
Abstract
Significance:
It is now clear that genetic changes underlie the basis of cancer, and alterations in functions of multiple genes are responsible for the process of tumorigenesis. Besides the classical genes that are usually implicated in cancer, the role of noncoding RNAs (ncRNAs) and reactive oxygen species (ROS) as independent entitites has also been investigated.
Recent Advances:
The microRNAs and long noncoding RNAs (lncRNAs), two main classes of ncRNAs, are known to regulate many aspects of tumor development. ROS, generated during oxidative stress and pathological conditions, are known to regulate every step of tumor development. Conversely, oxidative stress and ROS producing agents can suppress tumor development. The malignant cells normally produce high levels of ROS compared with normal cells. The interaction between ROS and ncRNAs regulates the expression of multiple genes and pathways implicated in cancer, suggesting a unique mechanistic relationship among ncRNA-ROS-cancer. The mechanistic relationship has been reported in hepatocellular carcinoma, glioma, and malignancies of blood, breast, colorectum, esophagus, kidney, lung, mouth, ovary, pancreas, prostate, and stomach. The ncRNA-ROS regulate several cancer-related cell signaling pathways, namely, protein kinase B (AKT), epidermal growth factor receptor (EGFR), forkhead box O3 (FOXO3), kelch-like ECH-associated protein 1 (Keap1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), nuclear factor erythroid 2-related factor 2 (Nrf2), p53, phosphatase and tensin homologue (PTEN), and wingless-related integration site (Wnt)/glycogen synthase kinase-3 beta (GSK3β).
Critical Issues:
To date, most of the reports about ncRNA-oxidative stress-carcinogenesis relationships are based on cell lines. The mechanistic basis for this relationship has not been completely elucidated.
Future Directions:
Attempts should be made to explore the association of lncRNAs with ROS. The significance of the ncRNA-oxidative stress-carcinogenesis interplay should also be explored through studies in animal models.
Introduction
RNA
Since the first discovery of silencing RNA in Caenorhabditis elegans, a substantial number of ncRNAs involved in genomic stability and regulation have been discovered. There is no clear denotation between the classes of ncRNAs. However, the ncRNAs can be divided into two major groups based on their length: small noncoding RNA (sncRNA) and long noncoding RNA (lncRNA) (56) (Fig. 1). While sncRNAs contain <200 nucleotides (nt), lncRNAs are >200 nucleotides long in length. The sncRNAs can be grouped into three major families, microRNAs (miRNAs), endogenous small interfering RNAs (endo-siRNAs), and piwi-interacting RNAs (piRNAs) (28). The sncRNAs regulate the gene expression through two mechanisms: (i) through RNA induced silencing complex, resulting in inhibition of messenger RNA (mRNA) translation, and (ii) by degrading the target mRNA through the Trf4/Air2/Mtr4p polyadenylation complex (TRAMP) pathway (55). Of the three classes of sncRNAs, miRNAs (∼22 nt) comprise the most extensively studied sncRNAs. The miRNAs negatively regulate gene expression. More than 2000 miRNAs have been discovered to date in different organisms. Their regulatory functions in cell differentiation, development, apoptosis, and proliferation have been well documented (58, 121, 125). The piRNAs comprise the second-most extensively studied sncRNAs. They are generally 24–30 nt long and are specifically enriched in germ line tissues of the organism. The piRNAs interact with the piwi protein and form the RNA-protein complex that silences the transposon posttranscriptionally. More than 21,000 piRNAs have been identified to date. The piRNAs play a crucial role during development, reproduction, and metabolic processes in higher eukaryotes (144). The endo-siRNAs (20–24 nt) are also involved in silencing the activity of transposable element in and out of germ line tissues (50, 81).
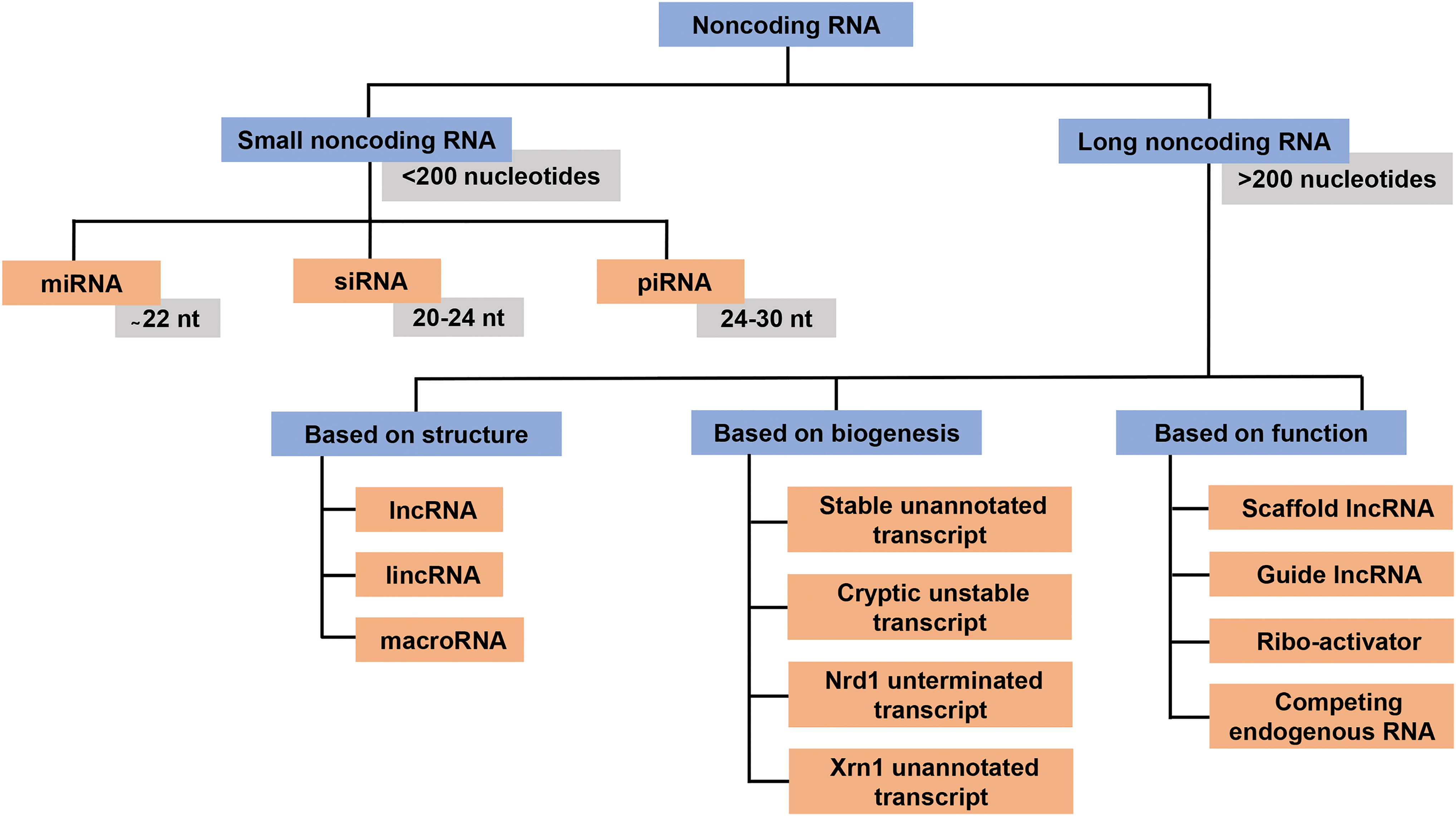
The lncRNAs show cell-specific expression and are often localized in a specific organelle in a cell. The lncRNAs have been reported from plants and animals (22, 73) and likely to play a vital role in maintaining cellular homeostasis (51, 75, 124, 179). According to the GENECODE project, humans have more than 17,000 lncRNAs, while the total number in mice is more than 13,000 (5). The lncRNAs are gouped into three major categories based on their biogenesis, structure, and mode of action (Fig. 1). The role of ncRNAs in the pathology of chronic diseases is extensively investigated. More specifically, the role of ncRNAs in tumorigenesis is notable. As on July 9, 2019, the keywords “Cancer and noncoding RNA” in PubMed database (hosted by the National Institutes of Health) produced ∼73,136 articles, of which, almost 80% were published in the last decade. The ncRNAs are reported to regulate many aspects of malignant tumorigenesis, including survival, proliferation, invasion, angiogenesis, and metastasis (57, 102).
