
Abstract
Significance:
Over the past several years, oxidative post-translational modifications of protein cysteines have been recognized for their critical roles in physiology and pathophysiology. Cells have harnessed thiol modifications involving both oxidative and reductive steps for signaling and protein processing. One of these stages requires oxidation of cysteine to sulfenic acid, followed by two reduction reactions. First, glutathione (reduced glutathione [GSH]) forms a S-glutathionylated protein, and second, enzymatic or chemical reduction removes the modification. Under physiological conditions, these steps confer redox signaling and protect cysteines from irreversible oxidation. However, oxidative stress can overwhelm protein S-glutathionylation and irreversibly modify cysteine residues, disrupting redox signaling.
Recent Advances:
The latest studies of glutaredoxin-1 (Glrx) transgenic or knockout mice demonstrate important distinct roles of Glrx in a variety of pathologies. Endogenous Glrx is essential to maintain normal hepatic lipid homeostasis and prevent fatty liver disease. Further, in vivo deletion of Glrx protects lungs from inflammation and bacterial pneumonia-induced damage, attenuates angiotensin II-induced cardiovascular hypertrophy, and improves ischemic limb vascularization. Meanwhile, exogenous Glrx administration can reverse pathological lung fibrosis.
Critical Issues:
Glutaredoxins mainly catalyze the removal of protein-bound GSH and help maintain protein thiols in a highly reduced state without exerting direct antioxidant properties. Conversely, glutathione S-transferase (GST), peroxiredoxins, and occasionally glutaredoxins can also catalyze protein S-glutathionylation, thus promoting a dynamic redox environment.
Future Directions:
Although S-glutathionylation modifies many proteins, these studies suggest that S-glutathionylation and Glrx regulate specific pathways in vivo, and they implicate Glrx as a potential novel therapeutic target to treat diverse disease conditions. Antioxid. Redox Signal. 32, 677–700.
Introduction
Oxidative post-translational modifications (OPTM) of protein cysteines (Cys) have emerged as a cellular signaling mechanism to regulate physiology, and they become dysregulated in pathophysiology. Reactive oxygen species (ROS) oxidize thiols (R-SH) to reversible sulfenic acid (R-SOH), and further oxidation forms sulfinic acid (R-SO2H) and irreversible sulfonic acid (R-SO3H), which may permanently alter the function of the protein. Reactive nitrogen species also interact with thiols to form reversible S-nitrosothiols (R-SNO), commonly referred to as S-nitrosylation. R-SNO and R-SOH are intermediates with a fast turnover rate known to enable redox signaling. Other thiols may react with these reversible oxidation forms and result in either an intra- or intermolecular disulfide, or a mixed disulfide with a small thiol glutathione (reduced glutathione [GSH]), a process termed S-glutathionylation (100, 103, 146, 185). The reductive cellular environment or catalysis by glutaredoxins will remove the S-glutathionylation and restore the protein cysteine. This reversible process permits redox signaling and may protect proteins from irreversible oxidation. S-glutathionylation is reported on a large number of proteins and regulates a wide variety of cellular functions, including signal transduction, transcription, cytoskeletal assembly, cell survival, and apoptosis (127, 146).
In this article, we review redox signaling by focusing on the chemical background of thiol modification, the enzymatic regulation of S-glutathionylation, and the biological roles of glutaredoxin-1 (Glrx). Thiol modifications can also occur through radical-mediated pathways, which have been extensively discussed elsewhere. Glutathione S-transferase (GST) and glutaredoxins are key enzymes that regulate the redox signaling cycle. Glrx may exhibit some overlapping function with other reducing proteins such as thioredoxins (7, 51, 66, 78, 87, 144). However, studies of Glrx transgenic or knockout mice elucidate the unique in vivo abilities of Glrx to regulate metabolism, angiogenesis, inflammation, and fibrosis (11, 17, 62, 129, 130, 163, 180).
Chemistry of Redox Signaling
Biology and chemistry of cysteines
Cysteine is a semi-essential amino acid that is structurally similar to serine (Fig. 1A). Instead of oxygen, cysteine contains sulfur in its side chain, forming a thiol. A thiol is a functional group consisting of sulfur and a hydrogen atom and, thus, is also referred to as a “sulfhydryl.” The thiol or thiolate (deprotonated negatively charged thiol) group confers unique chemical properties to cysteines, including extreme nucleophilicity, high-affinity metal binding to form zinc fingers and iron-sulfur clusters, and the ability to create structural and regulatory disulfides (149). Another closely related amino acid is selenocysteine, which harbors selenium instead of sulfur. Both proteinogenic amino acids play an indispensable role in enzyme catalysis, redox signaling, and cellular redox status.
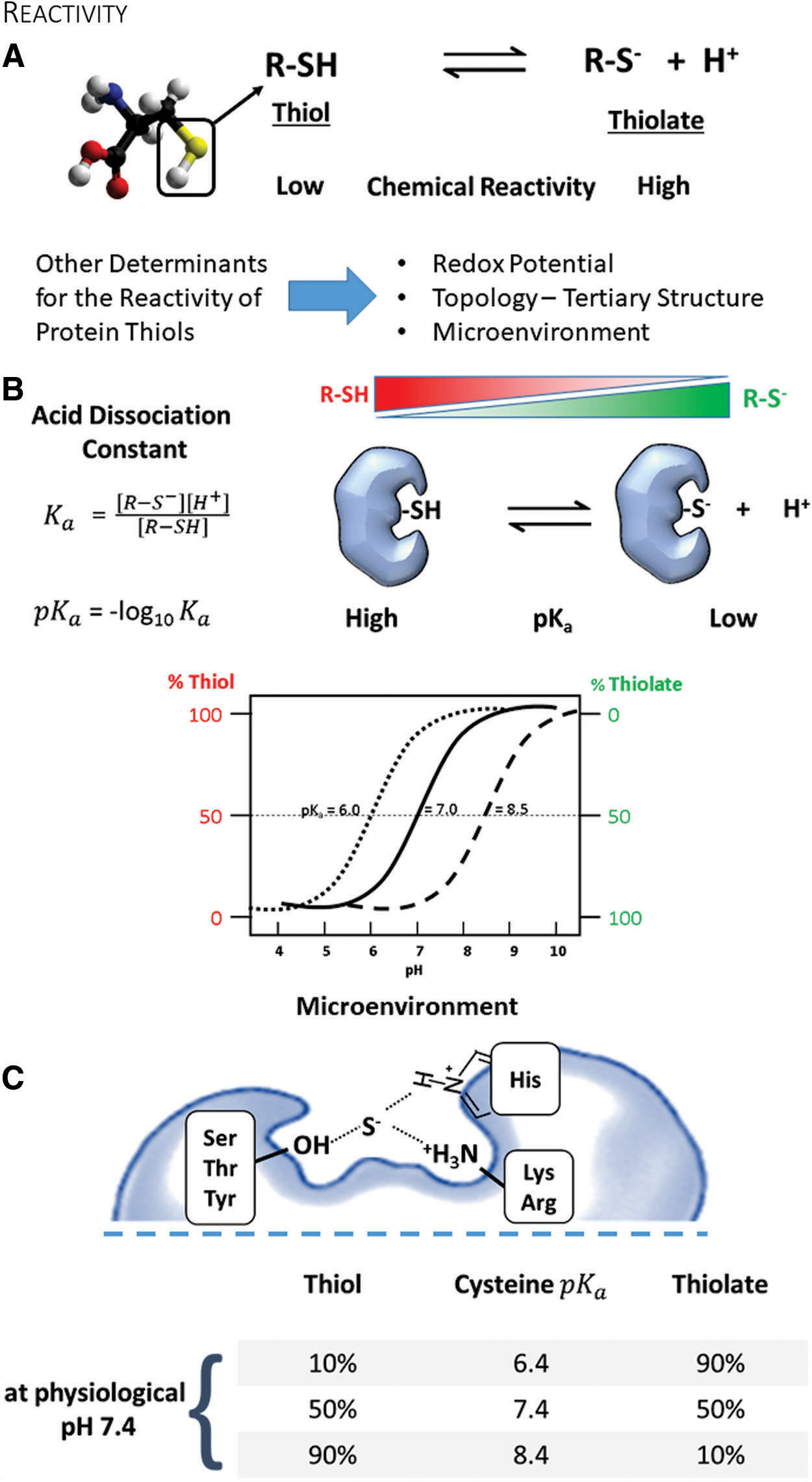
Although cysteine is the least abundant amino acid in proteins, 90% of the cysteines are highly conserved within protein sequences among species (53). Surface-exposed cysteines occur less frequently than other amino acids. One explanation is that cysteines fall under a general classification of highly hydrophobic amino acids, which prevents solvent exposure (156). The basis of the hydrophobicity index of amino acids, including cysteine, is mostly derived from the analysis of three-dimensional structures. However, this classification has been recently revised, resulting in cysteine becoming grouped with polar amino acids together with serine (53). Thus, it is possible that evolution selected and preserved cysteine due to its function in redox signaling, rather than its general physicochemical properties (118). This notion is further supported by bioinformatic analysis of human genetic diseases, demonstrating that mutations of cysteine occur more frequently relative to its abundance in proteins (188). Intriguingly, cysteine appears to be a later addition to the genetic code, and it may still accumulate in the genome of present-day organisms (60).
Cysteine is an amino acid with a unique chemistry. It exists in two forms, the thiol and its deprotonated ionized form, the thiolate (Fig. 1A). Both types contain a lone pair of electrons (nonbinding) and thus are chemically nucleophiles. Although the thiol has low reactivity, conversion to a thiolate renders cysteine one of the most reactive intracellular nucleophiles that can readily undergo alkylation or redox reactions. Because the dissociation of the thiol to thiolate occurs in the context of an acid-base reaction, the negative base-10 logarithm of the acid dissociation constant—pKa—quantitatively relates to its reactivity (Fig. 1B) (149). Briefly, the pKa denotes the pH at which cysteine is equally present in the thiol and thiolate form. The pKa of the unperturbed cysteine thiol side chain is ∼8.25 (104). However, neighboring amino acids may create a milieu that perturbs the pKa of cysteine in a protein and hence changes its reactivity (31). Due to this alteration in the microenvironment, reactive cysteines can often have a lower pKa and exist predominantly in the thiolate form that is stabilized by vicinal amino acids through hydrogen bonds or positive charges (68). Thus, cysteines participating in redox processes are usually in the vicinity of either arginine (Arg, R), lysine (Lys, K), or histidine (His, H), providing an electropositive setting, or serine (Ser, S), threonine (Thr, T), or tyrosine (Tyr, Y) promoting hydrogen bonding. Often, these amino acids are organized as pairs (dyad) or triplets (triad) around the cysteine to create a particular microenvironment and enhance the thiolate stabilizing effect (Fig. 1C).
