
Abstract
Significance:
Acute myocardial infarction (AMI) is a leading cause of death worldwide. Post-AMI survival rates have increased with the introduction of angioplasty as a primary coronary intervention. However, reperfusion after angioplasty represents a clinical paradox, restoring blood flow to the ischemic myocardium while simultaneously inducing ion and metabolic imbalances that stimulate immune cell recruitment and activation, mitochondrial dysfunction and damaging oxidant production.
Recent Advances:
Preclinical data indicate that these metabolic imbalances contribute to subsequent heart failure through sustaining local recruitment of inflammatory leukocytes and oxidative stress, cardiomyocyte death, and coronary microvascular disturbances, which enhance adverse cardiac remodeling. Both left ventricular dysfunction and heart failure are strongly linked to inflammation and immune cell recruitment to the damaged myocardium.
Critical Issues:
Overall, therapeutic anti-inflammatory and antioxidant agents identified in preclinical trials have failed in clinical trials.
Future Directions:
The versatile neutrophil-derived heme enzyme, myeloperoxidase (MPO), is gaining attention as an important oxidative mediator of reperfusion injury, vascular dysfunction, adverse ventricular remodeling, and atrial fibrillation. Accordingly, there is interest in therapeutically targeting neutrophils and MPO activity in the setting of heart failure. Herein, we discuss the role of post-AMI inflammation linked to myocardial damage and heart failure, describe previous trials targeting inflammation and oxidative stress post-AMI, highlight the potential adverse impact of neutrophil and MPO, and detail therapeutic options available to target MPO clinically in AMI patients.
Introduction
Coronary artery disease (CAD) is generally asymptomatic for the first four decades of life; thereafter, the risk of the onset of clinical complications caused by acute cardiac ischemia or myocardial infarction (MI) increases. The primary cause of CAD is the accumulation of coronary artery atherosclerotic plaques that narrow the lumen, thereby decreasing blood flow and cardiac function. Myocardial ischemia induces hypoxic and oxidative responses in both cardiac cells and the vasculature, which together promote myocardial tissue damage (21, 121), often presenting as angina or fatigue on physical exertion, as the heart continues to work under a limited oxygen supply. Often, the first indication of ischemia is a heart attack or acute coronary syndrome (ACS) triggered by coronary artery occlusion or acute rupture of an atherosclerotic plaque. Myocardial ischemia and infarction increase the risk of subsequent heart failure by weakening or irreversibly compromising the function of the affected myocardium. Together, CAD and heart failure are leading causes of mortality and morbidity worldwide (133).
The pathogenesis of atherosclerosis is characterized by a chronic inflammatory response within the arterial intima that facilitates excessive lipid accumulation (both native and oxidized) in the arterial sub-endothelium and atheromatous plaque formation (175). A key early atherogenic event precipitated by cardiovascular risk factors such as hyperlipidemia, hyperglycemia, smoking, or hypertension is endothelial dysfunction, which manifests as impaired bioactivity of the vasoprotective signaling molecule nitric oxide (NO), as well as increased vascular permeability and endothelial-leukocyte interactions (184) (Fig. 1A). These events facilitate the infiltration of activated immune cells and low-density lipoprotein into the sub-endothelial space (175).
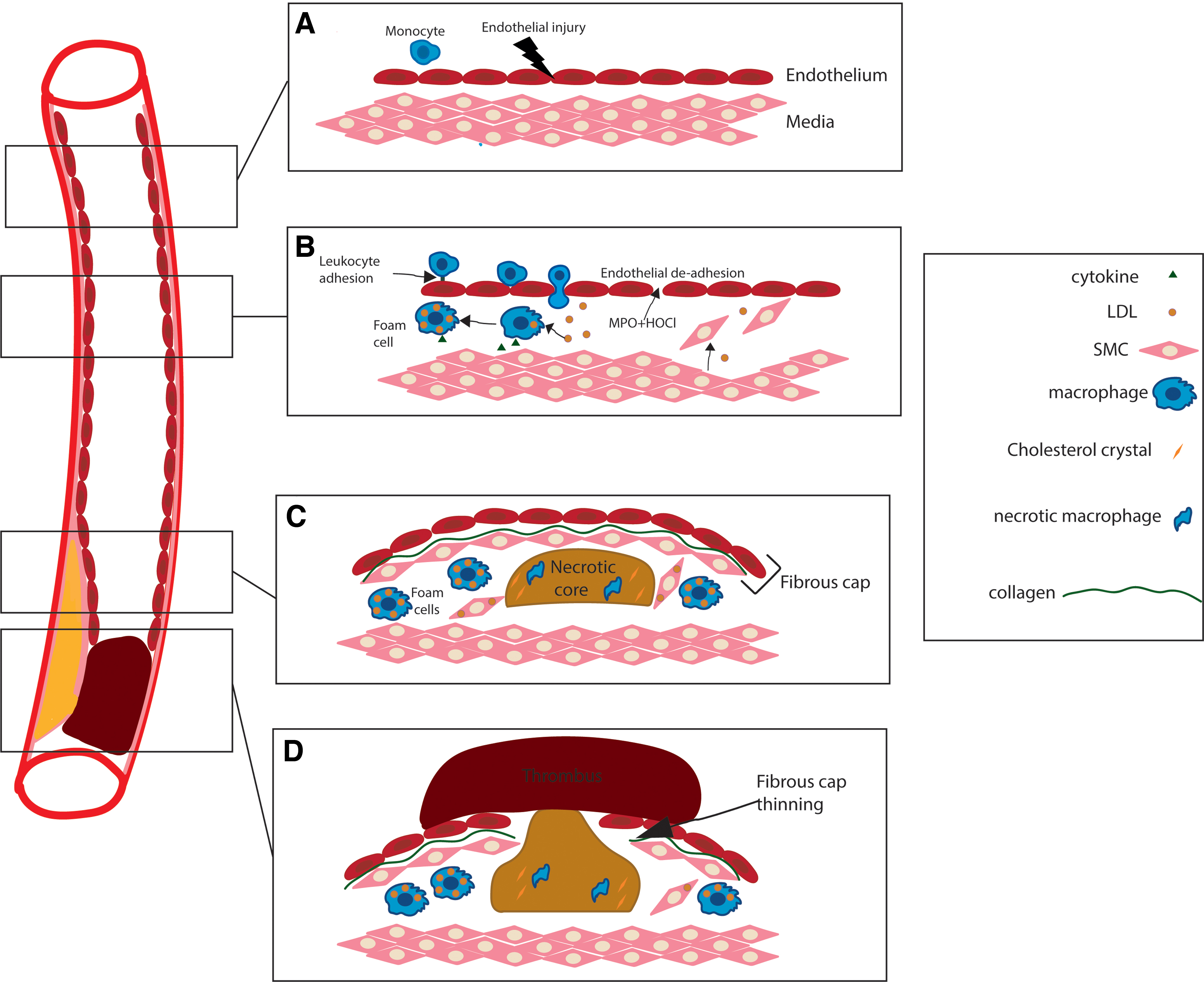
This initial innate immune response is likely aimed at negating the atherogenic stimuli (93). However, the continued presence of cardiovascular risk factors manifests as a chronic inflammatory/immune response that promotes cycles of vascular insult, inflammation, and remodeling (92). Thus, monocytes recruited by the inflamed endothelium into the sub-intimal space differentiate into macrophages that accumulate excessive amounts of lipid to form foam cells; the hallmark of early vascular lesions (152, 175) (Fig. 1B). Continued intimal inflammation underpins the transition of these early lesions to more complex atheromatous plaques characterized by a fibrous cap and a necrotic core, derived from dying cells and the extracellular accumulation of cholesterol crystals (Fig. 1C) (152, 175). Progressive inflammation at the shoulder regions of advanced plaques can signal for smooth muscle cell apoptosis, increased proteolytic matrix metalloproteinase (MMP) production, and fibrous cap thinning (Fig. 1C); this series of events may lead to acute plaque rupture and formation of occlusive luminal thrombi in the coronary artery to precipitate an acute myocardial infarction (AMI), resulting in ischemic cardiomyocyte death (121) (Fig. 1D).
Cardiomyocyte death in the sub-endocardium occurs within minutes of AMI onset and progresses to the epicardium with prolonged ischemia. Ischemia facilitates cardiomyocyte ATP deficiency and alterations in intracellular ion gradients (especially Ca2+), thereby promoting mitochondrial dysfunction and decreased cell viability (121). These events manifest as the pathological production of reactive oxygen species (ROS) and stimulation of the innate immune response, which, in turn, further promote tissue damage including cardiomyocyte death (126).
Prolonged ischemia within the infarct region may expand after the initial event and induce cardiac dysfunction in the adjacent myocardium. Loss of cardiac contractility after AMI is a common cause of chronic heart failure (35). To circumvent this complication, clinical intervention aims at rapidly restoring blood flow and preventing further infarction by rapidly opening occluded coronary arteries, which has led to a decrease in mortality rates (72). Paradoxically, reperfusion therapy that is critical to recovery of the affected myocardium also results in subsequent myocardial dysfunction that correlates with increased heart failure rates within the same patient cohort (196). It is now recognized that reperfusion after myocardial ischemia elicits inflammation that also yields oxidative host-tissue injury, which together adversely affects myocardial cells and instigates cardiac complications after AMI (38). This review discusses the implications of the post-AMI inflammatory response in driving subsequent heart failure with a focus on the adverse role of neutrophils and their potential as therapeutic targets to limit chronic myocardial dysfunction.