The equilibrium of redox species in the cellular compartment is of utmost importance for maintaining cellular functions. The imbalance in the redox status is caused either due to enhanced generation of reactive oxygen species (ROS)/reactive nitrogen species or because of a malfunctional antioxidant defense system. This results in the damage to biomolecules, including lipid, protein, and DNA (104). The ROS are generated as a consequence of oxidative metabolism inside the cell. The ROS are also generated as a part of the signal transduction/cellular defense system in response to physical, biological, and chemical stress. The imbalance in redox status is repaired by an antioxidant defense system that can be both enzymatic (superoxide dismutase [SOD], catalase, and glutathione peroxidase) and nonenzymatic (reduced glutathione [GSH], vitamin A, C, E, and so forth) in nature (87). The ROS also act as signaling molecules to control numerous cellular activities such as growth, apoptosis, and metabolism (21). Environmental factors such as tobacco, smoke, pollutants, and xenobiotics are the major extrinsic activators of ROS. The nicotinamide adenine dinucleotide phosphate reduced (NADPH) oxidase complex from cell membrane, mitochondria, peroxisomes, and endoplasmic reticulum (ER) forms the major intrinsic source for ROS (105, 113).
During early stages of cancer development, intracellular ROS can activate pro-oncogenic signaling pathways such as receptor tyrosine kinase (RTK), phosphatidylinositol 3-kinase (PI3K)/AKT, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), thereby aiding in cancer growth. In contrast, in the later stages of carcinogenesis, excessive ROS may counterintuitively lead to apoptosis. However, cancer cells possesses mechanisms that counter the increase in ROS to prevent cell death. Cancer cells show a metabolic shift by deviating the glycolytic metabolites to the pentose phosphate pathway to produce NADPH and GSH, which helps in combating oxidative stress (9). Similarly, cells with elevated ROS activate nuclear factor erythroid 2-related factor 2 (Nrf2) antioxidant signaling that alleviates the ROS-mediated effects (90). In particular, ROS play multiple roles in cancer progression and development. For example, ROS help to generate self-proliferation characteristics in cancer cells by decreasing RTK activation (99). The loss of contact inhibition through ROS makes cancer cells insensitive to antiproliferative signals (158). The ROS can promote tissue invasion and metastasis through the activation of metalloproteinase and overexpression of epithelial-to-mesenchymal transition (EMT)-associated factors (70). The ROS are also capable of driving tumorigenesis through genetic alteration or genomic instability (35, 98).
Besides their role in tumorigenesis, ROS also function in the biogenesis of ncRNAs. Conversely, ncRNA can also modulate the redox status of cells. Thus, it is conceivable that there exists a tripod relationship among ncRNA, cancer, and ROS, and thorough evaluation of the published literature provides a strong basis for the existence of this relationship. Since, signaling pathways are tightly controlled for maintaining cellular homeostasis, alteration in these signaling molecules can initiate the process of malignant transformation. Both healthy cells and cancer cells have identical signaling pathways. However, the mutations of key players in these signaling pathways result in constitutive activation of these signaling pathways in cancer cells. Accumulating evidence suggests that the ROS-ncRNA-cancer triad relationship can affect many aspects of tumor development (Table 1). Here, we focus on the relationship between ncRNAs, ROS, and cancer. We further highlight how ncRNAs and ROS modulate the cancer-related pathways.
Summary of Signaling Pathways Associated with Noncoding RNA-Reactive Oxygen Species-Cancer Axis
AKT, protein kinase B; AML, acute myeloid leukemia; ASK1, apoptosis signal-regulating kinase 1; Bcl-2, B cell lymphoma 2; CRC, colorectal cancer; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; EMT, epithelial-to-mesenchymal transition; ERK, extracellular signal-regulated kinases; FGF2, fibroblast growth factor 2; FOXO3, forkhead box O3; G6PD, glucose 6-phosphate dehydrogenase; GSK3β, glycogen synthase kinase-3 beta; HCC, hepatocellular carcinoma; HK2, hexokinase 2; HUVEC, human umbilical vein endothelial cells; IMPDH, inosine monophosphate dehydrogenase; JAK, janus kinases; JNK, c-Jun N-terminal kinase; Keap1, kelch-like ECH-associated protein 1; KRAL, Keap1 regulation-associated lncRNA; K-ras, kirsten rat sarcoma 2 viral oncogene homologue; ncRNA, noncoding RNA; Nf-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear factor erythroid 2-related factor 2; NSCLC, nonsmall-cell lung carcinoma; PDK2, pyruvate dehydrogenase kinase 2; PDK4, pyruvate dehydrogenase kinase 4; PFK-1, phosphofructokinase-1; PKM2, pyruvate kinase muscle isozyme M2; PRXL2A, peroxiredoxin-like 2A; PTEN, phosphatase and tensin homologue; ROS, reactive oxygen species; RTKs, receptor tyrosine kinases; SCAL1, smoke- and cancer-associated lncRNA-1; SIRT6, sirtuin 6; TNF, tumor necrosis factor; Trx1, thioredoxin-1; TUG1, taurine-upregulated gene 1; TUG2, taurine-upregulated gene 2; UCA1, urothelial carcinoma-associated 1; VEGFA, vascular endothelial growth factor A; Wnt, wingless-related integration site.
ROS, ncRNA, and p53
p53 is the most important tumor suppressor known to date. More than 50% of human cancer carry mutations in p53 gene; in the remaining, the pathways that p53 controls are defective. Among the various pathways regulated by p53, those related to cell cycle arrest and cell survival are important (89). The loss of p53 function results in defective transactivation and breakdown of downstream effector genes involved in cell cycle control, apoptosis, and DNA repair (86, 133, 146). p53 is nicknamed as the guardian of the cell by virtue of its ability to control the expression of multiple genes. It is well known that p53 is mutated in many cancers and several hotspot point mutations lead to the gain of function in p53 (mutant p53). Aggressive tumor progression and emergence of drug resistance are major outcomes of these gain-of-function mutations. Thus, such mutations represent potential targets of cancer therapy. Inhibitors of heat shock protein 90 (HSP90) such as 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), which stabilizes mutant p53, have been shown to degrade mutant p53. Similarly, ganetespib, which binds to and inhibits HSP90, shows >50% of 17-AAG activity (6, 177). Nutlin-3, an mdm2 antagonist, was found to exhibit an antitumorigenic and antimetastatic activity (117). p53 reactivation and induction of massive apoptosis 1 (Prima 1), another small molecule, have been shown to restore the conformation of a mutant p53 to the wild-type form, thereby inducing apoptosis (71). Another example is 4,5-diphenyl-2-methyl picolinate (DMP), which induces DNA damage and brings about senescence in gastric cancer cells. (172). Besides these chemically synthesized compounds, several naturally occurring plant-based compounds are known to either modulate mutant p53 or restore wild-type p53, thereby affecting several alternating survival pathways (1).
During the past decade, mounting evidence suggests that the interplay among p53, ncRNA, and ROS helps in controlling p53-regulated genes that are directly or indirectly linked to tumorigenesis (Fig. 2). miR-16, which has a tumor-suppressive function, is predominantly downregulated in several cancer types. Sanguinarine was found to induce miR-16 that further activates p53 (165). The activated p53 suppresses cell growth and induces ROS-mediated apoptosis in the hepatocellular carcinoma (HCC) cells. A rank aggregation analysis study identified several miRNAs, namely, miR-34a-5p, miR-1915-3p, miR-638, and miR-150-3p, which respond to oxidative stress in HCC cells (137). Of the four miRNAs, miR-34a-5p and miR-1915-3p are regulated by p53 under oxidative stress situation. Proline oxidase (POX) is a tumor suppressor regulated by p53 that reduces the proliferation and induces ROS-mediated apoptosis involving the mitochondria. miR-23b, which is upregulated in renal cancer, can directly bind to POX mRNA and inhibit its tumor suppressor activity (80).