Mutating cysteines for functional redox studies
Classical consensus sequences that identify reactive cysteines are minimally helpful, and they may only apply to proteins within the same evolutionarily conserved group. Various tools and strategies (53) developed to identify reactive protein cysteines further highlight that the pKa alone is a limited predictor of reactivity, and other factors such as the solvent exposure, reduction potential, protein topology, and the microenvironment around the cysteine contribute to amino acid reactivity and redox modifications. Because of these difficulties, biological studies often involve experiments with mutant proteins to confirm the gain or loss of function regarding a particular cysteine residue.
Due to their related redox properties, the exchange of redox-active cysteine for selenocysteine, the 21st amino acid in the genetic code, occurred during evolution within groups of related enzymes (13). Fomenko et al. applied this concept to database searches and identified redox-active cysteines in several proteins selectively (53). A few studies substituted cysteine with selenocysteine, changing the enzyme's pKa and redox properties (160). Generating these mutations is technically not straightforward, and selenium supplementation can be challenging (64, 108).
On the other hand, serine and alanine are common substitutions to neutralize the thiol function of cysteine while minimizing changes in the protein's structural integrity. The role of choosing a cysteine to alanine or serine substitution should be dependent on the role that the cysteine plays in the protein's function. Due to its size, polarity, and hydrogen bonding ability, serine is the closest substitution (149). However, serine also has the potential to become a target of phosphorylation or glycosylation, which adds additional complexity to the analysis. In contrast, alanine or sometimes valine is better matched to cysteine in terms of hydrophobicity relative to serine (73), and notably, mutation to alanine enables better evaluation of any side-chain interactions.
Other strategies exchange cysteine for very polar (aspartic or glutamic acid) or aromatic amino acids (phenylalanine or tryptophan) to recapitulate the polarity of the oxidized thiol (R-SOH, R-SO2H, and RSO3H) or induced structural perturbations, respectively (179).
In summary, these approaches are not perfect, and careful biochemical and functional characterization of each mutant is required to minimize artefactual results. Although the mutants can provide insights about the biological function of cysteine, they are unsuitable for identifying the specific post-translational modification (PTM).
PTMs of cysteines
Protein cysteines, based on their nucleophilic and redox properties, can have diverse functions and undergo numerous PTMs (Fig. 2A). These functions often require ionized cysteines to act as active site catalysts such as in phosphatases, as part of zinc fingers to coordinate bivalent zinc, as components of iron-sulfur clusters for electron transport, as reactive sites for nonoxidative modifications for intracellular protein trafficking (65), or as semiconductors in the P450 enzymes to control phase I biotransformation reactions (e.g., hydrolysis, reduction, oxidation).
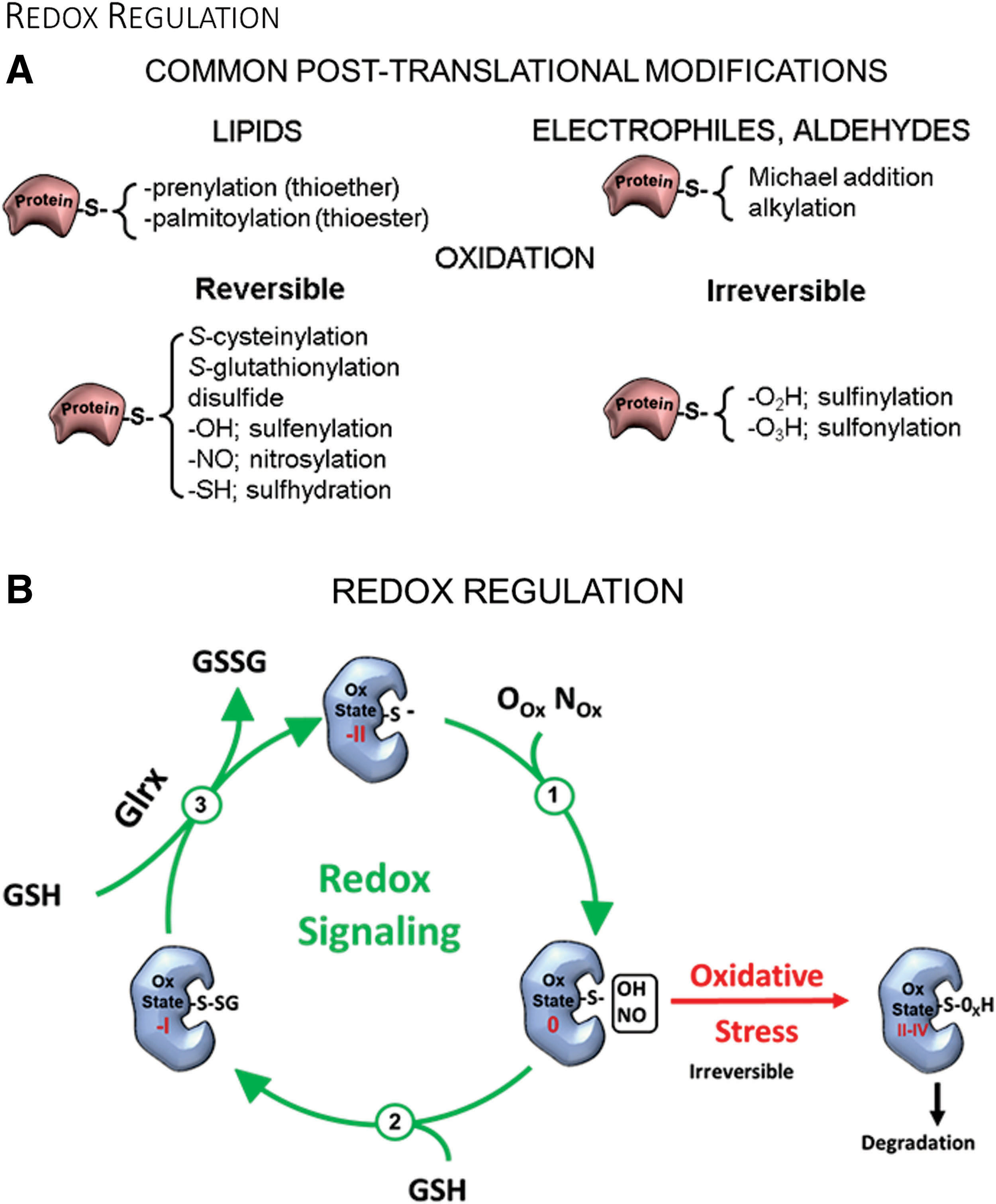
Reactive cysteines are a prerequisite for protein prenylation (thioether bond) and acylation (thioester bond) reactions catalyzed by prenyltransferases and palmitoyl acyltransferases, respectively. Both modifications anchor proteins in biological membranes and regulate trafficking between intracellular membranes such as endosomes and the plasma membrane (65). Oxidants and redox signaling, including S-glutathionylation, may compete with these modifications and interfere with membrane trafficking and activity, as demonstrated for H-Ras (33). Acylation also includes shorter modifications such as acetylation and succinylation that impact epigenetics and mitochondrial function.
Thus, oxidants and OPTMs may compete with other modifications on an affected amino acid. In the next few paragraphs, we will briefly outline these to emphasize how S-glutathionylation and glutaredoxins can impact other cysteine modifications.
Redox reactions of cysteine thiols are exceptionally dynamic processes based on complex redox chemistry. Cells can generate oxidants that preferentially react and oxidize protein thiolates (R-S−) (Fig. 2B). The most common reactions are reversible R-SOH and R-SNO, which can not only possess signaling functions but also serve as intermediates for S-glutathionylation, S-cysteinylation, and sulfhydration (R-SSH). Owing to highly abundant intracellular GSH levels, S-glutathionylation (R-SSG) is the predominant modification in the cell. Meanwhile, cysteine is the most prevalent small-molecule thiol in circulating plasma rather than glutathione, and, thus, protein S-cysteinylation is the most common thiol modification (157).
Detection of thiol modifications and S-glutathionylation
Experimentally, cysteine modifications such as S-glutathionylation can be evaluated by direct quantification with mass spectrometry (MS) (72) to more qualitative methods using antibodies, switch assays (26, 32, 72), or in situ labeling (5, 67). MS is the most direct means to identify and evaluate protein modifications and encompasses label-free analysis as well as labeling with cysteine-reactive mass tags (71). An inherent limitation to the detection of most PTMs is their low abundance. On average, the site occupancy, the percentage for which a specific amino acid residue in a protein is modified, lies within approximately 5%–20% for most modifications with a signaling function (18, 21, 131, 132, 189). Thus, MS analysis often necessitates affinity enrichment to increase sensitivity. However, in cells exposed to oxidative stress, the site occupancy of OPTMs can be much higher and occurs randomly in less reactive amino acids.
Bioinformatics presents another bottleneck for analysis, because most existing search algorithms perform poorly in identifying multiple OPTMs, including S-glutathionylation, and even fail to detect crosslinks or unknown OPTMs. Various groups have developed novel strategies to improve MS detection and analysis of OPTMs such as the “selectively excluded mass screening analysis” (SEMSA) and the search algorithms MODi (97), MODMap, MODa (133), and DBond (41). These approaches led to the discovery of additional cysteine modifications that fall into several categories: oxidation (e.g., Cys to Ser or dehydroalanine conversion, thiosulfonic acid, sulfonamide), acylation (palmitoylation, succinylation, acetylation, diacylglycerol), prenylation (e.g., geranyl-geranylation or S-farnesylation), S-ubiquitination, S-guanylation, and chemical modifications (e.g., 15d-PGJ2) (84, 137). The biological significance for some of these modifications and competition with OPTMs remains elusive.
S-guanylation is an understudied modification that is formed after peroxynitrite-mediated nitration of the guanine base in the nucleotide to 8-nitroguanosine, an electrophile that irreversibly adducts to cysteine thiols. For example, this modification inhibits KEAP1 from binding to nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (55), activates the GTPase H-Ras by allowing greater translocation (135), and induces 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) opening (153). S-guanylation, however, is not enzymatically reversed and can theoretically occur at any thiolate in the presence of 8-nitroguanosine. Thus, further studies need to determine whether this modification occurs randomly or participates in directed signaling changes (95).
