metabolic processes, clearance and labeling of unintended proteins (off-target effects). Good drugs accordingly have k inact/K i above 104 M −1s−1 (49), although most modern covalent kinase inhibitor anti-cancer drugs have k inact/K i 's approaching close to the diffusion-controlled limit, which is around 108 M −1s−1. It is typically assumed that as the rate of the on-target interaction(s) increase(s), the specificity of the inhibitor will increase. However, there is a large amount of proteomic evidence showing that off-target labeling of reactive drugs, even those with high second-order rates of reaction with their chosen targets, can be significant. For drugs such as Ibrutinib, these unintended targets can contribute to negative effects in human patients. Thus, even “highly selective” binders have to run the gauntlet when it comes to avoiding inadvertent labeling. Nevertheless, rapid on-target labeling is still an “ideal” property of covalent drugs, especially when it comes at the expense of off-target reactions.
We and others have shown that even ostensibly simple biologically derived electrophiles can regulate cellular information flow in a similar way to covalent drugs. Because of their simple size and often higher implicit reactivity than approved drugs as well as their rapid clearance, reactive lipid-derived electrophiles (LDEs) must perform a delicate balancing act between rapid on-target labeling, minimizing off-target labeling, and ensuring that on-target labeling can compete against clearance (29, 40). Although there are many constraints on this system, we and others have provided evidence that several critical signaling processes, including ubiquitination and phosphorylation, are subject to electrophile regulation. The time period in which a labeling reaction must occur to be able to trigger cell signaling is difficult to define, and likely depends on what downstream processes are being intercepted.
For signaling modes involving protein upregulation, time frames likely have to be considerably faster than the time taken for downstream signaling to occur, such as transcriptional upregulation, which takes a few hours. For signaling modes such as apoptosis, which involves activation of pre-existing proteins, the process likely has to be faster (44). However, duration or half-life of reactive electrophiles may well be rate limiting for signaling, as reactive small molecules appear to show metabolic conversion and clearance mechanisms much faster than approved drugs (as may be expected given that drugs are usually optimized to be metabolically stable) (29). The half-lives of electrophiles such as α,β-unsaturated carbonyls are likely no more than minutes in most cells (29, 40).
Assuming that the interaction between the α,β-unsaturated carbonyl and a specific enzyme is biologically relevant (i.e., leads to a significant selective modulation of a specific pathway), it will also have to happen in such a way that there is selective buildup of the modified protein under conditions where other proteins are ostensibly unaffected. But as the covalent bonding energy is so dominant over noncovalent interactions, at equilibrium every protein cysteine and small-molecule thiol would be modified, assuming there were enough electrophile molecules to go around!
Fortunately, the rate of reaction of free cysteine (and by extension, most unhindered, unactivated protein cysteines) with β-substituted α,β-unsaturated aldehydes, such as the prototypical LDE, 4-hydroxynonenal (HNE), is slow, around 1 M −1s−1 (40), that is, considerably below the theoretical limit. Further, with concentrations of β-substituted α,β-unsaturated aldehyde and enzyme set to 20 μM, the half-life for labeling of a cysteine of standard reactivity (regardless of the cysteine being incorporated into protein or being free) is ∼10 h. For comparison, with a rate constant of 106 M −1s−1 [similar to the k inact/K i for the approved covalent kinase drugs afatinib and neratinib (49)] at the same concentration, the half-life becomes 0.05 s (Note: 20 μM is a relatively high concentration for an enzyme, and these bimolecular half-lives will increase in length as the enzyme concentration decreases, as shown in Fig. 2B).
Our considerations may be mitigated somewhat by HNE concentration, which has been proposed to be able to reach up to 500 μM (40). However, we stress that this would require sustained elevation of such concentrations (which are known to be toxic, at least when administered from outside of the cell) and, hence, are unlikely to be relevant. In any case, the pseudo first-order half-life for protein labeling under these conditions is ∼25 min, significantly longer than the half-life of HNE in most cells (∼2 min).
It is commonly argued in the literature that tweaking the acidity of a thiol such that it is exclusively a thiolate at neutral pH can cause such an increase in reactivity that these “acidic” or “hot” thiols will be inherently able to act as “biologically relevant” nucleophiles. Unfortunately, the complete deprotonation of small-molecule cysteine (and by extension, a nonactivated cysteine on a protein) can increase the second-order rate constant by maximally 20-fold relative to the equilibrium mixture of cysteine and cysteine thiolate that exists under close to biological conditions (40). In absolute terms, for a reaction with a signaling molecule such as HNE reacting with a cysteine, this represents a jump of second-order rate constant from around 1 to 20 M −1s−1. As we see from Figure 2B, these second-order rate constants are unlikely to enable a significant amount of product to build up, even if the proteins and the electrophile can each reach 20 μM.
Indeed, if we assume that for a labeling process to be “biologically relevant” kinetics should occur around the rate of a typical enzyme reaction, one can see that a threshold of 1000 M −1s−1 is needed to be meaningful (4). As noted earlier, HNE is also not particularly stable in cells (29), and hence the time frame to react under signaling conditions may well be short (60), perhaps minutes. This constraint may make the rate threshold for HNE signaling higher than some enzymatic processes.
In fact, real physical processes also make matters worse for the “acidic thiol” hypothesis. This is because as pK a decreases, the nucleophilicity of the thiolate consequentially decreases, all other factors being equal, rendering a 20-fold increase in rate not readily attainable by pK a decreases alone. Thus deprotonation alone is also not likely to lead to signaling output, and further, given that the total cellular protein-cysteine content is around 10 mM (40), such minimal rate enhancements are unlikely to lead to either significant occupancy or selective buildup of the modified cysteine over the other background cysteines in the cell in sufficient time to relay a signal. Throughout this mini review, we will evaluate this hypothesis in more detail, and ultimately conclude that it is likely correct. However, we will also give some evidence that indicates that thiolate formation could well play some role in this process.