High-grade serous ovarian carcinoma (HGSC) is a type of ovarian cancer that originates from the secretory epithelial FTSE cells located at the fimbriated end of the fallopian tube. Ovulation and associated inflammation are local factors that contribute to HGSC. Cultured primary FTSE cells show increased ROS and upregulation of a subset of miRNAs, and p53 regulates the function and expression of these ROS-mediated miRNAs in FTSE tumor cells (82). Among various stress-related miRNAs, miR-200 is regulated by p53, and overexpression of miR-200 suppresses EMT. p53 has been shown to upregulate miR-200c in hydrogen peroxide (H2O2)-exposed cells, which can be reversed by p53 deficiency (84), thus suggesting a triad relationship among miRNA, ROS, and p53. The tumor suppressor miR-34 is a transcriptional target of p53. Capsaicin-induced oxidative damage leads to activation of p53 in nonsmall-cell lung carcinoma (NSCLC) cells. Upregulated p53 leads to enhanced expression of miRNA-34a, which in turn inhibits B-cell lymphoma 2 (Bcl-2) expression thereby enhancing cell death. In another study, the function of miR-30 in gastric neoplasia was examined (138). The downregulation of miR-30 in HGC 27 upregulates p53, which further promotes ROS-mediated apoptosis. Ferroptosis is a mode of apoptosis characterized by iron-dependent lipid peroxidase accumulation. P53RRA lncRNA plays a tumor-suppressor role in cells by enhancing ferroptosis, cell cycle arrest, and apoptosis through maintaining p53 in the nucleus (85). Thus, ncRNAs, p53, and ROS interplay in tumor cells.
ROS, ncRNA, and Nrf2
Nrf2 is a transcription factor and a universal master regulator for cellular resistance to oxidants and cell survival in physical, biological, and chemical stress (132, 136). Nrf2 is normally expressed in the cytoplasm. Its activity is negatively regulated by kelch-like ECH-associated protein 1 (Keap1). During the oxidative stress conditions, Nrf2 stabilizes and translocates into the nucleus due to the loss of negative regulation by Keap1 (136). In the nucleus, Nrf2 interacts with its target genes through the antioxidant response element (a cis-acting element) and transcribes almost 200 genes associated with cytoprotection/detoxication processes. Nrf2 has been shown to play a role in cancer management through its ability of controling the expression of ncRNAs (62). Nrf2 acts as a double-edged sword; it can be both oncogenic and tumor suppressive in function. A number of agents, namely, luteolin, apigenin, and chrysin, have been shown to reduce Nrf2 mRNA levels and sensitize cancer cells to antitumor drugs (37, 38, 129), leading to a reduction in tumor size (18). Conversely, luteolin and apigenin have also been shown to possess cytoprotective activities (78). Other modulators of Nrf2 signaling include flavonoids such as wogonin, 4-methoxychalcone, and epigallocatechin-3-gallate (EGCG) (178).
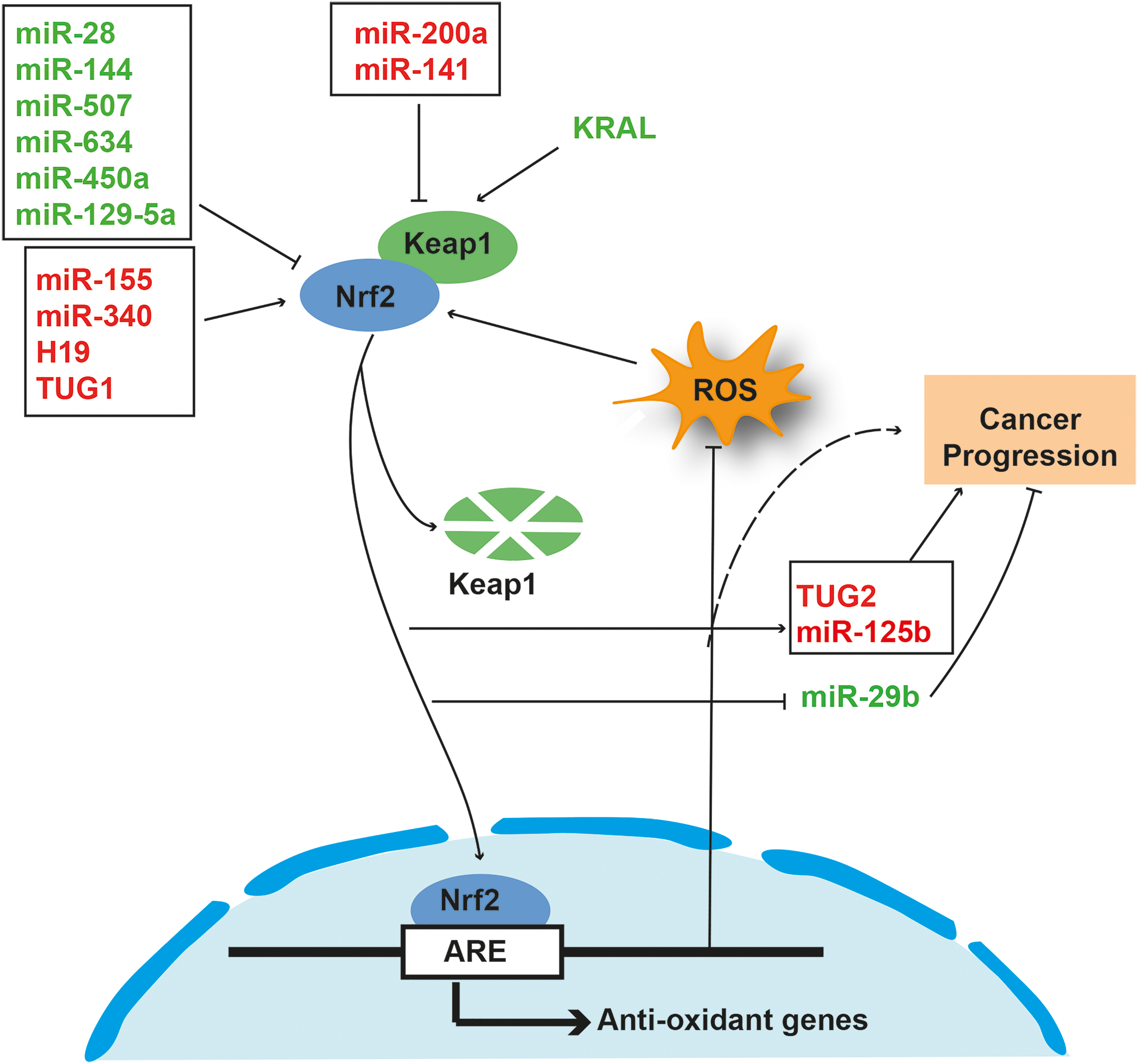
Nrf2 plays a role in the biogenesis of several tumorigenic ncRNAs. Conversely, ncRNAs can also regulate the Nrf2 pathway. It has been reported that in human breast cancer cells, an miRNA-based new regulation of Nrf2, which is independent of Keap1, is present (157). The study further demonstrated that miR-28 degrades the Nrf2 mRNA by binding to its 3′-untranslated region (3′UTR). Similarly, miR-507, -634, -450a, and -129-5p are known to attenutate Nrf2-mediated oncogenic pathways (153). Nrf2 is normally overexpressed in multiple cancer types, including breast cancer. In MCF-7 breast cancer cells, overexpression of Nrf2 promotes cancer progression by enhancing the glycolysis process (166). The connection among miR-200a-Keap1-Nrf2 in breast carcinoma has been reported (33). miRNA-200a is downregulated in breast cancer. However, re-expression of miRNA-200a triggers Keap1 mRNA degradation by binding to 3′UTR of Keap1, thereby releasing Nrf2 from Keap1 leading to its translocation into the nucleus. miR-141 that targets Keap1 is upregulated in cisplatin-resistant ovarian cancer and 5-fluorouracil (5-FU)-resistant HCCs (120, 135). Peroxiredoxin-like 2A (PRXL2A) is an antioxidant protein commonly upregulated in oral squamous cell carcinoma (OSCC). Recently, miR-125b was reported an upstream regulator of PRXL2A and Nrf2 in OSCC cell lines (16). The group concluded that the miRNA-125b-PRXL2A-NRF2 axis protects cancer cells from oxidative stress. In another study, Nrf2 was found to regulate the expression of miR-125B1 and miR-29B1 positively and negatively, respectively, in acute myeloid leukemia by binding to their regulatory region (116). The ability of arsenic trioxide (ATO) to induce oxidative and cell death has been utilized as a strategy for cancer treatment. However, relatively higher doses of ATO are required for cancer treatment. It has been reported that overexpression of miR-155 in lung cancer cells can lead to the development of resistance to ATO partly through activation of Nrf2 (46). In another study, it was shown that inhibition of miR-155 can control the NF-κB-oxidative damage-associated malignant transformation in arsenite-exposed human bronchial epithelial cells (14).
