
Abstract
Significance
: The growing incidence of neurodegenerative diseases significantly impacts the individuals who suffer from these disorders and is a major health concern globally. Although the specific mechanisms of neurodegenerative diseases are still far from being acknowledged, it is becoming clear that oxidative stress and neuroinflammation are critical contributing factors to the progression of neurodegeneration. Thus, it is conceivable that the inhibition of oxidative stress and neuroinflammation may represent promising therapeutic targets for the treatment of neurodegenerative diseases.
Recent Advances
: Recently, the strategy for neurodegenerative disease therapy has shifted from the use of antioxidants and conventional anti-inflammatory targets to upstream mediators due to the failure of most antioxidants and nonsteroidal anti-inflammatory drugs in clinical trials. Nicotinamide adenine dinucleotide phosphate oxidases (NOXs), a family of superoxide-producing enzyme complexes, have been identified as an upstream factor that controls both oxidative stress and neuroinflammation. Genetic inactivation or pharmacological inhibition of NOX enzymes displays potent neuroprotective effects in a broad spectrum of neurodegenerative disease models.
Critical Issues
: The detailed mechanisms of how NOX enzymes regulate oxidative stress and neuroinflammation still remain unclear. Moreover, the currently available inhibitors of NOX enzymes exhibit nonspecificity, off-target effects, unsuitable pharmacokinetic properties, and even high toxicity, markedly limiting their potential clinical applications.
Future Directions
: This review provides novel insights into the roles of NOXs in neurodegenerative pharmacology, and indicates the types of NOX enzyme inhibitors that should be identified and developed as candidates for future applications, which might reveal novel neurodegenerative disease therapies based on NOXs.
Introduction
Neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), are recognized as multifactorial disorders characterized by progressive damage of neuronal populations in distinct brain areas. The growing incidence of these disorders significantly impacts the individuals who suffer from these disorders and is a major health concern globally. Neurological disorders are the leading cause of disability and the second leading cause of death worldwide (59a). Currently, no effective therapies aimed at halting progressive neurodegeneration are available. Although the specific mechanisms of neurodegenerative diseases are still far from being acknowledged, many studies have shown that oxidative stress and neuroinflammation play important roles in the progression of neurodegeneration (49, 135).
Early epidemiological and animal studies support the notion that antioxidants and nonsteroidal anti-inflammatory drugs (NSAIDs) reduce the risk of acquiring AD and PD (28, 163). However, clinical trials for antioxidants (such as vitamin C, vitamin E, and coenzyme Q10) and NSAIDs (most of which inhibit the activity of cyclooxygenase-1 and/or -2) have failed to demonstrate efficacy for treating neurodegenerative diseases, and some of these treatments have even been shown to be harmful (155), highlighting the need for better therapeutic strategies and targets in developing therapies. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are a family of superoxide-producing enzyme complexes in both phagocytic and nonphagocytic cells. It is well documented that excess activation of NOX enzymes, especially NADPH oxidase 2 (NOX2), is involved in a variety of neurodegenerative diseases (79). Indeed, several comprehensive review articles of NOXs and neurodegenerative disorders have been published (20, 107, 116, 155, 162). However, most of them only focused on one or two specific aspects, such as how NOXs regulate oxidative stress, NOX2 isoforms in blood samples as a biomarker, or the treatment efficacy of currently available inhibitors of NOX2 in neurodegenerative disease. In this review, we summarize the recent exciting progress regarding the crucial role and underlying mechanisms of NOX enzymes in both oxidative stress and neuroinflammation in neurodegenerative diseases. Furthermore, we discuss the beneficial effects and potential drawbacks of inhibitors of NOX enzymes in combating the progression of neurodegenerative diseases, as well as the types of NOX enzyme inhibitors that should be identified and developed as candidates for future clinical trials.
