
Abstract
Significance:
The geological record shows that as atmospheric O2 levels increased, it concomitantly coincided with the evolution of metazoans. More complex, higher organisms contain a more cysteine-rich proteome, potentially as a means to regulate homeostatic responses in a more O2-rich environment. Regulation of redox-sensitive processes to control development is likely to be evolutionarily conserved.
Recent Advances:
During early embryonic development, the conceptus is exposed to varying levels of O2. Oxygen and redox-sensitive elements can be regulated to promote normal development, defined as changes to cellular mass, morphology, biochemistry, and function, suggesting that O2 is a developmental morphogen. During periods of O2 fluctuation, embryos are “reprogrammed,” on the genomic and metabolic levels. Reprogramming imparts changes to particular redox couples (nodes) that would support specific post-translational modifications (PTMs), targeting the cysteine proteome to regulate protein function and development.
Critical Issues:
Major developmental events such as stem cell expansion, proliferation, differentiation, migration, and cell fate decisions are controlled through oxidative PTMs of cysteine-based redox nodes. As such, timely coordinated redox regulation of these events yields normal developmental outcomes and viable species reproduction. Disruption of normal redox signaling can produce adverse developmental outcomes.
Future Directions:
Furthering our understanding of the redox-sensitive processes/pathways, the nature of the regulatory PTMs involved in development and periods of activation/sensitivity to specific developmental pathways would greatly support the theory of redox regulation of development, and would also provide rationale and direction to more fully comprehend poor developmental outcomes, such as dysmorphogenesis, functional deficits, and preterm embryonic death.
Introduction
Developmental programs in animals can be disrupted by oxidative stress, and early research in developmental toxicology shows that oxidants cause malformations due to macromolecular damage to oligonucleotides, proteins, lipids, and glycans. Recent research shows, however, that oxidants also beneficially function as signaling molecules, and that oxidative stress disrupts the signaling process. Over the past 20 years, this research has advanced through studies of redox signaling, and an improved understanding is beginning to emerge through formulation of principles governing the use of electron transfer reactions in living organisms, termed here, redox theory. Knowledge of these underlying principles of beneficial and harmful use of oxidants and O2 will enable more precise redox systems biology methodologies to test hypotheses and predict outcomes of environmental and chemical exposures during specific developmental windows. Such models are also expected to be useful coupled with new redox measurements, which enable detailed study of specific cell populations within relatively precise spatial and temporal sequences.
The purpose of the present article is to review research on oxidants during development within the context of emerging redox theory. This builds upon the earlier free radical theory of development described by Allen and Balin (4) and further elaborated by Hitchler and Domann (80). Allen and Balin reviewed evidence for O2 effects on development and increased abundance of antioxidant systems during development. They postulated that the effects of metabolic gradients that occur during development result from differential O2 supplies and that metabolically generated oxidants direct the initiation of certain developmental events. Hitchler and Domann extended these principles by linking glutathione (GSH) production and O2 sensing to establish the epigenotype during development. They ascribed a role for increased GSH production in epigenetic processes through limiting the availability of S-adenosylmethionine, the cofactor utilized during epigenetic control of gene expression by DNA and histone methyltransferases. They purport a role for O2 through histone demethylases, using O2 as a required cofactor.
In consideration of the central tenets of the free radical theory of development and current understanding of redox biology, we suggest that an updated interpretation warrants reformulation of the free radical theory into a redox theory of development. The most critical issue is that nonradical oxidants are more abundant than free radicals and appear to be quantitatively more important in oxidation/reduction reactions during development of complex organisms. A second critical issue is that nonradical redox systems involving NADH/NAD+ and NADPH/NADP+ maintain stable nonequilibrium systems to organize energy metabolism and maintenance of the redox proteome. These systems are counterbalanced in the steady state by relatively stable oxidant pools, including hydrogen peroxide (H2O2) and other nonradical oxidants. They are not dependent upon relatively short-lived, low-abundance free radical species.
As elaborated in the redox code (see Redox Theory, Oxidative Metabolism, and Oxidative Stress section), the nonradical NAD+, NADP+, and oxidant systems support a series of switching mechanisms, which link bioenergetics and metabolism with conformation and function of macromolecules. Since most of these macromolecules contain thiols (i.e., cysteine [Cys] residues) that are reversible under physiological conditions, this review primarily focuses on thiol switching, although other couples are likely of equal importance in the context of development (16, 37, 158). Finally, the free radical theory places O2-dependent radical generation directly in causal pathways for development, counterbalanced by antioxidant free radical scavenging systems. In redox theory, O2 is viewed as a critical determinant in the evolution of differentiation programs for complex organisms and as a required intermediate to maintain bioenergetics, the redox proteome, and supply of H2O2, nitric oxide, oxidized lipids, and other redox signaling systems. The redox theory does not exclude the function of superoxide anion radical or other free radicals in developmental mechanisms, but rather abandons the principle that O2-dependent free radical production is a central guiding principle for multicellular development.
We present this initial draft of a redox theory for development with an expectation that modifications and improvements will be needed as efforts are made to translate this into improved fertility, obstetric, and pediatric medicine. We start with concepts of redox theory, oxidative metabolism, and oxidative stress that are particularly relevant to development. We follow this with review of the O2 shifts during development, metabolic reprogramming, and resulting shifts in central hubs controlling thiol redox systems, associated developmental programming, and detailed considerations of redox circuitries for redox signaling and transcriptional control. This enables definition of a paradigm for redox signaling in directing changes in multicellular organization and allows formulation of an initial redox theory of development.
Redox Theory, Oxidative Metabolism, and Oxidative Stress
Life has four general characteristics, including capabilities to (i) extract energy from the environment, (ii) maintain physical and chemical organization, (iii) delineate and defend self from environment, and (iv) reproduce (Fig. 1A). The genetic code provides a basis to define and preserve the encoded definition of an individual. Animals use their genetic information within the context of pre-existing nuclear and cellular structures to faithfully generate complex multicellular progeny. Thus, the concept of reproduction in animals extends well beyond that of replication of the genetic code in single cells to generation of a spectrum of reproducing cell types in complex communities of interacting cells in organ systems. While the genetic code provides understanding of the molecular bases for replication and information transfer, the genetic code provides little help in understanding the principles for use of that genetic information in development of complex multicellular progeny.
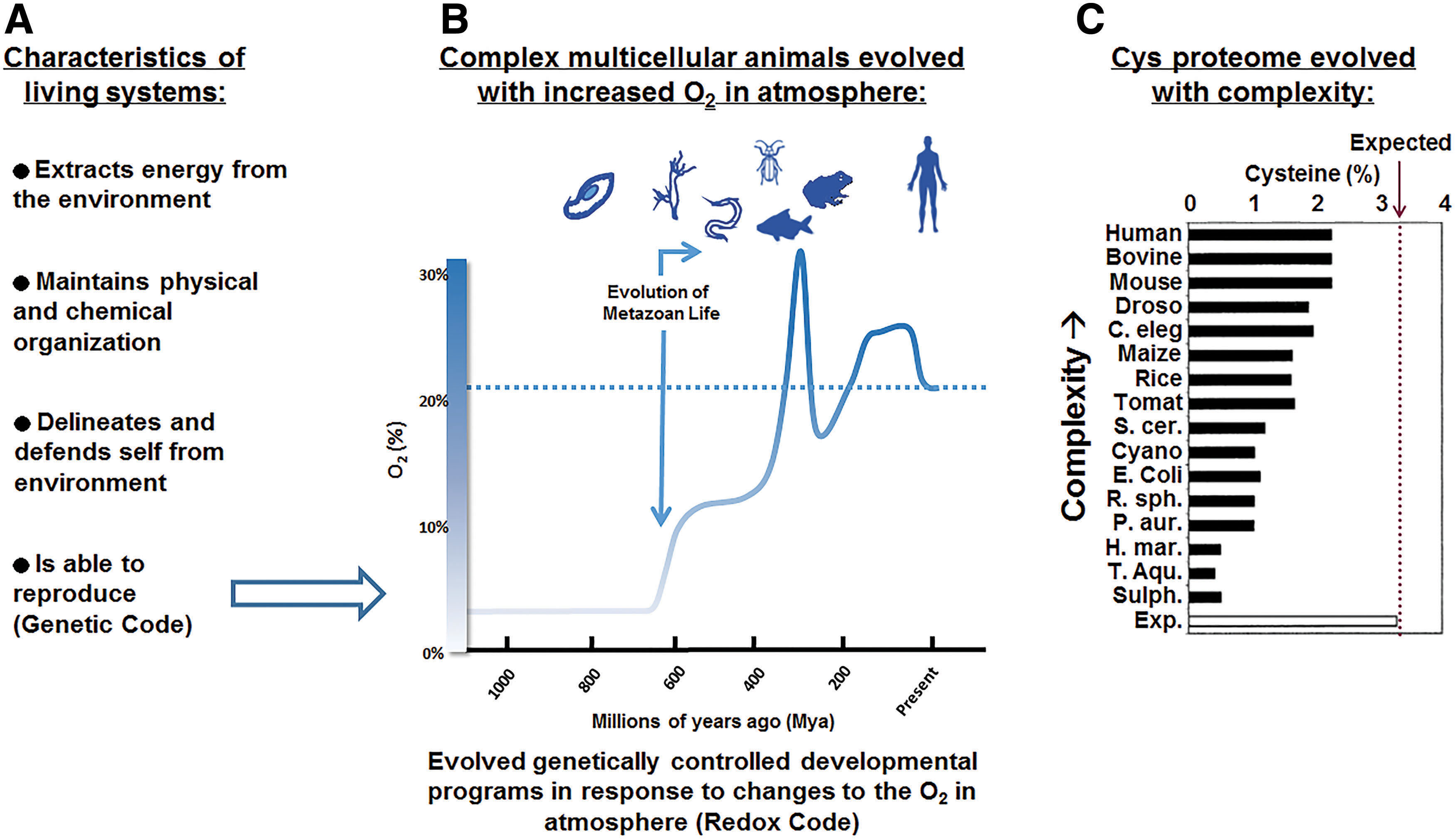
The redox code provides logic for characteristics of life (i.e., energy extraction, molecular organization, delineation of self, and reproduction) to complement the information storage and transfer provided by the genetic code (97). All aspects of life depend directly or indirectly upon oxidative reactions. Extraction of energy involves high-flux oxidative reactions of glycolytic and mitochondrial systems that maintain cellular bioenergetics and metabolism. Cellular energetics and metabolism are coupled to macromolecular structure and function through many types of reversible switches in proteins; some directly involve oxidation and reduction of cysteine or methionine residues in proteins, while others involve post-translational modifications (PTMs) such as phosphorylation/dephosphorylation, methylation/demethylation, and other covalent alterations that are indirectly dependent upon oxidative metabolism. Activation/deactivation cycles of these switches support temporal and spatial organization, allowing metabolism and other functions to be coordinated within and between cells. Finally, the entire system of bioenergetics, metabolism, and macromolecular structures functions together as an integrated network to support an individual's interaction with the environment.
The principles of the redox code were previously outlined to contain four basic principles, including (i) bioenergetics, (ii) macromolecule structure and activities regulated through kinetically controlled sulfur switches in the redox proteome, (iii) H2O2 production to control spatiotemporal redox signaling, and (iv) redox networks regulated in microcompartments to form an adaptive system to respond to the environment (97). The redox code has been considered within the context of aging (59, 96), environmental exposures (58), and nutrition (28), but not within the complex framework of mammalian development. Unlike the processes of aging, environmental exposures, and nutrition, which can be considered in holistic models, the developmental programs require specific temporal and spatial cues to use genetic information to elaborate multiple cell types within tissues and organ systems. The redox code provides simple and straightforward logic for this process within our understanding of the evolution of multicellular animals with the dramatic rise in O2 in the Earth's atmosphere (Fig. 1B).