In HCC cell lines, miR-340 contributes to cisplatinum or cis-diamminedichloroplatinum(II) (CDDP) resistance by regulating the Nrf2-dependent antioxidant pathway (119). Conversely, miR-144 downregulates the Nrf2-dependent antioxidant pathway and can reverse chemoresistance in HCC cells (176). In addition to sncRNAs, lncRNAs are also associated with Nrf2 during tumorigenesis. The interaction between H19 and Nrf2 contributes to resistance in ovarian cancer cells (174). Nrf2 has been shown to activate smoke- and cancer-associated lncRNA-1 (SCAL1), interacting with oxidative stress machinery in lung cancer cells (131). Through transcriptomic analysis, Ashouri et al. identified Nrf2-regulated lncRNAs in nuclear factor erythroid 2-like 2 (NFE2L2) gain-of-function condition. Furthermore, long intergenic nonprotein coding RNA 942 (LINC00942) was found as a new direct target of Nrf2 involved in antioxidant response mechanism (8). In another study, it was reported that Nrf2 activates lncRNA taurine-upregulated gene 2 (TUG2), which can contribute to tumor progression and development of adriamycin resistance in bladder carcinoma (126). In a recent report, lncRNA TUG1 was shown to upregulate Nrf2 and contribute to TE-1-derived cisplatin resistance in esophageal squamous cell carcinoma (170). In another recent study, it was shown that lncRNA Keap1 regulation-associated lncRNA (KRAL) inhibits the Nrf2 expression by regulating Keap1, which leads to development of 5-FU resistance in HCC cells (147).
Taken together, this evidence suggests that the interplay among ncRNA-Nrf2-ROS is crucial for the regulation of tumorigenesis both in a positive and a negative manner (Fig. 3). Future studies should be focused on deciphering the underlying mechanism involved in the ncRNA-Nrf2-ROS axis.
ROS, ncRNA, and NF-κB
Persistent inflammation is a major cause of several chronic medical conditions, including cancer (91). The discovery of proinflammatory transcription factor NF-κB has provided a link for the role of chronic inflammation in cancer. NF-κB is a transcription factor known to regulate more than 500 antiapoptotic, metastatic, proangiogenic, and cell cycle regulatory genes (49). Thus, NF-κB is a positive regulator of inflammation and tumorigenesis (151). The NF-κB activation contributes to the protumorigenic microenvironment through accumulation of cytokines in cancer tissue (148). Inflammation in cancer tissue is mainly controlled by its microenvironment and NF-κB signaling has a prominant role in this process.
NF-κB signaling is essential for cancer progression. Despite the crucial role in cancer development, NF-κB inhibitors have not been very successful in cancer patients. A study by Demchenko et al. (26) demonstrated that NF-κB inhibitors such as AM-0216, AM-0561, and AM-0650 are cytotoxic in nature and can induce apoptosis in cancer cells. However, poor pharmacological properties led to their failure in clinical trials. Evidence suggests that a crosstalk between ROS and NF-κB can contribute to tumorigenesis (Fig. 4). The toll-like receptor is directly linked to invasiveness and metastasis in pancreatic cancer, which is one of the most aggressive cancers. Reports of a triad relationship among NF-κB-ROS-miR-29c in pancreatic cancer is well known (10). It has been shown that palmitic acid can trigger invasion in AsPC-1 pancreatic cancer cells through ROS-mediated NF-κB activation involving the secretory matrix metalloproteinase-9 (MMP-9). Furthermore, NF-κB downregulates miR-29c, which is a regulator of extracellular matrix proteins. The NF-κB-mediated downregulation of miR-29c is in concordance with an increase in MMP-9 activity that contributes to the metastatic nature of pancreatic cancer cells.
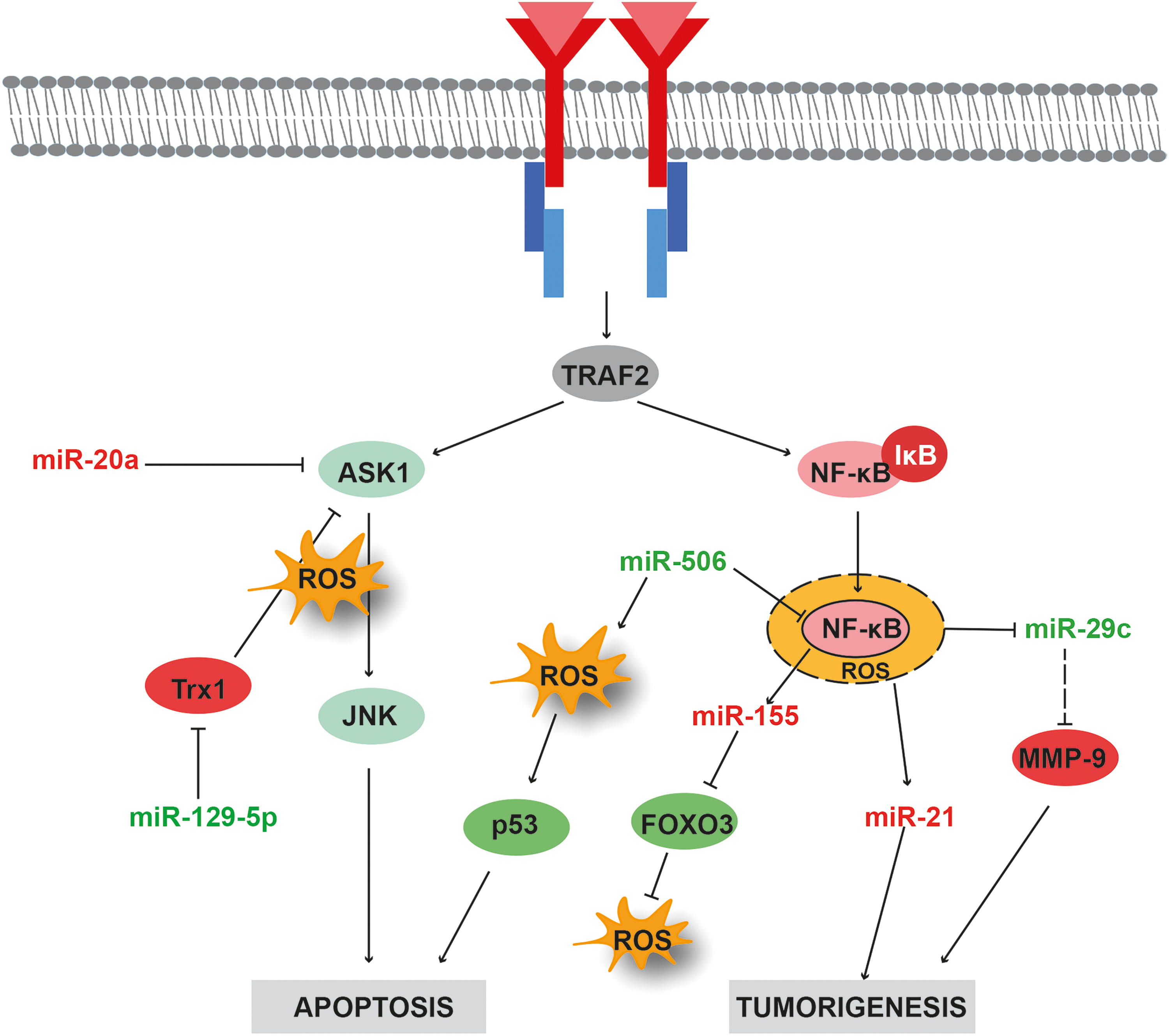
Kirsten rat sarcoma 2 viral oncogene homologue (K-Ras) is another oncogene upregulated in pancreatic cancer. K-Ras signaling activates NF-κB, leading to enhanced miR-155 expression that in turn represses forkhead box O3 (FOXO3) expression. Furthermore, FOXO3 repression leads to ROS accumulation, which may affect cancer cell proliferation and transformation (139). miR-506 is typically overexpressed in lung cancer. miR-506 can induce ROS accumulation via negative regulation of NF-κB(p65), resulting in enhanced p53-mediated apoptosis in lung cancer cells (162). The exposure of human lung embryo fibroblast cells to arsenic (an exogenous source of ROS) triggers the neoplastic transformation through increased expression of miR-21. Increased expression of miR-21 is also a downstream consequence of ROS-dependent NF-κB activation (79).