We have previously discussed the different ways in which a specific protein could elevate the reactivity of one (or some) of its cysteine(s) above the maximal rate of reaction possible for a common cysteine thiolate (29, 40). In general, these methods involve a rapid noncovalent prebinding interaction leading to positioning and/or activation of the electrophile before covalent bond formation (both of which can elevate reactivity many folds above background), for instance 4-oxononenal (ONE) reacts with cysteine with a rate constant of around 150 M −1s−1; further, numerous organocatalytic Michael reactions to enals proceed through preformation of an iminium ion from the enal, speeding up Michael addition due to the fact that the unsaturated iminium ion has a lower lowest unoccupied molecular orbital (LUMO) than the enal (37).
Mimicking these processes by activating the electrophile via LUMO lowering, positioning the electrophile in an orientation predisposed to undergo attack by a pendant thiol, or ideally stabilizing the intermediate formed along the Michael adduction pathway (or by raising the HOMO of the nucleophile) certainly would lead to increases in reactivity much higher than what occurs through cysteine deprotonation alone (31), as is quite clear from both examples of enzymes, and the specific examples just cited.
Similar to the proposals of Michaelis and Menten or the lock and key/induced fit models, this logic would predict that proteins with elevated reaction rates, that is, that are privileged sensors of electrophiles, should prebind their targets before reaction. There is evidence for HNE-binding noncovalently to its molecular targets; for instance, molecular modeling studies have indicated that HNE may have an affinity ∼16 kJ mol−1 (K d in the mM range) for its postulated target, Src kinase (22). It is worth noting that in the optimized binding mode published, the enone was in a high-energy staggered conformation that is unable to undergo conjugate addition, and the proposed nucleophilic residue was postulated to be involved in the recognition between HNE and the protein through H-bonding to the OH. Only time will tell as to what extent such models are realistic. Certainly, more experimental work needs to be done to understand the noncovalent aspects of protein–electrophile interaction. Experiments such as investigating blocking of modification of HNEylation by HNE-analogs that cannot undergo Michael adduction would be a fair start. But such experiments—unless carried out on proteins that are themselves kinetically highly reactive and under conditions where binding sites are not saturated by HNE—could lead to confounding results.
There is another simple way to resolve whether something other than the “brute force” of chemical reactivity is responsible for the reaction between α,β-unsaturated ketones and enzymes. One could just measure the rate of the second-order reaction between a specific protein and HNE to test whether the rate constant exceeds what is possible by a common thiolate reagent, such as glutathione. The higher the rate of reaction above the theoretical maximum, the more likely prebinding/enzyme behavior is at play. The enzyme glutathione-S-transferase (GST) that catalyzes conjugate addition of glutathione to Michael acceptors can give rate accelerations around three to five orders of magnitude above background rates (29, 40). This enzyme uses prebinding and other “enzyme tricks” to promote electrophile adduction. These tricks include: promoting correct substrate alignment, stabilization of the transition state for electrophile adduction, stabilization of the intermediate, and leaving group stabilization (where applicable) (12). Although glutathione deprotonation is also considered to be part of GST mechanism, we need to bear in mind that the maximal acceleration for deprotonation alone is minimal compared with an acceleration of five orders of magnitude. Unfortunately, second-order rates of enzyme labeling by electrophiles have proven to be quite difficult to assess quantitatively in vitro; cell-based experiments cannot lead to directly extractable kinetic parameters.
However, there is a corollary to the question of kinetics that can be answered; that is, how selective are different electrophiles for different proteins? Should certain protein cysteines display reactivities that are elevated above that which is possible for a canonical protein/small-molecule cysteine, at least some of these reactive cysteines would be expected to show selectivity toward a specific subset of reactive small molecules.
We stress that this trait is not absolutely necessary as some GSTs, for instance, are not particularly substrate discriminating. However, in cases of proteins that rapidly engage HNE and are primed to undertake biologically relevant cell signaling roles, which we have termed “privileged first responders” (29), such selectivity would be expected because noncovalent prebinding of the electrophile is likely employed by a subset of these proteins to promote fast kinetics. Such an interaction could, in effect, act as a filter priming the protein to favor a type, or in an extreme case, one specific electrophile, over others. Indeed, this question obviates thiol-specific tricks, such as thiol desolvation, that could potentially lead to an overall hyperactivity that is faster than expected than a thiolate in solution (40). These assays would be more poignant should the assayed cysteines be shown to have highly elevated reactivity compared with the global cysteine pool.
Mass Spectrometry Datasets Indicate Some Proteins Show Privileged Reactivity with Electrophiles
For many years, there was very little evidence showing that some proteins have elevated reaction rates with α,β-unsaturated ketones. However, there are now several groups investigating electrophile labeling that have provided good evidence for such phenomena. We will here review the work showing that proteins are highly discriminatory for various electrophiles. In many instances, this question has been approached from various different angles and similar results have been obtained. We will further discuss ways in which confounding results can occur.
The Cravatt laboratory has published numerous datasets that are highly useful. These are mass spectrometry data, profiling reactivity of a pool of protein cysteines toward various electrophiles. The main method used is a venerable chemical biology approach. However, several implicit limitations for the method are noteworthy and have been discussed in detail elsewhere (29, 31, 35, 40), such as: the method being built on loss-of-signal (and hence prone to artifactual readouts, especially when reactive ligands are employed); assumption that the cell is a homogeneous entity; and only detecting a small fraction [at most ∼4% (31)] of the total cellular cysteine content. Encouragingly for practitioners of this method, several variants have been recently published by other laboratories that have begun to address some of these issues, including, for example, derivatization of acyl group (10) and photocaged electrophiles (1). The datasets from the Cravatt laboratory and those from applying the acyl-group derivatization approach for the most part agree, arguing at least within the profiled data set, comparisons and conclusions are valid.