Oxidative Stress and Neuroinflammation in Neurodegenerative Diseases
Oxidative stress
Reactive oxygen species (ROS), including superoxide anion, hydrogen peroxide (H2O2), hydroxyl radical, ozone and singlet oxygen, are chemically reactive molecules due to unpaired electrons. ROS play critical roles in both physiological and pathological processes. In physiological conditions, the production of ROS is restricted and regulated by a series of oxidative and antioxidative mechanisms (23, 150). Once disruption of this redox hemostasis in favor of increased levels of ROS occurs, ROS will be excessively produced, resulting in oxidative stress, a condition that leads to oxidative modification of proteins, lipids, RNA, DNA, and other biomolecules (23, 150). In addition, excess ROS cause dysfunction of the ubiquitin–proteasome system and autophagy, two important protein degradation pathways, resulting in the accumulation of abnormal proteins and subsequent activation of the cell death cascade (140). The brain is extremely susceptible to oxidative damage due to its high oxygen consumption, relatively weak antioxidant mechanisms, and restricted capacity for neuron renewal and regeneration (142). Actually, increasing evidence indicates that oxidative stress is associated with the pathogenesis of multiple neurological disorders (7, 150).
Neuroinflammation
Neuroinflammation (inflammation in the brain) primarily involves the activities of two types of glial cells: microglia and astroglia. Microglia are the resident macrophages of the brain that arise from the embryonic yolk sac (61). In mature brains, resting microglia exhibit a characteristic ramified morphology and actively partake in immune surveillance. As the brain's first line of defense, microglia can be rapidly activated and undergo dramatic changes, metamorphosing into an amoeboid morphology in response to brain injury and immunological stimuli (122). Activated microglia respond to injurious signals by migrating to the site of injury, releasing factors to recruit more cells and phagocytizing foreign substances. Currently, it has been accepted that activated microglia can be polarized into two phenotypes; that is, classic M1 (proinflammatory status) and alterative M2 (anti-inflammatory status), to exert neurotoxic and neuroprotective functions, respectively (161). In addition to microglia, reactive astrocytes are induced in some pathological conditions in the brain and are involved in the neuroinflammatory response (103). Recent studies have shown that reactive astrocytes can also display toxic A1 and beneficial A2 subtypes (74, 104). However, the inflammogen lipopolysaccharide (LPS) fails to cause the appearance of A1 astrocytes in mice without microglia (colony stimulating factor 1 knockout mice) (104), suggesting that microglia exert phenotypic control over astroglial A1 activation. Growing evidence suggests that in normal or mild inflammatory states, glial activation serves the beneficial purposes of immune surveillance and noxious stimuli clearance, whereas in chronic inflammatory conditions, activated glial cells can be neurotoxic and significantly contribute to neurodegeneration.
Role of oxidative stress and neuroinflammation in neurodegeneration
A great amount of evidence has implicated oxidative stress and neuroinflammation as the potential common etiology of most neurodegenerative diseases. Malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and lipid peroxidation (LPO) are sensitive markers for oxidative stress. Elevated levels of MDA, 4-HNE, and LPO, as well as reduced glutathione content, have been detected in the brain tissues of AD and PD patients and in the serum or cerebrospinal fluid of patients with ALS (9, 79). The activation of glial cells into proinflammatory states and a large number of proinflammatory mediators (such as ROS, chemokines, and cytokines) in the central nervous system (CNS) are the main features of neuroinflammation. There is abundant evidence that chronic microglial activation, especially microglial M1 polarization, is highly associated with an increased risk of AD and PD, as demonstrated by the accumulation of activated microglia and proinflammatory factors in the lesion sites of the brain (27, 79, 105).
Abnormal aggregation of specific proteins is a remarkable pathological feature of neurodegenerative diseases. Oxidative stress and neuroinflammation participate in the process of pathological protein formation (57). In turn, aggregated proteins further facilitate ROS production and neuroinflammatory responses, forming a vicious cycle of pathogenesis. Recent studies indicate that neuroinflammation and ROS promote the expression and accumulation of β-amyloid (Aβ), the main component of senile plaques in AD, through increased activities of β and γ-secretases and/or decreased levels of Aβ-degrading enzymes, such as tissue plasminogen activator and ethylenediaminetetraaceticacid chelation-sensitive protease (41, 95). Aβ fibrils have been found to activate microglia to produce superoxide and multiple proinflammatory factors (40, 136). Moreover, Aβ contributes to oxidative stress in AD, and this stress has been confirmed in an AD transgenic mouse model expressing mutant amyloid precursor protein (APP) and presenilin-1, with increased levels of H2O2 and the peroxidation of proteins and lipids (95, 199). Oxidative stress can modify α-synuclein aggregation in dopaminergic neurons in PD pathology and α-synuclein further increases iROS (intracellular ROS) (142). α-Synuclein is considered a key protein that contributes to PD pathogenesis, and it can stimulate microglial activation to produce superoxide (76). In addition, α-synuclein has a mitochondria-targeted amino-terminal sequence that associates with the inner mitochondrial membrane and disrupts complex I function, thus generating oxidative stress (19). In Huntington's disease (HD), huntingtin aggregation is also responsible for increased levels of ROS (19, 66).