Photodynamic therapy (PDT) is a novel cancer therapy in which drugs promote cell death under certain light. PDT is reported to induce ROS production and unstable mitochondrial membrane potential, triggering an apoptotic cascade within mitochondria. It has been observed in glioma cells that upregulation of NF-κB positively correlates with the accumulation of ROS followed by the upregulation of hsa-miR-7641, hsa-miR-9500, hsa-miR-4459, hsa-miR-21-5p, hsa-miR-663a, and hsa-miR-205-5p (164).
ROS, ncRNA, and RTKs
Signaling through RTKs is one of the vital paths for maintaining cellular homeostasis. The RTK family consists of 58 receptors grouped into several specified subfamilies. These subfamilies are involved in several cellular processes such as growth, motility, differentiation, and metabolism (72). Dysregulation in RTKs is a feature in many diseases, including cancer (Fig. 5). During the past two decades, many RTKs have been targeted for the development of potential diagnostic and prognostic biomarkers. Most RTKs are activated by ligand-induced dimerization of the receptors.

Generally, autophosphorylation at the C-terminal region of the tyrosine kinase domain leads to the activation of secondary messengers such as PI3K, diacylglycerol, phospholipase C, and signal transducer and activation of transcription (STAT). This allows the activation of the downstream cascades such as AKT, extracellular-signal regulated kinase (ERK), mitogen-activated protein kinase (MAPK), and STAT (154). Since RTKs play a crucial role in the development and progression of cancer, some of the anticancer drugs were designed against RTKs in the last two decades. The anti-RTK agents block either the extracellular ligand binding domain or the intracellular tyrosine kinase domain and enhance overall survival and response. The current approaches to target RTKs involve tyrosine kinase inhibitors (TKIs) and monoclonal antibodies (mAbs). In epidermal growth factor receptor (EGFR) mutation-induced cancers, improved prognosis was observed in patients treated with first-generation TKIs such as gefitinib and erlitinib (69, 122). However, the emergence of resistance to first-generation TKI led to the discovery of second- and third-generation drugs such as osimertinib, olmutinib, and ociletinib, which are generally recommended for patients showing resistance to TKIs (123, 140, 143). Apart from TKIs, mAbs have also been developed against EGFR that target extracellular ligand binding domain leading to inactivation of EGFR signaling (134). Cetuximab (Erbitux) and panitumumab (Vectibix), which target the ligand binding domain, are some of the examples of EGFR-targeting mAbs. Transtuzumab is the most prominent mAb against human epidermal growth factor receptor 2 (Her2), and lepatinib is used for patients with transtuzumab resistance (93).
Recent advances have laid some solid basis for the association of RTKs with noncoding transcripts. Studies have shown that along with ncRNA, oxidative stress and RTKs help in the progression of cancer (30, 145). The role of EGFR signaling in cancer progression is well established. EGFR is known to regulate multiple survival pathways such as AKT and ERK. The suppression of EGFR signaling is an attractive approach for cancer prevention. Curcumin, the the major active ingredient in turmeric, is reported to suppress EGFR signaling (36). In colon cancer cells, administration of curcumin causes ROS accumulation, which in turn reduces the levels of specificity proteins (SPs; SP1, SP3, and SP4) and EGFR signaling. Further analysis revealed that oncomiRs such as miR-27a, miR-20a, and miR-17-5p are positive regulators of SP. Curcumin elicits ROS-mediated reduction in transcription of miR-27a/20a/17–5p and enhances the levels of SP repressors. In another study, it was shown that high ROS generation inhibits the expression of miR-199a and miR-125b by DNA hypermethylation (53). Moreover, inhibition of miR-199a and miR-125b activates ErbB2 and ErbB3 that promote tumor progression.
The development of resistance to apoptosis provides a major road block for cancer therapy. Mucin-1 (MUC1), an oncoprotein with multiple roles in cancer progression, is known to associate with endogenous ROS in rendering chemoresistance. Accumulation of ROS due to lack of catalase, a major antioxidant enzyme, is a significant reason for MUC1 overexpression in lung cancer cells (152). Upregulated MUC1 can activate EGFR-mediated survival signaling cascade such as AKT/cyclooxygenase2 (Cox2). miR-551b plays a crucial role in apoptosis resistance by regulating the catalase levels and by maintaining high ROS levels, leading to EGFR activation. Thus, the crosstalk among miR-551-ROS-EGFR provides clues for understanding chemoresistance in lung cancer cells.
Vascularization is a rate-limiting process in metastatic tumors. Once angiogenesis is completed, the mesenchymal transition of epithelial cells is initiated by several factors. Members of the vascular endothelial growth factor (VEGF) family play a crucial role in the vascularization of tumors. Multikinase inhibitors (MKIs) are now available for targeting VEGF receptors and other family members such as fibroblast growth factor receptors (FGFRs) and patelet-derived growth factor receptors, which are the prime mediators of angiogenesis. The drugs such as sunitinib, regorafenib, axitinib and cabozantinib, all MKIs, can target advanced and solid tumors. Other examples include bevacizumab and ramucirumab, which are prescribed for glioblastoma patients in combination with chemotherapy (154, 171). In addition, sorafenib (Nexavar) and sutinibhave have been reported to improve the overall progression-free survival of patients [reviewed in Regad (106)]. Nonsteroidal anti-inflammatory drugs (NSAIDs) are shown to induce apoptosis in colon cancer cells by inhibiting VEGF signaling (100). Similarly, GT-094, a nitric oxide-based NSAID (NO-NSAID), can induce mitochondrial dysfunction by reducing membrane potential, which in turn triggers ROS accumulation.
The accumulation in ROS downregulates oncogenic miR-27a, which represses the negative regulators of SP, thus blocking VEGF signaling and eliciting apoptotic pathway. In a similar study, curcuminoids were found to inhibit cancer progression by suppressing the ROS-miR-27a-specificity protein axis that mediates the blockage of VEGF signaling and increases the sensitivity of cancer cells to 5-FU (96). VEGF is a principal mediator of angiogenesis through its receptors. The VEGF signaling is involved in transcription of genes necessary for angiogenesis. While a large number of miRNAs act as oncogene by inhibiting the repressors of VEGF signaling, few of them block angiogenesis by inhibiting the function of vascular endothelial growth factor receptor (VEGFR) and/or their downstream proteins. Superoxide produced due to endoplasmic stress is often converted into peroxides by antioxidant enzyme SOD in a tumor necrosis factor-alpha (TNF-α)-mediated manner. miR-21 can inhibit TNF-α, which prevents the conversion of superoxide to peroxide. These superoxides activate the feedback loop by facilitating the binding of VEGF/fibroblast growth factor 2 (FGF2) to their receptors (168). Some antiangiogenic miRNAs such as miR-497, miR-503, and miR-126 are known to inhibit ROS-activated feedback loop by binding to 3′UTR of vascular endothelial growth factor A (VEGFA)/FGF2 (141, 169, 175).
Hypoxia induces a highly stereotypical cellular response within a cell for viability. Hypoxia inducible factor (HIF), being a master regulator, allows the cells to adapt in hypoxia stress. While HIF regulates many stress-related factors during hypoxia, miRNAs coordinate in both an HIF-dependent and HIF-independent manner to combat hypoxia stress (94). Hypoxia-miRNAs, exclusively involved in hypoxic condition, stabilize HIF in a positive feedback loop, while HIF directly regulates the transcription of these hypoxia-miRNAs (43). Accumulating data suggest that HIF regulates ROS levels, which directly inhibit the transcription of several miRNAs by blocking enzymes such as Drosha and Dicer (94). The increase in miR-210 expression in response to hypoxia is strongly correlated with poor prognosis in breast and pancreatic cancers. Accumulating evidence suggests that miR-210 is correlated to HIF-induced ROS by binding of HIF to hypoxia response element in proximity to miR-210 promoter (27, 65). Moreover, the role of miR-210, a hypoxia-induced miRNA, in the tumor microenvironment has been well studied (24). The process of EMT is most crucial in allowing malignant cells to move from the primary site of tumor to distant sites in the body. Thioredoxin is known to control ROS levels in cancer tissues, thereby blocking EMT. miR-373 binds to 3′UTR of thioredoxin interacting protein (TXNIP) and downregulates it, thus promoting metastasis (15). Interstingly, miR-373 is also known to positively regulate HIF1α via downregulation of TXNIP, which also initiates metastasis. The interplay between ROS and miRNAs in angiogenesis is well understood. HIF proteins under hypoxia stress induce the accumulation of ROS and initiate vascularization. However, miR-519c and miR-107 inhibit HIF proteins and suppress the transcriptional activation of angiogenic genes (142).