Given the costs of MS analysis, the development of thiol labeling using “switch techniques” has proven helpful (21, 26, 32, 194). The best-known method, the biotin switch assay, starts by blocking free cysteines with a thiol-reactive compound, unaffected by the subsequent chemical step to remove the targeted modification. In most cases, the removal step is a reduction (e.g., ascorbate for S-nitrosylation), but protocols may also use hydrolysis to cleave the thioether linkage (e.g., neutral hydroxylamine) as found in S-palmitoylation (29, 65). Recent revisions of the assay to analyze S-glutathionylation include enzymatic removal catalyzed by recombinant Glrx (170). A tagged labeling reagent then reacts with the newly liberated thiols, allowing detection with analysis techniques such as Western blot, MS, and histology. Burgoyne and Boivin (26, 32) have described the switch assays that are the most readily usable for single-protein evaluation. Switch assays are particularly useful if the modification is chemically labile, such as S-nitrosylation or S-sulfenylation, and unlikely to survive sample preparation for MS analysis. To perform proteome-wide analysis of thiol modifications, labeling with stable isotope mass tags combined with MS is ideal (21, 132, 194). However, this technique requires advanced bioinformatics, demanding sample preparation, and instrumentation, and it is often available only in specialized laboratories. A limitation of switch assays is the indirect detection of the thiol modification, and if performed incorrectly, switch assays may misrepresent cysteine modifications. Common pitfalls include insufficient degassing of buffers to limit artefactual thiol oxidation, incomplete removal of incompatible chemicals during the switch procedure, lack of specificity of the reagent to remove targeted modification (95), over-alkylation, and nonspecific labeling. Nevertheless, correctly executed and well-controlled switch assays produce meaningful results.
Other described methods to detect S-glutathionylation include specific antibodies (74, 165). Because GSH is a small peptide and universal biomolecule present in plasma, raising antibodies requires peptide- or protein-GSH conjugates (hapten). Various groups linked GSH with glutaraldehyde to bovine serum albumin and demonstrated the specific recognition of cytoplasmic and mitochondrial glutathione (74, 165). Söderdahl et al. also showed its usefulness for detecting protein S-glutathionylation in cultured cells (165). One report produced a specific GSH antibody against Cys141 of p53 (197). In collaboration with Millipore, our lab attempted to generate a site-specific antibody against Cys118 of H-Ras by using the same strategy. However, the affinity-purified antibody still recognized the Cys to Ser H-Ras mutant. Because S-glutathionylation is reversible, the modification may be reduced after injection of the hapten into the rabbit. Thus, it is not surprising that site-specific antibodies against S-glutathionylation are a rarity. We believe that the generation of site-specific antibodies is possible, but likely an antigen conjugate with a nonreducible GSH-like modification or screening against phage-display libraries (27) is necessary. In our hands, the best commercial anti-protein GSH antibody is from Virogen. However, we know from MS studies that not all S-glutathionylated proteins are recognized with equal sensitivity. Also, nonspecific binding to mouse proteins should be tested by the secondary antibody alone when a mouse monoclonal antibody is used for Western blot.
Metabolic labeling of cysteine modifications using radioactive or stable isotopes is another sensitive method for analyzing OPTMs, particularly S-glutathionylation. Biotin (171) or newer CLICK-chemistry labels (91) can be attached to the amino group of cell-permeable glutathione monoethyl ester, a cell-permeable version of glutathione, allowing direct and specific detection of S-glutathionylated proteins. The high costs and high concentrations required to load cells, however, currently make these compounds impractical for large-scale or in vivo experiments.
In summary, the available tools to detect a thiol modification or S-glutathionylation specifically are limited and slowly evolving. Better and more specific molecular tools with broader applicability are necessary to advance the field.
Glutathione
In 1888, De Rey-Pailhade originally described “philothion,” which Hopkins rediscovered in 1921 and renamed as glutathione (GSH) (8). GSH is a small tripeptide comprising glutamine, cysteine, and glycine (γ-
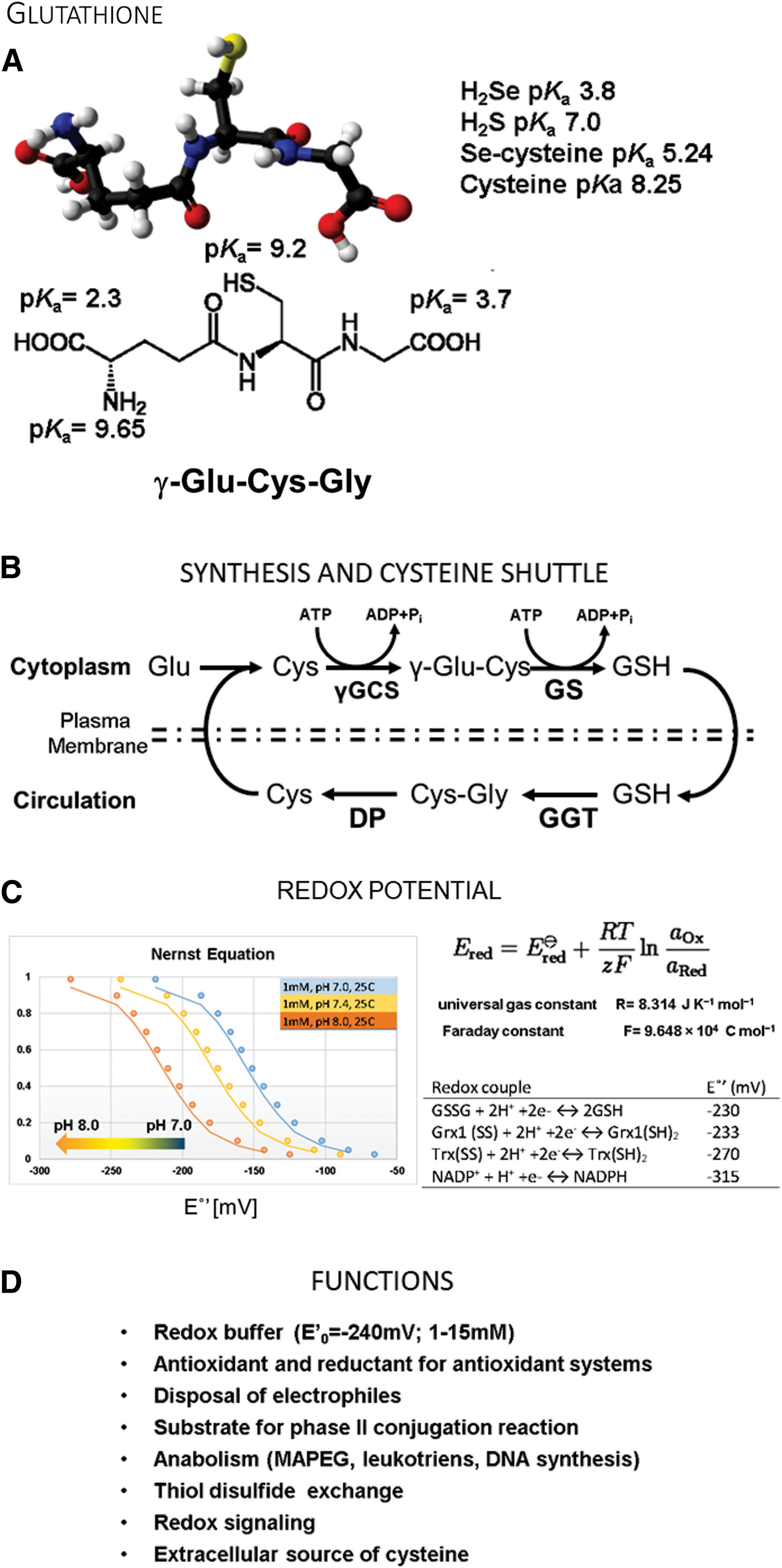
Antioxidant systems, including peroxiredoxins, glutaredoxins, and glutathione peroxidases, have active site thiolates or selenolates. For all these proteins, the enzyme reacts with oxidants or an oxidized target protein and becomes oxidized itself by direct or trans-S-glutathionylation. GSH serves as a reductant for the enzyme and forms GSSG (oxidized glutathione), restoring the protein's function. Glutathione reductase then recycles the oxidized GSSG back to GSH with the consumption of NADPH (nicotinamide adenine dinucleotide phosphate) and restores the cellular redox equilibrium. In situations when excess GSSG is formed, cells can also release GSSG through MRPs to restore the redox potential quickly (79).
Thus, the GSH and GSSG equilibrium constitutes the most critical cellular redox buffer, and the ratio determines the redox potential (159). The cell uses the GSH:GSSG ratio to create cellular compartments with defined redox conditions. For example, the cytoplasm has a redox potential between −220 and −260 mV; and a GSH:GSSG ratio of 30 to 100:1, which is actively reducing. Under these conditions, it is difficult to form intra- and intermolecular protein disulfides. The endoplasmic reticulum (ER), which contains the enzymatic machinery to catalyze protein disulfide formation, has a more oxidizing environment of −180 mV, established by a lower GSH:GSSG ratio. The Nernst equation allows researchers to calculate the redox potential and recreate redox buffers with defined redox potential to mimic physiological conditions (159) (Fig. 3C).
In summary, a lower pKa would render the GSH thiol too reactive and promote nonspecific reactions in the cell. The higher pKa of GSH allows it to act as the cellular redox buffer. This function, coupled with the actions of GSTs, provides a means to harness and control the chemical reactivity of the thiolate nucleophile.
The liver plays a unique role in GSH metabolism since it removes the major amount of resorbed cysteine from the portal circulation (57) and converts it into less reactive GSH, which is released back into the blood flow (168). Thus, GSH may also serve as a cysteine source for other tissues (111). The normal GSH turnover is about 40 mmol per day in a human adult and is estimated to be slightly higher than the protein cysteine turnover (168). All functions of glutathione are summarized in Figure 3D.
Cellular oxidant sources
Various isoforms of NADPH oxidases (Nox) generate oxidants at physiological levels. These enzymes predominantly release superoxide (•O2 −), which is quickly converted into hydrogen peroxide (H2O2). Nox 4 is the only isoform that has been reported to release H2O2 directly (28). NADPH oxidases contribute to growth factor signaling, cell differentiation, and cell migration but have also been linked to pathology, playing roles in vascular disease and inflammation. The inflammatory system, particularly immune cells such as neutrophils, contains high amounts of Nox 2, which participates in phagocytosis and produces an oxidative burst to neutralize pathogens. Rather than a defense mechanism, other organs and tissues have utilized this system for cellular signaling. For example, vascular smooth muscle cells express large amounts of Nox in hypertrophy, contributing to oxidative stress, endothelial dysfunction, and vascular remodeling (107, 181). Most tissues, however, express Nox at much lower levels, which allow for participation in cellular redox control. The pentose phosphate pathway, the primary metabolic pathway and the source of NADPH, regulates Nox-induced oxidant generation. Lower NADPH production in glucose-6-phosphate dehydrogenase-deficient mice causes decreased oxidant formation in the vasculature (121, 122). This effect is explained by the Km of NADPH, which is markedly higher for NADPH oxidases (172) than for the reductive system (186). Thus, even at low NADPH concentrations, the antioxidant system is fully active, whereas the oxidant sources are attenuated (121, 122).