In addition to abnormal protein aggregation, oxidative stress and neuroinflammation contribute to progressive neurodegeneration in neurodegenerative diseases. Oxidative stress injures the cell structure, lipids, proteins, and DNA, resulting in a cascade of events and finally progressive neuronal death, such as apoptosis and necrosis (11). Currently, multiple types of antioxidants, including vitamins, phenolic compounds, and carotenoids, have been demonstrated to mitigate neuronal damage in both in vitro and in vivo models of neurodegeneration (165). Recent findings indicated that oxidative stress also contributes to ferroptotic cell death in neurodegenerative disorders (157). Ferroptosis is a recently recognized and iron-regulated form of cell death, in which oxidative stress, especially LPO, is an important driving force (45). Free radical scavenger ferrostatin-1 attenuates ferroptotic cell death of dopaminergic neurons in a 1-methyl-4-phenylpyridinium (MPP+)-induced in vitro PD model (88). Furthermore, neuroinflammation has been shown to play an important role in neurodegeneration. The activation of the NLRP3 inflammasome is very common during neuroinflammation. It is well known that NLRP3 inflammasome activation mediates the pyroptosis of neuronal cells in neurodegenerative diseases through cleavage of the gasdermin D protein. Pharmacological or genetic inactivation of the NLRP3 inflammasome has been reported to protect against neurodegeneration in AD (71) and PD mouse models (63).
Indeed, oxidative stress and neuroinflammation are not independent processes and interact each other, collaterally resulting in neuronal damage. Neuroinflammation can be both a cause and a consequence of chronic oxidative stress. Overactivated microglia generate not only proinflammatory cytokines and chemokines but also copious amounts of ROS and reactive nitrogen species (RNS) (158). Oxidative modifications of macromolecules can also modulate the neuroinflammatory response via transcription and other signaling pathways. In oxidative stress conditions, reactive species can act as signaling molecules mediating the activation of signaling pathways in microglia and astroglia, leading to the increased release of proinflammatory cytokines and chemokines (83, 152). Free radicals, especially iROS, can activate certain transcription factors, including nuclear factor-κB (NF-κB) and activator protein 1, upregulating the expression of proinflammatory cytokines (99, 171). Interactions of oxidative stress and neuroinflammation exaggerate oxidative and proinflammatory responses in the microenvironment, exacerbating neuronal death. In turn, noxious endogenous ligands released by injured neurons, such as μ-calpain, α-synuclein, Aβ, and high mobility group box 1 (HMGB1), are thought to continually reactivate microglia (reactive microgliosis), resulting in additional neurodegeneration (13, 58, 94, 101). Consequently, a self-propelling vicious cycle is created between injured neurons and dysregulated microglia, inevitably resulting in delayed and progressive neurodegeneration (172), which might be one of the potential mechanisms for the progressive nature of neurodegenerative diseases (54).
Distribution and Physiological Function of NOX in the CNS
Distribution of NOX
The NOX family are a group of multisubunit enzyme complexes that catalyze the reduction of oxygen to superoxide radicals or H2O2 using NADPH as an electron donor (Fig. 1) (166). For many years, NOX has been synonymous with NOX2/gp91phox, the first identified NOX enzyme in the study of respiratory bursts (57). At present, seven members of the NOX family have been identified, including NOX1, NOX2, NOX3, NOX4, NOX5, and Dual oxidase 1 and 2 (DUOX1 and DUOX2). All these NOX isoforms are transmembrane proteins, but they differ in their regulation and tissue and subcellular localization. NOX isoforms are widely distributed throughout different tissues: NOX1 in the colon, NOX2 in phagocytes and B lymphocytes, NOX3 in the inner ear and some fetal tissues, NOX4 in the kidney and blood vessels, NOX5 in lymphoid tissue and the testes, and DUOX1 and DUOX-2 in the thyroid and lungs (4, 57, 107). In the brain, NOX1, NOX2, and NOX4 can be detected in multiple brain regions of mice/rats, including the cerebral cortex (34), hippocampus (72), cerebellum (38), hypothalamus (87, 186), midbrain (78, 79), and/or striatum (31, 64). NOX3 and NOX5 are located in the cerebral cortex (30) and glioblastoma (5), respectively. Notably, NOX5 is not expressed in rats and mice (4). The distribution of DUOX1 and DUOX2 remains to be investigated (107).