Transforming growth factor-beta (TGF-β) signaling is widely known for its dual role in tumor development. TGF acts as a tumor suppressor at early stages of cancer. However, at later stages, it functions in the role of an oncoprotein, thereby promoting the development and progression of cancer. EMT is the hallmark of TGF signaling. During the transition, TGF signaling enhances the expression of mesenchymal markers such as N-cadherin and vimentin and attenuates the epithelial markers such as E-cadherin (150). TGF signaling is temporally activated during cancer progression, making it more complex for targeting. A number of approaches can be used to target TGF-β in cancer. It is now clear that targeting TGF-β RII rather than TGF-β RI is more effective as TGF-β RII can be activated independent of TGF-β RI. Small-molecule inhibitors such as LY2109761, 1D11, gakunisertib, and SM16, which can inhibit metastatis, are known targets of TGF-β RI and TGF-β RII. Similarly, mAb such as freslimumab is a known inhibitor of TGF-β I and TGF-β II (20, 39). Besides small molecules and mAbs, antisense oligonucleotides have also been developed to target TGF-β. For instance, trabedersen and belagenpumatucel-L have been shown to target TGF-β 2 (52). Retaining the activity of endogenous inhibitors of TGF-β such as dickkopf-related protein 3 (Dkk-3) has been proposed as another indirect strategy for targeting TGF-β [reviewed in Ahmadi et al. (4)]. TGF signaling interacts closely with ROS and ncRNA in the mesenchymal transition of epithelial cells. It has been shown that TGF signaling can regulate ROS-mediated expression of miRNA (66, 127). TGF signaling and ROS activate miR-21, an oncogenic miRNA. The underlying mechanism of ROS-TGFβ-miR-21 axis has been studied by several investigators. Guo et al. (2015) unveiled the mechanistic link involving ROS-TGFβ-miR-21to be important for suppressing EMT. The group demonstrated that the heparin-binding site of kallistatin protein (a plasma protein) inhibits TGFβ signaling and ROS production, which further suppresses miR-21 and AKT signaling, thus blocking EMT (48). In another study, it was shown that enhanced SOD expression followed by quenching of ROS in tumors can block the TGF pathway, and inhibit MMP-2 and MMP-9expression (118). In support of these observations, TGF signaling was found to be upregulated in irradiated NSCLC (61). Irradiation results in accumulation of ROS, which leads to the activation of TGF signaling and subsequent overexpression of miR-21 leads to DNA damage.
ROS, ncRNA, and Metabolic Reprogramming
Reprogramming energy metabolism is one of the emerging hallmarks of malignant tumors. Unlike normal cells, cancer cells carry out glycolysis in the presence of oxygen. This phenomenon was first discovered by Otto Warburg in the 1920s. Since then, researchers have thoroughly investigated the possible links between metabolism and cancer progression. Over the past two decades, our understanding of the altered metabolism/cancer axis has significantly advanced. It has opened many diagnostic and therapeutic possibilities. Cancer cells could use alternative metabolic pathways for their survival. During the 1940s, Sidney Farber introduced antifolates as the inhibitors of single carbon transfer during de novo nucleotide biosynthesis. Since then, numerous antimetabolites have been introduced as anticancer agents. The main reason for these antimetabolites to emerge as the new-generation anticancer agents is because of the metabolic nature of malignant cells, where demand for nucleotide biosynthesis is ever increasing. Some of the most successful antimetabolites include methotrexate, which targets dihydrofolate reductase, 6-mercaptopurine, which targets phosphoribosyl pyrophosphate amidotransferase, and 5-FU, which blocks the activity of thymidylate synthase.
The concept of reprogramming energy metabolism by cancer cells was introduced by Otto Warburg, where he explained the ability of neoplastic cells in converting glucose to lactate instead of pyruvate in the presence of oxygen. The discovery of Warburg effect led to the discovery of drugs that inhibit glucose metabolism by inhibiting glycolysis. The 2-deoxyglucose (2-DG) is a well-known inhibitor of glucose metabolism. Once hexokinase converts 2-DG to 2-DH-6-phosphate, further conversion is inhibited. However, the inefficieny of the drug kept it out of the track (83). The renewed interest in cancer metabolism has widened the prospects of targeting this pathway and it has now moved beyond glucose metabolism. Glutamine metabolism is one such important part in amino acid metabolism, which has an extraordinary role in cancer progression. It has been shown that targeting the glutaminase (GL) enzyme can produce antitumor activity. CB-839, a rationally designed drug against GL, is currently in clinical trials (7, 45). However, evidence toward this hypothesis is currently limited (25). Redox signaling is closely associated with reprogrammed metabolism in cancer. Cancer cells exhibit high levels of ROS due to increased metabolic activity, genetic changes, and oncogenic signaling. They also have an adaptive mechanism to control the antioxidant system, which compensates oxidative damage to these cells. This modulates cellular signaling leading to metabolic reprogramming in cancer cells (64, 68). Besides ROS, ncRNAs have also been linked to metabolic reprogramming of tumor cells (155). Evidence also suggests a possible role for ncRNAs and ROS in the modulation of metabolic genes and associated intermediates (Fig. 6).
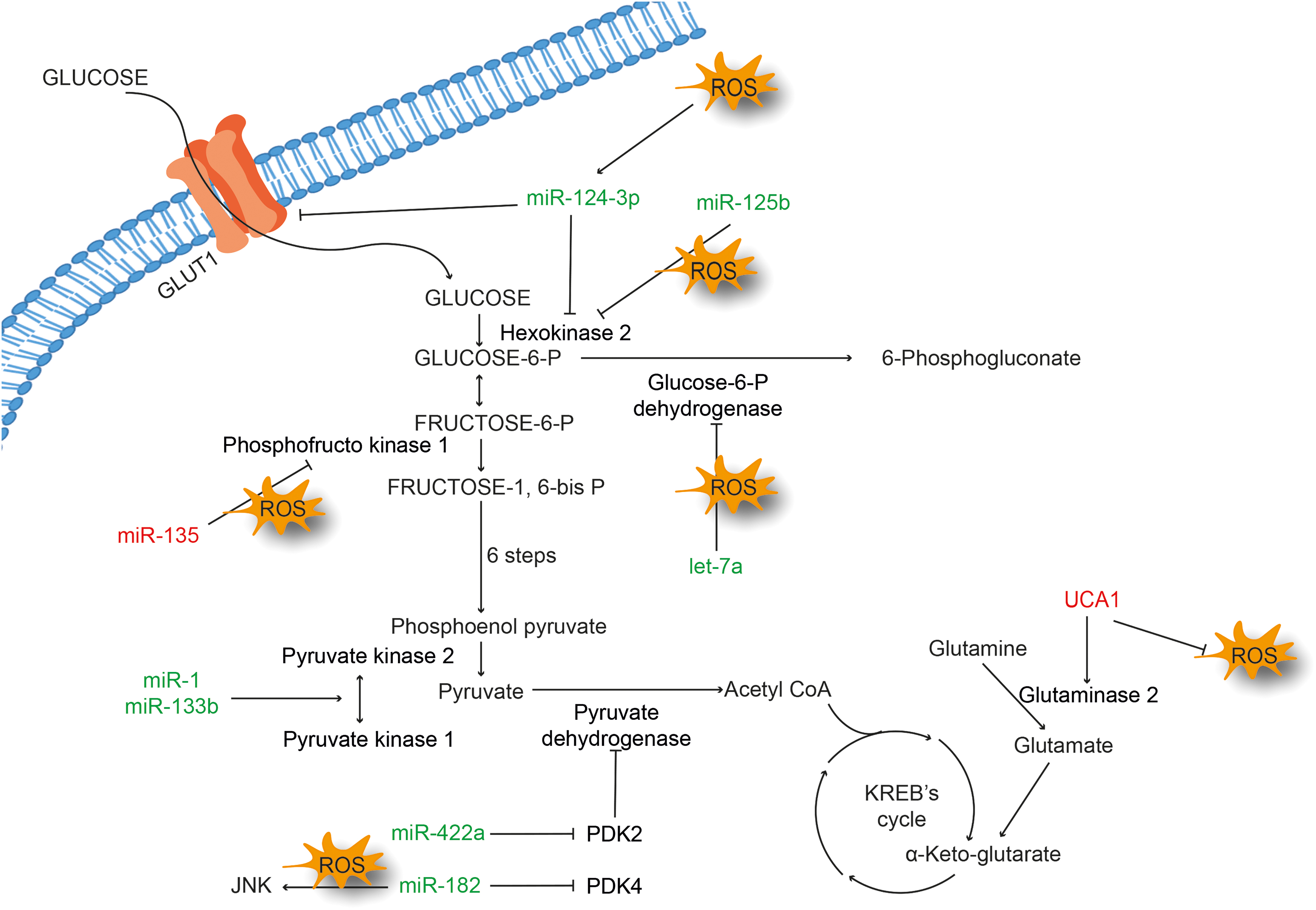
Pancreatic ductal adenocarcinoma (PDAC) is one of the dreadful cancers in humans with an ability to thrive under low-nutrient conditions. In a comprehensive study where the ability of PDAC to grow in a nutrient-scarce medium was examined, it was observed that glutamine deprivation accumulates ROS in MIA-PaCa-2 cells. The accumulated ROS activate mutant p53, which promotes miR-135 expression (160). The study further revealed that ROS-mediated upregulation of miR-135 suppresses glycolysis by directly targeting phosphofructokinase 1. Similarly, in gastric cancer cells, overexpression of miR-422a inhibits its target pyruvate dehydrogenase kinase 2 (PDK2) and shifts the energy metabolism toward oxidative phosphorylation (54). Glucose deprivation caused by oxidative stress leads to cytotoxicity in cancer cells. It has been shown that induction of ROS due to glucose deprivation results in increased acetylation of miR-466h-5p and reduced activity of histone deacetylase (HDAC) (31). miR-466h-5p has also been suggested to be involved in the accumulation of toxic metabolites (31). Astragalin (ASG), a nutraceutical present in a variety of foods, was shown to be cytotoxic in multiple cancer types.