Myocardial Ischemia and Reperfusion Injury
The myocardium becomes significantly altered both during and after an AMI, culminating in the loss of contractility and cell death in the infarct region (21). The biochemical and cellular events that facilitate these changes are specifically related to the disparate, yet inter-linked processes of ischemia and reperfusion (Fig. 2). Ischemia deprives the myocardium of nutrients and oxygen distal to the site of coronary occlusion, rendering cardiomyocytes unable to undergo aerobic metabolism to support their energy requirements (121). The low oxygen environment is rapidly sensed by homeostatic intracellular signaling systems, such as the hypoxia inducible factor (HIF) transcription factor cascade (87), which induces the upregulation of numerous compensatory genes involved in elevating anaerobic glycolysis and promoting angiogenesis and vascularization (71, 108). However, increased anaerobic metabolism yields lactic acid that provokes acidosis and myocyte death (99) and, therefore, expansion of the primary infarct area (Fig. 2).
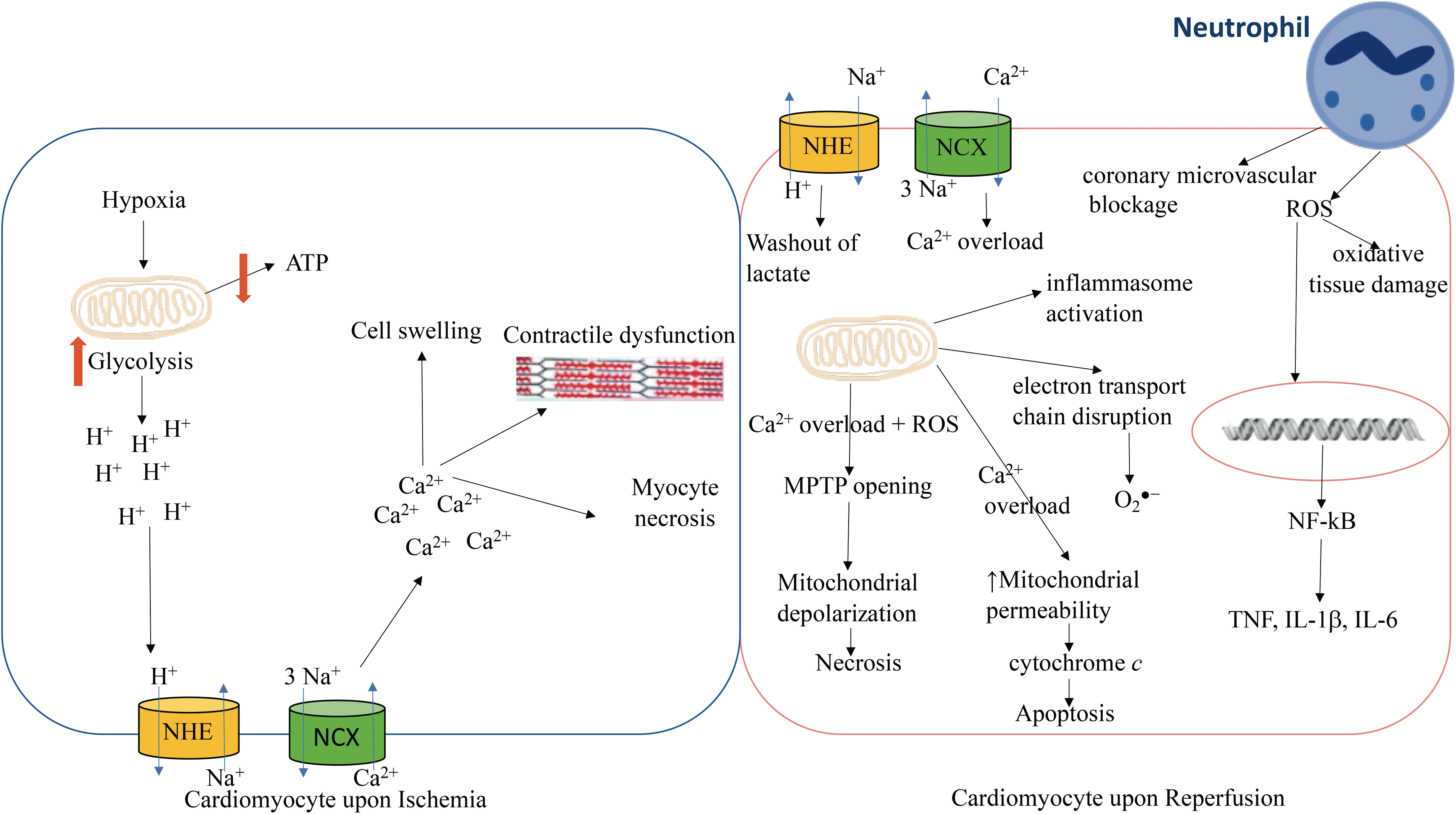
Considering the relatively high requirement of the myocardium for ATP, this shift in energy production to glycolysis results in the inefficient production of ATP and constitutes a pathological feature, which needs to be quickly reversed to protect against the progression toward detrimental complications such as heart failure and ischemia-induced ventricular fibrillation (11, 44). The uncoupling of glycolysis and glucose oxidation has recently been identified as an early contributing factor in the progression toward heart failure with preserved ejection fraction (44). Accordingly, restoration of coupling between glucose oxidation and glycolysis, by inhibition of fatty acid oxidation, has been reported to improve cardiac function in a rat model of AMI induced by the permanent ligation of the left anterior descending coronary artery (200). Decreased glucose oxidation and increased anaerobic glycolysis is also observed during postischemia ventricular fibrillation, with the addition of a stimulator of pyruvate dehydrogenase noted to decrease anaerobic glycolysis, induce glucose oxidation, and mitigate postischemic myocardial contractile dysfunction (11) (Fig. 2).
In addition, ischemia post-AMI decreases Na+/K+ pump activity that reduces ATP and alters Na+/H+ ion exchange via decreased intracellular pH, which drastically changes the intracellular ion balance by facilitating intracellular Na+ ion accumulation (27, 137) (Fig. 2). Multiple adverse events then ensue, including myocyte swelling due to altered osmotic gradients in response to elevated Na+ levels (21) and cellular Ca2+-overload via increased Na+/Ca2+ exchanger activity (80, 96), which manifests as contractile dysfunction (188), uncoupling of the mitochondrial electron transport chain, further ATP depletion in the damaged myocardium, cell swelling, and cardiomyocyte necrosis (Fig. 2) (50, 114).
Many modes of cell death are observed after AMI, including apoptosis (e.g., via intrinsic or extrinsic pathways), necrosis (e.g., via opening of the mitochondrial permeability transition pore; MPTP), and the more recently identified necroptosis. Necroptosis is a form of programmed necrosis that is elevated in failing human and rat hearts (94, 178) and can be induced by tumor necrosis factor (TNF) binding to the TNF Receptor 1 with a sequential activation of multiple intermediate mediators culminating in the activation of receptor-interacting protein 1 (RIP1), which activates receptor-interacting protein 3 (RIP3), which, in turn, activates mixed lineage kinase domain-like (MLKL). Thus, the RIP1-RIP3-MLKL axis is involved in necroptosis induction (219). It has been recently shown that RIP3 acts via Ca2+-calmodulin–dependent protein kinase (CaMKII), to activate MPTP opening and subsequent cell death (217). A key study reported that inhibition of RIP3 or CaMKII ameliorates ischemia reperfusion (I/R) injury in a mouse model of experimental AMI (217). Also, the administration of the micro-RNA, miR-105, a dual-inhibitor of RIP3-induced necroptosis and BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3)-induced apoptosis, decreased the levels of both apoptosis and necroptosis and reduced infarct size in a rat model of experimental AMI and reperfusion (165). On the other hand, TNF receptor-associated factor 2 (Traf2) has been recently shown to possess protective mechanisms in the heart by negatively modulating necroptosis through the regulation of the RIP1-RIP3-MLKL pathway (59). Knockout of Traf2 in mice resulted in adverse cardiac remodeling and heart failure, suggesting to be due to a feedforward mechanism that increases the levels of TNF to promote cell death in the absence of Traf2. However, deletion of the TNF receptor 1 largely but not fully rescued the development of adverse cardiac remodeling signaling that although TNF plays a role in adverse cardiac remodeling post-AMI, other TNF receptor 1 independent pathways might also be involved in this pathological pathway (59).
Another recently identified form of cell death is ferroptosis, which involves the uptake of iron into the cell and into the mitochondria prompting the excessive production of ROS (113) that mediates damage to the myocardium as discussed in this review. Notably, these ROS oxidize membrane lipids, resulting in the formation of lipid peroxides, a feature of ferroptosis (113). Thus, higher levels of cardiac non-heme iron were detected in mice subjected to experimental AMI with reperfusion. Blocking ferroptosis in these animals by using a pharmacologic inhibitor or an iron chelator decreased infarct size, levels of serum cardiac enzymes (lactate dehydrogenase, aspartate transaminase, and creatine kinase-MB isoenzyme), cardiac remodeling, and the extent of fibrosis (42). Hence, many cellular targets involved in initiating cell death pathways are potential therapeutic targets for the reduction of cardiomyocyte cell death post-AMI.