Another group of oxidant sources consists of uncoupled nitric oxide synthases (NOS). NOS usually produces nitric oxide, which regulates vascular tone, cell proliferation, platelet aggregation, immune response, and neurophysiology. When
Various P450 enzymes (including epoxygenases) and flavin-containing proteins (including xanthine oxidase) are additional oxidant sources that are capable of generating both superoxide and H2O2. Under physiological conditions, xanthine dehydrogenase is the principal form of the enzyme. Cysteine oxidation or proteolytic cleavage converts xanthine dehydrogenase into an oxidase that may play a role in pathobiology, as with ischemia
Yet another source of cellular oxidants is mitochondria. Various mitochondrial flavoproteins such as the 2-oxoadipate dehydrogenase, pyruvate dehydrogenase complex, and 2-oxoglutarate dehydrogenase complex (134) can produce oxidants, but the components of the electron transport chain, especially complex I (NADH ubiquinone oxidoreductase) and complex III (coenzyme Q cytochrome c oxidoreductase), are prominent sources of superoxide. However, more recent findings have also identified complex II as a significant producer of superoxide (61). Predominant fatty acid oxidation can generate conditions of a highly reduced ubiquinone pool combined with low succinate levels (perturbed anaplerosis), leading to an electron leak at complex II (61) and oxidant generation. These conditions often occur in metabolic disease and obesity when muscle cells, such as cardiomyocytes, predominantly metabolize fatty acids through β-oxidation (143).
The ER has a low GSH:GSSG ratio to enable proper protein folding and introduction of structural protein disulfide bridges. The ER oxidoreductin 1 (ERO1) system generates H2O2, oxidizing highly conserved thiolates of ERO1 to disulfides. Oxidized ERO1 then transfers the disulfide through a thiol-disulfide exchange reaction via protein-disulfide isomerases (PDI) to the nascent target protein. Misfolded proteins and GSSG can be redirected and reoxidize PDI as well. In the final step, correctly folded proteins are transported by vesicles to the Golgi apparatus for secretion or insertion into the plasma membrane.
A second independent mechanism from the ERO1-PDI system controlling ER protein folding involves peroxiredoxin 4 (PRDX4) and an unknown H2O2 source (114). In combination with the glutathione redox buffer, the ER-resident PRDX4 harnesses oxidation and scavenges excess H2O2 to prevent irreversible cysteine oxidation. Interestingly, neither ER-produced H2O2 nor cytosolic- or mitochondrial-produced H2O2 diffuses across the ER membrane, suggesting a very tight regulation of ER-specific oxidants. Thus, oxidants produced in the ER may be limited to ER function and stress, whereas redox signaling in the cytosol or other organelles such as mitochondria proceeds unaffected (16, 114). Whether the overproduction of H2O2 by perturbations of the ERO-1 system can lead to ER stress is unclear.
Lysosomes are other cellular organelles affected by ROS and can also produce oxidants. Lysosomes and related vesicles, including phagosomes and autophagosomes, have a membrane-associated NADPH oxidase that becomes activated during the degradation process. Lysosomal degradation of proteins such as ferritin releases iron that initiates Fenton chemistry, generating highly reactive hydroxyl radicals, which, subsequently, results in ferroptosis (102). This mechanism of inducing oxidative stress is being leveraged for cancer chemotherapy with the use of FDA-approved iron nanoparticles (98, 199). Specialized phagosomes also use myeloperoxidase for generating hypochlorite, a highly reactive and bactericidal species.
Lysosomes also seem to be directly activated by external or mitochondrial ROS via a redox sensor transient receptor potential cation channel 1 (TRPML1). Activation of the channel causes lysosomal calcium release that induces autophagy to remove damaged mitochondria and, thus, limits excess ROS formation by dysfunctional mitochondria (200).
Oxidative stress versus redox signaling
Oxidants at elevated levels can nonspecifically attack biological molecules, including purine bases of DNA, unsaturated lipids, or reactive side chains of proteins. Lipid peroxidation can start a chain reaction, also referred to as autoxidation, which results in various reactive end-products, including malondialdehyde and hydroxynonenal. These aldehydes preferentially modify cysteines but can also affect other amino acids. As a consequence, oxidative stress produces a broad spectrum of oxidized biological molecules and post-translational protein modifications. These oxidation reactions can damage biological macromolecules, interfere in cellular physiology, and are often observed in advanced diseases, such as heart failure, atherosclerosis, and nonalcoholic steatohepatitis (NASH), leading to cell death and tissue fibrosis.
In contrast, redox signaling is targeted and enabled by particular localized amino acids, in many cases reactive thiolates of proteins. These solvent-exposed thiolates can exhibit a thiol peroxidase-like activity to catalyze their modification (Figs. 1C and 2B).
Similar to protein phosphorylation, redox signaling requires two components: a catalysis agent (ATP vs. an oxidant source) and a reactive amino acid residue (serine, threonine, or tyrosine vs. a cysteine) that directs the specificity of the reaction. Enzymes catalyze the removal of the PTM but, unlike phosphorylation, deglutathionylation may also occur due to the reductive cellular environment.
Reversible Protein S-glutathionylation
Glutathione S-transferase
Oxidants initiate redox signaling through the reversible PTM of reactive amino acids, such as cysteine, which, in particular, leads to S-glutathionylation. In the presence of abundant cellular GSH, reactive cysteines become S-glutathionylated. This process can occur spontaneously, but it may also involve other enzymes that activate and transfer glutathione to the target protein. GSTs catalyze the conjugation of GSH to electrophilic substances in phase II detoxification reactions but may participate in S-glutathionylation as well. GSTs are dimeric and can form heteromers to adjust to different substrates and conditions. Among several classes of cytosolic GSTs, GSTπ is shown to promote S-glutathionylation both in vitro (116) and in vivo (177). GSTπ is an abundant protein in the lung and liver but is also present in tissues, not known for detoxification reactions, such as the placenta, brain, and heart. Certain tumor cells overexpress GSTπ, which may stimulate cell proliferation or confer chemoresistance (105). The expression of GSTπ markedly decreases during liver development, which might be a mechanism to minimize protein S-glutathionylation and prevent hepatic lipid accumulation (see Nonalcoholic Fatty Liver Disease section) (105).
Experiments in a clinically relevant disease model of lung fibrosis recently demonstrated the importance of GSTs for protein S-glutathionylation. GSTπ ablation in mice attenuates protein S-glutathionylation and lung fibrosis due to the mitigation of Fas ligand (CD95L) mediated cell death of lung epithelial cells (125). S-glutathionylation of the Fas receptor amplifies epithelial cell apoptosis (9, 12), and thus, using therapeutic strategies to diminish protein glutathionylation including airway delivery of Glrx (10, 11) or the GSTπ inhibitor TLK117 (125) attenuates lung fibrosis.
Interestingly, GSTπ itself is a redox-regulated enzyme. Glutathionylation of Cys47 and Cys101 inhibits GSTπ activity and binding to c-Jun N-terminal kinase (JNK) (177). Direct protein
In addition to GSTπ, the glyoxalase system may play a role in the enzymatic formation of glutathione adducts. The glyoxalase system removes dicarbonyl species, such as the reactive glucose-derived metabolite methylglyoxal, with high specificity by glyoxalase 1 and 2 in two concerted reactions. Glyoxalase 1 converts the spontaneously formed hemithioacetal (the product of methylglyoxal and GSH) to S-D-lactoylglutathione that is then subsequently metabolized by glyoxalase 2 to
The glyoxalase system has been implicated in vascular disease. Deletion of glyoxalase 1 can lead to diabetic nephropathy-like pathology, whereas the overexpression of glyoxalase 1 can prevent diabetic renal disease and microcirculatory perturbations in mice (58). It appears that glyoxalase 1 knockdown in endothelial cells (ECs) results in changes that are consistent with vascular damage and dysfunction (169). In obese and overweight patients, activation of glyoxalase by trans-resveratrol (tRES) and hesperetin (HESP) improved vascular function, decreased circulating methylglyoxal levels, and induced weight loss (193). Although glyoxalase 1 seems to be only modified to a mixed disulfide with lower enzymatic activity (25), glyoxalase 2 has been shown to induce S-glutathionylation of proteins in vitro (43). Whether these observations about glutathione and the glyoxalase system hold in vivo needs further investigation.
Glutaredoxins
Dithiol glutaredoxins—GSH-dependent oxidoreductases
In an Escherichia coli mutant lacking thioredoxin, A. Holmgren initially discovered a small GSH-dependent hydrogen donor for ribonucleotide reductase, which he named glutaredoxin (78). Ribonucleotide reductase catalyzes an essential step to produce deoxyribonucleotides, the DNA precursors for DNA synthesis. Thioredoxin was an established co-factor and hydrogen donor of ribonucleotide reductase, but the functions of glutaredoxin were beyond a simple substitute for thioredoxin (51).
In parallel, Mannervik and Axelsson biochemically isolated a thioltransferase in the liver that removed or formed glutathione protein adducts depending on the GSH:GSSG ratio (14, 15, 49). Other groups confirmed this activity and improved the purification methods (56, 69). After producing specific antibodies against purified pig liver thioltransferase, Gan and Wells discovered that the calf thymus (69) and E. coli glutaredoxin (101) were identical with the liver thioltransferase. Additional biochemical characterization identified several extra activities of Glrx, including thiol-redox properties, cystine conversion, dehydroascorbate reductase activity, and transhydrogenase activity. The thioltransferase activity, however, appears to be the most critical function for physiology in vivo (51, 150).
De-glutathionylation can occur in cells spontaneously because of the reductive environment and high concentrations of GSH. Glutaredoxin, however, has a specific binding groove for GSH (191) and is the prime catalyst (Fig. 4A). Experiments in liver homogenates demonstrated very rapid GSH transfer onto or removal from proteins. More importantly, Glrx caused a marked shift in the equilibrium of glutathionylated to de-glutathionylated proteins (117). Thus, under already reducing conditions in the cell, Glrx can further decrease the levels of glutathionylated proteins.