ASG exhibits antiproliferative activity by repressing hexokinase 2 via upregulation of miR-125b, with subsequent accumulation of ROS in HCC cells (77). In another study, amentoflavone (AF), a plant flavonoid, was shown to induce antitumor effects in glioma cell lines via upregulation of miR-124-3p. AF was also shown to activate apoptosis, and inhibit glycolysis and cell proliferation in a dose-dependent manner. The study showed an association between miR-124-3p overexpression and the activation of ROS/5′ adenosine monophosphate-activated protein kinase (AMPK) signaling pathway (173). Another example of a crosstalk between ncRNA and oxidative stress is the involvement of PDK4-miR-182 and ROS in lung tumorigenesis. miR-182 negatively regulates PDK4 and promotes carcinogenesis with increased ROS and c-Jun N-terminal kinase (JNK) signaling. However, miR-182 possesses both oncogenic and tumor suppressive functions (74). Overexpression of let-7a in triple-negative breast cancer cell line represses cell proliferation, increases ROS, and downregulates proteins involved in glucose metabolism, namely, glucose 6-phosphate dehydrogenase (G6PD) and inosine monophosphate dehydrogenase. Furthermore, let-7a generated ROS were used with doxorubicin to enhance the therapeutic efficacy (114).
Pyruvate kinase, a rate-limiting enzyme of the glycolysis, plays a crucial role during cancer metabolism. Colorectal tumors show overexpression of pyruvate kinase muscle isozyme M2 (PKM2), which is downstream target of polypyrimidine tract-binding protein 1 (PTBP1). These tumors also show reduced expression of miR-1 and miR-133b. It has been shown that ectopic expression of miR-1 and miR-133b silences PTBP1 in colorectal tumors and suppresses growth by converting an active PKM2 to an inactive PKM1 (130). An interaction among lncRNA IDH1-AS1-ROS-α-ketoglutarate was found to form a functional link between c-Myc and HIF1α, which in turn modulates the Warburg effect (149). The amino acid metabolism also plays a crucial role in metabolic reprogramming of cancer cells. It is well understood that uptake of glutamine and its breakdown are vigorous in cancer cells due to overexpression of two isoforms of GL, GLS1, and GLS2. It has been reported that a higher level of GLS2 mRNA correlates with increased expression of urothelial carcinoma-associated 1 (UCA1) in bladder cancer cells. UCA1 is known to control GLS2 expression by decreasing ROS and acting as miR-16 sponge (76).
ROS, ncRNA, and AKT
AKT is a crucial signaling pathway that controls growth, proliferation, apoptosis, and metabolism in cells (115). Deregulated AKT signaling is a signature of many cancers. During cancer progression, several related pathways converge at AKT, thus indicating it to be a master regulator. RTKs such as EGFR, VEGFR, FGFR, insulin-like growth factor receptor, and transforming growth factor receptor (TGFR) activate AKT through PI3K. Once activated, AKT promotes activation of several transcription factors such as mammalian target of rapamycin (mTOR), FOXO, GSK3, and p53 (2, 23, 109). Being the most frequently misregulated signaling pathway in cancer, PI3K/AKT signaling represents a wide window for developing targeted therapy. Although several classes of inhibitors have been developed, only few of them are approved by the U.S. FDA. Examples include vistusertib and sapanisertib, which are the inhibitors of mTORC. Similarly, alpelisib, taselisib, and duvelisibare inhibitors of PI3K and miransertib, ipatasertib, and uprossertib are known to target AKT. However, among many, only temsirolimus and everolimus that target mTORC, and idelalisib and copanlisib against PI3K have been approved for clinical use by the U.S. FDA (60). ROS generated by NOX act directly on AKT or its downstream targets such as FOXO and p53. ROS can also indirectly act on AKT through modulators such as phosphatase and tensin homologue (PTEN), GSK3, and Src (42). miRNAs directly or indirectly modulate AKT pathway components and lncRNAs show sponging properties to these miRNAs (97).
miR-210 downregulation can induce or prevent cell progression (19, 159). Overexpression of miR-210 in colorectal cancer (CRC) correlates with increased apoptosis, resulting from enhanced ROS levels and hypophosphorylation of AKT (128). The NOX-mediated ROS generation can induce the expression of onco-miRNAs. As seen in prostate cancer, ROS can enhance miR-21 expression through AKT signaling (59). The inhibition of aldose reductase, an oxidative stress-associated enzyme, controls the growth of colon cancer by suppressing miR-21 in association with ROS and AKT pathway (112). Programmed cell death 4 (PDCD4), a proapoptotic gene, is a target of miR-21. It has been reported that accumulation of ROS suppresses PDCD4 through AKT/mTOR and miR-21 in colon cancer (111). ROS induced in response to nanomaterials can also regulate ncRNAs. For example, copper nanowire (Cu-Nw) can induce ROS in breast cancer cells. Furthermore, Cu-Nw has been shown to enhance ROS formation in mitochondria and induce the NF-κB-mediated regulation of miR-425 with a concomitant increase in PTEN expression in MDA MB-231 cell lines. Overall, this study provides evidence that Cu-Nw induces ROS, regulates miR-425-PTEN axis, and induces apoptosis (3).
ROS, ncRNA, and Ras/Raf/MEK/ERK
Ras/rapidly accelerated fibrosarcoma (Raf)/MEK/ERK signaling is activated by growth factor receptors and is prominently deregulated in cancer. This causes changes in the gene expression of both pro- and anticancerous genes involved in cell proliferation, invasion,and angiogenesis (95). Ras is frequently mutated in cancers. Mutations in Ras allow uncontrolled activation of Raf leading to constitutive expression of MAPK signaling cascade thereby resulting in increased cell survival and proliferation (92). Ras/Raf/Erk/MAPK is one of the major pathways for targeted therapy in cancer. Considering the crucial role of this pathway, several inhibitors have been developed so far that either modulate the components of signaling pathway or target upstream activators. Small-molecule inhibitors such as PD-0325901, GSK1120212, and TAK-733 are known to target MEK without any side effects. Indeed, RO5126766 and RO5068760, which also target MEK, are already in clinical trials. Similary, a number of inhibitors have been developed against Raf, namely, sorafenif, LErafAON, Raf-265, GSK2118436, and many more [reviewd in Santarpia et al. (110)]. Germann et al. (40) observed that ulixertinib selectively inhibited Erk 1/2 and found it promising for the treatment of cancer.