The timely restoration of coronary artery blood flow after AMI halts progressive cardiomyocyte death from ischemia and potentially rescues 30%–50% of cardiac tissue in the area at risk surrounding the infarct (69). However, reperfusion per se imposes the risk of further myocardial injury via the interlinked processes of inflammation and oxidative stress that in the long term can lead to chronic heart failure (121). Thus, acute reperfusion reintroduces oxygen, inflammatory mediators, and various ions (e.g., Na+ and Ca2+) that instigate a second wave of damaging oxidant formation; alters metabolic function(s); and/or activates immune responses that further damage cardiomyocytes (147). Accumulating ROS are involved in direct oxidative tissue damage and the redox activation of cell signaling pathways, including the key redox-sensitive transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulates several key inflammatory genes such as TNF, IL-1β, and IL-6 (122, 135). Augmented innate immune responses increase the local recruitment of activated leukocytes that can promote coronary microvascular blockage and impair NO bioactivity, resulting in endothelial dysfunction and heightened vascular inflammation (Fig. 2) (184).
Functional mitochondria are essential for cardiomyocyte viability, producing >90% of the ATP required for cardiac output (58). However, both ischemia and reperfusion induce mitochondrial damage and dysfunction that impact cardiomyocyte function and viability (88). For example, Ca2+ overload causes MPTP opening, leading to mitochondrial depolarization, ATP depletion, and myocyte necrosis (114). Also, excess Ca2+ and unregulated mitochondrial depolarization increase mitochondrial permeability, facilitating the release of cytochrome c and other cell death factors, thereby triggering myocyte apoptosis (162). Myocyte apoptosis can activate inflammasome signaling and proinflammatory cytokine production, for example, IL-1β (164). Another consequence of ischemia-induced mitochondrial dysfunction is electron transport chain disruption, which leads to a decrease in oxidative phosphorylation and an increased leakage of electrons that react with molecular O2 to form the superoxide radical anion (O2 •−) (Fig. 2). This increase in mitochondrial ROS can induce cellular oxidative and/or mitochondrial DNA (mtDNA) damage (166). Reperfusion after ischemia triggers a secondary phase of ROS production (88), which further limits ATP production and augments cellular ROS generation, events that, together with myocyte mitochondrial Ca2+ overload (114), provoke post-AMI cardiac dysfunction and eventual progression to heart failure (17).
Progression to Cardiac Dysfunction and Heart Failure After AMI
Although reperfusion of the ischemic myocardium is critical for patient survival post-ACS (193), AMI remains a leading cause of left ventricle (LV) dysfunction and cardiac-related death, with ∼25% of survivors subsequently progressing to chronic or congestive heart failure with 12 months after suffering an AMI (196). This secondary phase of pathology is characterized by cellular responses to the initial myocardial ischemia and, in particular, to the extent of cardiomyocyte necrosis within the infarct (47, 67). Adaptive ventricular remodeling after AMI involves the reorganization of supporting structures, myocyte hypertrophy, and collagen deposition (leading to scar formation). This latter feature is primarily the result of resident cardiac fibroblast activation, which responds to inflammatory stimuli by differentiating into a myofibroblast phenotype, proliferating, and subsequently causing maladaptive extracellular matrix (ECM) turnover; these adaptive processes lead to adverse matrix remodeling (111) and infarct expansion that is conducive to heart failure (191, 204).
Role of Innate Immune Cells After AMI and Reperfusion
The innate immune response after ischemic myocardial injury is aimed at facilitating beneficial tissue remodeling and repair of the damaged cardiac muscle (48). Neutrophils are the predominant early responder cells recruited into the myocardial infarct zone during the first 24–48 h post-AMI and reperfusion (Fig. 3). Neutrophils aggregate around the infarcted tissue in the area at risk, yielding a definitive hyperemic border, where these leukocytes play a role in phagocytosing necrotic cells, which can further augment the inflammatory response (209). Notably, neutrophils contain potent ROS-generating and proteolytic enzyme systems involving the coordinated activation of NADPH oxidase 2 (NOX2) and myeloperoxidase (MPO), as well as the release of MMPs and other proteolytic enzymes that initiate tissue remodeling and repair (116). In addition, the presence of cytokines (e.g., TNF, IL-1, IL-6) (132) and alarmin binding to neutrophil receptors activates signaling pathways involving mitogen activated protein kinases (MAPK) and NF-κB, further enhancing the local production of inflammatory cytokines (e.g., TNF, IL-1; IL-8) that recruit circulating Ly6Chi monocytes into the injured site from days 1 to 3 postinjury (118, 209) (Fig. 3). The population of infiltrating Ly6Chi monocytes then differentiate into macrophages of the M1 phenotype (between 3 and 5 days post-AMI) that secrete proteolytic enzymes and inflammatory cytokines such as IL-6 and IL-1β in an attempt to remove dying neutrophils and necrotic debris (118, 209).

Changes in monocyte and macrophage phenotype under the influence of neutrophils occur after the 5th day from AMI onset and help in the transition from the innate immune response to the repair phase of the acute response (47, 209). Thus, Ly6Clo monocytes, characterized by high expression levels of the chemokine receptor CX3CR1, predominate from day 5 post-AMI and differentiate into macrophages of the reparative M2 phenotype, which are an important source of anti-inflammatory mediators such as IL-10 and transforming growth factor beta (TGF-β) (209) (Fig. 3). These reparative macrophages further modulate ECM deposition and metabolism via regulating the expression of matrix-degrading MMPs and tissue inhibitors of MMPs (TIMPs) while stimulating the parallel activation, proliferation, and differentiation of resident fibroblasts into myofibroblasts. Recent studies have reported that similar to monocytes, different neutrophil populations are mobilized into the infarcted myocardium. Myocardial tissue injury stimulates the release of endogenous agents known as damage-associated molecular patterns (DAMPs), which activate TLR4 expressed on neutrophils and which promote the mobilization of inflammatory N1 neutrophils and the release of inflammatory cytokines (TNF and IL-1β) and matrix degrading enzymes such as MMPs that contribute to thinning of the LV wall. This front-line population of neutrophils can persist in the infarcted region over the first 5 days after injury, although their levels decrease over days 1–5 (Fig. 3). Another population of neutrophils that elicit anti-inflammatory characteristics is reported to increase around day 7 (101) (Fig. 3). As such, these changes in neutrophil population phenotype from inflammatory to anti-inflammatory parallel the changes observed in the monocyte phenotype post-AMI.
Fibroblasts make up ∼27% of all cells in the heart, with this number greatly increasing during injury (15). They also release angiogenic mediators such as vascular endothelial growth factor (VEGF), which initiates angiogenesis (118). These tissue repair processes are important in promoting scar formation and preventing cardiac rupture, adverse LV remodeling (208), and progression to heart failure (204). Further, TGF-β stimulates myofibroblasts to deposit collagen type I/III to yield scar formation [reviewed in Czubryt (30) and Van Amerongen et al. (192)], which acts to replace dying cardiomyocytes within the infarct zone with connective tissue that leaves a firm but thinned, noncontractile scar (118).
Studies with animal models have highlighted the importance of Ly6Clo monocytes in the resolution of inflammation and ventricular remodeling; for example, atherosclerosis-prone ApoE−/− mice characterized by a Ly6Chi monocytosis displayed elevated inflammation and deteriorated ventricular function 5 days post-AMI (134). Therefore, a balance is required between inflammatory Ly6Chi and reparative Ly6Clo monocytes to control the resolution of inflammation, myocardial healing, and repair (52). Neutrophils influence the recruitment and polarization of monocyte/macrophage populations post-AMI, and therefore, they can be involved in governing tissue repair and fibrosis. For example, an aberrant wound-healing response in neutrophil-depleted mice was characterized by a reduction in the removal of dying cardiomyocytes [a critical precursor for wound healing and collagen deposition (197)], excessive fibrosis, and ventricular dysfunction post-AMI, which were linked to a decreased recruitment of inflammatory M1 macrophages and reparative M2c macrophages and an increase in classic anti-inflammatory M2 macrophages (70).
These data suggest that neutrophils play dual roles, both positive and detrimental, in the post-AMI innate immune response involved in infarct wound resolution. This complexity of action indicates that neutrophils are a functionally and phenotypically heterogenous population of innate immune cells that depending on the local environment are capable of exhibiting a longer lifespan, which permits interaction with multiple immune cell types to shape innate and adaptive immune responses (167). Therefore, rather than broadly targeting neutrophils per se to limit post-AMI tissue damage, it may be more important to selectively target tissue-damaging neutrophil proteins, for example, the homodimeric and oxidative heme protein MPO.
Given the limited regenerative capacity of cardiomyocytes, fine-tuning of the reparative innate immune response by heterogeneous populations of neutrophils, dendritic cells, monocytes, and macrophages is vital for the successful restoration or preservation of adequate cardiac function (168). Endogenous anti-inflammatory signals and processes that suppress an excessive inflammatory response are important for transitioning away from further tissue injury both within and surrounding the infarct zone (47). A prolonged proinflammatory phase and/or limited reparative anti-inflammatory phase can exacerbate reperfusion injury and contribute to adverse tissue remodeling post-AMI, thereby increasing the risk of subsequent heart failure (7).