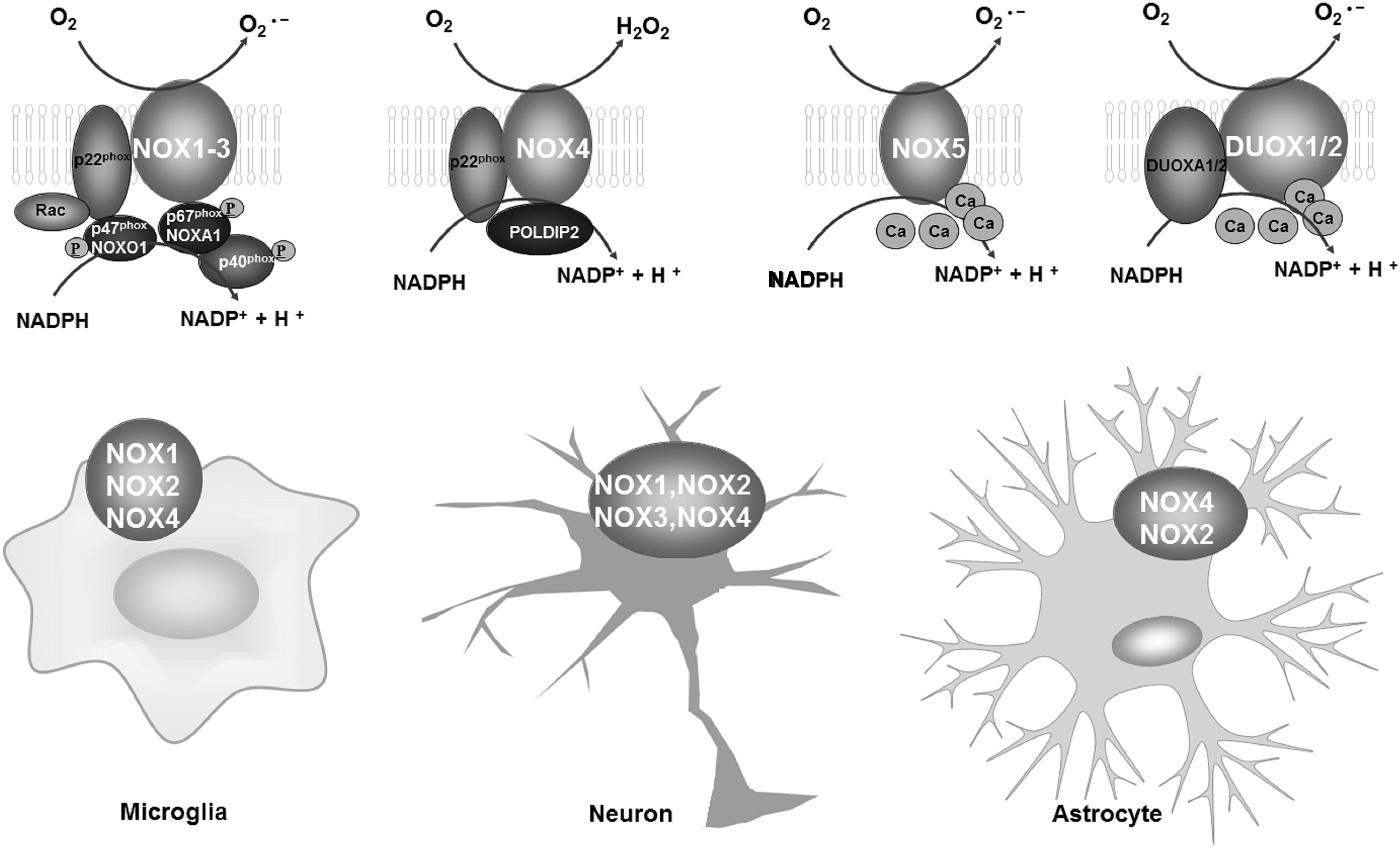
Although the distribution of NOX isoforms is relative clear, the cellular distribution of NOX isoforms remains unclear. Recent studies using a combination of quantitative polymerase chain reaction (qPCR) and double-immunofluorescence analyses in the brain or in different cell types isolated from the mouse brain have indicated, to some degree, the cellular location of NOX family members in the CNS. In addition to NOX2, the microglial expression of NOX1 was confirmed by Cheret et al. by qPCR and immunofluorescence staining analyses in microglia purified from wild-type or NOX-deficient mice (31). Cooney et al. performed double-immunofluorescence staining in a rat brain injury model to determine the cellular and temporal expression of NOX isoforms in the brain. They found that NOX2 and NOX4 were expressed in microglia, and that this expression was dependent on the postinjury time (37). NOX2 was also found to be expressed in neurons, while NOX4 was expressed in both neurons and astroglia (37). In contrast, NOX3 has been positively identified in neurons only (37). Although a specific cell type can express different NOX isoforms, the levels of expression are different. For example, the expression of NOX2 in microglia is ∼10-fold higher than that of NOX1 (31, 37). Similarly, different levels of specific NOX isoforms have been observed in different cell types. For example, NOX2 is expressed in microglia at a much higher level than in neurons and astroglia (37).