Unicellular predecessors survived excessive O2 by developing sensors for O2 and oxidative stress that incorporated abundant atmospheric and terrestrial materials such as sulfur and iron into iron/sulfur clusters (26, 106). It is also certain that prokaryotes used O2-sensing HIF-1α (hypoxia-inducible factor) systems and their PAS (Per-Arnt-Sim) domains because of their similarity to the PAS domains used in bacterial systems for locomotor behavior in response to O2 and redox environments (201). The concordance of form and function is well recognized for organ systems functioning in O2 transfer and delivery. Similarly, at the cellular and subcellular level, density and distribution of mitochondria determine intracellular O2 gradients and responses to tissue O2 availability. Responses to oxidative stress are also adaptive, and evidence is available indicating that the master regulator of adaptive response to oxidative stress, nuclear factor erythroid-derived 2-like 2 (Nrf2), also evolved with the rise in O2 in the Earth's atmosphere (49). Thus, the available evidence suggests that redox theory extends to development and, perhaps more importantly, that O2 availability was likely a central driving force in the emergence of redox mechanisms in developmental programs.
These observations converge to enable the construction of a model, in which the rise in atmospheric O2 drove multicellular evolution to a reliance on O2 for energy production, structural and functional organization, defense against environmental insults, and the regulation of reproduction. At the molecular level, these adaptations are observed in parallel with the increased percentage of Cys residues in proteins (Fig. 1C). Miseta and Csutora (136) showed that the percentage of Cys residues in protein does not achieve the expected percentage (3.2%) in any organism based upon the percentage of codons. They also showed that the percentage of Cys residues increased with evolution of complexity. Obviously, the complexity of the developmental program is directly proportional to the complexity of the organism. Thus, multicellular evolution is explicitly related to the evolution of mechanisms for multicellular development. Because O2 was the driving force and O2-sensing and oxidative stress responses were available to provide mechanisms for both spatial and temporal sequences required for multicellular development, it follows that redox theory can provide a central logic to understand developmental programs and ultimately develop models for systems biology of development.
Oxygen Gradients During Metazoan Development
An essential characteristic of life involves the ability to reproduce and therefore pass along critical genetic information to successive generations. The information embedded in the DNA of each species and life-form constitutes its respective genetic code, including the instructions for how environmental signals are to initiate and regulate the controlled expression of the genetic developmental program. Following the combination of male and female genetic material at fertilization, metazoans and many other complex life-forms temporarily abdicate their direct physiological reproductive responsibilities by physically isolating the early developing conceptus (embryo proper and its associated extraembryonic membranes interstitially implanted into the maternal endometrium or egg buried in the sand or released into a stream) into a more hypoxic environment, where fluctuations in atmospheric O2 are still possible. Mammals, birds, amphibians, reptiles, and fish, all situate their developing embryos in states of sustained hypoxia to complete the most formative and sensitive steps of early organogenesis/embryogenesis (38, 112, 59a). Of these, embryos growing in aquatic environments seem to have a wider range of tolerance to changes in O2 concentrations (149). Organisms found at lower levels of the phylogenetic tree such as the round worm, Caenorhabditis elegans, can complete their entire developmental program in near complete anoxia, around 0.1% O2 (46, 148). Atmospheric normoxia equates to O2 concentrations of 20.8% (160 mmHg). Normal or physoxic levels in cells and tissues of a given organism usually range from 5% to 7.4% O2 (38–65 mmHg) but can be quite heterogenous depending on the type of tissue, density, diffusion distances, and other factors (62). In physiological hypoxia, O2 concentrations drop to 2%–6% (15–45.6 mmHg), with pathological hypoxia occurring below 4.2% O2, from 0.3% O2 to 4.2% O2 (2–32 mmHg).
To best describe the relationships between metazoan development and atmospheric O2, we mostly use rodent models as examples, the mouse specifically, but also include some references to chick, human, and general metazoan development. Maturation of the oocyte, its subsequent expulsion from the follicle (ovulation), and fertilization in the ampulla/fallopian tubes of the uterine tube (oviduct) take place in maternal tissues that are physoxic and, in general, adequately perfused. O2 concentrations in the ampulla are around 4.2%–5% (32–38 mmHg) and increase up to 6.8% (52 mm HG) by day 3 day postcoitum (45) (Fig. 2).
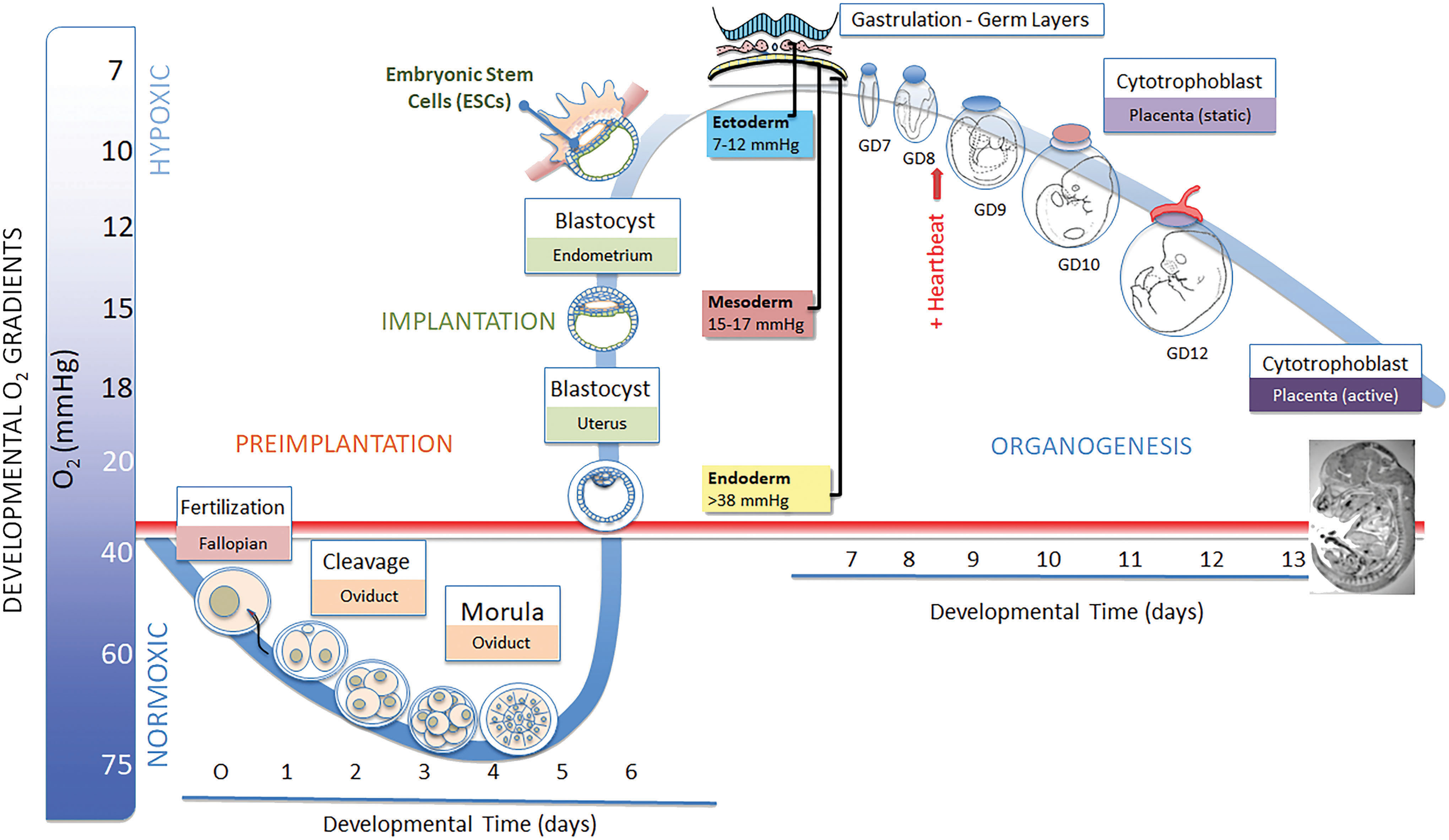
Around day 4, the blastocyst is expelled into the uterine fluid, sheds the zona pellucida (hatching), and prepares to attach to the uterine endometrial wall (implantation). In the uterine environment, O2 concentrations range from 1% to 5% O2 (0.5–38 mmHg) (145a). Within the blastocyst proper, O2 concentrations are maintained below 2% (15 mmHg). This physical removal of the conceptus from a physoxic environment and its placement in a hypoxic environment allow atmospheric O2, or the lack thereof, to instruct expression of the developmental program. This phase of severe hypoxia has numerous developmental ramifications because it provides the ideal conditions to maintain totipotency and pluripotency and facilitate colony expansion of the blastocyst inner cell mass, the source of embryonic stem cells (ESCs). This relates to why O2 concentrations are kept low (1.0%–0.2% O2) during in vitro fertilization procedures to increase positive developmental outcomes (103). The juxtaposition of fluid-filled cavities, such as the chorioallantoic and amniotic cavities, that are positioned opposite of embryonic tissue layers (epiblast and hypoblast) helps to preserve the extreme hypoxic state by increasing diffusion distances and establishing distinct environmental barriers. Once the conceptus (embryo proper and its associated extraembryonic membranes) interstitially implants into the endometrial mucosa, contact with the uterine fluid is lost and no further direct contact with perfused, active O2 sources is available until the chorioallantoic placenta becomes active somewhere near gestational day (GD) 11.5 (in mouse) and throughout the completion of organogenesis on GD 15.
As the implanted embryo expands, begins to establish its dimensional growth axes, and undergoes gastrulation to form the three primary germ layers, ectoderm, mesoderm, and endoderm, spatial internal O2 gradients are established to direct selective signaling where the ectoderm (7–12 mmHg) remains extremely hypoxic, mesoderm (15–17 mmHg) slightly less so, and the underlying endoderm (>38 mmHg) approaches physoxia. Spatially distinct foci of hypoxic cells and tissues remain throughout organogenesis GD 9.0–15 and, although reduced in size, persist throughout gestation (Fig. 2 inset) (38, 192). Trophoblast differentiation into the definitive yolk sac and cell types necessary for placental remodeling require O2 concentrations of >2% as is clearly illustrated for the invasive cytotrophoblast cell populations (20, 54, 192). Hematopoiesis, the appearance of blood islands, and angiogenesis, in the visceral yolk sac (VYS) and embryo proper, require low O2 (48, 192, 228), as do cardiac progenitor cells (81), myocardial progenitors, myocardium (46), angiogenesis (48), neural crest cells (182), chondrocytes, chondrogenesis (179), neurogenesis (163, 211), and myogenesis (132); in essence every aspect of embryo- and fetogenesis requires acutely hypoxic conditions and subsequent HIF-mediated regulation to develop properly. The focal, punctate regions of hypoxia remaining in the fetus as it approaches parturition are believed to represent pockets of various stem cells requiring the low O2 conditions to maintain stemness, undergo colony expansion and proliferate when instructed (38).
So how does the developing embryo manage to thrive under conditions of pathological hypoxia? The answer lies in the sensors and processes that appear to have been recapitulated from evolutionary experience with fluctuating O2 and overall atmospheric hypoxia. Prokaryotes and early life-forms have overcome the potential dangers of increasing and fluctuating O2 levels by first developing means to sense changes in O2 and then subsequently develop protective/adaptive measures for their survival (172, 202). During the process of evolution of complex organisms, an interface was established between atmospheric O2 and the susceptible oxidizable proteins, DNA, lipids, and other biomolecules in the live organism to sense O2, activate protective measures, and control expression of the genetic code (49, 138, 162, 197). The process of metazoan development recapitulates the hypoxic conditions and reprograms the developing embryo to utilize the full complement of these signals and controls during each reproductive cycle. This concept was first introduced in a different context in the early 1800s as the Meckel–Serres law, and was most famously articulated by Ernst Haeckel as “ontogeny recapitulates phylogeny.” Haeckel's oft criticized and now rejected dictum postulated that: the embryonic development of an animal from fertilization to parturition or hatching (ontogeny) progresses through stages that resemble or recapitulate the structural forms from successive stages seen in the evolution of the animal's remote ancestors (phylogeny) (147). In the current discussion, we see a more defensible recapitulation of atmospheric sensing, metabolism, and developmental regulation through a specific redox interface in which the normoxic embryo/conceptus at fertilization is physically removed to a hypoxic environment and essentially repeats the stages (phylogeny) represented by the evolution of adaption to increasing O2 (38, 202).