Inflammation Mediates Reperfusion Injury Post-AMI
A multitude of studies have shown that the degree of inflammation, both locally within the injured myocardium and systemically, is upregulated after AMI, with the latter response in some cohorts being predictive of adverse clinical outcomes, including a recurrent heart attack, heart failure, and death (10, 191). Various proinflammatory chemokines and cytokines are elevated in heart failure patients, suggesting that endogenous cytokines exert deleterious effects on the heart and peripheral circulation (105). Accordingly, preclinical studies support the fact that antagonism of innate immune receptors, signaling pathways, or proinflammatory cytokines attenuates adverse cardiac alterations post-AMI (52, 105). For example, IL-6 levels are elevated in heart failure patients and inversely correlate with LV function (140), whereas an IL-6 receptor antagonist alleviates inflammatory damage in patients with non-ST-elevation myocardial infarction (NSTEMI) (82).
Excessive or dysregulated reperfusion-induced inflammation promotes aberrant ECM degradation and deposition to yield excessive tissue fibrosis. Thus, increased levels of active MMPs, coupled with a downregulation of TIMP levels, are found in failing human hearts, which facilitate dysfunctional ECM remodeling (91, 190). Although coordinated ECM turnover is necessary for the removal of necrotic debris, an augmented or prolonged inflammatory response leads to excessive ECM degradation by MMPs that elevates fibrotic scar formation (36) that is associated with elevated immune cell infiltration in heart failure patients (204). Preclinical studies show that antifibrotic therapy affords a decrease in total and nonscar fibrosis, which improves LV function post-AMI (124).
ROS and Mitochondrial Dysfunction Contribute to Inflammation in Reperfusion Injury
The coordinated production of low levels of ROS, in particular hydrogen peroxide (H2O2), can act as endogenous redox cell signaling molecules that regulate immune and inflammatory responses (e.g., via activation of redox-sensitive transcription factors such as NF-κB and nuclear factor erythroid-2 related factor 2 [Nrf2]) (184). However, excessive or aberrant ROS production can promote dysfunctional signaling responses and directly mediate oxidative damage to proteins, lipids, and nucleic acids in inflammatory tissues, thereby adversely affecting cell function and stimulating the further production of oxidants that can result in positive feedback to augment inflammation (114).
A key factor orchestrating the postreperfusion inflammatory response is the elevated production of ROS and associated redox signaling. In the infarcted myocardium, a principal source of ROS is the oxidative burst of infiltrating activated leukocytes (neutrophils and monocytes) that involves the concerted activation of NOX2 and MPO (47). In addition to the phagocytic oxidative burst, local parenchymal cells also represent a significant source of ROS in response to I/R injury. For example, activated cardiomyocytes, myofibroblasts, or endothelial cells can produce ROS via various endogenous sources, including activation of several NOX isoforms including NOX 1, 2, 4, and 5, electron leakage from the mitochondrial respiratory chain, uncoupled NOS, or xanthine oxidase (57, 184).
Accumulation of ROS and resultant oxidative stress are implicated as drivers of chronic diseases characterized by a prolonged dysregulated inflammatory response, including atherosclerosis (136, 175), and they are associated with the severity of reperfusion injury post-AMI and progression to heart failure (103, 189, 201). For example, 8-iso-prostaglandin F2α, a commonly measured marker of in vivo oxidative stress, is elevated in patients with severe heart failure (103). Further, the tissue-specific overexpression of superoxide dismutase (SOD) in cardiac myocytes protects against myocardial reperfusion injury and results in a decrease in infarct size through a mechanism involving enhanced NO bioavailability (128). These studies support attempts at bolstering the myocardium's antioxidant capacity to provide cardioprotection in AMI.
Studies with mice deficient in p47phox (an essential subunit for NOX enzymes) displayed decreased fibrosis, apoptosis, and ventricular dilation in response to MI, identifying a pathogenic role for NOX-derived ROS post-AMI (40). Interestingly, highlighting their complex role in AMI, other studies report that NOX-derived ROS can also signal for protective adaptive responses to I/R injury, involving the activation of the HIF1α and Nrf2 transcription factor pathways (107) and signaling for the polarization of macrophages into a reparative M2 phenotype (117). The studies just cited indicate that the role of NOX-derived ROS in AMI is complex and that targeted, temporal NOX inhibition rather than global, persistent NOX inhibition is a more viable therapeutic value.
Unregulated ROS production during I/R injury can act to promote cardiac dysfunction in several ways. For example, ROS can selectively oxidize amino acid residues or active-site metal centers to target proteins that modulate cardiomyocyte function such as myoglobin (201) and electrical conductance such as the sarco-endoplasmic reticulum Ca2+ ATPase that are essential for the maintenance of intracellular Ca2+-homeostasis (28). Interestingly, hypochlorous acid (HOCl), an oxidant produced by the neutrophilic enzyme MPO on reaction with Cl− ions and H2O2, affects the peroxidase activity of myoglobin but does not impact its catalase-like activity (3). Dysregulated ROS production can also promote cardiomyocyte death via activating proapoptotic signaling kinases such as the MAPK p38 and jun N-terminal kinase (202, 211), inducing mitochondrial dysfunction involving the opening of the MPTP and/or through oxidatively damaging DNA that can activate poly(ADP-ribose) polymerase-1, which is intricately involved in cell death (189).
In addition to the NOX enzymes, mitochondria also yield ROS in response to I/R injury post-AMI. During ischemia, succinate from the citric acid cycle accumulates and is oxidized by succinate dehydrogenase on reperfusion, producing mitochondrial ROS by reverse electron transport at complex I (26). The ketoglutarate complex, an enzyme in the citric acid cycle, is inactivated in reperfusion, resulting in a decline in NADH-linked respiration (158). However, it is not studied widely in the field of myocardial I/R injury, with some studies reporting that although I/R injury involves mitochondrial damage, the damage is primarily attributed to changes in the mitochondrial electron transport chain and not the citric acid cycle enzymes (68). However, other studies have reported that progressive changes to the citric acid cycle can occur in the longer term after MI, as a maladaptive response to MI-induced metabolic alterations. These changes are proportional to the degree of impairment in the myocardium as a result of MI (39). Nonetheless, it is not a focus of current preclinical or clinical trials in AMI research.
Moreover, it is increasingly recognized that ROS produced by different NOX isoforms and mitochondria cross-talk to augment overall cellular production of ROS (37). A paradigm linking oxidative stress, mitochondrial dysfunction, and chronic inflammation is supported by recent studies reporting that mitochondrial-derived ROS stimulate inflammation by triggering proinflammatory redox-sensitive signaling events such as NF-κB activation or directly activating the NLRP3 inflammasome (164, 222). This is relevant to resident cardiac fibroblast cells; early inflammasome activation in cardiac fibroblasts has been shown to contribute to early I/R injury, and animal knockout studies targeting various components of the inflammasome system alleviated injury (77, 159). NLRP3 inflammasome activation has also been associated with atrial fibrillation (AF) in human patients (210), and NLRP3 inhibitors have alleviated AF and I/R injury in in vivo and ex vivo mouse models of experimental acute myocardial infarction and reperfusion (AMI/R) and AF (120, 210).
Mitochondrial dysfunction is a key event in AMI and resulting complications such as ventricular dysfunction, increased ROS production, and loss of ATP are features of failing hearts (19) (Fig. 2). On reperfusion, increased mitochondrial levels of ROS and Ca2+, together with the associated changes to pH, stimulate the opening of the MPTP channel (162), resulting in membrane swelling, subsequent mitochondrial release of cytochrome c into the cytoplasm, and eventual cell death (60). Blockage of the mitochondrial electron transport chain with reversible inhibitors, such as amobarbital, during ischemia reduces reperfusion-induced Ca2+-overload and ROS generation, thereby protecting against cardiomyocyte injury (24, 180). Further, damaged mitochondria and necrotic cells release DAMPs, such as DNA and ATP (215, 218), that stimulate sterile immune responses, the inflammasome, and the time- and stage-dependent activation of neutrophils and their polarization into inflammatory N1 neutrophils (7, 101, 139) (Fig. 3). Preclinical animal studies have linked mitochondrial DAMP production with cardiac inflammation and heart failure (119).
Therapies Targeting Inflammation and ROS in Reperfusion Injury Post-AMI
Current interventions for AMI focus on timely restoration of coronary flow via percutaneous coronary intervention (PCI). However, PCI stimulates reperfusion-tissue injury in the local coronary artery wall and adjacent cardiac tissue, which is associated with poor clinical outcomes (142). This supports the need for adjunct therapies that target the local inflammatory and oxidative response to reduce the accompanying reperfusion-stimulated injury subsequent to PCI.
Current data show that limiting the degree of inflammation, ROS production and mitochondrial dysfunction can ameliorate I/R injury and preserve cardiac function in both the early and late phases post-AMI (55, 104, 160). However, adjunct therapies that have proven beneficial in preclinical models of reperfusion injury have not yielded translational benefits in the clinic, especially in terms of reducing infarct size. Of particular note is the relative failure of translating positive phase I/II trial outcomes for interventions, such as cyclosporine A (CsA) and the MPTP inhibitor TRO40303, into long-term clinical benefit in large, multicenter trials (63, 65). Targeting inflammation or ROS-mediated oxidative damage through the use of broad-spectrum antioxidant therapies, such as vitamins E and C, has also proven largely unsuccessful in improving cardiovascular endpoints in large and long-term follow-up human trials in nondiabetic CAD patients (75, 163). Reasons for these disappointing clinical outcomes include the underlying incongruity between animal disease models and human pathology, reflecting differences in the timing of drug delivery, that is, prophylaxis delivery in animal models versus treatment of established disease in humans.