After some thought-provoking work showing that iodoacetamide (IA)-derived ligands have a kinetic preference for reaction with a subset of protein cysteines, out of a profiled dataset of around 1000 protein cysteines (58), Cravatt lab's attention turned to investigating the interaction preference of natural electrophiles (56). HNE and 15-deoxy-delta (12, 14)-prostaglandin J2 (15d-PG J2) were chosen. From our perspective, this choice was very insightful. The principal reactive motif is, in each instance, an ene-carbonyl. HNE possesses an enal; the prostaglandin possesses two enones, one being endocyclic to the five-membered ring, the other being exocyclic and conjugated to both the ketone and another olefin (Fig. 3A). Given the subtle differences in terms of electronic properties of the two structures, it is difficult to predict which of the two molecules would be more reactive.
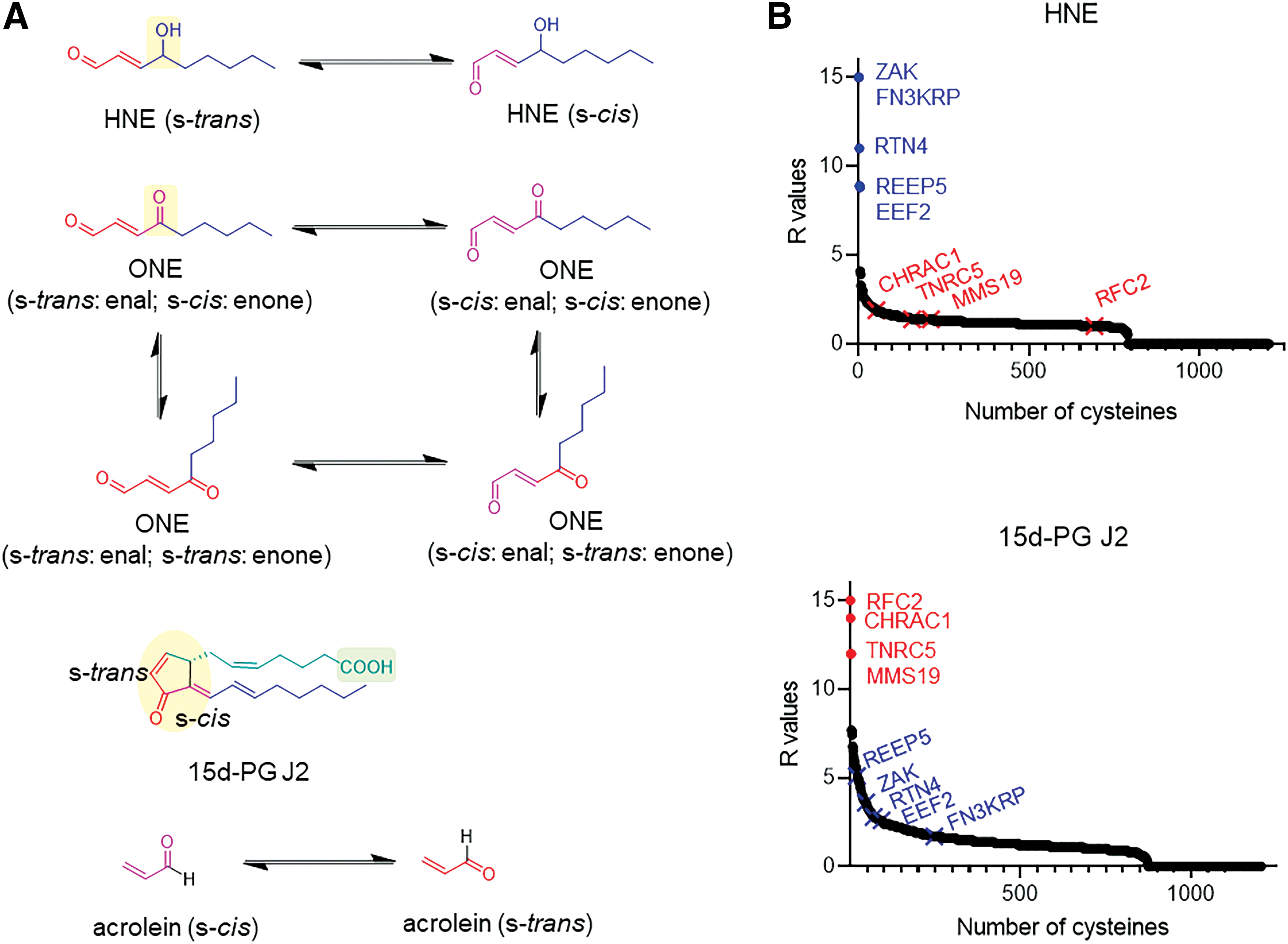
Although the rate constants for reaction with model nucleophiles has not been systematically disclosed, both are known to be depleted by glutathione on addition to cells (3), and both react with serum albumen in vitro (28). However, these two molecules have similar reactive chemotypes and prostaglandin possesses enones in both the s-cis and s-trans configurations, which can mimic the two reactive, low-energy conformations of HNE (Fig. 3A). Thus, one may naively predict that there would at least be a “decent” level of overlap between the adducted proteins in cells treated with these two small molecules, if only reactivity and olefin orientation were considered.