O2 Sensing and Regulation by Basic Helix Loop Helix/PAS Transcription Factors
Early prokaryotic life evolved under environmental conditions that were substantially deplete of O2 but also subject to increasingly frequent fluctuations in its concentrations. When O2 levels increased, affected organisms became susceptible to oxidative damage due to metabolic generation of reactive O2, exacerbated by a geologic abundance of ferrous iron and atmospheric ionizing radiation, along with their collective abilities to catalyze the generation of reactive oxygen species (ROS) such as O2 •−, H2O2, and 1O2 (26, 152). Mounting an effective response to these uncertain environmental conditions involves at least three critical components: (i) a means to sense the threat (e.g., an environmental sensor), (ii) an affector/regulator to direct adaptive changes, and (iii) activation of target pathways to neutralize threats and adapt to restore cellular homeostasis.
Primitive prokaryotic proteins that evolved to act as direct sensors and regulators of O2 belong to the Fnr (fumarate and nitrate reductase) regulon (26, 106). In the absence of O2, the Fnr protein is able to homodimerize, translocate to the nucleus, and bind to its DNA response element, regulating the expression of genes involved in anaerobic energy metabolism. Under aerobic conditions, O2 binds to the prosthetic surface-exposed Fe-S cluster ([4Fe-4S]2+) of Fnr converting it to [Fe2-S2]2+ and leading to its dissociation into monomers, thus preventing DNA binding. This is an example of direct transcriptional redox control where the Fnr protein acts as the primary O2 sensor and the DNA-binding transcriptional regulator for over 100 target genes related to anoxia (168). Other systems coevolved to sense ROS (O2 •− and H2O2) and mediate necessary metabolic changes conscripting the service of reactive intermediates themselves as signaling molecules. In this context, the hydrogen peroxide-inducible gene activator (OxyR) is the master peroxide sensor that responds to very low levels of H2O2 in cells and tissues through its Cys199 cysteine switch. SoxR and SoxS (superoxide response regulon) also recognize ROS and other oxidants to protect the organism and mediate adaptation (184). In eukaryotes, nuclear respiratory factor 1 (NRF1) expression can be maintained via activation of redox-sensitive elements (30), where NRF1 activities can support mitochondrial biogenesis, expression of mitochondrial components involved in respiration, and indirectly the regulation of mitochondrial genome (i.e., Tfam [transcription factor A, mitochondrial] expression) (177).
As atmospheric O2 concentrations continued to rise and fluctuate over evolutionary time, organisms acquired multiple means to regulate O2 homeostasis. Preserved mechanisms of environmental surveillance and response pathways include those mediated by mTOR (mammalian target of rapamycin), associated with autophagy, and the endoplasmic reticulum (ER) stress response (51, 165, 192, 221). Perhaps the most important developmental regulators belong to the bHLH (basic helix loop helix)/PAS transcription factor superfamily members of HIFs (27, 38, 42, 192). Characterization of HIF's roles and functions has revealed the extent to which O2 regulates embryonic development across the phylogenetic scale from nematodes to humans. The role of O2 (or the lack thereof) in directing the growth, differentiation, and cell fate is so integral to successful expression of the genomic developmental program that it has been classified as a developmental morphogen similar to classically termed morphogens (i.e., secreted growth factors) (43, 192).
The family of bHLH/PAS transcription factors can activate hundreds of different genes with enhancer regions containing the hypoxia response element (HRE; 5′-RCGTG-3′). Three genes encode the HIF-α subunits (HIF-1α, HIF-2α, and HIF-3α). HIF-1α is ubiquitously expressed in all the developmental tissues, but its function as a transcription is regulated by O2 concentrations. HIF-2α is localized to the developing early embryonic vasculature and neural crest cells and later expressed in the lung, liver, and kidney. HIF-3α is expressed in various other restricted organs and compartments. An additional HIF subunit, HIF-1β (also known as ARNT; aryl hydrocarbon receptor nuclear translocator), is constitutively and ubiquitously expressed and serves as the heterodimerization DNA binding partner for HIF-1α-HIF-3α. All HIF-1 subtypes contain bHLH/PAS domains, yet none binds any known ligand and is reserved for dimerization and DNA binding (27). The HIF-1β/ARNT subunits are constitutively expressed and translated at a very high rate but are rapidly degraded by the 26S proteasome in the presence of normal concentrations of O2.
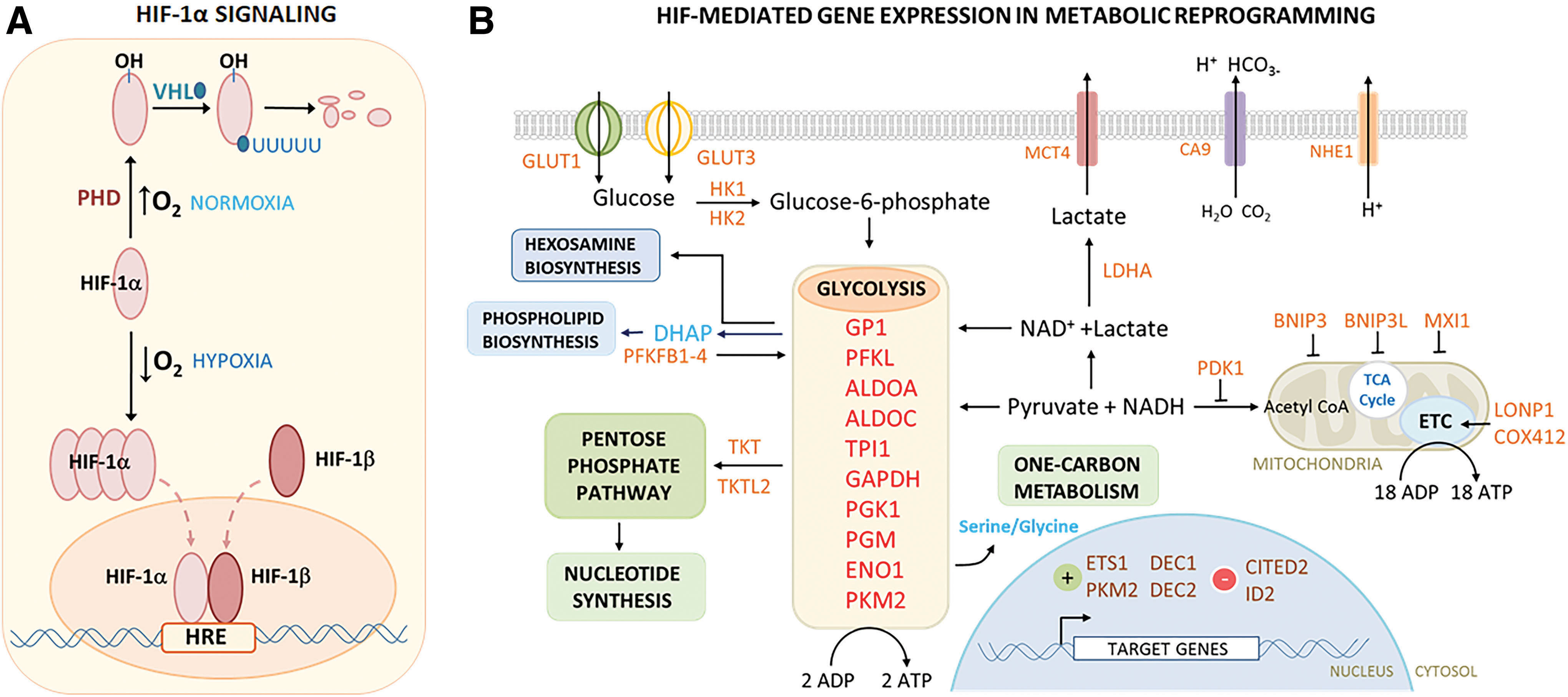
Oxygen sensing is accomplished through reversible hydroxylation of proline residues in the highly conserved O2-dependent degradation domain (ODD) of HIF-1, catalyzed by HIF-specific prolyl hydroxylases (PHD1–3) (181). Under normoxic conditions (>6% O2), the HIF prolines are hydroxylated by PHDs and bound by the von Hippel–Lindau tumor suppressor (pVHL) gene product which, as an ubiquitin ligase complex, polyubiquitinates the HIF-1α protein and sends it to the proteasome for degradation. When O2 concentrations drop into the hypoxic range (1%–5% O2), the hydroxylation activity of PHD is inhibited, and pVHL association is prevented so that HIF-1α subunits are no longer degraded and accumulate rapidly, translocate to the nucleus, and combine with HIF-1β/ARNT to activate the constellation of HIF target genes that mediate cellular and metabolic adaptations to moderate-severe hypoxia (99) (Fig. 3A).
Metabolic Reprogramming and Mitochondrial Remodeling in a Hypoxic Environment
The spatial and temporal fluctuations in ambient O2 concentrations that occur throughout the early stages of development influence bioenergetic processes, which meet energy demands through central carbon metabolism, and also regulate the metabolic signaling functions (137) that enable adaptation and protection from potentially harmful metabolites and metabolic by-products. Re-examination of the roles of metabolism during development has also helped to reinforce how metabolism serves to regulate specific biological processes during embryogenesis that direct cell proliferation, cell migration, differentiation, and cell signaling (137). Thus, the dual functions of bioenergetics and metabolic signaling are dependent on the process of metabolic reprogramming within the conceptus and are initiated in response to environmental cues, specifically the transition from physioxic to increasingly more hypoxic oxygenation states through the activity of O2 sensors and adaptive response networks. This pattern of metabolic reprogramming is not only unique to mammals but is found in birds, fish, amphibians, and other species as well (99, 172). The redox interface involved in O2 sensing and metabolic activity is not concerned only with oxidation events but is also affected by the corresponding reductions as part of their regulation (Fig. 3B). Considerable recent research has focused on this pattern of O2-dependent reprogramming because of the striking parallels between changes seen during development and those observed in cancer biology (114, 140, 194). Both circumstances use O2 sensing, metabolic reprogramming, and developmental controls on growth, proliferation, differentiation, and regulation of O2 supply to adapt to atmospheric conditions. This is the Warburg effect as described for metabolic changes in cancer cells (114).
The net consequence of metabolic reprogramming in a developmental context is the pronounced shift from an early reliance on oxidative phosphorylation (OXPHOS) to supply bioenergetic needs, to a strict dependence on glycolysis as embryogenesis proceeds from early cleavage stages to postimplantation organogenesis. Accommodation of these changes involves significantly altered gene expression, shifts in carbon utilization (glycolysis), as well as physical and functional changes to the mitochondria that are driven by bHLH/PAS transcription factors such as HIF/ARNT and their associated O2 sensing capacity.
Shortly after fertilization and during its transit through the uterine duct, the newly formed conceptus exists in a relatively O2-rich environment (Fig. 4), having access to a variety of high-energy substrates that include glucose, pyruvate, lactate, and glutamine (36). During the first few cleavage stages of the newly formed embryo and before the eight-cell stage, OXPHOS predominates, glucose is not well utilized for energy production, and glycolysis is largely suppressed due, in part, to improper glucose transport (82, 153, 166). As seen in bovine embryos, much of the available glucose at this stage is shunted through the pentose phosphate pathway (PPP) (93), which would increase the synthesis of nucleotides and NADPH, in preparation for rapid growth, and aid in the reduction of upcoming periods of oxidative stress. NADPH production does not require lamellar cristae but rather would be supported by a large matrix, suggesting low, sustained ATP output through OXPHOS (36). Pyruvate and glutamine remain as the principle ATP substrates during these early developmental stages (12). Uterine duct fluids contain high levels of lactate, which in itself can be used as a potential ATP generating substrate, but its use during early development was initially unclear (35). Cultured mouse zygotes showed that lactate could be converted to pyruvate but this was not used for ATP production. However, external pyruvate sources and glutamine could be consumed for eventual mitochondria-mediated ATP synthesis. Together, the observations that decreased glycolysis and increased pyruvate-derived ATP synthesis through OXPHOS demonstrate an increasingly important role of the mitochondria early in the preimplantation stages of development. Only during later stages, such as the morula and midblastocyst, do glycolytic processes begin to gradually increase and lead to a decreased ATP/ADP ratio (9, 164).