Physiological function of NOX
ROS generation is the main function of different NOX enzymes, although the activation mechanisms of NOX isoforms are different. Most NOXs, including NOX1, NOX2, NOX3, and NOX4, interact with p22phox in the membrane, which is essential for their maturation, stabilization, and heme incorporation (10). In addition, the activation of NOX1, NOX2, and NOX3 requires the phosphorylation and subsequent translocation of cytosolic subunits (p47phox/NOXO1, p67phox/NOXA1/NOXA2, p40phox, and the G-protein Rac1/2) to the membrane-bound subunit p22phox, whereas the activation of NOX4 needs p22phox only (10, 155). Conversely, elevated cellular Ca2+ concentrations, but not alterations in p22phox and cytosolic subunits, are required for the activation of NOX5, DUOX1, and DUOX2 (10, 155).
NOX enzymes possess various physiological functions, such as host defense and inflammation, as well as neuronal development and survival in the CNS (154). Strong evidence suggests that ROS derived from NOX2 are key regulators of host immune responses and the removal of debris from the brain (108, 191). Microglia deficient in NOX2 display reduced ROS production capacity and phagocytic activity in response to respiratory burst stimulants (69, 185). The proliferation and development of microglia are also regulated by NOX2-derived ROS. Mander et al. found that H2O2 produced by activated NOX2 mediates interleukin-1β (IL-1β) or tumor necrosis factor α (TNFα)-induced proliferation of microglia since inhibition of NOX2 by diphenyleneidonium (DPI) or apocynin blocks, while activating NOX2 enhances, microglial proliferation in primary rat glial cultures (110). Furthermore, mutations in genes that encode NOX2 have been reported to result in chronic granulomatous disease (CGD) in humans and CGD-like phenotypes in mice (118, 154). The inability of NOX2-deficient phagocytes to kill ingested bacteria and fungi results in recurrent, persistent infections and granuloma formation in many organs of CGD patients (96). In contrast to NOX2 deficiency, genetic deletion of NOX1 and 4 is not associated with any significant spontaneous pathologies (21, 22).
In addition to immune regulation, NOX enzymes have been shown to regulate neuronal function, plasticity, and survival. NOX2 is the main enzyme in neurons responsible for superoxide production in response to the activation of N-methyl-
In addition to microglia and neurons, astroglia are essential to the integrity and function of the brain. Recent evidence has begun to unravel the function of NOX enzymes in astrocytes. Yang et al. reported that NOX2-derived ROS contribute to astrocyte migration induced by metalloproteinase-9 (182), a fundamental phenomenon for astrocytes to monitor normal functions of the CNS (192). NOX enzymes are also found to be able to regulate the survival of astrocytes. VAS2870, a pan-NOX inhibitor, restores cell viability, mitochondrial inner membrane potential, and levels of ATP in rat astrocyte cultures treated with staurosporine (151), suggesting that NOX activation is detrimental for astrocyte survival. In response to immunologic challenges or brain injury, astroglia also become activated (93). Activated astroglia produce a host of proinflammatory cytokines and neurotrophic factors (74, 82). We recently found that microglial NOX2-derived H2O2 is a key factor mediating the balance between the proinflammatory and neurotrophic functions of astrocytes in inflammatory conditions through a JAK-STAT1/3-dependent pathway (82). Altogether, the function of NOX enzymes in neurons, microglia, and astrocytes is important for the integrity and physiological function of the CNS.