Conversely, the connectivity and topology of the two molecules is quite distinct. HNE possesses a pendant hydroxyl group, which is not present in prostaglandin. Further, we know, for instance, from Von Hippel Lindau (7) and studies from Src (22) that the hydroxyl function within HNE may be important for binding in some instances. On the other hand, the ring core of prostaglandin and its pendant carboxylic acid are standout differences in the carbon
The profiling experiments showed that fast-reacting proteins for HNE and 15d-PG J2 did not overlap at all (56). Indeed, many of the proteins that were efficiently labeled by HNE did not react appreciably with 15d-PG J2, and vice versa (Fig. 3B). Unfortunately, the kinetics of binding/labeling of the electrophiles for their respective targets were not calculated. Thus strictly speaking, we have no clear idea as to how reactive the proteins labeled selectively are, or how much of these rate enhancements could be explained by thiol deprotonation alone; however, given the duration of the experiment (1 h), and that permeation of reactive electrophiles into cells is relatively slow, it seems unlikely to us that deprotonation alone is sufficient to explain the data. (This is because, based on the data discussed earlier, with deprotonation alone, the absolute maximal second-order rate achievable would be 20 M −1s−1; and at this rate, reaction half-life would be too long [∼100 min in the second-order limit; or 30 min assuming a first-order reaction in HNE at a constant concentration of 20 μM] for 4–20 μM protein/electrophile concentrations and incompatible with this experiment's timescale.) Of course, the use of cells to probe these labeling interactions may complicate interpretation, and indeed, no direct in vitro comparisons between labeling of sensitive proteins by the different electrophiles were shown. However, some hits were inhibited on addition of the electrophiles used, at least validating the general points of the assay.
The Wang lab developed a different version of activity-based protein profiling (ABPP) originally established by the Cravatt laboratory (Fig. 4A). The Wang lab's method makes use of the carbonyl function integrated into the protein post
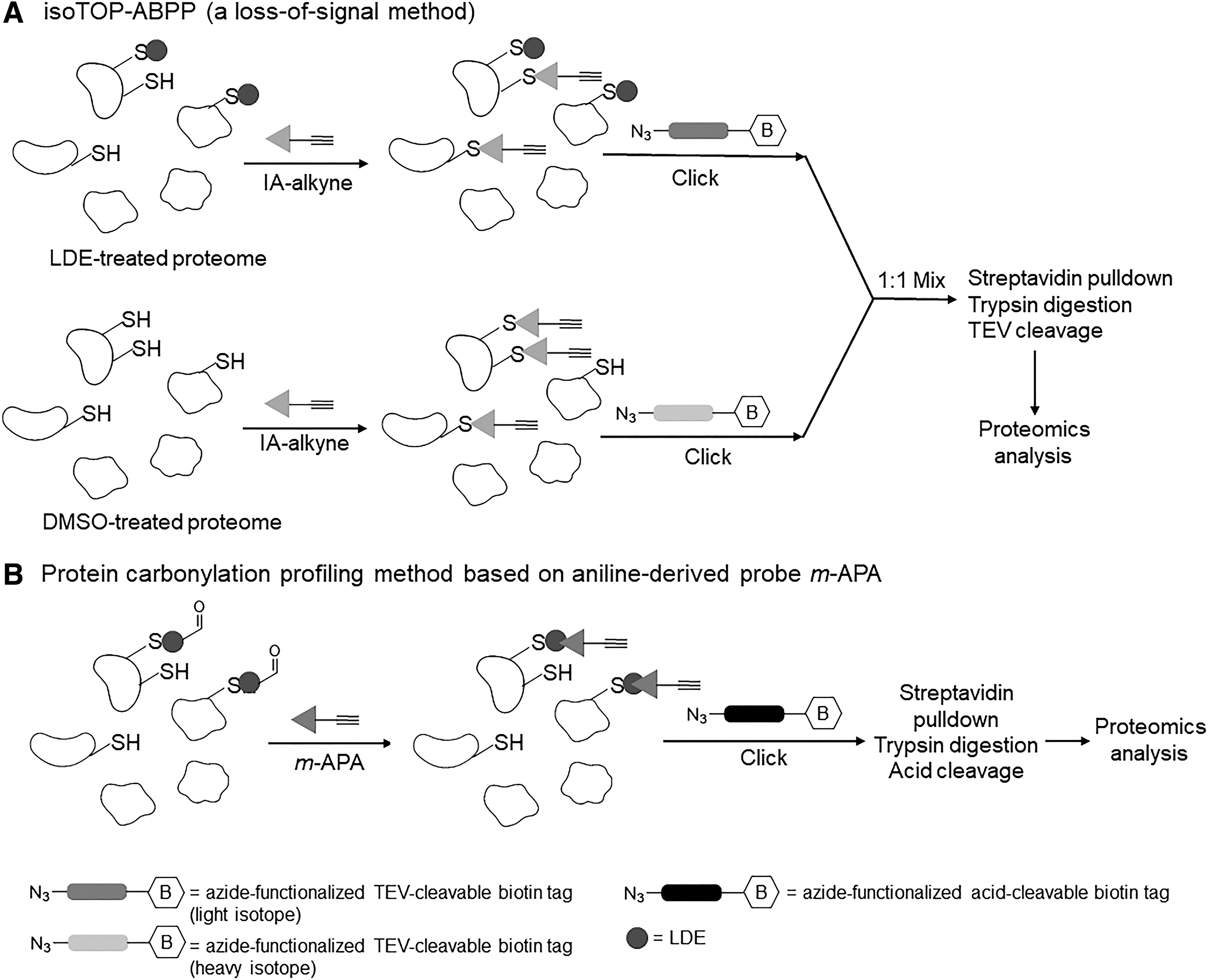
One of the clear issues with the approaches described earlier is that modified proteins were identified under conditions where excess of electrophile was administered to the cells for periods likely longer than “signaling conditions” persist. The Wang lab proceeded to investigate proteins affected by electrophile modification under ferroptosis (a process known to involve lipid oxidation) (10). Because the Wang method is based on enrichment of specific, electrophile-modified peptides, the method was able to be applied not only to target ID but also to identifying specific modifications [in this case, HNE and ONE] (18). Once again, such comparisons are instructive because ONE is 100-fold kinetically more reactive than HNE to cysteines, even though ONE and HNE have very similar connectivities. Thus, one would expect the targets of HNE and ONE to overlap.