Several ncRNAs are known to target Ras/Raf/MEK/ERK pathway. A slight deregulation in ncRNAs can result in abnormal activation of Ras/Raf/MEK/ERK pathway (88). The Ras/Raf/MEK/ERK signaling can also be activated by oxidative stress. Let-7 miRNA is well known for its anticancer activity due to its ability to block the expression of Ras and Myc. The underlying mechanistic phenomenon involving let-7/ERK signaling was uncovered in a radioactive element-based approach using radon exposure (17). Radon exposure results in downregulation of let-7 miRNA in rat lung tissue with reciprocal regulation of K-ras levels. Transfection of let-7a-3p/7b-3p results in a negative correlation with the levels of K-ras, suggesting that let-7a-3p can potentially bind to the 3′UTR of K-ras. It has been shown that radon exposure could result in enhanced ROS production, which is closely associated with K-ras. miR-155 is another oncomiR that is known to have a link with the Ras/Raf/MEK/ERK pathway.
According to an miRNA array-based study, miR-155 expression is linked with K-ras activation via the involvement of ERK signaling. Once activated, miR-155 blocks FOXO3 and negatively regulates antioxidant genes such as SOD and catalase, which enhance ROS-dependent pancreatic cell proliferation (139). H19 is a well-characterized lncRNA known for its role in embryonic development and regulation. Its role in regulating oxidative stress has also been defined. A ternary relationship among H19/ROS/ERK-MAPK was reported in HCC (29). H19 was shown to be upregulated in HCC along with ERK/MAPK signaling, and was responsible for the chemoresistance nature of CD133+ stem cells. Blocking of H19 downregulates ERK signaling and results in ROS enrichment with increased antioxidant activity of SOD and GST, resulting in reduction in the chemoresistance of CD133+ cells (29).
ROS, ncRNA, and Other Signaling Pathways
Wingless-related integration site (Wnt) pathway controls development and homeostasis of stem cells. Wnt pathway is known to be inherently conserved across evolution. Cancer stem cells show aberrant Wnt signaling (32). Besides its role in stem cell maintentance, Wnt signaling also plays a role in repair of oxidative DNA damage. Wnt is known to regulate several miRNAs at levels such as ligands, receptors, receptor-associated proteins, catenin, and transcription factors (63, 103). It has been reported that ER stress upregulates miR-346, which reduces ROS level through mitophagy (47). ER stress also enhances glycogen synthase kinase-3 beta (GSK3β) expression, thus suggesting the involvement of GSK3β in diminishing ROS. Indeed, GSK3β inhibition reverses the effects of miR-346 on ROS generation thus providing the evidence for the role of Wnt pathway in regulating cell viability through the miRNA-346-ROS-GSK3β axis.
Another important signaling pathway that shows a link with ROS and ncRNA is the janus kinase (JAK)/STAT, which is triggered by cytokines and is primarily known for its role in inflammation and immunity. Activation of cytokine signaling enhances cell survival, proliferation, and inflammation (67, 91). Cytokine signaling is activated by the binding of ligands that results in the dimerization and autophosphorylation of JAK leading to STAT activation. After activation, STAT translocates into the nucleus and regulates the transcription of proinflammatory molecules (101). In the context of inflammation, JAK/STAT signaling is crucial in the progression of cancer. Sevaral inhibitors have been developed against JAK and STAT. However, these inhibitors could not advance due to poor pharmacokinetics and efficacy (44). JAK/STAT signaling underpins the role of inflammation in carcinogenesis. Cytokine signaling plays a crucial role in the activation of pro-oncogenic molecules, while ROS produced by these signaling act as secondary messengers in cancers. The ncRNAs are also known to modulate the cytokine signaling pathway. Some lncRNAs and miRNAs directly modulate the JAK/STAT signaling, while some modulate the regulators of JAK/STAT. Glioma and glioma-derived cell lines show upregulation of miR-33a when compared with healthy tissues. Sirtuin 6 (SIRT6) is the potential target for miR-33a. It has been shown that overexpression of SIRT6 reduces cell survival and initiates apoptosis by enhancing the ROS level. In parallel, it was reported that inhibition of JAK2 and STAT3 phosphorylation downregulates cytokine signaling. Overall, this study suggests that miR-33a acts as oncogene through modulation of SIRT6-ROS-JAK/STAT axis (13).
JNK, a member of the MAPK family, plays a crucial role in human health and diseases (107). In a recent study, JNK1 was reported to enhance tumor growth and induce chemoresistance (41). The role of JNK in cancer pathogenesis has been investigated. A variety of inhibitors, namely, indazoles, aminopyrazoles, and pyridine carboxamides, are known targets of JNK signaling by preclinical studies [reviewed in Bubici and Papa (11)]. JNK pathway is reported to be highly susceptible to oxidative stress. ROS result in the activation of JNK by phosphorylation and subsequent upregulation of p53and apoptosis (156). Moreover, evidence suggests that miRNAs regulate JNK signaling directly or indirectly and affect the cellular processes such as apoptosis and chemoresistance. An interplay among miR-20a-ROS-JNK was reported in CRC cells upon cisplatin exposure (167). Knockdown of miR-20a enhances the sensitivity of CRC to cisplatin. This was attributed to increased expression of miR-20a target genes such as apoptosis signal-regulating kinase 1 (ASK1), ROS, and phosphorylated JNK. In another study, suberoylanilide hydroxamic acid, an HDAC inhibitor, was found to induce apoptosis in lung cancer cells through modulation of ASK/JNK pathway (163). The mechanism involved miR-129-5p, which downregulated thioredoxin-1 (Trx1) by binding to 3′UTR of Trx1 mRNA, leading to elevated ROS levels. Increase in ROS levels triggered the activation of ASK/JNK signaling, which induced apoptosis.
Conclusion
For decades, it has been known that ROS play a pivotal role in the pathogenesis of several chronic diseases, including cancer. ROS are known to exhibit both oncogene and tumor suppressor functions and thus act as a double-edged sword. As discussed in this review, ncRNAs and ROS functionally interact with several tumor suppressor and oncogenic signaling pathways during the course of tumorigenesis. The interaction can be during ncRNA biogenesis or during the transduction of signal across the cell. The interaction can also take place at the epigenetic level. Majority of the ncRNA-based studies showing the relationship of oxidative stress during cancer pathogenesis involved miRNAs. Although the lncRNAs play crucial role during cancer pathogenesis, studies exploring their association with ROS have been limited. Although several prime candidates have been identified and the pathways they target are known, many of these candidates fail due to their poor pharmacokinetics. Thus, targeting ncRNAs could be significant in developing novel cancer treatments. Moreover, in this era of technical advancement in cellular and molecular biology, namely, nanopore-based genome sequencing, protein expression analysis, and gene modification techniques, targeting ncRNAs would be a prototypic intervention in cancer therapy. In addition, an in silico approach could be used to screen large numbers of molecules, which show association with ncRNAs. However, this needs a bioinformatic advancement to envisage secondary and three-dimensional structural infomation of ncRNAs. Besides their therapeutic interventions, oxidative stress-associated ncRNAs could be used as a predictive biomarker for cancer development. However, tissue-specific expression of ncRNAs and the variety of off-targets are major challenges.
Thus, the ROS-ncRNA-cancer axis has provided hope for early cancer detection and therapeutic intervention. However, most of the studies carried out to date are from cell lines. Future studies should explore the significance of this axis in animal models of cancer to effectively translate the results for clinical applications.
Footnotes
Funding Information
This work is financially supported by University Grants Commission [No. F.30e112/2015 (BSR)] and DST-Science and Engineering Research Board (ECR/2016/000034) awarded to S.C.G. A.S. (Anurag Sharma) and A.C. like to acknowledge DST-Science and Engineering Research Board; ECR/2016/001863 and ECR/2016/000798, respectively, for the research grants. S.M. and L.C.D. thank ICMR New Delhi (3/1/3/JRF-2016/LS/HRD-65-80388) and Directorate of Minorities, Karnataka (DOM/FELLOWSHIP/CR-33/2018-19), respectively, for the fellowship to pursue their doctoral program.