After the eight-cell stage, metabolically active mitochondria are replaced by immature, poorly formed mitochondria containing few lamellar cristae with low OXPHOS activity, low ATP production, and low O2 consumption (186, 207, 208). In the mouse, from early stages of development to the late blastula, total ATP levels continually decrease and synthesis is significantly slowed (56). Hypoxia-induced expression and activity of HIF transcription factors result in the subsequent expression of a battery of target genes in a number of diverse pathways that mediate metabolic needs (Fig. 3B). To optimize nutrient and O2 consumption during hypoxia and also to minimize unintended ROS production in mitochondria, HIF orchestrates the significant metabolic shift away from high O2 demand (TCA cycle/OXPHOS) toward anaerobic metabolism with activation of glycolysis and PPPs (47, 234). Glucose transport is significantly increased, ion and other metabolic pathways are upregulated, and all 10 enzymes of the glycolysis pathway are upregulated through HIF activity in conjunction with regulation that inhibits or limits TCA/OXPHOS, decreases mitochondrial number and volume, and maintains redox homeostasis (27, 173) (Fig. 3B). The redox interface involved in O2 sensing and metabolic activity is not concerned only with oxidation events but is also affected by the corresponding reductions as part of the regulation.
The higher ATP yields of active OXPHOS that existed in preimplantation development are exchanged for the additional biological benefits of sustained dependence on glycolysis/aerobic glycolysis throughout organogenesis. The shift to glycolytic dependence is a hallmark of highly proliferative cells with the subsequent consequence of increased biomass accumulation that is necessary for proper development and morphogenesis. The conversion to glycolytic dependence and its consequences have been well characterized as the Warburg effect of cancer cells and have been shown to be nearly identical to the reprogramming event that occurs during development (114). Under the reducing conditions of hypoxia, bioenergetics, and carbon metabolism, glycolysis/aerobic glycolysis is not only able to meet the needs for energy (ATP) demands but is also able to address the other metabolic needs for development by providing adequate reducing equivalents (NADPH), substrates for nucleic acid (serine/glycine), phospholipid biosynthesis (DHAP; dihydroacetone phosphate, a precursory substrate), and substrates for one-carbon metabolism (137).
Much underappreciated is also the role of specific glycolytic metabolites in the direct regulation of developmental signaling and control. Amino acids and other sources of carbon necessary for biosynthesis during development come from glycolytic by-products. Recent reviews and reports have also highlighted the direct nonbioenergetic roles of specific glycolytic metabolites in the compartmentalized signaling and modulatory functions during development (15, 137). Segregated in the cytosol, away from mitochondrial functions, many glycolytic reactions appear to be localized to subcellular sites (glycosome) where the highest demands for energy and the greatest need for sensing and rapid physiological adaption are found. A prime example is seen in the lamellipodia during the process of actin remodeling and processes necessary for cell motility (25, 84). Of considerable interest are the observations that several enzymes are active in the glycolysis pathway, such as pyruvate kinase M2 (PKM2), which is the major embryonic isoform of PK and is responsible for the alternative flux of glycolysis pathway, intermediates to the PPP, C1, hexokinase, and nucleotide biosynthesis pathways. Both PKM2 and GAPDH are also found in the nucleus where they are known to function as nuclear transcriptional coactivators and redox regulators (233). GAPDH and OCT1 are both modulated through the NAD+/NADH redox state (233). The consequence of all this is that pyruvate is produced from PEP without transferring a phosphate to ADP and, therefore, eliminating the production of an ATP. In this scenario, overall glucose uptake and phosphorylation are increased where the glucose metabolites are diverted to the PPP for the increased production of NADPH. Interestingly, PKM2 also acts directly as a transcription factor/coactivator to regulate the expression of a number of developmentally relevant genes (Oct4, HIF, p53, SLC2A1, Myc, KRAS, TFAM, STAT3) (116, 137, 225, 226).
After the developing embryo reaches the blastula stage and implants into the endometrial wall, mitochondria have few lamellar cristae, energy production through OXPHOS is greatly diminished, and a high proportion of available ATP is produced through glycolytic processes (anaerobic glycolysis) as a result of metabolic reprogramming (83, 207). Thus, O2 availability is dramatically decreased and glucose consumption and glycolysis are high (187). In the hypoxic state of rodent gastrulation-stage embryos (∼24 h postimplantation), the morphological maturation of mitochondria resumes, although OXPHOS activities remain low. In early somite stages (two to four somites), embryonic mitochondrial cristae become more numerous but have a blebbed morphology and are not yet lamellar, suggesting a gradual progression toward a more OXPHOS competent embryo (36, 186).
As organogenesis proceeds toward completion, and in anticipation of the major shift in available O2 that will accompany the activation of the chorioallantoic placenta, mitochondrial morphology continues to change where cristae become increasingly more lamellar and blebbing decreases to minimal levels. These changes occur at the time of completion of neurulation (186). In the mouse, from GD 8–10, embryonic metabolism radically switches back from anaerobic glycolysis to OXPHOS, again driven by O2 concentration gradients (129, 130, 186, 187). Reliance on mitochondrial function during this “switching” period has been demonstrated in numerous studies utilizing various knockout mouse models involving mitochondrial proteins.
For instance, knocking out a subunit of succinate dehydrogenase (complex II in the electron transport chain) resulted in early embryonic death evident at GD 7.5 (161). In another study, cytochrome c, which shuttles electrons to complex IV in the electron transport chain, was deleted and development stalls on GD 8.5 (120). Thioredoxin 2 (Trx2) is a mitochondrial protein reductase that is involved in the regulation of apoptosis (229). Loss of Trx2 results in dysmorphogenesis, including exencephaly, embryonic death, and increases ROS production on GD 10.5 (145). Mitochondrial thioredoxin reductase 2 (TR2) reduces Trx2 using NADPH as a cofactor. Loss of TR2 is also embryonic lethal, occurring around GD 13.5 (24). Together, these studies [and others reviewed in Baker and Ebert (8)] show that disruption of mitochondrial components tends to largely result in cessation and disruption of development during periods of metabolic reprogramming, where embryos switch from anaerobic glycolysis to OXPHOS for the majority of their ATP needs. As such, it is during these early periods of organogenesis and metabolic switching that redox signaling may support varying aspects of normal development, such as proliferation, differentiation, and apoptosis. Redox signaling and control are a core component of these key metabolic pathways, although relatively little molecular or biochemical detail is currently available to elucidate their mechanisms.
Developmental Programming Through HIF Expression
Having successfully rewired conceptal metabolism to adapt to hypoxic conditions, the HIFs continue to exert significant direct influence on development by also directing the expression, proliferation, differentiation, cell fate, and cell death events for the biogenesis of nearly every cell type and structure in the developing conceptus (developmental programming). Complete deletion or conditional knockout of HIF-1α or HIF-1β/ARNT results in embryonic lethality by GD 10.5 in the mouse and blocks the formation and function of all major organs and structures within the conceptus, affecting every stage of embryogenesis/organogenesis (Fig. 5) (38, 192).
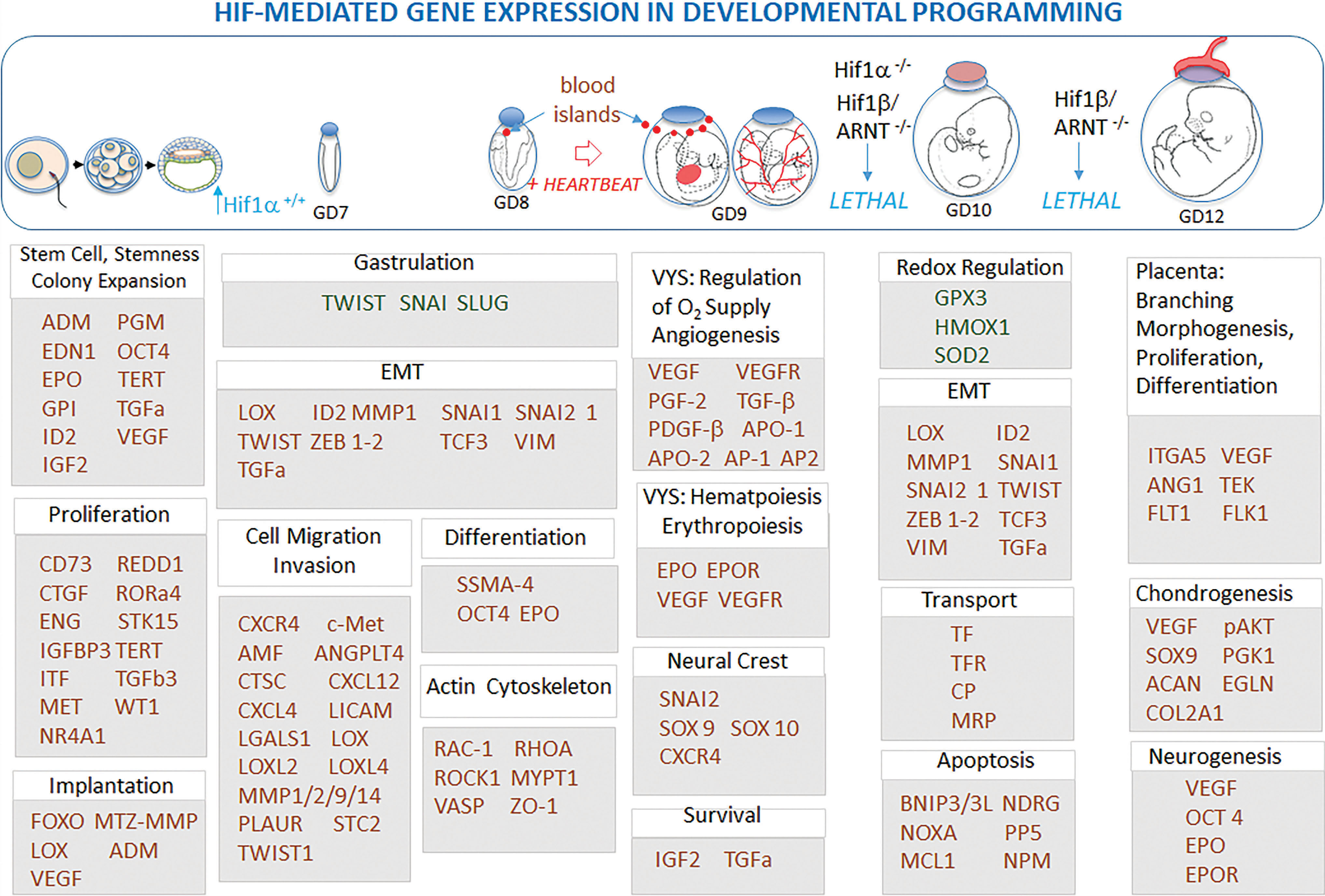
Concentrations of atmospheric/environmental O2 and the juxtaposition of tissues and fluid compartments within the conceptus create niches of hypoxia that vary in severity both spatially and temporally, suggesting that various HIF targets may be regulated differentially in a context-specific manner. The HIF-1s are known to regulate general pathways and processes during development that range from overall O2 and cellular homeostasis, to autophagy, inflammation, cell motility, apoptosis, and redox homeostasis (27, 183, 215). Beginning with the earliest stages of cleavage division until birth, HIF transcription factors regulate critical developmental processes such as maintenance of stemness (OCT4) and the control of colony expansion, pluripotency, proliferation, and fate determination (EPO, TGFb3, TERT, VEGF, ID2, IGF2, ADM, PGM) of ESCs in the early blastocyst, neural crest cells, and numerous other progenitor cells (192, 220). Often reiterated developmental process such as epithelial to mesenchymal transitions (TWIST [gastrulation, dorsoventral patterning, cell proliferation, morphogenesis, cell migration], Snail, Slug, SIP1, E47, Zeb1) used in gastrulation, angiogenesis, neural crest migration, and fate determination limb morphogenesis, and in nearly every other structural transformation during development, are all regulated by HIFs (227). Dynamic processes such as the determination of cell shape and cell migration through changes in the actin cytoskeleton are mediated through targets regulated by HIFs (214, 235). Critical to all aspects of the developing embryo is HIF control of hematopoiesis, vasculogenesis, and angiogenesis as part of the establishment of an active cardiovascular system to regulate O2 and distribute nutrients (46, 117, 118). Ultimately, many of the structural aspects of O2 sensing and developmental programming relate directly to the preparation of the conceptus to regulate future increases in O2 concentrations through the genesis and maturation of vasculature within a mature cardiovascular system.