Intriguingly, in spite of the similarities between the two electrophiles, the Wang lab identified several proteins that were labeled selectively by HNE and ONE, again indicating that there are some very efficient and specific protein sensors of electrophiles. We recognize: (i) that it is possible that HNE adducts can be converted to ONE adducts postadduction and vice versa (even during downstream processing); (ii) we are cognizant of the fact that many HNE adducts do not give free aldehydes for conjugation as required to be detected by this method, but ONE modifications do not have the ability to form cyclic structures and hence are more likely to be detected by the method; and (iii) we do not strictly know whether there are different compartments of formation for ONE and HNE. Given the differences in reactivities, however, we can speculate that the diffusion distances of the two molecules are quite different that could lead to different protein labeling spectra. Nevertheless, these data are far from expected and further indicate that proteins may well be very discerning when it comes to electrophile reactivity, and further can countermand reactivity (ostensibly by 100s of fold) even between electrophiles of seemingly similar structures.
Data from REX Technologies are Also Consistent with Protein-Ligand Recognition
We ourselves have focused on protein-specific modification events. To achieve this goal, we developed the first protein-specific electrophile modification strategy that functions in cells and whole organisms, called targetable reactive electrophiles and oxidants (T-REX) (Fig. 5A) (14, 27, 30, 32, 33, 38, 39, 42, 51, 54, 61). These experiments have told a similar story to that of the Cravatt and Wang labs' experiments, but from the perspective of protein-specific modifications by specific electrophiles. We stress that because of the way our experiments are set up, many of the caveats of the Cravatt and Wang experiments are assuaged by T-REX. Thus, the data sets are, in fact, very complementary.

As recently discussed at length (29, 39, 40, 42), T-REX does have its own caveats, which are principally limited to the fact that an ectopic fusion protein is required to perform T-REX, although careful implementation of built-in technical T-REX controls (42), and the use of “non-fusing protein” control (40, 42) and hypomorphic mutants (32, 61), altogether allow any off-target responses/potential mis-interpretations to be ruled out rigorously in T-REX-experiments. Notably, the combination of T-REX and genome-wide profiling of sensors responsive to reactive electrophiles and oxidants (42) (a system to release low concentrations of a specific reactive electrophile unfettered into the cellular milieu to identify the best native sensors) (29) has assuaged the concerns underlying overexpression of fusion proteins quite considerably.
We first compared how different electrophiles were able to trigger downstream signaling on Keap1 labeling (27, 30, 38, 39). Keap1 is one of the master regulators of the linchpin antioxidant response (AR) regulator, Nrf2 (19). Keap1 typically binds Nrf2, but on electrophile adduction, this binding is impaired, leading to an increase in Nrf2 and upregulation in AR, which can typically be read out by reporter assays, quantitative polymerase chain reaction, or Western blot (39).
Expecting that Keap1 may prove differentially susceptible to electrophiles under protein-specific adduction, we compared the extent of labeling of Keap1 and downstream AR triggered under Keap1-specific modification in living cells with unstimulated levels and bolus dosing levels for a number of α,β-unsaturated electrophiles (27). Perhaps, disappointingly, both the extent of electrophile labeling and the overall AR upregulated was similar for all electrophiles (aside for electrophiles that transpired to be incompatible with the T-REX strategy). One surprise was that AR upregulation on T-REX did not differ markedly from AR upregulation observed under bolus dosing (27, 38), implying that both positive and negative regulation of AR may be incurred on bolus electrophile treatment conditions. We found such antagonistic behavior for numerous other systems in subsequent studies (30).
We elected to investigate the molecular underpinnings of Keap1-specific versus global AR more closely by identifying residue occupancy under the different conditions. At this level of scrutiny, the outputs were quite different. T-REX, delivering a cyclohexenyl electrophile, 6-(prop-2-yn-1-yl)cyclohex-2-en-1-one (CHE, Fig. 5B) in cells, hit C613 and "//172.16.100.5/aps3b2-share/tiger/THYROID/30_10/3B2/THY-2019-0648-ver9-Stephenson_4P.3d"C489; whereas on bolus electrophile dosing of cells, CHE hit C513/C518 and C273 (Fig. 6A) (27). On the other hand, for HNE, T-REX in cells labeled C513 and C518; bolus HNE treatment (100 μM) of cells labeled C23, C226, C273, and C368. But only C23 and C368 were hit when cells were treated with 25 μM HNE (38). In vitro, C77, C151, C226, C273, C319, and C368 were hit by HNE on bolus dosing (38). Just as in the data from ABPP cited earlier, the mass spectrometry analyses cannot give a complete picture of the different residues modified, so we recognize that it is possible that some of these conclusions are biased by ionization propensities, solubilities/stabilities of the product peptides, or resolution of the mass spectrometer. Nonetheless, these data suggest that different cysteines within Keap1 have different preferences for different electrophiles.