NOX Enzymes Are Important Regulators of Oxidative Stress and Neuroinflammation in Neurodegenerative Diseases
NOX enzymes regulate oxidative stress in neurodegenerative diseases
Emerging experimental evidence suggests that many NOX isoforms are involved in CNS oxidative stress and neurodegeneration. Postmortem studies illustrate increased activities of NOX enzymes in the brains of patients suffering from neurodegenerative disorders. For instance, elevated expression, activity, and mRNA transcript levels of NOX1, NOX2, and/or NOX4 have been observed in the brains of patients with AD, PD, and ALS, which are associated with high levels of ROS compared with healthy controls (10, 148, 155, 177, 178, 189). Furthermore, in an AD mouse model overexpressing mutant APP, pharmacological inhibition of NOX2 by a membrane-permeable inhibitor NOX2ds-tat or genetic ablation of NOX2 mitigates oxidative stress and cognitive dysfunction (162). Inhibition of NOX by apocynin and 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) also reduces ROS production and neurodegeneration in cell cultures treated with Aβ (26, 85), highlighting the critical role of NOX-derived ROS in oxidative damage in AD.
Similarly, structurally diverse NOX inhibitors, AEBSF, apocynin, DPI, or NOX2 knockdown also markedly attenuate ROS production, peroxynitrite-mediated protein radical formation, and dopaminergic neuron cell death induced by α-synuclein (98), the main component of Lewy bodies in PD, and PD-inducing neurotoxicants, including rotenone, paraquat, LPS, and MPP+ (39). Moreover, in a mouse model of PD generated by 6-hydroxydopamine (6-OHDA), adeno-associated virus-mediated NOX1 knockdown or Rac1 inhibition reduces 6-OHDA-induced ROS production and the levels of the DNA oxidative stress marker, 8-oxo-dG, in the brain (33). Remarkably, genetic deletion of NOX1 or NOX2 mitigates NOX activity and ROS production, which are associated with dampened neurotoxicity, delayed disease progression, and increased life span in an ALS mouse model that carries mutant superoxide dismutase 1 (SOD1) (111, 113), further supporting a prominent role of NOX complexes in oxidative stress and neurodegenerative diseases.
In addition to NOXs themselves, there is evidence that the activation of NOX enzymes may regulate mitochondrial ROS production in neurons (Fig. 2). Mitochondria are usually considered the prime source of ROS in the CNS and age-related neurodegenerative diseases (100). However, in a mouse PD model induced by combined paraquat and maneb, increased mitochondrial ROS content and reduced mitochondrial potential are observed, and they are significantly prevented by preadministration of the NOX inhibitor apocynin (149). PINK1, a mitochondria-targeted serine/threonine kinase, is capable of protecting neurons from oxidative stress by maintaining mitochondrial Ca2+ handling (168). Gandhi et al. demonstrated that the reduction of NOX2 expression in PINK1 knockdown neuroblastoma cells significantly attenuates ROS production (53). Similarly, NOX4 knockdown also attenuates mitochondrial superoxide production in angiotensin II-stimulated neurons (24). These results suggest that blocking the activation of NOX may dampen mitochondrial stress.