GSH During Early- and Mid-Developmental Periods
Much work has been performed to better understand the role of GSH and its redox status during development. Perhaps some of the most supportive evidence for the importance of GSH during development is the generation of mice lacking GSH synthesis enzymes. Generation of homozygous knockout γ-glutamate cysteine ligase catalytic subunit (GCLc), the enzyme responsible for catalyzing the initial and rate-limiting step of GSH synthesis (glutamate+cysteine→glutamylcysteine), conceptuses did not produce any viable pups carried to term, showed cessation of development, and produced high levels of lethality before implantation. In addition, GCLc knockout embryos failed to undergo proper gastrulation, did not form mesoderm, and failed to develop exocoelomic and amniotic cavities, all occurring concomitantly with increases in apoptosis, demonstrating a disruption in developmental programs (188). In addition, proliferation in mutant embryos occurred but was region specific.
Before fertilization, murine oocytes contain high concentrations of GSH (6–7 mM) (52). Following fertilization, GSH decreases slightly, but declines sharply beginning at the two-cell stage and continues descent through the blastocyst stage to a low of 0.7 mM (Fig. 6). During these early preimplantation stages, bovine zygotic ROS production progressively decreases as cleavage is initiated (119), which may be a result of metabolic shifts from OXPHOS to glycolysis. Supplementation of vitrified murine embryos with glutathione ethylester (GSH-EE) artificially increases intracellular GSH levels, significantly increasing fertilization rates and promoting positive developmental outcomes through increased blastocyst viability and decreased ROS (123). Although decreasing concentrations of GSH during implantation are not entirely understood, they may be a result of poor Cys intracellular availability. Cysteine is the rate-limiting precursor to GSH synthesis, and low intracellular concentrations can compromise GSH synthesis. In the oviductal and uterine fluids, total Cys (cysteine+½cystine [CySS]) concentrations are 1.4 and 0.55 mM, respectively (40). Two factors may cause limited intracellular cysteine concentrations: (i) poor transporting capabilities and (ii) highly oxidized Cys:CySS ratios. While initially low, Cys transport into preimplantation embryos does increase from the one-cell stage to the blastula stage (198), but overall increase is relatively modest. In addition, in bovine embryos, adding reductants to preimplantation embryo media resulted in an increase in intracellular Cys concentrations, promoting an increase in GSH levels and further growth (198, 199). The authors conclude that reduced Cys is more readily transported, whereas CySS is poorly mobilized. Together, these may be contributing factors to decreases in GSH concentrations in preimplantation embryos.
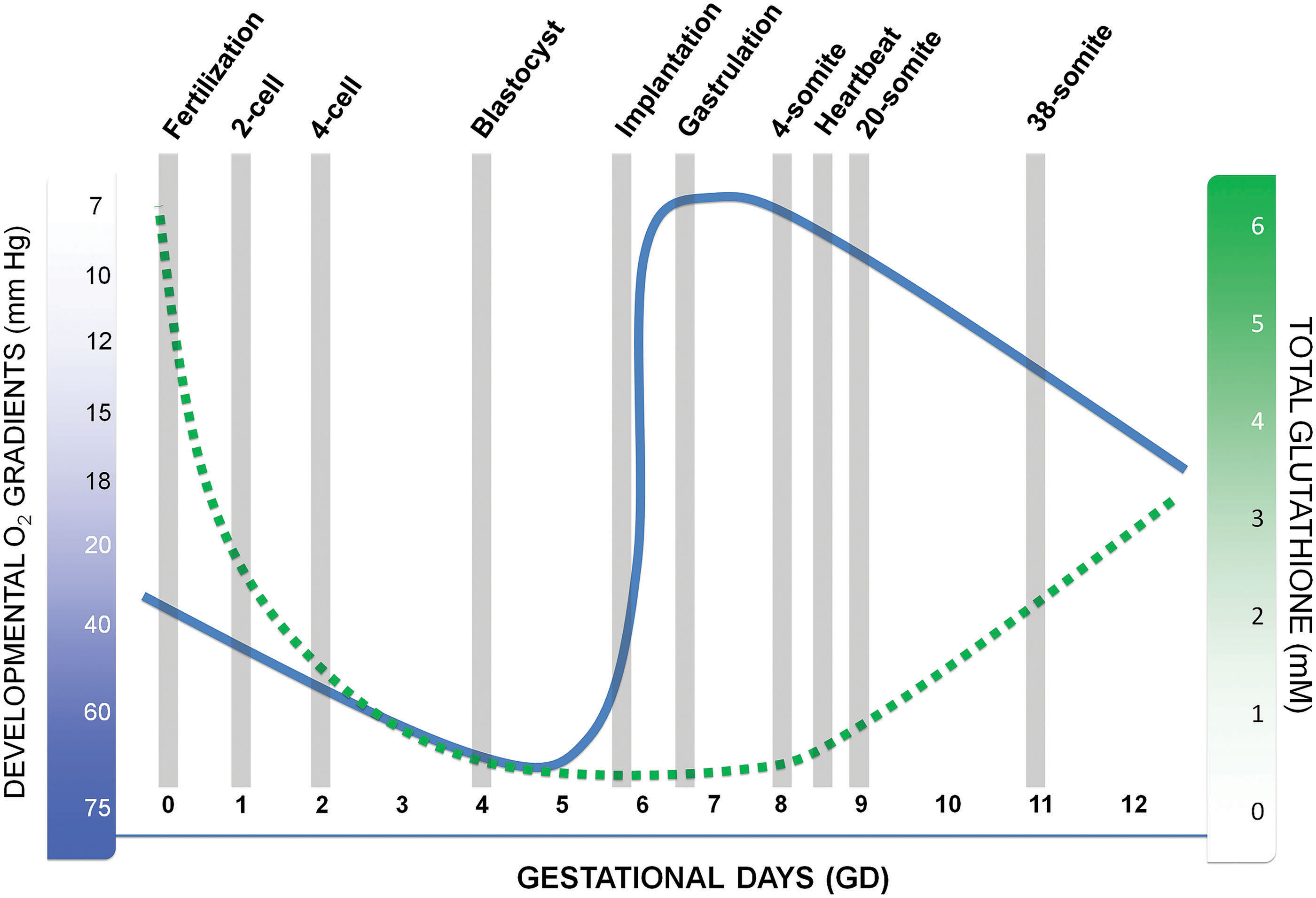
While concentrations of GSH dramatically decrease in the preimplantation blastocyst-stage embryo, little is known about GSH levels during implantation. This is clearly a critical time for embryonic development, especially in the context of nutrients, O2 availability, and metabolic reprogramming toward an increasing dependence on OXPHOS.
Postimplantation embryos are introduced into the uterine environment during a time where total GSH concentrations are relatively low (∼0.4–0.7 mM), but this demarcates the low point in total GSH levels during early/midgestation. Beginning on GD 8, postimplantation mouse embryos gradually increase their total GSH concentrations, reaching 1.7 mM by GD 11 (71). In rats grown in whole embryo culture, the switch to 95% O2 on GD 11 caused a significant increase in total GSH (78), demonstrating a correlation between O2 availability and GSH content. GD 8–11 encompass early organogenesis-stage processes such as neurulation, establishment of a heartbeat, and axial rotation/flexion, and coincide with relatively lower concentrations of GSH. This occurrence may contribute to the organogenesis period of development, being the most sensitive stage for chemical-induced disruption and dysmorphogenesis.
Redox Circuitry During Cellular Differentiation
Development is characterized by change. Cells undergo reprogramming to change their biochemistry, three-dimensional shape, and orientation, and physiological function. As such, undifferentiated cells undergo differentiation to establish cellular mechanisms to cope with extrauterine life and promote homeostasis as a multicellular organism. Undifferentiated cells (i.e., ESCs) have the plasticity to differentiate into a wide variety of phenotypes to accomplish this purpose (Fig. 7A).
Reduction/oxidation (redox) potentials (Eh) are defined as the ratio of interconvertible oxidized and reduced forms of a specific redox couple (i.e., GSH and glutathione disulfide [GSSG], Cys and CySS, etc.) and reported as a voltage, where the more negative the value, the more electrons are available (178), By definition, a more negative Eh represents a greater reducing capacity and the more positive Eh, a more oxidizing capacity. Typical intracellular Eh states, specifically focused on the GSH/GSSG redox couple, are known to correlate closely with cellular functions such as proliferation, differentiation, and apoptosis (178).
In seminal work in this area, proliferation was evaluated in the context of GSH redox states, using normal and transformed fibroblasts. At low density, normal proliferating fibroblasts had a GSH Eh of −220 mV, yet as these cells became increasingly confluent, proliferation slowed due to contact inhibition and the GSH Eh shifted to −185 mV at confluence (87). Comparatively, transformed fibroblasts under similar confluence demonstrated no GSH Eh shift even at maximum confluence. Supplementation with Cys precursors potentiated a reductive redox environment, which supported sustained proliferation (87). Conversely, treatment with a GSH synthesis inhibitor, buthionine-S,R-sulfoximine (BSO), oxidized the GSH Eh by +24 mV and reduced proliferation by 50%. Other studies using fibroblasts showed that ROS levels were lower in M phase than in S phase of the cell cycle and corresponded with increased levels of GSH during M phase, suggesting a more reducing environment during M phase (121).
Perhaps just as important as proliferation during development, studies in cellular differentiation also support an important role of redox states during development. Cellular models are useful in terms of better understanding redox shifts during differentiation. Although there appear to be exceptions (myogenesis in H9c2 cells), the vast majority of studies using these models describe a GSH Eh shift toward a more oxidizing state regularly occurring during differentiation (Table 1). These exceptions may be due, in part, to the interplay between mitochondria and myogenic differentiation, independent of ATP synthesis, but are more centered on redox changes, although to a more reducing state (167).
Redox State Shifts in Various Models of Cellular Differentiation
Comparisons are shown as changes of Eh (more positive numbers = more oxidizing environments and more negative numbers = more reducing environments), ratios (lower ratios = more oxidizing environments and higher ratios = more reducing environments), or changes in GSH/GSSG concentrations between undifferentiated and differentiated cells.
Eh, redox potential; GSH, glutathione; GSSG, glutathione disulfide; hMSC, human mesenchymal stem cell.