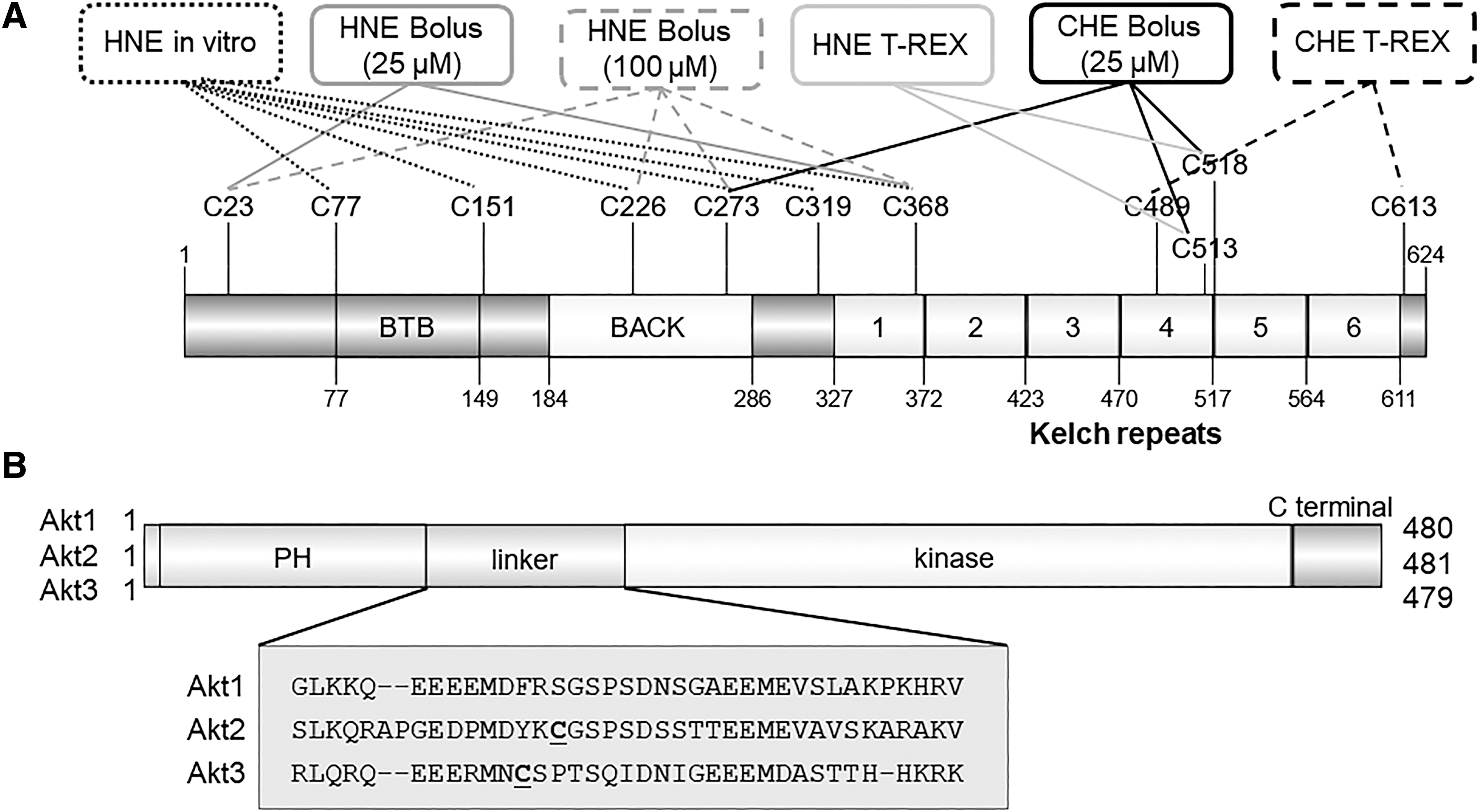
Pleasingly, these data agree with other reports that different reactive electrophiles and oxidants interact with specific subsets of Keap1 cysteines (8). However, in our case, the electrophiles are quite similar and in the case of T-REX are delivered from a similar position relative to Keap1; the electrophiles are also delivered at the same concentrations, under conditions where off-target effects and negative impacts, permeation, and other issues are side-stepped. Notably, irrespective of whether HaloTag is fused to N- or C-terminus of Keap1 protein, the same residue is modified (C613) on T-REX-mediated CHE targeting of Keap1 in live cells (39). It is also critical to bear in mind that labeling efficiency of Keap1 by CHE and HNE under T-REX conditions was similar (27). Thus, the T-REX data imply that reactive cysteines in Keap1 are highly sensitive to chemical change.
We also investigated how different isoforms of different proteins sense HNE. After a medium-throughput screen, we identified Akt, an enzyme that has three isoforms, as an interesting model system for further study (32). These proteins share >75% similarity overall, and the pairwise similarity comparisons are shown in Figure 6B.
It quickly became apparent that Akt1 is insensitive to HNE. On the other hand, Akt2 is weakly sensitive to HNE, but Akt3 is one of the most sensitive HNE sensors we have characterized. Mass spectrometry showed that a specific residue (C119) that is unique to Akt3 sensed HNE. Mutation of this residue to serine (C119S) ablated Akt3 HNE sensing.
Intriguingly, the sensor residue, C119 in Akt3, lies on a flexible linker region between the two principal domains of Akt, the PH and kinase domains (Fig. 6B). This linker is highly divergent between the three isoforms. Further, Akt2 also houses a reactive cysteine (C124) in this linker region, which is believed to react with peroxide (57). Although we have shown that the primary structures surrounding these two sensor cysteines [Akt3(C119) and Akt2(C124)] are different, the question remains unanswered whether or not primary sequence differences alone are sufficient to cause such a divergence in selectivity. However, it is likely that reactivity induced by primary sequence changes is promoted by pK a decreases, increasing the amount of thiol anion formed. Such changes favor interaction with both oxidants and electrophiles similarly, and the margin for improvement for cysteine is relatively small (∼20-fold) (40) in each case. Thus, given that Akt3 was at least fourfold more reactive to HNE than Akt2, and Akt2 is significantly more reactive to peroxide than Akt3, it seems unlikely that only pK a effects are at work.
Of course, the background labeling of Akt2 by HNE could certainly represent a contribution from increased thiol anion, although the comparison in this instance is that Akt1 does not possess a cysteine and hence is not a fair comparison. In other work, we have mutated a putative general base that is spatially close to the privileged cysteine in HSPB7 (51). This led to a significant decrease in HNE sensing, arguing that deprotonation may be part of the story, likely if other important aspects of activation are also present.