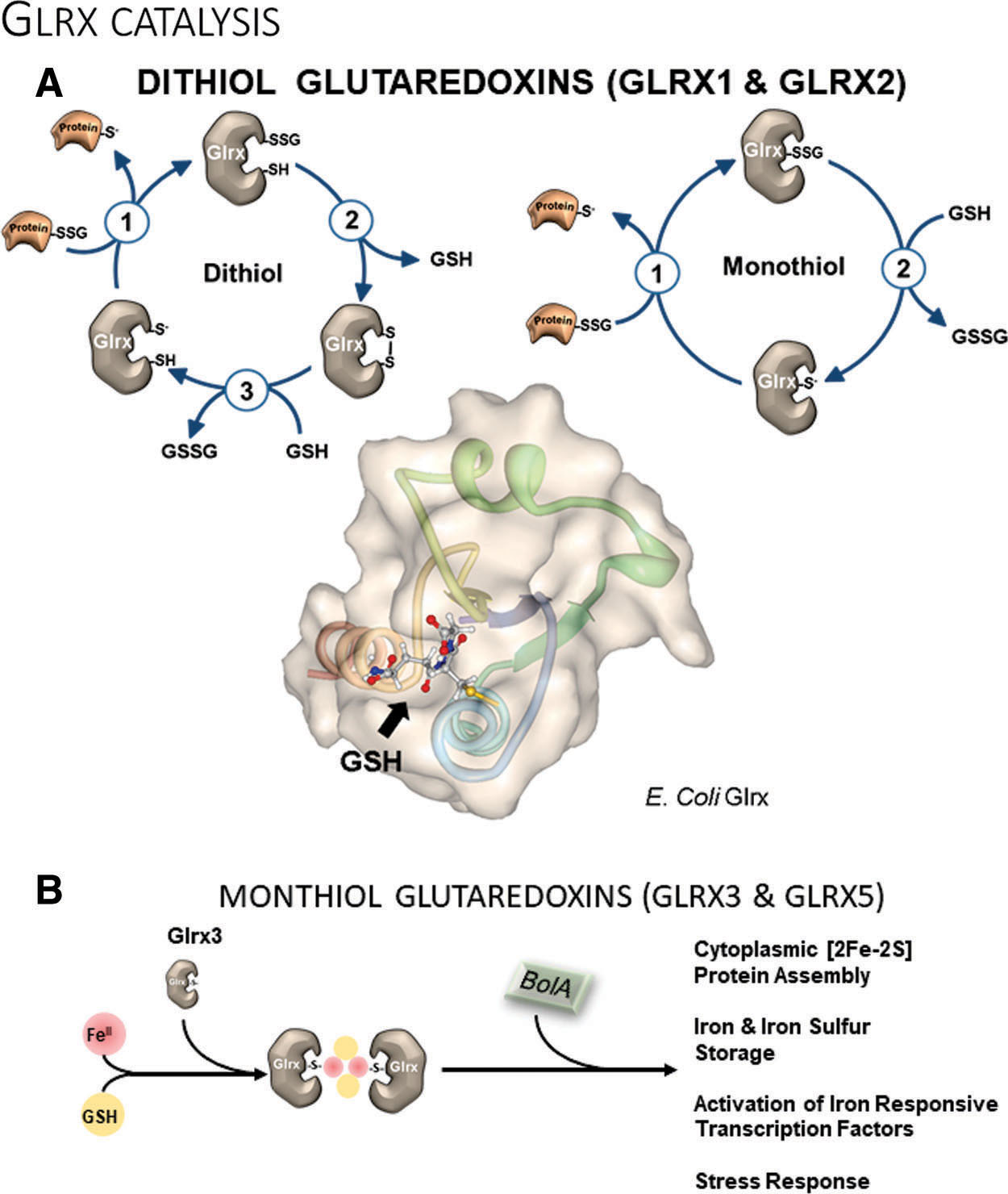
Because the catalysis occurs in both directions, Glrx can also amplify protein S-glutathionylation under oxidative stress. Also, Glrx may have a preference for targets in a specific setting. This complexity causes both favorable and unfavorable outcomes in in vivo genetic mouse models. For example, Glrx deletion shows favorable (130, 180) consequences for revascularization, and unfavorable repercussions for hepatic lipid metabolism (163), which we will discuss in more detail later.
Glutaredoxin contains the conserved Cys-X-X-Cys active site motif to reduce protein-bound GSH (R–SSG) using GSH, glutathione reductase, and NADPH. Unlike thioredoxin, Glrx does not catalyze the reduction of sulfenic acid or intra- and intermolecular disulfides (24, 42). Catalysis of deglutathionylation can occur by two distinct catalytic mechanisms: the dithiol or monothiol pathways. Monothiol catalysis depends solely on the active site thiolate and is a simple thiol exchange reaction (Fig. 4A, left). Glrx binds to the glutathionylated protein, and its active site thiol reacts with the sulfur of the GSH, which forms a new bond between GSH and Glrx. In the second step, Glrx reacts with free GSH to restore its thiolate and releases GSSG. The initial reaction of the dithiol pathway is identical to the monothiol mechanism, but the next step involves the second cysteine of Glrx, which releases GSH in the process of forming an intramolecular disulfide (Fig. 4A, right). Two molecules of GSH then reduce the oxidized Glrx and form GSSG. The biological significance of the dithiolic pathway is still unclear but may explain some of the overlapping functions with thioredoxin. Other monothiol members of the Glrx family are also involved in cellular iron homeostasis, but their participation in redox chemistry is controversial. Future in vivo studies with Glrx mutants may elucidate the biological role of the second cysteine at the active site.
Mammalian cells encode two principal dithiol isoforms of Glrx. Glrx1 mainly exists in the cytoplasm, and Glrx2 localizes to mitochondria or the nucleus depending on gene splicing. Human Glrx1 contains Cys-Pro-Tyr-Cys, and Glrx2 contains Cys-Ser-Tyr-Cys as the active site motifs. Human Glrx2 is only ∼34% homologous to Glrx1, and the specific activity is <10% of Glrx1 (59, 114). Glrx2 forms an enzymatic inactive iron-sulfur cluster with reduced GSH (two protein monomers and two glutathione molecules). During oxidative stress, the iron-sulfur cluster likely serves as a highly sensitive oxidant sensor, as disruption of the cluster activates Glrx2 (22). Glrx2 KO mice develop cardiac hypertrophy and fibrosis (88, 115). Maintenance of mitochondrial iron-sulfur clusters and protein glutathionylation are critical for the functioning of mitochondrial oxidative phosphorylation and energy fluxes in the heart.
Glrx1 is redox sensitive and is reversibly inactivated by S-glutathionylation or S-nitrosylation at its active site cysteine thiol (11, 59). Importantly, Glrx1 also catalyzes the formation of S-glutathionylation in the presence of thiyl radicals (GS•). Due to the low pKa of the active site Cys, Glrx1 may form a radical intermediate that reacts with GSH to generate Glrx-SSG, which mediates protein-SSG formation (127). Anaerobic conditions increase GAPDH-SSG formation by Glrx1 in vitro (167).
Monothiol glutaredoxins
Monothiol glutaredoxins with their active-site motif Cys-Gly-Phe-Ser are evolutionarily highly conserved from bacteria to mammals. Of the initially discovered three monothiol glutaredoxin isoforms in yeast (ScGrx3, 4, and 5), only two—Glrx3 and Glrx5—exist in mammals. Across organisms, monothiol glutaredoxins are essential for intracellular iron homeostasis and actively catalyze the assembly of iron-sulfur [2Fe-2S] clusters (Fig. 4B). Glrx3 and Glrx5 also regulate iron trafficking and gene expression of proteins involved in intracellular iron homeostasis. Other functions include the protection of proteins from oxidation, signaling, cell growth, apoptosis, and proliferation that will be explained later in more detail.
Glrx3
Several groups originally identified Glrx3 with screening assays or bioinformatics and referred to it as a human sequence similar to yeast (HUSSY-22) (166), thioredoxin-like 2 (Trxl2), or protein kinase C-interacting cousin of thioredoxin (PICOT) (184). Glrx3 can form a stable iron–glutathione complex with BolA-like proteins, whose function and structure are highly conserved among species. BolA proteins were originally identified in E. coli as stress-responsive transcriptional regulators involved in the regulation of iron homeostasis, bacterial shape, and biofilm formation. In mammalian cells, the Glrx3 and BolA-like 2 protein (BolA2) complex is essential for the assembly of iron-sulfur ([2Fe-2S]) clusters of a subset of cytoplasmic iron-containing proteins (54). Oxidative stress alters these complexes, and Glrx3 translocates to the nucleus (145), regulating iron-regulatory proteins (IRP1 and 2), cell cycle progression (40), and stress-induced DNA-damage responses (141).
Because of its conserved role in cellular iron homeostasis, the ablation of Glrx3 impairs hemoglobin maturation in zebrafish erythroid cells and upregulates molecular markers of cellular iron starvation in HeLa cells (70). Murine Glrx3 knockout (KO) is embryonic lethal after 12.5 days of gestation (40), likely due to effects on cell cycle progression and fetal hematopoiesis.
In contrast, Glrx3 overexpression protects the mouse heart from pressure overload-induced cardiac hypertrophy (83), ischemia
Glrx3 affects various intracellular signaling cascades, including PKC isoforms, JNK, AP-1, NFAT, and nuclear factor-κB (NF-κB) (90, 136), influencing immune function, neuronal cell differentiation (30), and tumorigenesis (109, 113). However, the mechanisms involved remain to be elucidated.
Glrx5
Similar to the function of Glrx3, Glrx5 is essential for the assembly of iron-sulfur clusters in the mitochondria, but it also affects cytoplasmic iron regulation through IRP1 and 2. Deficiency of Glrx5 results in iron overload and sideroblastic anemia in zebrafish and humans (35). Glrx5 deletion is embryonic lethal in mice (183). Glrx5 is indispensable for normal regulation of hemoglobin synthesis by the iron-sulfur protein aconitase 1 (ACO1) (195), aminolevulinic acid synthase ALAS2, and ferrochelatase (46). Lipoic acid biosynthesis also depends on the iron-sulfur cluster containing enzymes, and defects in Glrx5 lead to lipoic acid deficiency (123). Mutations of Glrx5 may also cause a variant of nonketotic hyperglycinemia in humans and cause childhood-onset spastic paraplegia, spinal lesion, and optic atrophy (19). However, other direct effects on intracellular signaling are understudied.
Molecular studies of Glrx3 and Glrx5 reveal that hemoglobin biosynthesis is regulated through [2Fe-2S] cluster assembly, particularly in red blood cells.