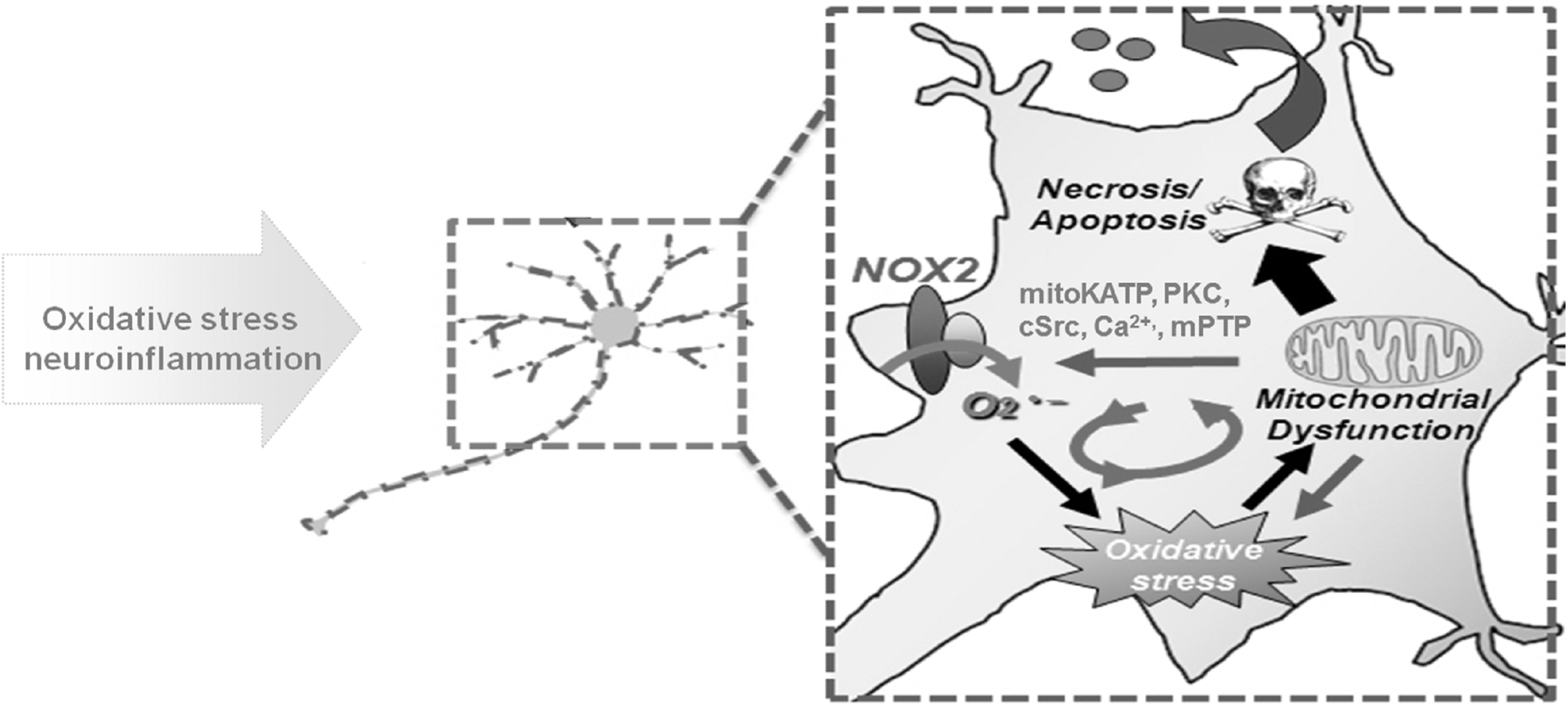
Of course, mitochondria are not only a target for NOX-generated ROS but also a significant source of ROS, which may in turn stimulate the activation of NOX (Fig. 2) (43). Increased mitochondrial superoxide production in response to inhibitors of the electron transport chain, such as rotenone or antimycin, increases the expression of NOX1 (51). Nazarewicz et al. reported that the activation of mitochondrial ATP-sensitive K+ channel increases NOX2-derived superoxide production in human aortic endothelial cells, which can be blocked by supplementation with a mitochondria-targeted SOD mimetic (mitoTEMPO) or a mitochondria-targeted glutathione peroxidase mimetic (mitoEbselen) (120). The molecular mechanisms for the regulatory effects of mitochondria on the activation of NOX enzymes are complicated since mitochondrial ROS-induced NOX activation can be prevented by inhibitors of mitochondrial permeability transition pore (mPTP), protein kinase C, tyrosine kinase cSrc, or an intracellular Ca2+ chelator, and is absent in leukocytes with cyclophilin D deficiency (regulates mPTP) (97). Taken together, crosstalk between NOXs and mitochondria may represent a feed-forward cycle of ROS production in the CNS, contributing to the oxidative damage and subsequent neurodegeneration observed in neurodegenerative diseases.
Notably, in contrast to the CNS, the activities of NOX isoforms in the peripheral nervous system of patients with neurodegenerative disorders remain unchanged, although the levels of NOXs in the serum of patients are upregulated (70). No meaningful difference in NOX2 activity and ROS production is observed in fresh whole blood prepared from patients with PD and ALS and corresponding controls (113, 114), indicating a major involvement of NOX2 in the CNS, especially oxidative stress (154).