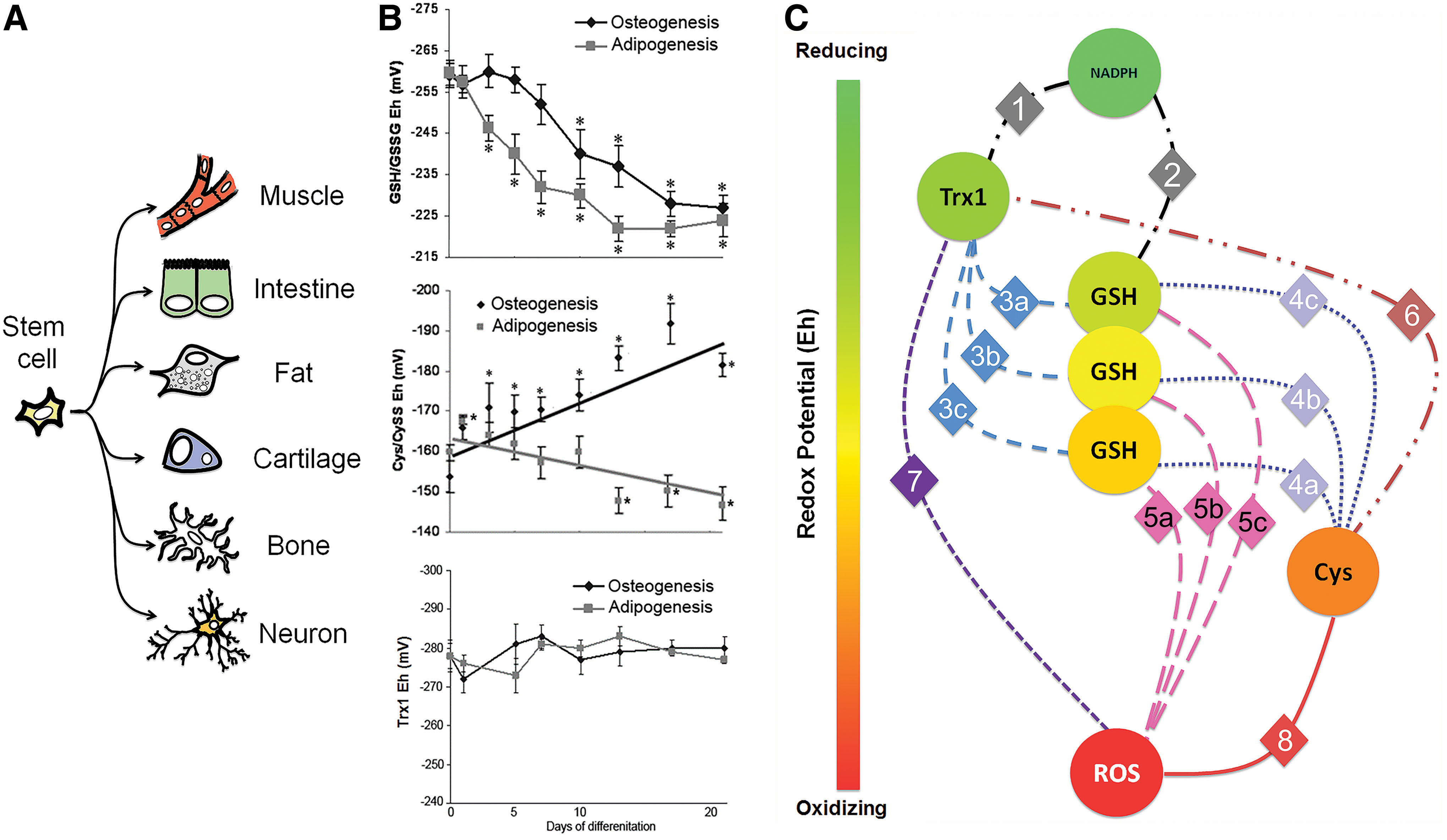
Redox shifts appear regardless of terminal cell phenotype, although one drawback in these approaches is that many of these cell lines do not have the plasticity of ESCs, which have a high degree of pluripotency. However, in a more plastic cell, the human mesenchymal stem cell (hMSC), these cells can be differentiated into multiple phenotypes of mesenchymal lineages, including adipocytes and osteocytes. Using hMSCs, cells were differentiated into either osteocytes or adipocytes over a 21-day period. GSH Eh in undifferentiated hMSCs was measured to be −259 mV and upon differentiation, GSH Eh shifted to −219 and −226 mV in adipocytes and osteocytes, respectively, and were not statistically different from one another (92). However, the rate by which the GSH Eh changed was unique to each cell type, where adipogenesis coincided with a much more rapid oxidation than a slower oxidation observed during osteogenesis (Fig. 7B). Moreover, the Cys/CySS Eh was also calculated over this same differentiation period. Unlike GSH Eh, the Cys/CySS Eh during osteogenesis became increasing reduced as differentiation ensued, but became increasing oxidized during adipogenesis. Assessment of thioredoxin-1 (Trx1) Eh showed that there was no change during either adipogenesis or osteogenesis. These data show that these thiol redox couples are not in redox equilibrium, suggesting that there are potentially unique, temporal redox signatures that exist to promote a specific developmental program toward distinct phenotype. As development is a highly dynamic process where O2 availability, redox states, morphology, and differentiation are relatively conserved, it would suggest that contributions by these determinants may serve as a means to direct differentiation, both in a temporal and spatial manner.
As the disequilibrium between various thiol redox couples increases, redox control of redox-sensitive pathways becomes more distinct and compelling as a means to regulate development. Original definitions of oxidative stress describe global oxidation of reducing equivalents, and at extreme levels, macromolecule damage and cell death (191). Newer paradigms and definitions of oxidative stress address this disequilibrium as a focal point to more finely regulate specific cellular pathways within specific redox couples (95). Since redox disequilibrium is a hallmark for differentiation, logically, it would support the rationale that redox regulation is an important regulator of development.
In light of the disequilibrium of redox couples, a newer, more complete view of oxidative stress can account for specificity in response and outcome. Previous views of oxidative stress focused on shifts in reducing and oxidizing equivalents, where oxidizing equivalents prevailed (191). As a consequence, the classical definition of oxidative stress would support oxidizing environments to cause macromolecule damage, such as DNA oxidation, protein oxidation, and lipid peroxidation, and would lead to cell death. This particular view is likely to occur under extreme conditions of oxidation, but under lesser, nontoxic levels, shifts in specific redox couples may promote responses through redox-sensitive pathways to support cell survival until redox homeostasis can be restored. As specific redox couples are in disequilibrium, this may account for more specificity in redox-sensitive processes during oxidative stress. For example, the Trx1 Eh is primarily maintained at −280 mV, but GSH Eh is maintained at −250 mV (Fig. 7C). Theoretically, a subset of redox-sensitive proteins may be reduced by Trx1, but oxidized by GSH under these conditions. Oxidation of GSH Eh, as is observed during differentiation or even in chemical insult, would result in a shift to the protein redox state favoring an oxidative-type PTM that would (de)activate the protein's normal function. Proteins that are not regulated by either Trx1 or GSH Eh would be unaffected. As such, a specific subset of proteins could be (de)activated to yield a more specific response as specific couples are altered during periods of redox disruption. As outlined in Table 1, cellular GSH Eh is shifting from more reducing to more oxidizing conditions as proliferation slows and differentiation increases and implicates GSH as an important driver of both proliferation and differentiation. More broadly, redox-sensitive elements may be (de)activated accordingly to support specific developmental programs and transitions.
General Paradigm for Redox Signaling in Development
Taking into consideration the newer definition of oxidative stress, in terms of disruption of redox-sensitive pathways and the potential role of specific redox couples, a general paradigm can be formed to understand the role that redox states play during development and how these relate to biochemical, cellular, and embryonic changes during developmental programming. Specific couples, such as GSH or NADPH (Fig. 8), can be modified with the introduction of an oxidative stimuli and/or hypoxia. These shifts in redox states can affect protein thiol targets to promote various PTMs. With thiol-based PTMs, many of these are reversible, including protein S-glutathionylation (Pr-SSG) and protein S-cysteinylation (Pr-SSCys). Even direct oxidation of protein thiols to a sulfenic acid (Pr-SOH) or a sulfinic acid (Pr-SO2H) is reversible through a variety of mechanisms, with oxidoreductases such as Trx1 or sulfiredoxins (Srx). Reversible PTMs can affect protein function to allow or disallow normal functionality, but being reversible permits the flexibility of an “on-off” switch. As such, during redox shifts, proteins are modified via PTMs until redox states can return to homeostasis after which they can be switched back to their original configurations. Temporary switching is likely involved in outcomes that favor cell survival until redox states are normalized, thereby protecting cells during periods of imbalance.
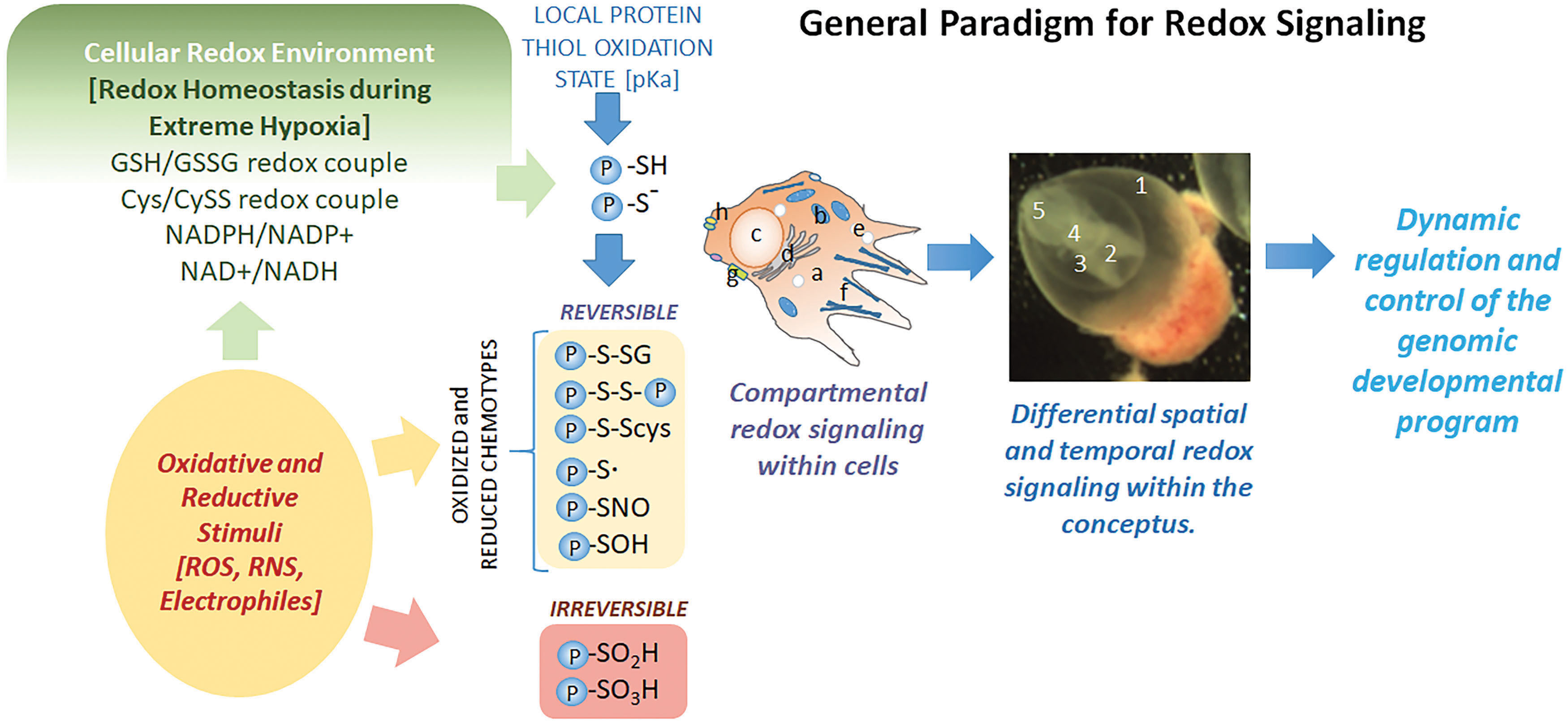
Another layer of complexity exists beyond specific redox couple control of thiol protein targets, namely signaling and control, within subcellular compartments. Data show that in response to oxidative stimuli, redox couples in different subcellular compartments act independently and are not in subcellular equilibrium. For example, in THP1 monocytes, cytoplasmic Trx1 was maintained at −280 mV, but nuclear redox states were at −300 mV, constituting 4.6-fold change in the ratio of oxidized to reduced Trx1 between these compartments (213). In the ER, GSH:GSSG ratios have been suggested to range from 1:1 to 5:1 (11, 31, 89), whereas in the cytosol, GSH:GSSG ratios range from 30:1 to 100:1 (57). As such, compartmental redox signaling and control can increase the complexity and control of redox-sensitive elements to better fine-tune activity and signaling. However, subcellular compartmentation of redox states is relatively unstudied as a function of development or differentiation. One ontogenic study of Trx2, mitochondrial thioredoxin, during early- to midstage organogenesis, showed a shift to a more oxidizing state on GD 10, yet no such shifts were observed in cytosolic Trx1 redox states over the same periods of development, suggesting that subcellular compartmental redox states may be repositioned during development based upon stage-specific programming (65).