The In Vivo Role of Glutaredoxin
Y-S. Ho at Wayne State University created genetically deleted Glrx mice to examine the role of Glrx in pathophysiology (75). Unlike the in utero lethality of thioredoxin deletion (119), mice lacking Glrx appear grossly normal. Although some reports consider Glrx as an antioxidant enzyme, global Glrx deletion in mice does not cause any apparent susceptibility to oxidative stress (75). Table 1 lists published studies using Glrx KO or transgenic mice.
Phenotypes of Mice with Genetically Modified Glrx Expression
Macrophage-specific Glrx2 TG.
Lung epithelial-specific Glrx TG.
AR, aldose reductase; Glrx, glutaredoxin-1; HFD, high-fat diet; HFHS, high-fat high sucrose; HIF, hypoxia-inducible factor; KO, knockout; LDL, low-density lipoprotein; LPS, lipopolysaccharide; LV, left ventricle; NASH, nonalcoholic steatohepatitis; NF-κB, nuclear factor-κB; Rac1, Ras-related C3 botulinum toxin substrate 1; SirT1, sirtuin-1; TG, transgenic; WD, western diet.
Cardiovascular system
Antioxidant therapy and direct inhibition of NADPH oxidase enzymes attenuate systolic cardiac dysfunction and hypertrophy caused by aortic banding and Ang II (194). In contrast, Glrx ablated mice infused with Ang II develop less cardiac and vascular hypertrophy than wild-type mice. As shown by nitrotyrosine staining in the aorta, the vasculature of Ang II-infused Glrx KO mice exhibits less oxidative stress likely due to impaired activation of NADPH oxidase (21). However, Glrx overexpression protects cardiac function after chronic ischemia in association with upregulated anti-apoptotic pathways and improved neovascularization (3). Thus, the functions of Glrx are much more far-reaching and beyond a simple cellular antioxidant.
Angiogenesis and ischemic revascularization
Peripheral artery disease (PAD) presents with clinical symptoms caused by arterial occlusion in the lower extremities. In severe cases, lasting arterial insufficiency results in critical limb ischemia associated with tissue necrosis, requiring amputation (187). PAD is associated with atherosclerosis and is potentiated by other risk factors such as age, smoking, and diabetes. The pathogenesis involving impaired angiogenesis and reduced microcirculation in the skeletal muscle accelerates to intractable limb ischemia in PAD patients (81). Various therapeutic approaches to improve vascularization with angiogenic factors, including vascular endothelial growth factor (VEGF), have shown promising effects in animal models (164) but failed in clinical trials (154). Surprisingly, patients with critical limb ischemia have elevated plasma VEGF-A levels (52), suggesting that effector molecules downstream of VEGF may impair angiogenesis in severe PAD.
Femoral artery ligation to induce hindlimb ischemia in the mouse has been used as a PAD model to identify molecular mechanisms and develop treatments to stimulate vascularization (Fig. 5A). After femoral artery ligation, arteriogenesis (collateral formation) and angiogenesis (new blood vessel formation) gradually restore blood flow to the affected muscle. Studies using genetically modified mice demonstrated an essential role of oxidants in these processes. Nox2 deletion impaired (38) and Nox 4 overexpression improved (44) ischemic limb vascularization.
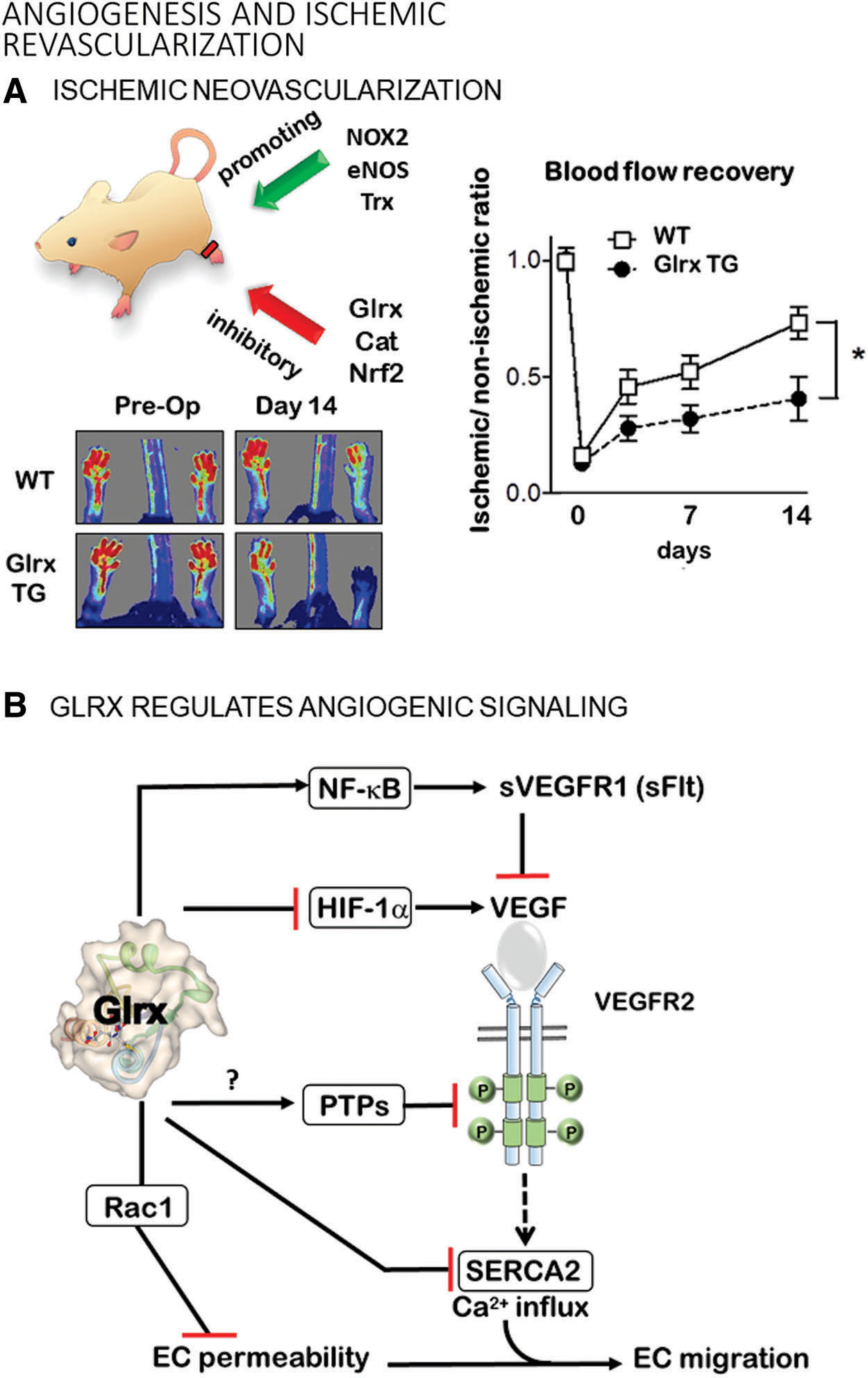
Angiogenic signaling by S-glutathionylation
S-glutathionylation occurs on a large number of proteins with redox-sensitive cysteines in vitro, but due to technical limitations, the in vivo significance is unclear (127). Oxidant-induced angiogenesis or vascularization is attributed, at least in part, to S-glutathionylation and regulation by Glrx. Thiol modifications are known to functionally control several signaling pathways in the angiogenic pathway in vivo (120), which we will explain in the next few paragraphs (Figs. 5A, B, and 6).
Tyrosine phosphatases
Growth factors such as VEGF bind to receptor tyrosine kinases, initiating signaling through phosphorylation cascades. Protein tyrosine phosphatases (PTPs) dephosphorylate the kinase substrates and cease signaling. Oxidants can target the active site Cys215 of PTP1B, an abundant PTP, and inhibit its activity via GSH adducts (20). A PTP inhibitor augmented phosphorylation of VEGF-Receptor 2 (VEGFR2) and blood flow recovery in ischemic rat limbs (37). PTP-1B (106) and other phosphatases, including low molecular weight (LMW)-PTP (80), inhibit phosphorylation of VEGFR2 in ECs. VEGF-induced S-glutathionylation of LMW-PTP also activates VEGF signaling, and reductive stress (increased GSH) inhibits this process (1), suggesting that excess GSH may potentiate the Glrx system. PTP1B ablation improves angiogenesis and cardiac function after coronary artery ligation in mice (23). EC-specific PTP1B deletion also enhances vascularization after femoral artery ligation in mice (106). Therefore, inhibiting PTPs by S-glutathionylation might be one of the targets by which oxidants promote vascularization. However, oxidized PTP1B can also form a sulfenyl-amide intermediate (158), which can be reduced by thioredoxin. The thioredoxin system can activate PTP1B (45) and inhibit VEGF-mediated angiogenesis (142). Whether Glrx can modulate the activity of PTPs remains unexplored.
Sarcoplasmic-endoplasmic reticulum calcium ATPase
S-glutathionylation of Cys674 of the sarcoplasmic-endoplasmic reticulum calcium ATPase 2 (SERCA2) activates the enzyme and triggers calcium influx (2). Overexpression of Cys674Ser mutant SERCA2 or Glrx inhibits VEGF-induced EC calcium influx and migration (50). The Cys674Ser SERCA2 mutant also impairs developmental angiogenesis in homozygous knock-in mice and is embryonically lethal (173). Similarly, the Cys674Ser SERCA2 mutation impairs blood flow recovery after hindlimb ischemia in heterozygous knock-in mice, and the mutation decreases VEGF-induced angiogenic responses in ECs isolated from these mice (126, 173). In summary, S-glutathionylation-mediated SERCA2 activation contributes to EC migration and angiogenesis.
NF-κB signaling
S-glutathionylation inhibits multiple components of the NF-κB pathway, including TRAF6 (39), IKK-β (nuclear factor-kappa-β kinase subunit beta) (155), p50 (147), and p65 (130, 151) subunits; whereas Glrx overexpression enhances NF-κB transcriptional activity (62, 130) (Fig. 6). Glrx overexpression attenuates limb revascularization after femoral artery ligation in association with elevated soluble VEGF receptor 1 levels (sVEGFR-1, also known as soluble fms-like tyrosine kinase-1s; sFlt-1) in the muscle and plasma of mice (129, 130). VEGFR-1 (Flt-1) has a higher affinity to VEGF-A, but the signal transduction is stronger through VEGFR-2. The soluble splice variant (sVEGFR-1, sFlt-1) acts as a decoy receptor competing with VEGFR2 for binding of VEGF-A (94).