The poor outcomes in large cohort trials to date may arise from inadequate pharmacological targeting of the most relevant sources and specific types of ROS that induce cardiovascular dysfunction. Thus, although mitochondrial aberrations and activation of different NOX isoforms are pivotal in driving myocardial ROS production/inflammation post-I/R, nutritional and therapeutic antioxidants may not be accessible to the relevant intracellular sites or organelles to effectively target the locally produced ROS. In support of this, mitochondrial-targeted antioxidants, such as mitochondrial-targeted forms of coenzyme Q (MitoQ) or the nitroxide tempol (Mito-TEMPO) (2, 100), have been shown to provide cardioprotection (32) through limiting oxidative damage and inflammation, as well as maintaining mitochondrial function in animal models, although their efficacy remains to be verified in humans.
Studies have also characterized the changes to mitochondrial function/structure during reperfusion injury (65). For example, CsA, a natural product and immunosuppressant, binds to cyclophilin D in the MPTP, inhibiting its opening (157). Preclinical studies show improved ventricular functioning with CsA administration after myocardial I/R (212). However, these promising preclinical findings have to date not been translated into the clinical setting. Thus, the recent double-blind randomized CIRCUS trial reported that patients presenting with ST-elevated myocardial infarction (STEMI) who received intravenous CsA before reperfusion (PCI) therapy showed no alteration in circulating creatine kinase levels (a marker of cardiomyocyte death), LV ejection fraction, end diastolic volume, or heart failure incidence (29), an outcome validated by the subsequent clinical trial (132).
Another molecular target for ameliorating reperfusion injury post-MI that has received recent interest is the inflammasome that is activated by DAMPs released on myocardial damage (164). For example, an NLRP3 inflammasome inhibitor decreased infarct size in a mouse model of postischemic injury (106, 187). Studies have also investigated targeting IL-1, the major product of the activated inflammasome. For example, the VCU-ART2 trial assessed the potential of an IL-1 receptor antagonist, Anakinra, to alleviate inflammation and adverse events after AMI. Although Anakinra failed to decrease infarct size, the drug improved LV end-volume indices together with decreasing inflammation and rates of heart failure 48 months post-AMI (1). Nonetheless, this study employed a small cohort (n = 10) and, as such, the results may be difficult to interpret and extrapolate to the broader community, with a phase II trial currently underway (194). The recent CANTOS study utilized another agent (Canakinumab) to neutralize the pro-inflammatory IL-1 in a long-term, large patient cohort with previous MI. The trial authors reported positive reductions in the levels of the inflammatory marker C-reactive protein (CRP) and the incidence of cardiovascular events and death, although the trial was primarily designed to test outcomes of immunomodulation on atherosclerosis and did not explore the direct impact on cardiac function and health outcomes (149).
Modulation of proinflammatory signaling pathways also showed promise in preclinical trials. For example, a p38 MAP kinase inhibitor decreased infarct size in an adult rat model of ischemia, although cardioprotection was only observed when the drug was administered before reperfusion, thereby questioning the drugs' clinical utility (177). The SOLSTICE phase II trial reported that another p38 MAPK inhibitor, Losmapimod, attenuated inflammation markers and improved LV function after PCI in NSTEMI patients (123). However, these cardioprotective outcomes were not observed in the larger and more recently conducted phase III randomized LATITUDE–TIMI trial that evaluated Losmapimod in AMI before reperfusion therapy, whereby the drug did not reduce the risk of major ischemic cardiovascular events despite having a positive impact on reducing circulating levels of inflammatory (hsCRP) and heart failure (NT-proBNP) biomarkers (129).
Beta-blockers have been successful in early clinical trials (56, 131). The METOCARD-CNIC randomized clinical trial reported a significant decrease in infarct size and a significant increase in LV ejection fraction in patients who received an oral or intravenous dose of metoprolol before reperfusion (74). However, decreased infarct size was only significant in patients with no or incomplete perfusion beyond the site of occlusion and infarct size was not improved significantly in patients with partially or completely restored blood flow in the distal regions beyond blood flow occlusion (74). Conversely, the EARLY-BAMI trial reported no significant change in infarct size in patients who received intravenous metoprolol before PCI (150), although differences in drug dosage and timing between the two studies may explain these apparent discrepancies (69). The mechanism underlying β1-adrenergic-receptor cardioprotection in reperfusion remains unclear, as the majority of these studies were performed before PCI was implemented as a routine clinical treatment for AMI. However, adrenergic signaling is linked to inflammation, in particular the activation of the neutrophil oxidative burst, adhesion to endothelium, and tissue migration (161). Interestingly, metoprolol's ability to reduce the infarct size in a pig model of AMI was reported to be independent of the drug's effect on heart rate and to instead correlate with a reduction in neutrophil infiltration (measured as MPO activity) and apoptosis in the postischemic myocardium (73). More recently, metoprolol has been demonstrated to reduce infarct size by altering neutrophil activity and neutrophil-platelet interactions in AMI patients (51). Hence, targeting neutrophil function and ROS production is a promising strategy in limiting the degree of chronic inflammation and reperfusion injury occurring after AMI. The next section discusses the role of the abundant neutrophil-derived oxidative enzyme MPO in promoting cardiac dysfunction post-MI and the extent to which it represents a useful therapeutic target.
Myeloperoxidase
Neutrophils are the first immune responders to tissue injury and infection. Neutrophil activation stimulates the release of MPO from its azurophilic storage granules into the phagosome or extracellular environment where MPO employs its co-substrate H2O2 (from the plasma membrane NOX2 enzyme) to catalyze the halogenation and peroxidase redox cycles (81, 206). On reaction with H2O2, the heme-iron undergoes a 2-electron oxidation to yield MPO-compound I, a ferryl-oxo (FeIV = O) species, and a heme porphyrin free radical (Fig. 4, Reaction A). MPO-Compound I catalyzes the two-electron halide oxidation to yield their respective hypohalous acids (predominantly HOCl) (Fig. 4, Reaction B). The potent oxidant HOCl reacts with a large array of biomolecules, including protein amino acids and amines, heme proteins, thiols, iron-sulfur complexes, nucleic acids, and lipids (34). Due to its pKa of 7.53 (6), HOCl is primarily protonated and membrane permeable under the postischemic acidic conditions in the myocardium. HOCl chlorinates biomolecules forming chloroamines, chlorinated lipids, or chlorinated amino acid residues including tyrosine to form 3-chlorotyrosine (3-Cl-Tyr), which is commonly assayed by using quantitative mass spectrometry and employed as a specific in vivo biomarker for MPO-derived production of HOCl (34).

The porphyrin radical of MPO-compound I can also mediate the one-electron oxidation of various low-molecular-weight substrates (e.g., NO, nitrite, tyrosine, urate, ascorbate) (Fig. 4, Reaction C) to form compound II (referring to the remaining ferryl-oxo species) and the one-electron oxidation product of the substrate. Compound II can mediate a second one-electron oxidation reaction to cycle the enzyme back to its native state (Fig. 4, Reaction D). A notable biological reductant for MPO-compound II is O2 •− that yields ferric MPO, which is then able re-enter halogenation/peroxidation cycles. Also, O2 •− can react with ferric-MPO to form MPO-compound III (formerly a ferrous iron-oxygen [FeII-O2] complex) (Fig. 4, Reactin E). In addition to the complexity of MPO's catalytic cycle, O2 •− can also react with MPO-compound III to re-form native ferric-MPO, while generating oxygen and H2O2 (78) (Fig. 4, Reaction F). Compound III is also a substrate for serotonin (HT), which also reduces it back to ferric MPO (207) (Fig. 4, Reaction G).
With respect to the physiologically relevant peroxidase substrates, nitrite is of considerable significance. On reaction with MPO compound I or II, nitrite is converted into the highly reactive nitrogen dioxide radical (•NO2), which is capable of initiating lipid peroxidation (216) and reacting with tyrosines to form 3-nitrotyrosine; a biomarker of oxidative tissue damage in disease (170).
Neutrophils and MPO in Cardiac Dysfunction Post-AMI
As indicated earlier, neutrophils are recruited early into the infarcted myocardium post-AMI (209), where they respond to DAMPs derived from damaged mitochondria and dying cardiomyocytes and release mediators that augment inflammation to resolve the ongoing tissue injury (139). Activated neutrophils also secrete an array of proteolytic and oxidative substances, including proteinases and MPO, with the potential to further promote myocardium injury, DAMP production and establish a chronic, low-grade inflammation (61). Hence, there is a potential for a pathogenic role of neutrophils in CAD and cardiac inflammation (127). Consistent with this, neutrophil recruitment into the infarct zone and surrounding areas is a prominent feature of ischemic cardiac injury (209) and neutrophils are causally linked to the microvascular dysfunction occurring during reperfusion injury and the microvascular disturbances associated with the no-reflow phenomenon, defined as the incomplete reperfusion of blood to areas of the myocardium (95). Also, poor prognosis of CAD patients is associated with elevated systemic neutrophil levels (8, 53), MPO (182) and neutrophilia correlates with the subsequent development of chronic heart failure post-AMI (86).