Proteins May Show Enantioselectivity, Although This Is Far from Clear
Cellular studies comparing (R)- and (S)-HNE have indicated that the two enantiomers have different biological properties (40). One study, assessing the effects of the two compounds in cultured mouse hepatocytes, found that (S)-HNE was better able to initiate apoptosis and several pro-apoptotic signaling pathways than its enantiomer. However, other studies in colorectal cells have found very little difference between the two enantiomers (59). Of course, such different behaviors could reflect different targets expressed in the two cells, or different metabolizing enzymes present. Nevertheless, such data are interesting, if somewhat preliminary.
Enantioselective modification of different proteins by HNE has been reported. One of the best examples is glyceraldehyde-3-phosphate dehydrogenase. This is inactivated more than three times faster by (S)-HNE than (R)-HNE (20). However, it should be ceded that these rates of reaction are quite slow (6 M −1s−1 vs. 2 M −1s−1) and the difference in energy for these two rate constants is not much greater than the thermal energy (∼3 kJ mol−1 * vs. ∼2.5 kJ mol−1, assuming T = 298 K). For instance, this pales into insignificance when we consider enantiospecificity of approved drugs. (S)-ibuprofen is more than 100-fold more potent an inhibitor of cyclooxygenase than its antipode (corresponding to a difference in energy of more than 11 kJ mol−1 for the different bound states).
One may predict that as the reactivity of different cysteines increases (i.e., the rate of reaction is increased above the background rate, and hence a larger interaction between electrophile and protein may be expected to be required), the enantioselectivity increases. Unfortunately, this is certainly not always true. Thioredoxin possesses two HNE-reactive cysteines, C73 and C32. C73 displays faster labeling kinetics than C32. However, C73 displays little preference for different enantiomers. On the other hand C32 displays significant preference for (R)-HNE (55). However, since no kinetic parameters were discussed in the article and it is possible that neither cysteine is particularly reactive, these data do certainly give a cautionary tale that study of electrophile interactions with proteins is likely to be complex and throw up some confounding information. It is thus worthwhile to ask whether we have chosen the correct proteins for these in vitro assays. Indeed, most of the proteins we discuss in this section, and the larger list from the literature of in vitro studied protein modifications have been carried out on proteins that are not detected as represented as HNE selective in any of the HNE profiling assays cited earlier (40). It is possible that these specific proteins have little to teach us about how HNE interacts with privileged sensor proteins as they do not have the mechanisms that lead to meaningfully enhanced rates of HNE adduction. Hopefully, more modern methods can be applied to these problems, and more downstream analysis can be used to help shed light on these problems.
Conclusion
We are a long way from understanding how LDEs interact semi-selectively with proteins. Our tools to study these processes are just on the verge of having the ability to scrutinize these systems well enough to ask meaningful questions that need to be answered. However, the data from the laboratories we have discussed earlier are starting to show that there is huge diversity in terms of lipid sensing ability and, indeed, selectivity among reactive-lipid-sensing proteins. Indeed, there is evidence that proteins not only exhibit kinetic selectivity for specific reactive ligands but also that these proteins can buck the trend of typical reactivity. The signs are encouragingly pointing to the fact that genuine enzyme-like sensing of electrophiles may be occurring. As we have also argued, if sensing occurs between specific proteins and specific electrophiles with defined molecular architectures, that is, through interaction with/occupancy of a specific binding pocket, this opens the doors for gain-of-function events, amplification of downstream signaling events, and other properties that have been proposed to occur on electrophile modification.
As previously argued, to really understand these processes, there is really a need for more quantitative and rigorous analysis of these data. It is apparent that gleaning this level of understanding will require a more diverse skillset than is currently applied to most papers dealing with reactive molecule signaling. As usual, resolution will ultimately need raising of the bar for the way the field, in general, goes about characterizing electrophile-protein interactions. However, in our opinion, this extra effort will be worth it. Our thoughts are summarized later.
From our introduction, it is clear that understanding kinetics of labeling is essential. Cell-based experiments unfortunately do not offer anywhere near precise enough measurements of sensing to give us a good platform to rank and understand sensing quantitatively. We have, for instance, measured externally administered HNE labeling of the soluble cellular proteome. There was a significant lag phase (around 5–10 min) between identification of labeling and HNE dosing. However, when HNE was released from inside the cell, maximal labeling of the proteome occurred in no more than 3 min (61) (Fig. 7). This labeling was similar to what was observed after bolus HNE treatment (30 min). Thus, permeation is almost totally rate limiting in this situation, although admittedly the labeled proteomes were not directly compared.