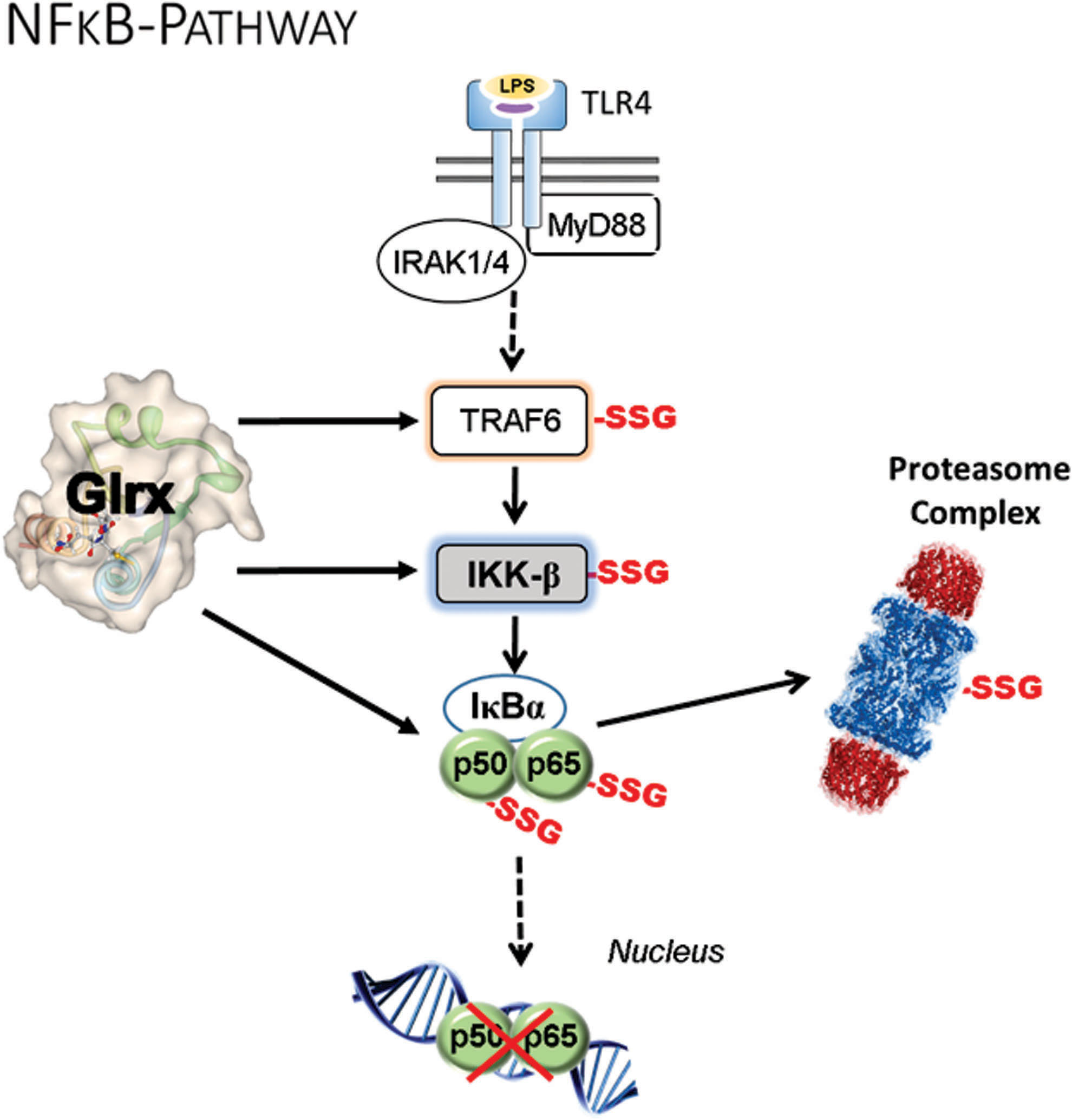
Overexpression of Glrx augments NF-κB activity and induces the anti-angiogenic factor sVEGFR-1 in ECs (129, 130). Since Glrx is an NF-κB-dependent gene (6), activation of NF-κB in inflammation or diabetes (36, 62) may increase Glrx expression. Subsequently, Glrx further activates NF-κB and may positively provide feedback and amplify the inflammatory response. Inhibition of NF-κB by mutant IκBα, which serves as a repressor, increases disorganized vasculature, decreases functional arteriogenesis in the ischemic mouse muscle (176), and enhances tumor vascularization and growth (99). These data suggest that either hyper-activation or inhibition of NF-κB activity can result in dysregulated angiogenesis.
Hypoxia-inducible factor-1α
Hypoxia-inducible factor (HIF)-1 is a major transcriptional factor that responds to hypoxia by activating angiogenic and glycolytic pathways to deliver oxygen and nutrients into hypoxic tissues (Fig. 7). HIF-1 is composed of an O2-regulated HIF-1α, and a constitutively expressed HIF-1β subunit (161). Protein expression levels of HIF-1α control HIF-1 activity. Therefore, HIF-1α stability and the regulation of its stability by PTM are critical for transcriptional gene activation. Under normal oxygen levels, HIF-1α is hydroxylated at proline 402 and 564 in the oxygen-dependent degradation (ODD) domain by prolyl-hydroxylases (PHD) (138). The proline hydroxylation facilitates the ubiquitination of HIF-1α by the E3 ubiquitin ligase von Hippel Lindau protein (pVHL) and causes rapid proteasomal degradation of HIF-1α. Hypoxia limits PHD activity and HIF-1α hydroxylation and subsequent degradation, resulting in stabilized HIF-1α protein. NO (nitric oxide)-mediated S-nitrosylation at Cys533 (mouse, human Cys520 equivalent) in the ODD domain prevents degradation of HIF-1α (110). Longer lasting S-glutathionylation occurs at the same cysteine (180). Glrx inhibition or increased protein S-glutathionylation stabilizes HIF-1α in vitro by preventing its interaction with pVHL. Glrx deletion in mice enhances protein S-glutathionylation, which stabilizes and transcriptionally activates HIF-1α and increases HIF-1α and VEGF expression in the ischemic muscle, promoting revascularization and blood flow recovery of the ischemic limb after femoral artery ligation (180) (Fig. 5). Our molecular mechanism can explain the ROS-dependent HIF-1α activation (38) and may play an important role in other biological processes. In addition to the direct modification of HIF-1α, mitochondrial oxidants and various other factors may affect PHD activity (140).
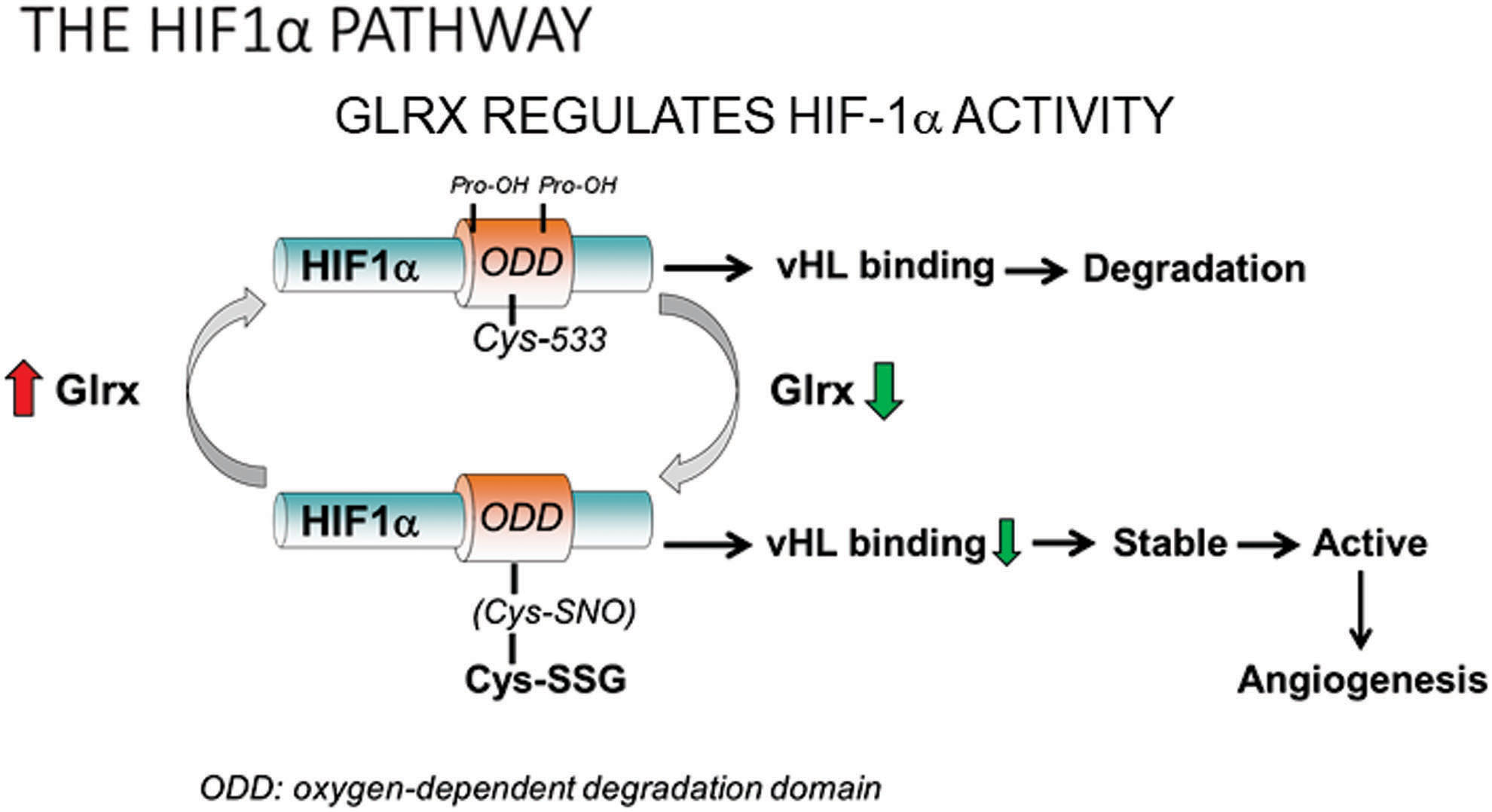
Other targets
S-glutathionylation inactivates the small Rho GTPase Rac1 (Ras-related C3 botulinum toxin substrate 1) under metabolic stress, and Glrx overexpression maintains Rac1 activity leading to protection of the EC barrier function, lowering of vascular permeability, and inhibition of angiogenesis (67). Therefore, upregulated Glrx may protect ECs from atherogenic insults at the expense of decreasing EC migration and angiogenic processes. Aside from Rac1, many other S-glutathionylated proteins may inhibit vascularization, but endogenous Glrx mostly has antiangiogenic effects, as shown in Glrx KO mice. The mechanism by which Glrx activates specific signaling pathways likely depends on the context of the pathology.
Nonalcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD) starts with simple, fully reversible hepatic fat accumulation, referred to as steatosis (Fig. 8A), that is not caused by alcohol. In most cases, NAFLD is asymptomatic. Although steatosis in NAFLD is fully reversible, the disease may advance to a more severe form that involves inflammation with or without fibrosis, referred to as NASH (Fig. 8B). Glrx KO mice develop obesity, hyperlipidemia, and fatty liver by 8 months of age on standard chow, mimicking NAFLD (163). The liver disease in the Glrx KO mice follows the “two-hit hypothesis” for NAFLD (163) (Fig. 8C); the fatty liver is present, and a second event leads to liver inflammation and progression to NASH. The second hit, a high-fat diet, induces steatohepatitis and advances to NASH in the Glrx KO mice.

However, in clinics, the exact reason or timing of NAFLD progression to NASH is often unclear and based on a complex and multifactorial etiology. Hence, the “multiple parallel hit” hypothesis (Fig. 8D) better reflects the NAFLD pathogenesis in patients.
Reconstitution of Glrx in the Glrx-deficient liver suppressed hepatic steatosis and lipid levels in the short term (163), indicating that the upregulation of Glrx might be beneficial to control lipid metabolism. Even though fibrosis is usually more difficult to produce in mice, Glrx-deficient mice fed a high-fat diet developed fibrosis, starting from the hepatic triad.
The role of Glrx and protein S-glutathionylation in the pathogenesis of NAFLD remains vastly unexplored. The high-fat diet-fed mice develop NAFLD and show increased levels of S-glutathionylated proteins. One of the target proteins is the central metabolic regulator sirtuin-1 (SirT1), an NAD+-dependent class III histone deacetylase (162). Thiol modifications of SirT1 modulated lipid metabolism via acetylation of key transcription factors. S-glutathionylation inactivates SirT1 and promotes hyperacetylation and activation of downstream target proteins such as p53 and sterol regulatory element-binding protein (SREBP) (Fig. 8E). p53 activation is a cellular response to stress that changes metabolism and halts cell cycle progression. NAFLD livers also express increased levels of p21, a downstream target gene of p53, which may interfere with liver regeneration by inhibiting hepatocyte proliferation. Hepatocytes experiencing grave oxidative stress may use p53 to initiate controlled cell death involving p53 upregulated modulator of apoptosis (PUMA), also known as Bcl-2-binding component 3. SirT1-mediated S-glutathionylation is reversed by Glrx, which can prevent the activation of p53 in hepatocytes. Consistent with this finding, overexpression of SirT1 improves NAFLD (47), whereas an inoxidizable cysteine to serine mutant SirT1 has an even stronger effect and blocks p53 activation. Conversely, SirT1 knockout mice develop NAFLD (196).
Glrx KO mice fed a standard chow diet spontaneously develop hepatic steatosis, and feeding a high-fat diet advances the disease to NASH, beginning with mild fibrosis (163). Glrx ablation increases S-glutathionylated SirT1, causing hyperacetylation and transcriptional activation of SREBP. Two isoforms of SREBP exist: SREBP-1, which regulates the de novo fatty acid synthesis, and SREBP-2, which controls cholesterol metabolism. Both of these pathways are upregulated in Glrx KO livers. Viral Glrx replenishment of the steatotic Glrx KO mouse liver attenuates de novo fatty acid synthesis and decreases hepatic lipids.
Overexpression of the cysteine to serine mutant SirT1 in steatotic Glrx KO mouse liver also reverses NAFLD (163). Active SirT1 not only attenuates hepatic lipid synthesis but also promotes fatty acid oxidation in peroxisomes and mitochondria. Clinical trials with SirT1 activators show little effect on insulin resistance and glucose metabolism, but they, undoubtedly, improve plasma lipids and likely modulate hepatic lipid homeostasis. Thus, increasing Glrx or SirT1 activity in NAFLD is an effective therapy to normalize hepatic lipid homeostasis in mice that appears to translate into clinical medicine.
In addition to lipid-lowering effects, Glrx protects endothelial barrier function (67) and inhibits monocyte chemotaxis under metabolic stress (178). These data imply that upregulation of Glrx may safeguard vessels from atherosclerotic insults.
Brain
Oxidative stress contributes to the pathogenesis of neurodegenerative diseases such as Alzheimer's and Parkinson's disease. Glutaredoxins, together with the thioredoxin system, assist in maintaining a reduced environment and protecting neuronal cells from apoptosis. The two reductive systems likely mediate amyloid neurotoxicity. Human brains affected by Alzheimer's disease increase Glrx expression levels, whereas thioredoxin expression decreases (7), indicating a distinct role of each system.
Glrx may also play a role in Parkinson's disease. The Parkinson's disease-like illness induced by the neurotoxin MPTP increases striatal Glrx expression, which is essential for the maintenance of mitochondrial complex I function in mice (93). Likewise, female mice have higher constitutive expression of Glrx than males and do not suffer the MPTP-induced loss of dopaminergic neurons. By using an estrogen receptor antagonist, Glrx activity decreases in the female mouse brain (but not in the liver), and MPTP-induced complex I dysfunction increases (92). Similarly, the loss of homologs to Glrx exacerbates neurodegeneration in Caenorhabditis elegans models of Parkinson's disease (86). Lastly, Parkinson's disease patients have decreased Glrx levels in the midbrain (86).
Therefore, the upregulation of Glrx is expected to be beneficial for Parkinson's disease. However, Glrx overexpression in microglia causes hyperactivation of NF-κB (129), increases inflammatory cytokines, and decreases tyrosine hydroxylase in the brain of Glrx-overexpressing mice fed a high-fat high-sucrose diet (85). These data indicate that higher Glrx expression in microglia induces inflammation and dopaminergic neuronal degeneration, outweighing the neuroprotective effects of overexpressed Glrx in dopaminergic neurons (63). In summary, balanced Glrx activity appears essential to maintain a healthy brain and cell-selective therapies are required to target Glrx.
Airway and lung
The respiratory system is easily exposed to environmental oxidative stress, and Glrx and S-glutathionylation significantly regulate lung pathology. In acute inflammatory states, elevated GSH adducts in Glrx KO mice suppress lung inflammation by inhibition of the NF-κB pathway. The administration of lipopolysaccharide (LPS) induces Glrx and inflammatory cytokines in the lung, but Glrx KO mice exhibit attenuated inflammatory responses (4). Glrx-deficient macrophages produce fewer oxidants and cause less phagocytosis (6, 17). Thus, Glrx induction can activate NF-κB (Fig. 5), which further induces Glrx (6) and accelerates the inflammatory responses in a feed-forward manner.
Glrx inhibits the apoptosis pathway by activating NF-κB as well as by inhibiting the pro-apoptotic receptor Fas. GSH-adducts of Fas amplify Fas-mediated apoptosis (6, 12). In Pseudomonas aeruginosa–induced pneumonia, inhibition of Glrx is protective since Fas-mediated apoptosis promotes pathogen clearance, whereas Glrx overexpression in lung epithelial cells increases mortality in infected mice (10).
In contrast, inhibiting apoptosis by Glrx overexpression in alveolar epithelial cells is protective against lung fibrosis (11). Thus, the redox regulation of Fas plays opposite roles in the lung, depending on pathological models. Interestingly, lungs from patients with idiopathic pulmonary fibrosis show lower Glrx activity and higher S-glutathionylated proteins (10). Glrx KO mice have exacerbated lung fibrosis induced by bleomycin or TGFβ, whereas Glrx overexpression in alveolar epithelial cells inhibits apoptosis and attenuates collagen accumulation and fibrosis. Intra-tracheal administration of recombinant Glrx also suppresses fibrosis in mouse lungs, suggesting the potential therapeutic use of Glrx to treat pulmonary fibrosis (11). Decreasing S-glutathionylation by inhibiting GSTπ, such as using GSTπ inhibitor TLK117, is another strategy to treat lung fibrosis (125).
Conclusions
The field of redox regulation has progressed slowly due to complex redox biochemistry and imperfect analytical tools. Recent improvements in genetically encoded redox biosensors that visualize live redox processes coupled with CRISPR-Cas9 and adeno-associated virus-mediated transduction will continue to accelerate our understanding of in vivo redox regulation.
Interconnected redox functions of antioxidant systems, such as thioredoxins and glutaredoxins, often complicate our understanding of systems. The built-in redundancies partially distract us from the real physiological importance of these enzymes. Thus, glutaredoxins are often perceived as another layer of the cellular antioxidant system and have found an entry in common protein databases as such.
However, protein cysteine modification with GSH adducts, S-glutathionylation, is controlled by oxidants and enzymes, including GSTπ and Glrx, contributing to redox regulation. In particular, the Glrx-mediated reversal of modified proteins modulates signaling cascades in vivo and controls pathological phenotypes. Upregulated Glrx inhibits ischemic vascularization of the hindlimb, whereas it improves lung fibrosis and liver steatosis in mice. This complexity in phenotypes likely derives from the hierarchical control of S-glutathionylated and Glrx-regulated proteins as well as tissue and cell type-specific regulation. Thus, cell-specific manipulation of Glrx will further elucidate Glrx-mediated redox signaling in vivo.
Further, recent experimental data suggest that redox regulation involves transglutathionylation between proteins, that is, redox relays that define novel signaling cascades between proteins. Transglutathionylation, in particular, may enable redox signaling across membranes and between cellular compartments. However, additional research is required to elucidate the significance of these redox relays further. For these reasons, Glrx can be a potential therapeutic target to modulate redox regulation and treat conditions such as fatty liver, PAD, and lung fibrosis.
Footnotes
Acknowledgment
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the awarding offices.
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
This work was supported by National Institute of Health grants R01 DK103750, R01 HL133013, R21 AG058983, R21 AA026922, R01 HL137771, and R03 AG 051857, American Heart Association “Grant in Aid” 16GRNT27660006, European Cooperation in Science and Technology (COST Action BM1203/EU-ROS), and the Metabolic Clinical Research Collaborative. This work was also supported by NIH/Boston University Clinical & Translational Science Institute grant 1UL1TR001430 to M.M.B. and R.M. M.M.B. was also supported by the Evans Junior Faculty Research Award by the Department of Medicine of Boston University. B.F. was supported by NIH T32 HL007224 Multidisciplinary Training in Cardiovascular Research through the Whitaker Cardiovascular Institute at Boston University.