Neutrophil-derived MPO has received considerable attention as a mediator of cardiovascular disorders. MPO is mechanistically linked to several key processes in the pathogenesis of CAD, including atherosclerosis, myocardial injury, endothelial and microvascular dysfunction, and plaque formation and vulnerability (33, 125). Clinical studies have shown that elevated circulating extracellular MPO is a valuable prognostic index for adverse events after AMI (79, 115), chest pain (18), and chronic heart failure (181). Notably, leukocyte MPO content significantly decreases on traversing the coronary circulation in ACS patients with unstable angina, consistent with intraluminal leukocyte degranulation and extracellular MPO release (20). Also, MPO levels in AMI patients increase acutely and remain elevated 4 h after AMI onset in patients, thereby supporting the acute activation of neutrophils in response to ischemia and before myocardial damage (54).
Importantly, MPO and biomarkers indicative of its oxidants (e.g., 3-Cl-Tyr, 3-NO2-Tyr, HOCl-oxidized and carbamylated proteins) are detected in diseased tissue, including atherosclerotic lesions of CAD patients and the atrium of AF patients (33, 66, 125, 155, 220). Further, infarcted myocardium exhibits increased levels of MPO-derived oxidation products, including chlorinated plasminogen activated inhibitor 1 (PAI-1) (9), amino acid-derived aldehydes (195), chlorinated lipids (185), and oxidized myoglobin (201).
Preclinical studies with MPO genetically modified mice indicate a role for leukocytes and MPO-generated oxidants in the pathophysiologic consequences of atherosclerosis (109), plaque rupture (143), and myocardial remodeling after ischemia (9, 195). With respect to the myocardial ischemia, MPO's pathogenic actions have been linked to the elevated infiltration of myocardial leukocytes, oxidative inactivation of PAI-1, and impaired LV function and potentially heart failure post-AMI (9) involving the MPO-dependent formation of cytotoxic aldehydes (195). Interestingly, the significant LV dysfunction induced by MPO-derived oxidants post-AMI is independent of changes in infarct size (195). Human AF, which is associated with ongoing inflammation and cardiac dysfunction occurring after an AMI, is also linked to increased levels of MPO in the plasma and cardiac tissue of AF patients (155). Moreover, in a mouse model of angiotensin II-induced atrial fibrosis and fibrillation, wild-type mice exhibited elevated MPO deposition, HOCl production, MMP activity, and atrial fibrosis/fibrillation, events abrogated in MPO gene-deficient mice (116, 155).
Mechanistically, MPO-derived oxidants react with bystander cells in the heart (including endothelial cells, resident cardiac fibroblasts, and cardiomyocytes) to potentiate cardiac dysfunction and promote endothelial/vasomotor dysfunction, adverse tissue remodeling, high-density lipoprotein (HDL) dysfunction, and cytotoxicity, all components underlying progression to heart failure (49, 145, 154, 203, 220).
A characteristic of CAD and ACS is the increased intravascular release of MPO by circulating leukocytes that has the potential to bind to and undergo transcytosis (13) into the vascular endothelium of local infarct-related coronary arteries (12) and cardiac tissue of atrial fibrosis patients (155). Within arteries, the transcytosed MPO accumulates in the sub-endothelium (12, 13), where it is positioned to catalyze local oxidative reactions that can promote endothelial dysfunction and impair vasomotor tone (145). For example, various studies have shown that MPO negatively modulates vascular tone by binding to the endothelium and impairing vasorelaxation by decreasing NO bioavailability (41, 154). Mechanistically, endothelial-transcytosed MPO can impair endothelial-derived NO bioactivity by producing HOCl or via its NO oxidase activity that limits bioavailable NO within the vessel wall (184). With respect to HOCl, exposure of the endothelium to this oxidant promotes the uncoupling of electron flow through endothelial nitric oxide synthase (eNOS), which stimulates endothelial O2 •− production that rapidly reacts with NO to impair its bioactivity (174). HOCl also limits NO production by oxidatively inactivating soluble guanylate cyclase that transmits/amplifies NO signaling in the vessel wall (25).
The NO oxidase function refers to the catalytic consumption of NO via the peroxidase cycle of MPO that can occur via direct NO oxidation by MPO compounds I and II or indirectly by the MPO-catalyzed one-electron oxidation of physiological substrates tyrosine, ascorbate, and urate that form NO-consuming substrate-derived radicals (41, 102, 110, 146). A role for the NO oxidase function as a cause of impaired NO bioactivity has been provided by experimental and clinical studies. For example, Eiserich et al. documented that endothelial-sequestered MPO in the presence of H2O2 catalytically consumed NO to compromise endothelial-dependent vasorelaxation of isolated arteries (41). Rudolph et al. reported that nicotine infusion into humans induced the intravascular release of MPO by circulating leukocytes and the acute impairment of endothelial-dependent vasomotor function (154). Notably, the nicotine-induced endothelial dysfunction was abrogated in MPO-deficient humans. The increased propensity for human AMI patients to consume NO has also been attributed to the NO oxidase activity of circulating MPO, which correlated with impaired endothelium-dependent microvascular dysfunction in patients with CAD (14).
Neutrophil-MPO-catalyzed oxidation reactions can also promote endothelial dysfunction via mechanisms independent of their effects on NO bioactivity. For example, HOCl derived from sub-endothelial MPO oxidatively modifies ECM proteins, such as fibronectin, which activates a novel mode of redox-sensitive “outside-in” signaling that activates focal adhesion and actomyosin signaling pathways to promote endothelial cell contractile responses and increased endothelial permeability (Fig. 1) (145). Also, exposure of endothelial cells to low, nontoxic concentrations of HOCl stimulates the expression of the potent thrombogenic tissue factor protein (176).
Chlorinated lipids result from the HOCl-mediated modification of plasmalogen phospholipids, which contain a vinyl ether bond and are present in endothelial cells and cardiomyocytes (171). Chlorination of the vinyl ether yields α-chloro fatty aldehydes such as 2-chlorohexadecanal (2-ClHDA) (4). Elevated levels of 2-ClHDA have been reported in the heart of rat models of experimental AMI and absent in infarcted hearts of neutropenic rats (185). These modified lipids can react with endothelial cells to promote endothelial cell dysfunction that contributes to atherogenesis by increasing (i) the expression of P-selectin to elevate endothelial-leukocyte interactions, (ii) the release of von Willebrand factor that can promote platelet adhesion, and (iii) angiopoietin-2 that increases endothelial permeability (Fig. 1B) (62). Hence, chlorinated lipids present another potential mediator of post-AMI and I/R cellular injury and dysfunction that is linked to the activity of MPO.
In addition to HOCl, MPO can produce hypothiocyanous acid (HOSCN) that promotes endothelial inflammation and dysfunction by upregulating the NF-κB-dependent expression of endothelial tissue factor or leukocyte adhesion molecules (198, 199), as well as causing mitochondrial dysfunction and release of proapoptotic factors, including cytochrome c, apoptosis-inducing factor, and endonuclease G (97).
Neutrophil-derived MPO is also linked with adverse tissue remodeling through the ability of HOCl to oxidize critical amino acid residues of proteins. For example, HOCl oxidizes cysteine thiol residues within the cysteine switch domain of MMPs, resulting in activation of the enzyme's proteolytic activity (49). Conversely, HOCl inhibits TIMPs by oxidizing the N-terminal cysteine, thereby impairing their inhibitory activity toward MMPs (203). Together, MPO-derived HOCl results in the unregulated activation of MMPs (Fig. 5), thereby facilitating dysregulated ECM turnover and potentially contributing to excessive fibrosis and tissue remodeling within the inflamed heart. Consistent with this, an increased colocalization of atrial MPO and the HOCl-biomarker 3-Cl-Tyr in human AF patients is linked to aberrant tissue remodeling and fibrosis, as well as electrical dysfunction in the heart atrium (155).