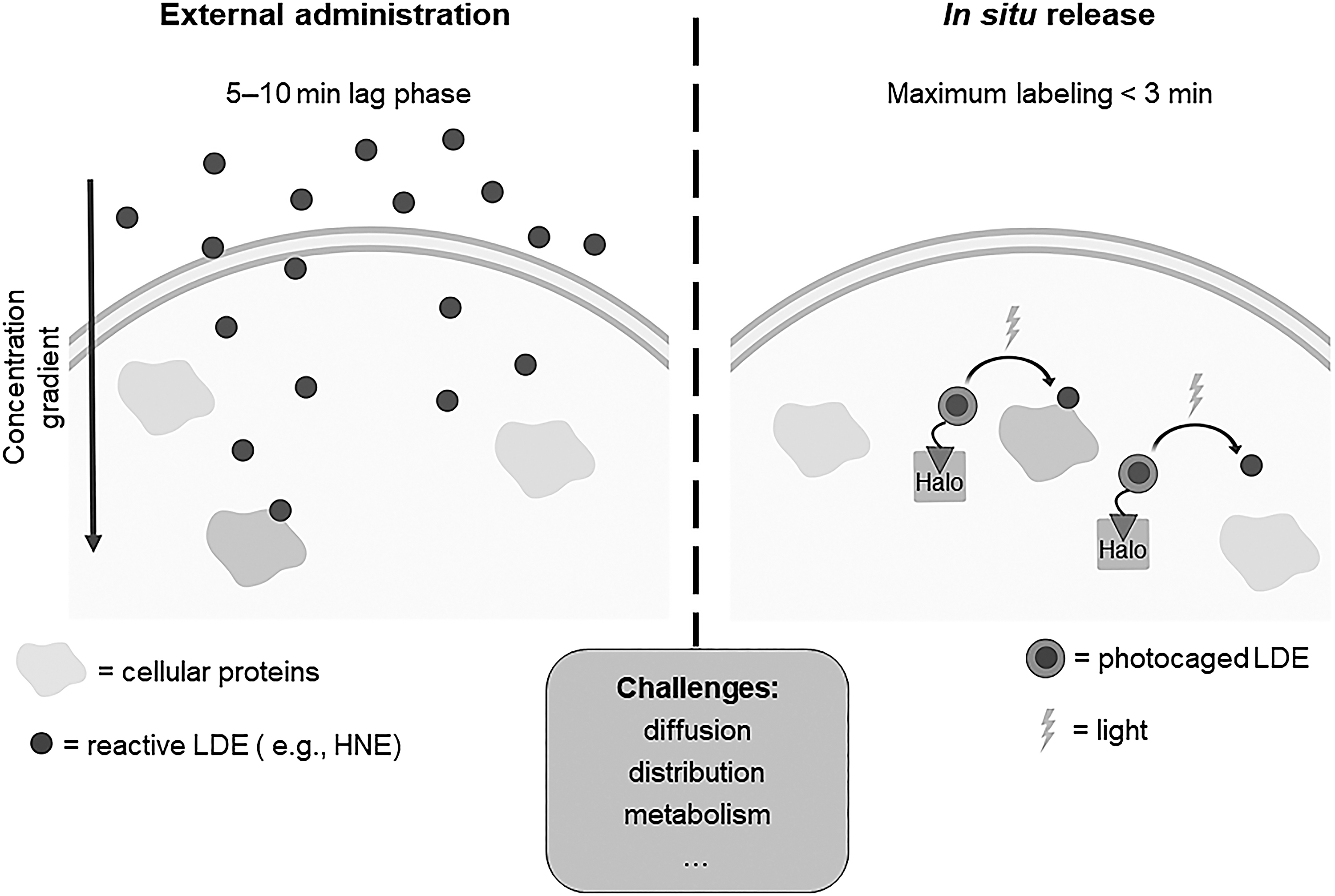
Indeed, it is known that under bolus dosing conditions, the cell experiences a gradient of reactive molecules (29), with concentrations of the reactive molecule being highest at the membrane and falling off toward the nucleus. Further, it is believed that for this gradient to be overcome some of the detoxification machinery needs to be compromised. Our experiments indicate that intracellular release of the electrophile may not be so subject to such issues. Regardless, it is important to question just what is leading to selectivity under bolus dosing; is it really genuine HNE sensitivity; or is it proximity, diffusivity, stability, or (an)other parameter(s) that dominate(s) over kinetics and masks the key sensors.
It is also worth noting that it is unlikely that external bolus dosing models particularly model endogenous signaling conditions well, which likely arrive from intracellular cues/electrophile release, unless significant stress is levied on the system. Thus, more time should be given to careful, and traditional characterization of labeling, or inhibition (k inact/K i) kinetics of identified proteins by a low concentration of electrophile on purified isolated sensor proteins. Such experiments should be performed at the expense of performing time-independent experiments with excess of electrophile and protein, which give little indication of kinetics or ability of proteins to react at a reasonable (or any) rate.
Of course, such considerations do not explain why different proteins show differential reactivity/susceptibilities to subtly different electrophiles. It is, indeed, this unexpected observation that is particularly exciting. But again, in vitro validation and quantitative assessment of such parameters need to be carried out, as confounding factors such as differential diffusion, distribution, or potentially metabolism could confound our interrogations. Conversely, as suggested elsewhere, T-REX may be able to better serve this purpose. This is because in vitro analysis is complex and T-REX is ostensibly an unbiased measure of a protein's sensitivity to electrophiles.
One issue where we have yet to really cover decent ground is enantioselectivity. Such a gap in our understanding is significant, as it is enantioselectivity, and the “natural” resolution of racemic products that is most indicative of biological processes. Hence, one could imagine that enantioselective reaction of different proteins with an electrophile would be most indicative of an “enzyme-like” recognition process occurring. Unfortunately, this area is also not immune to artifacts caused by metabolism. For instance, rat GSTα4-4 metabolizes (S)-HNE four-times faster at Vmax than (R)-HNE; the K m was threefold higher for (R)-HNE than the (S)-enantiomer. This corresponds to a 12-fold increase in the second-order rate of reaction for the (S)-enantiomer than for the (R)-enantiomer (20). To further confound matters, it seems that although many GSTs show S-selectivity, other GST isoforms may show the opposite sensitivity and other HNE detoxification processes such as NAD+-dependent oxidation may show (R)-HNE metabolism preference. Thus, administration of single enantiomers of HNE could lead to different enantiomers being enriched within different locales, etc (53).
We, thus, hope the field will not continue to neglect the quintessential aspects of biochemistry and recognition in its continued efforts to understand redox signaling. We feel that careful, and controlled experiments, such as those offered by several techniques available these days, can be used to interrogate electrophile sensing more accurately, and to generate data sets that are more transposable between laboratories. It is our feeling that the data hint that electrophile sensing is incredibly nuanced, and we hope that further experimental proof can be provided in the near future.
Footnotes
Funding Information
Swiss National Science Foundation project funding (310030_184729); National Centre of Competence in Research (NCCR): Chemical Biology; Novartis Foundation for Medical-Biological Research (Switzerland); Swiss Federal Institute of Technology Lausanne (EPFL); and NIH DP2 New Innovator Award (1DP2GM114850).