In the intestinal cell line, HT29, differentiation occurs over 3 days. While compartmental GSH Eh is difficult to measure as the GSH pool can leak during collection, confounding GSH measurements. An alternative means to determine GSH compartmental Eh is measuring Pr-SSG in specialized compartments. In differentiated HT29 cells, cytoplasmic Pr-SSG concentrations increased by ∼20% (64), but nuclear Pr-SSG levels decreased by nearly 50%. While not measuring GSH Eh directly, these data suggest a different regulation of redox-sensitive proteins in the cytosol versus the nucleus and may contribute to control of cellular differentiation.
Beyond subcellular compartmental control, macrocellular redox control can also occur in the developing embryo. The developing conceptus contains many different larger fluid-filled compartments and extraembryonic membranes that either directly or indirectly interact with the embryo proper. In rat embryos, total GSH concentrations varied in the major conceptal compartments, where VYS GSH was very high compared with that in the embryo (94), and in the visceral yolk sac fluid (VYF; fluid between the VYS and the amnion), GSH was much greater than in the amniotic fluid itself. These differences translated to basal differences in Eh as well, where GSH Eh in the VYS was nearly −223 mV, but −206 mV in the embryo. In the amniotic fluid and VYF, GSH Eh was −93 and −116 mV, respectively. As such, these compartments are unique, but specific redox regulatory components may exist to support each tissue/compartment independent of each other. For example, in conceptuses treated with diethylmaleate (DEM) to deplete reduced GSH, initial GSH depletion was similar in both the embryo and the VYS, where by 45 min postexposure, both tissues were below 30% of their original GSH concentrations (75). However, following maximum depletion, VYS GSH immediately began to recover, but embryonic GSH recovery was delayed for an additional 3 h, until the VYS had fully recovered. Again, these basal and exposure-based differences alone illustrate the unique nature of redox states in the various embryonic compartments and tissues during early organogenesis-stage embryos.
As we interconnect the several layers of redox control, the specificity and disequilibrium of redox couples, subcellular compartmentation of redox couples, redox-sensitive targets, and macrolevel redox regulation of Eh in larger developmentally relevant embryonic compartments, fine control of potential redox-sensitive pathways that are supportive of normal, healthy development is needed to fully understand redox regulation of the development of complex organisms.
Redox Control of Transcription Factors
Essential developmental signaling cascades include the well-known transcription factors NF-κB (nuclear factor kappa B) and Nrf2, which have critical roles in limb development and embryonic protection, respectively (18, 23, 63, 76). Indicative of the multiple levels through which Cys-based transitions can regulate genomic expression and transcription, the IκB (nuclear factor of kappa light polypeptide gene enhancer in B cell inhibitor)/NF-κB and Keap1 (Kelch-like ECH-associated protein 1)/Nrf2 systems possess dual activation and repression properties with regard to protein Cys oxidation and reduction.
The NF-κB pathway is an important regulator of cytokines, immune responses, and mitochondrial activity in the mature organism (14, 206) but has extended roles in development. During organogenesis, growth factors (Fgf8, Fgf10) secreted first from the primitive pronephros and subsequently from the lateral mesoderm trigger the induction of NF-κB in the lateral plate mesoderm of presumptive limb fields (18, 63). NF-κB transcriptional activity is essential for activating expression of genes (Fgf10, TWIST, Msx-1) required for limb outgrowth and patterning (18, 101). Secretion of these growth factors along with others (Fgf8) signals the maturation of the apical ectodermal ridge of the limb bud to form a feedback loop needed for limb morphogenesis. Regulation involves the IκB kinase-mediated deactivation, a repressor protein, which is bound to the two NF-κB subunits (p50, p65/Rel) to yield transcriptional activation (Fig. 9A).
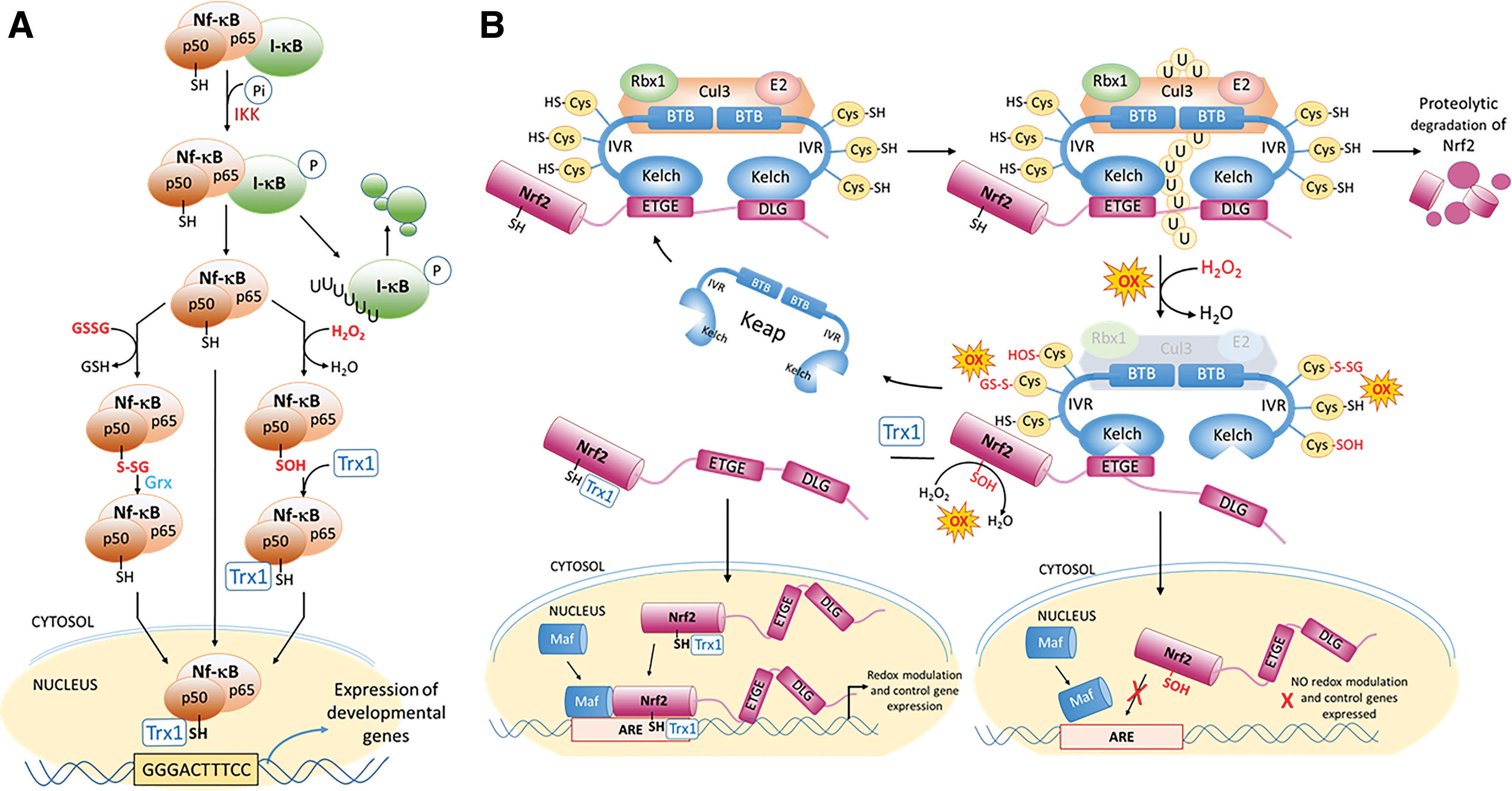
The generation of ROS, one of a number of potential chemical activators, activates the IκB kinase (IKK) that phosphorylates I-κB, releasing I-κB and activating an E3 ubiquitin ligase, causing polyubiquitination and degradation of the inhibitory IκB subunit. NF-κB can then translocate to the nucleus and bind to the κB promoter sequence, transcribing multiple NF-κB target genes (98, 151). Translocation of NF-κB to the nucleus does not, however, insure it will bind to is designated response element. The p50 subunit of NF-κB contains the DNA binding domain, which has a redox-active cysteine (Cys62) that must remain reduced for productive binding to DNA and successful transcriptional activation (205). The ROS activation of NF-κB can also have the result of oxidizing Cys62 and preventing DNA binding (98, 133). This second level of redox control requires reduction of the oxidized Cys62 by Trx1, which acts as a nuclear chaperone and oxidoreductase to keep the Cys reduced and increase DNA binding (133).
The second example of dual redox regulation centers on the homeostatic adaptation of cellular redox systems in response to environmental changes and involves the control of the Keap1/Nrf2 system (Fig. 9B). Under normal unstressed conditions, the Keap1 complexes with its active Cul3 (Cullin-3) ubiquitinase (BTB domains [BTB bric-a-brac, tramtrack, and broad complex domains]) and is anchored to the cytoskeleton (ring box 1 [Rbx1], E2) with the respective Kelch domains, attached by IVRs (intervening regions), sequestering Nrf2 through its ETGE and DLG domains. With multiple regulatory Cys in the IVRs reduced, the constitutively expressed Nrf2 is constantly ubiquinated and degraded to prevent its activation (111, 222). Increased ROS or electrophile generation selectively oxidizes multiple Keap1-IVR Cys, resulting in the exposure of nuclear translocation domains, dissociation of the Cul3 complex to stop polyubiquitination and allow the buildup of cytosolic Nrf2, which is now able to translocate to the nucleus and bind to the antioxidant response element (ARE) to actively transcribe antioxidant and regulatory genes (128).
Dual redox regulation comes into play as the Nrf2 transcription factor also contains a redox-sensitive nuclear exporting signal (122) and a redox-active Cys (Cys506) in its DNA binding domain that must be reduced before productive transcriptional activation can occur (13). If Trx1 is present and active, it acts as a nuclear chaperone to maintain Cys506 in its reduced state and promotes active DNA binding and transcriptional activity (109). In terms of developmental Nrf2 functions, Nrf2 expression is not required as knockout pups are phenotypically normal, but likely serves a more response-related function during the perturbation of redox states following exposure to chemical or environmental insults. Nrf2 activation in embryos prevents prolonged periods of imbalance following oxidant treatments and decreases the frequency of dysmorphogenesis, suggesting an important role for redox adaptation during periods of oxidative disruption (33, 76). Each of these two examples has been shown to be relevant to successful embryogenesis, although the dual regulation is not unique to these examples and is likely to be important for many other transcription factors, such as activator protein-1 (AP1).
Redox Dysregulation in Teratogenesis
In the United States and in most developed countries, birth defect rates are ∼1 in every 33 live births (131). Internationally, rates are even higher in regions where health care availability and nutrition are serious issues, approaching 1 in every 12 live births. Thus, a conservative worldwide estimate of total individuals born with a birth defect is nearly 4 million/year. This statistic is likely even higher if undiagnosed functional deficits and preterm pregnancy losses were included. Unfortunately, our current understanding of the causes and mechanisms of the majority of birth defects is lacking.
Supportive of a redox theory of development are the numerous studies that have shown that nutritional, environmental, or physical insults that cause untimely shifts in redox potential can disrupt developmental programs to yield poor outcomes, including spontaneous abortion, structural malformation, or functional deficits. One of the basic tenets of teratogenesis/developmental toxicology centers on the timing of toxicant exposure during a period of developmental susceptibility. In mammals, including humans, organogenesis is a period where developmental toxicants can promote dysmorphogenesis at the highest rate (Fig. 10). During organogenesis, individual organ systems can become selectively vulnerable to disruption and damage based upon timing of exposure and susceptibilities characteristic of that system at a given point in development (70, 216). Exposures before organogenesis yield low malformation rates, but are more likely to promote embryonic death rather than dysmorphogenesis. Similarly, after weeks 8–10 in human development, the rate of congenital abnormalities drops dramatically, but is more likely associated with functional deficits than structural malformations.