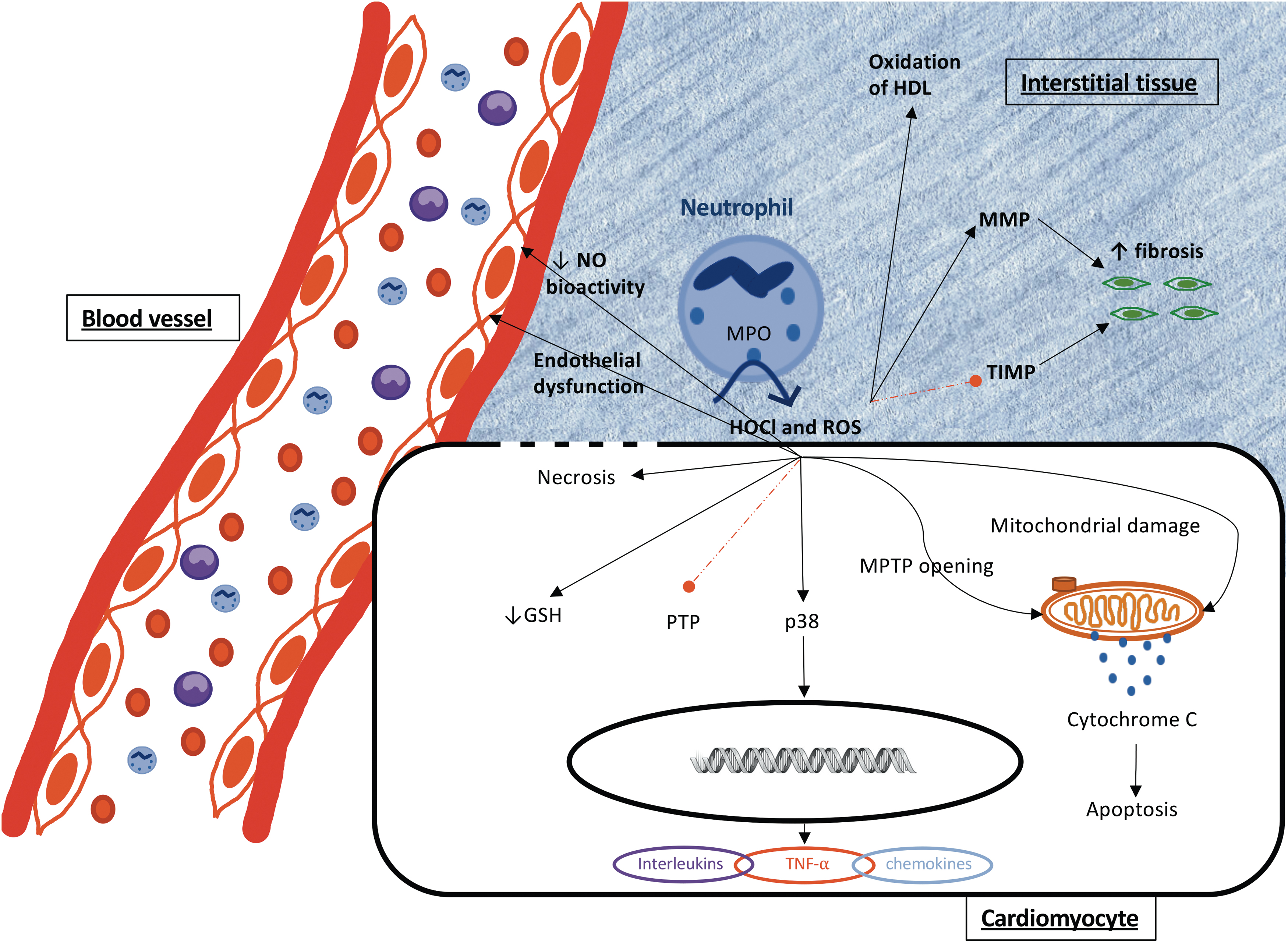
Characteristically, CAD and ACS/AMI patients display increased conversion of HDL into a dysfunctional form that exhibits a loss or impairment of the cardioprotective actions of HDL concomitant with a gain of specific proinflammatory properties (151, 172). During cardiovascular disease, apolipoprotein A-1 (ApoA-1; the major apo-protein component of HDL) is a major binding partner and oxidation target of MPO and oxidation/nitration of specific amino acid residues in ApoA-1 is a significant cause of impaired cholesterol efflux, antioxidant and anti-inflammatory activities of HDL (16, 220). Therefore, the MPO-catalyzed oxidation of HDL and loss of HDL cardioprotective actions represents a further mechanism by which MPO may exacerbate local ongoing cardiovascular inflammation in ACS/AMI patients.
Potent oxidants generated by MPO can contribute further to tissue damage during AMI by oxidizing intracellular thiols and antioxidants and promoting redox cell signaling that alters the function/phenotype of cardiomyocytes and non-cardiomyocyte bystander cells such as endothelial cells and promote cytotoxicity (206). Thus, HOCl readily oxidizes reduced glutathione (GSH) (Fig. 5), yielding oxidation products including GSH disulfide (22) and GSH sulfonamide (64). Major intracellular targets for HOCl also include thiol-containing enzymes, including GAPDH, peroxiredoxins (97, 141, 173), and iron-sulfur and heme-containing enzymes (179), including those present in the mitochondria.
The ability of HOCl/HOSCN to alter the redox status of labile amino acid residues (e.g., cysteines, tyrosines, methionines) and active-site metal centers also modulates the activities of signaling proteins, including phosphatases, protein kinases, and transcription factors (23, 112) (Fig. 5). Specifically, the modulation of the cellular phosphoproteome involves promoting phosphorylation by activating MAPK (e.g., p38) and deactivating protein tyrosine phosphatases (PTPs); the modulated phosphoproteome then shifts to a hyperphosphorylated state that ultimately decreases cell viability (Fig. 5). For example, the exposure of cardiomyocytes to exogenous HOCl inhibits phosphatase activity concomitant with the activation of kinase activity and promotion of apoptosis in a co-culture model of cardiomyocytes and activated HOCl-producing neutrophils (23). In endothelial cells, exposure to HOCl induces MAP kinase signaling involving the activation of Erk1/2 and p38, but not JNK, with the activation of Erk1/2 being a protective signaling response to the elevated oxidative stress (112). Activation of the Nrf2-ARE axis and AP-1 transcriptional responses have also been identified as protective intracellular signal transduction responses on exposure to MPO-derived oxidants (148).
The decrease in intracellular levels of low-molecular-weight and enzymatic thiol-based antioxidants together with the induction of aberrant redox signaling and impairment of fundamental metabolic processes (mitochondrial respiration) leads to unregulated cytotoxicity. Exposure of cardiomyocytes to HOCl and HOSCN increases cytosolic calcium, depletion of protein thiols and GSH, alters mitochondrial membrane potential, and increases necrosis (148). In endothelial cells, exposure to HOCl or HOSCN promotes oxidation of intracellular thiols and antioxidants and loss of cell viability (97, 176). Clinical data indicate that circulating MPO is strongly associated with a higher incidence of endothelial erosion in coronary atherosclerotic plaques, supporting that MPO induces loss of endothelial viability in vivo (43).
Pharmacological Inhibition of MPO
In light of MPO's pathogenic potential in various inflammatory disorders, there is currently significant interest in the discovery and development of new potent and selective MPO inhibitors. Two major classes of inhibitors have received most attention: irreversible or suicide inhibitors and reversible inhibitors.
Irreversible MPO inactivators (186) act by rapidly reacting with MPO compound I to form a drug-derived radical that subsequently forms a covalent drug-heme adduct with the MPO active site to irreversibly inactivate the enzyme. Overall, 2-thioxanthines are a potent and selective class of irreversible MPO inhibitors (186) (Fig. 6). This nontoxic class of pharmacologic inhibitors of MPO potently inhibited the chlorination (186) and NO oxidase (102) activities of purified MPO, mitigated the formation of HOCl by activated neutrophils in a mouse model of peritonitis (186), and provided protection against endothelial dysfunction (25) and atherosclerotic plaque instability in mice (143). Due to these promising outcomes, 2-thioxanthines have entered phase I clinical trials for COPD and multiple sclerosis and phase IIA trials in Parkinson's disease (31).
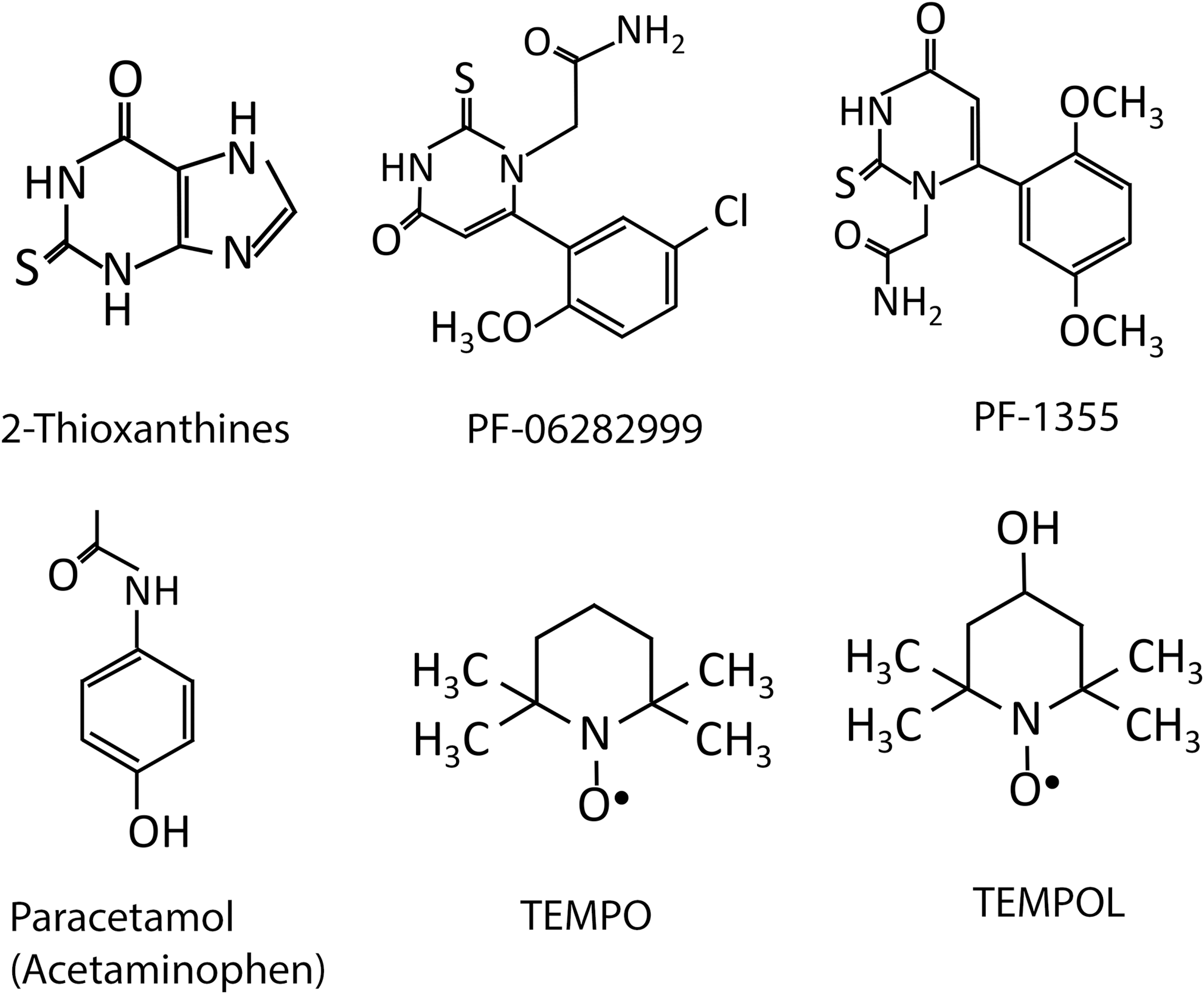
Other irreversible mechanism-based MPO inhibitors that have received recent interest are thiouracil derivatives, of which PF-06282999 and PF-1355 are lead candidates (156) (Fig. 6). Inhibition of MPO by PF-1355 is reported to provide protection in murine models of immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis (221). Recent work has reported that although administration of PF-06282999 to atherosclerosis-prone mice did not slow lesion formation, it did reduce the extent of lesion inflammation and necrotic core size, a finding indicative of enhanced plaque stability (153). Notably, oral PF-1355 treatment also reduced MPO-mediated tissue damage in a mouse MI model, reducing the inflammatory cell infiltrate and improving myocardial function as judged by improved LV ejection fraction and reduced end-diastolic volume (5). This study, together with studies using MPO gene-deficient mice (9, 116, 195), highlight that targeting MPO is a viable and promising therapeutic option for treating post-AMI cardiac dysfunction.