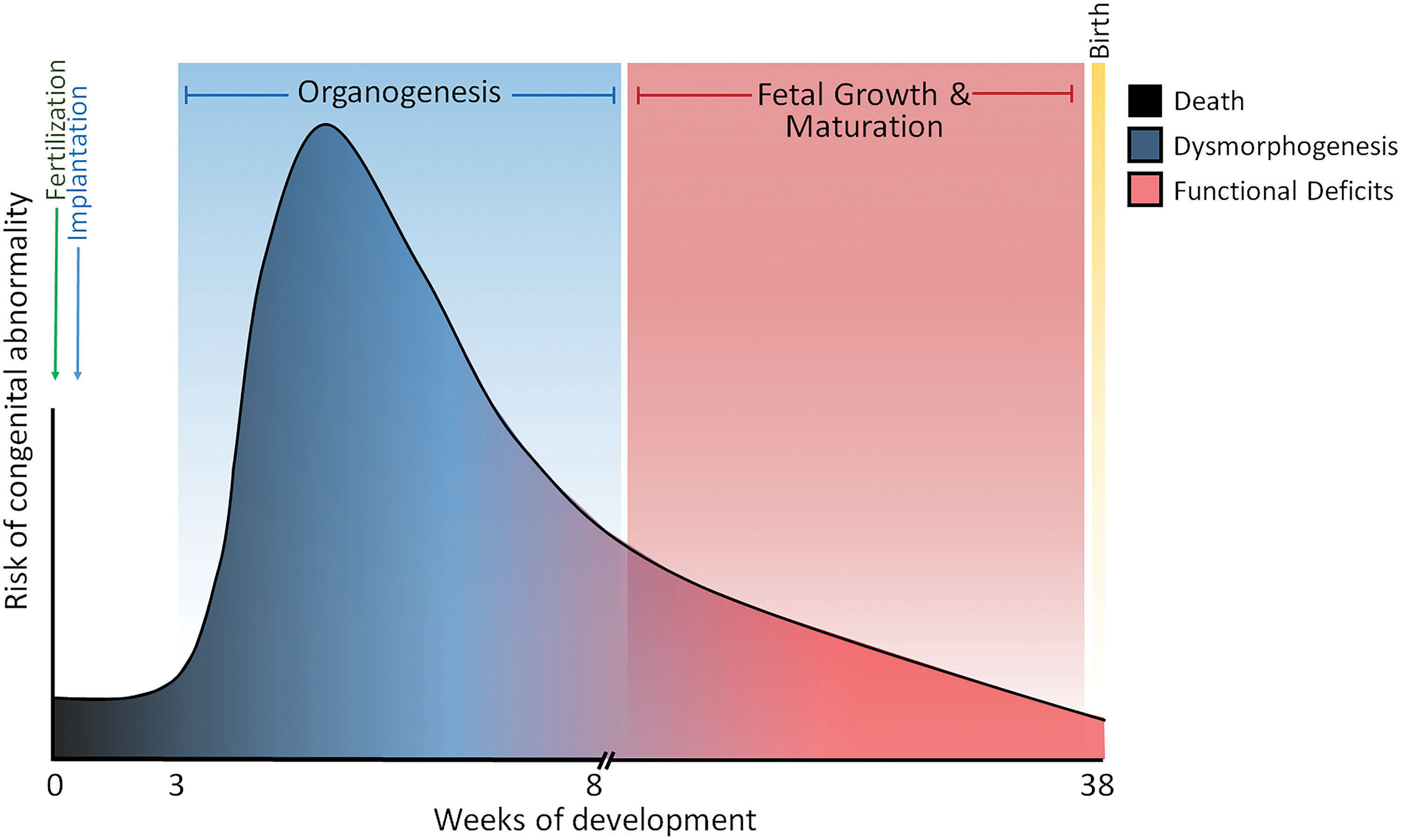
A number of different environmental and therapeutic chemicals have been identified as developmental toxicants (see abbreviated list in Table 2). These chemicals vary significantly in their structure and use and, as such, are expected to have different toxicological modes of action. Interestingly, many of these and other chemicals (64) can act as oxidants, by directly or indirectly generating ROS or other types of reactive compounds. Although the consequences of oxidative damage produced by these compounds have not been clearly demonstrated in developmental models, the literature still promotes the notion that oxidative stress disrupts redox-sensitive pathways and other oxidation targets. In the context of developmental toxicology, most of the studies that have reported deleterious outcomes and toxicities are due to increased generation of oxidants such as ROS and/or decreased antioxidant capacity as being caused by “oxidative stress,” and are based on the assumptions drawn from the binary oxidant/antioxidant paradigm. Developmental targets of excessive oxidant loads include DNA, lipids, and proteins that have different structural and functional roles. A series of seminal studies have shown that ROS generating developmental toxicants such as phenytoin, hydroxyurea, ethanol, and thalidomide, all cause significant oxidation of genomic DNA as measured by the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) (126, 135, 154). The broader but not less significant implications of 8-OHdG formation include its impact on DNA repair mechanisms (185), cell cycle regulation (158), tumor suppressor gene function (p53), and the resulting dysregulation of the developmental program (39). In consideration of this ever-growing body of evidence supporting oxidative disruption of the developmental program as a mechanism for causing birth defects, oxidative damage of this type is primarily toxicological/pathological and extends beyond the roles of the redox code as described earlier for regulation of the normal developmental programs in the redox theory of development. Somewhere between these two roles lies an area of redox regulation and control where consequences of oxidative stress and normal physiological redox regulation likely intersect to produce a more complete and inclusive theory.
An Abbreviated List of Known Developmental Toxicants
? denotes unclear data, where some studies have shown the opposite to be true.
One example may suffice to show how oxidative developmental toxicants can disrupt redox signaling and control to produce dysregulation, malformation, and other defects. Thalidomide is a human developmental toxicant that has been extensively studied to determine its mechanism of action. Thalidomide primarily induces limb reduction defects and also has been described to produce other birth defects as well in offspring of mothers who were exposed during pregnancy. When approved for human use, nearly 100,000 babies were affected worldwide.
The failure to identify thalidomide as a developmental toxicant in animal models contributed to its widespread use in humans (107), as it is not teratogenic in rodents (mice, rats), but is in rabbits. Exploring this species susceptibility difference, studies compared mouse and rabbit embryos treated with thalidomide during organogenesis and showed that sensitive rabbits had increased markers of oxidative stress (155). Treatment with free radical traps decreased limb bud malformations, suggesting that thalidomide causes oxidative stress as a central teratogenic mechanism. Thalidomide preferentially induced ROS in rabbit limb buds (66), which correlated to a preferential decrease in reduced GSH in the whole rabbit embryo compared with whole rat embryos following treatment in vitro (66) and specifically in limb bud culture as well (72). As another possible contributor, Trx1 abundance in the limb of the rabbit is considerably lower than the rat, suggesting that Trx1-mediated protein reduction in the rabbit limb may be compromised allowing for prolonged periods of protein oxidation in the rabbit but not the rat, which may further explain these species differences (67). These studies provide strong evidence that cellular redox states are becoming more oxidized following thalidomide exposure in the rabbit, the thalidomide-sensitive species.
The mechanism by which thalidomide causes limb reduction has produced a wide variety of potential hypotheses (195), including oxidative stress (69). However, the link between oxidative shifts in the embryo and specific embryopathies has been mechanistically difficult to establish. The disruption of redox signaling and control, as suggested in the redox theory of development, stems from developmental biology studies showing a critical role for the transcription factor NF-κB in establishing normal limb development. Thalidomide exposure or molecular disruption of NF-κB produced similar limb reduction defects (18, 101).
Early embryonic activity of NF-κB establishes and directs the normal signaling pathways involved in limb bud outgrowth. However, NF-κB, being redox sensitive (Fig. 9A), could be disrupted leading to the failure of these pathways to be properly instigated, including those involving important limb developmental genes, such as Fgf-8, Fgf-10. Comparing rat and rabbit embryos, rat embryos showed no alteration in the developing limb following thalidomide exposure, but rabbit embryos show disruption (68). Normal expression patterns could be effectively re-established in rabbit embryos with cotreatment of a free radical trap (PBN), which was previously shown to be effective in reducing the frequency of limb malformation in these same species (155). NF-κB binding was inhibited with thalidomide but could be restored through free radical trap cotreatments (68). Together, these findings support a mechanism where thalidomide induces oxidative stress, which alters the NF-κB signaling pathway and thus fails to establish a normal limb developmental signaling pathway, leading to limb reduction. Other studies have shown that shifting GSH Eh toward more oxidizing conditions causes S-glutathionylation of the DNA binding domain in the p50 subunit of NF-κB, resulting in the inhibition of its binding (160). As thalidomide causes shifts in GSH states, it is hypothesized that prolonged thalidomide-induced oxidation is likely to lead to PTMs (i.e., Pr-SSG) that disrupt developmental signaling. As such, prevention of untimely shifts in redox states in the limb would support normal signaling pathways, allowing for normal limb bud outgrowth. Disruption of redox states and dysregulation of redox signaling and control by toxicants such as thalidomide and phenytoin are likely to be mediated through multiple control interfaces such as the roles of catalase in phenytoin exposures (1) and the thalidomide-induced degradation of potential antioxidant proteins such as SALL4 (34, 88). The paucity of data available for redox mechanisms in development does not currently allow reconciliation of the more narrowly constructed redox theory of development with the large number of other oxidative reactions that are observed during development.
Conclusion
The concept of a “redox theory of development” has come about through the convergence of ideas and information spanning the evolutionary history of living organisms through our emerging understanding of internal molecular and biochemical signaling and the external cues that regulate function through an environmental interface. In essence, the theory is an attempt to reconcile the expression and control of the genomic metazoan developmental program with the myriad of atmospheric and environmental signals that are temporally and spatially generated throughout each reproductive cycle. The hypoxic conditions that prevail during metazoan development stimulate HIF expression to direct metabolic reprogramming and developmental programming in a manner that incorporates and utilizes the redox code as the guiding logic for viewing O2 as a developmental morphogen and in directing the expression of the genomic developmental program. The working concept of a redox theory of development rests on the organizing principles of redox theory, which describes the general transfer of electrons in living systems and also includes the redox code, which provides the logic for using thiol steady states (GSH, Cys), nonradical redox couples (NADP+/NADPH), oxidoreductase and antioxidant enzymes, reversible cysteine switches, and post-translational oxidative modification of proteins to regulate developmental pathways and process environmental signals, including proliferation, differentiation, and apoptosis. The identification of a majority of chemicals, developmental toxicants known to cause anatomical and functional birth defects, as agents that produce “oxidative stress” suggests how perturbation of cellular redox states may lead to prenatal death and malformation. The redox theory of development seeks to explore the mechanisms that lie beyond the assumptions of global oxidant and antioxidant functions as broadly defined by the oxidative stress paradigm and to focus on principles proposed in the “redefinition of oxidative stress,” where redox states promote proteomic PTMs and alter cellular signaling and control (95). In this context, altered cellular control and signaling from environmental- and chemical-induced oxidative influences are capable of disrupting timing of developmental signaling/cell function that can lead to poor developmental outcomes. With regard to the broader implications of redox chemistry and biology in developmental organisms, we regrettably acknowledge the omission of a great deal of important work related to the general and perceived consequences of “oxidative stress” and radical/nonradical redox reactions, but hope that the redox theory of development, as presented, stimulates the additional research that will fill the knowledge gaps and stimulate meaningful mechanistic studies in developmental toxicology.
Footnotes
Funding Information
D.P.J. was funded by National Institutes of Health, and National Institute of Environmental Sciences (ES023485). C.H. and J.M.H. received no funding for this article.