Reversible MPO inhibitors are enzyme substrates that are able to competitively react with MPO's active site heme to reversibly inhibit enzyme activity. These inhibitors can act in several ways: (i) by reversibly binding to ferric-MPO to occupy the enzyme's active site and effectively inhibit access of physiological substrates (e.g., aromatic hydroxamates) (46), (ii) effectively competing with physiological substrates for reaction with MPO compounds I and II (e.g., paracetamol also known as acetaminophen) (84) (Fig. 6), (iii) by competitively reacting with MPO compound I, but not MPO compound II, thereby promoting the build-up of compound II in the peroxidase cycle (e.g., nitroxides) (144), and (iv) promoting the formation of MPO compound III (e.g., isoniazid) (45).
Promising classes of reversible MPO inhibitors with therapeutic potential include aromatic hydroxamates and cyclic nitroxides. With respect to the aromatic hydroxamates, these agents selectively dock into the active-site pocket of native MPO to retard subsequent substrate binding and potently inhibit the enzyme's halogenation and peroxidase cycles (46). This reversible mode of inhibition has a potential advantage over substrate-based irreversible inhibitors, as MPO's oxidative capacity is selectively blocked without permanently inactivating the enzyme. To date, aromatic hydroxamates have not been trialed in preclinical inflammatory models for their therapeutic potential (46).
Cyclic nitroxides are stable free radicals characterized by a backbone containing a five-membered pyrrolidine, pyrroline, or oxazolidine or a six-membered piperidine ring with a nitroxyl group (89) (Fig. 6). Several classes of nitroxides have received wide interest as a versatile group of therapeutic antioxidant drugs that are capable of reducing the extent of inflammation and reperfusion injury in various tissues (205) such as the kidney (214), brain (76), intestinal (183), and ovarian tissue (138). Notably, cyclic nitroxides are a class of therapeutic radical scavengers that also exhibit potent MPO inhibition via a reversible mechanism (144). These compounds are cell permeable, display relatively low toxicity, with low-millimolar concentrations achievable in vivo with no apparent acute or long-term toxic effects (169). Accordingly, nitroxides are currently used clinically in the treatment of radiation-induced alopecia and macular degeneration (213). Nitroxides exhibit multiple antioxidant properties, including an ability to directly scavenge free radicals and act as SOD mimetics (89, 130) with catalytic self-regeneration.
Nitroxides have been identified as reversible MPO inhibitors that inhibit HOCl production by rapidly reacting with MPO compound I to form the corresponding nitroxide oxoammonium cation and MPO compound II, thereby diverting the enzyme from its halogenation cycle (144). As nitroxides are poor substrates for compound II, this redox intermediate of MPO accumulates and HOCl production is effectively limited with an IC50 of ∼1 μM reported for 4-amino-TEMPO (144). A potential drawback for nitroxides as MPO inhibitors is that physiological small-molecule substrates (e.g., ascorbate, O2 •−) can reduce MPO compound II back to native MPO, which can limit the inhibitory capacity of competitive substrate MPO inhibitors such as nitroxides in complex biological systems (144).
Despite this, nitroxides effectively inhibit the production of HOCl by activated neutrophils that also produce elevated levels of O2 •−, with an IC50 of ∼6 μM reported for 4-amino-TEMPO (144). The ability of nitroxides to maintain their inhibitory capacity in the presence of O2 •− likely reflects their actions as SOD mimetics. As such, the pleiotropic antioxidant actions of nitroxides that include radical scavenging, SOD mimetic activity, and reversible MPO inhibition mean that they have greater potential in alleviating oxidative tissue damage within complex inflammatory tissues compared with selective MPO inhibitors.
Of the nitroxides, 4-aminoTEMPO and 4-hydroxy-TEMPO (tempol) represent the most potent inhibitors of HOCl production by MPO (144). Another nitroxide, 4-methoxy TEMPO, attenuated inflammatory damage in a cardiomyocyte cell line incubated with activated neutrophils (23). In this coculture system, exposure of cardiomyocytes to activated neutrophils as a cellular source of MPO-derived HOCl increased oxidative stress, redox cell signaling, and cardiomyocyte apoptosis. The addition of 4-methoxy TEMPO decreased the formation of 3-Cl-Tyr, indicating the successful inhibition of MPO catalytic activity, restored the balance of the intracellular phosphoproteome and protected cardiomyocytes against apoptosis (23). Nitroxides have also been reported to protect cardiomyocyte plasma membranes against rigidification in vivo in mice treated with the cardiotoxic anthracycline drugs aclarubicin and doxorubicin (83).
A limitation for use of nitroxides as pharmacological agents is the short half-life of the active nitroxide moiety that is reduced by biological reductants to the corresponding hydroxylamine, which may exhibit variable pharmacokinetics (85, 90). Therefore, current studies of nitroxides have focused on designing approaches to ensure the longevity of the drug within the desired tissue. For example, an injection of 4-amino TEMPO covalently bonded to thermally responsive hydrogels (that enhance the nitroxide in vivo lifetime) at different infarct zones after coronary ligation/reperfusion in a rat model of AMI reduced the levels of apoptotic cells in the infarct and border regions and decreased oxidative stress in the infarcted hearts, resulting in improved overall cardiac functioning to a greater extent compared with an injection of 4-amino TEMPO alone (223). Although hydrogels mitigated oxidative heart damage, a number of limitations impact their use, including inability to enter cells and scavenge intracellular ROS. Further, studies are required to assess whether the introduction of hydrogels disturbs cardiac electrical conductance or activates immune cascades in response to foreign material (223). Another approach to enhance the half-life of tempol in vivo is its administration in conjunction with polynitrated albumin, which ensures the recycling of the hydroxylamine, that is, conversion of the biodegraded form of tempol back into the active tempol form. This approach enhanced tempol's half-life to 30 min in an in vivo rat model of I/R injury and subsequently decreased infarct size post-AMI/R, whereas the administration of tempol alone failed to replicate this protective effect (90).
Together, what has been cited earlier identifies nitroxides as a promising drug class to target oxidative tissue damage mediated by activated neutrophils post-AMI. Further studies are required to determine the efficacy by which novel, tissue-targeted nitroxides protect the heart post-AMI and to determine the extent to which this cardioprotective activity can be attributed to targeting MPO versus other documented antioxidant activities.
Concluding Remarks
After AMI, neutrophils, the first immune cell infiltrators into the damaged myocardium, degranulate and release MPO. MPO catalyzes the production of various oxidants, including HOCl, which are capable of inducing oxidative stress, mitochondrial dysfunction, and decreased cardiomyocyte viability and activating MMPs, leading to increased ECM turnover, fibrosis, and infarct expansion. Neutrophils and MPO levels are associated with a worse prognosis in patients post-AMI, and studies with MPO gene-deficient mice and irreversible MPO inhibitors establish a pathogenic role for the enzyme during AMI. As such, targeting neutrophil-derived MPO by pharmacological inhibitors may be a promising therapeutic strategy to limit tissue damage post-AMI (particularly in the myocardial area at risk) and subsequently diminish heart failure rates in humans. For example, nitroxides show significant potential in combating I/R tissue injury as they are capable of simultaneous ROS scavenging, anti-inflammatory actions, and MPO inhibition and, therefore, represent a promising class of versatile redox-active drugs in treating post-AMI injury and preventing heart failure. Further studies are warranted to determine the efficacy by which novel MPO-targeted inhibitors such as cyclic nitroxides with improved cardiac tissue half-life can ameliorate post-AMI tissue injury and the therapeutic window for the administration of this drug class as an adjunct therapy to PCI.
Footnotes
Funding Information
The authors acknowledge funding from the Australian Research Council (DP0878559 and DP160102063 grants to P.K.W.), NHMRC Project Grant 1125392 awarded to S.R.T. and P.K.W., and CSIRO SIEF STEM+ Business Fellowship to B.S.R.