NOX enzymes regulate neuroinflammation in neurodegenerative diseases
Currently, increasing evidence indicates a prominent role of NOX complexes, especially microglial NOX2, in neuroinflammatory responses in neurodegenerative diseases (Fig. 3). Microglia play key roles in neuroinflammation, and their activation can release several inflammatory and cytotoxic factors, such as chemokines, cytokines, proteases, and ROS (54). NOX2 has been shown to modify microglial activation and related neurotoxicity. LPS fails to elicit the production of ROS in NOX2-deficient neuron–glial cultures, and this failure is associated with a marked reduction of microglial activation, TNFα release, and dopaminergic neurodegeneration (133). Furthermore, reduced microglial activation and the gene expression of proinflammatory cytokines are observed in the brains of LPS-treated NOX2-deficient mice compared with WT controls (132). Moreover, the inhibition of NOX2 using an ultralow dose of DPI mitigates microglia-mediated neuroinflammation in response to LPS (172). Similar regulatory effects of NOX2 on neuroinflammation are observed in models of AD and PD. In a TgCRND8 mouse model of AD (141) and paraquat- or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mouse PD models (81, 172), pharmacological inhibition or genetic inactivation of NOX2 markedly reduces microglial activation as evaluated by Iba1 immunoreactive cell morphology, intensified CD11b staining, or proinflammatory cytokine expression.

NOX2 can also regulate microglial polarization (Fig. 3). Choi et al. investigated the role of NOX2 in microglial activation using p47phox-deficient mice, as well as the NOX inhibitor apocynin, and found that the pharmacological or genetic inactivation of NOX2 reduces the gene expression of proinflammatory factors such as TNFα, chemokine ligand 2, and chemokine receptor 2 and simultaneously elevates the mRNA levels of M2 markers, including Ym1, CD163, and MARCO in the brains of mice treated with LPS or Aβ1–42 (intracerebroventricular injection) (36). We recently found that in paraquat- and maneb-induced mouse PD models, the inhibition of NOX2 by apocynin also reduces classic microglial activation by showing reduced gene expression of M1 markers, such as inducible nitric oxide synthase (iNOS), TNFα, and IL-1β (79, 81). The underlying mechanisms for the NOX2-regulated microglial phenotype remain unclear. Taetzsch et al. reported that microglia exposed to steady fluxes of H2O2 show altered NF-κB p50 protein–protein interactions, decreased NF-κB p50 DNA binding, and augmented late-stage TNFα expression, indicating that H2O2 impairs NF-κB p50 function and prolongs amplified M1 activation (27, 79, 160). We found that inhibition of NOX2 can also suppress activation of the JAK-STAT pathway (27, 77, 82), another signaling pathway capable of regulating the gene expression of microglial M1 markers (124). These reports suggest that NOX2 is critical for controlling the balance of microglial M1 and M2 phenotypes in inflammatory conditions.
In agreement with the findings in microglia, the activation of astroglia can be regulated by microglial NOX2. Pharmacological or genetic inactivation of NOX2 markedly attenuates astroglial activation in LPS- and MPTP-injected mice by showing reduced immunostaining of glial fibrillary acidic protein, a marker for astrocytes (82). Mechanistically, microglial NOX2-derived H2O2 serves as a direct signal to regulate astrogliosis, in which H2O2 diffuses into the cytoplasm of astrocytes and subsequently enhances the phosphorylation of the transcription factors, STAT1 and STAT3, therefore regulating the gene expression and immunological functions of astrogliosis (Fig. 3) (82).
In addition to NOX2, NOX1 has been illustrated to play a role in mediating neuroinflammation. Compared with controls, shRNA-mediated knockdown of NOX1 in microglia mitigates LPS-induced activation of iNOS and the release of inflammatory/neurotoxic factors, such as IL-1β (31). Comparisons of microglia purified from WT and NOX1-KO mice further indicate that NOX1-derived superoxide is required to optimize microglial production of toxic factors (31), suggesting that NOX1 activation promotes proinflammatory responses.
Activated microglia are not only the downstream targets of NOXs but can also feed-forward the signal activities of NOX enzymes. A variety of proinflammatory cytokines and chemokines released from activated microglia can stimulate the activation of NOX2 in microglial cells. For example, TNF-α, a common cytokine released from activated microglia, stimulates the activation of NOX2 through p47phox membrane translocation in human colorectal cancer HCT116 cells (188) and rat embryonic heart-derived H9c2 cells (183). Similarly, proinflammatory cytokines (TNFα, IL-1β, and IL-6) also elevate the activity of NOX enzymes in hippocampal neurons (169). Thus, the reciprocal interactions between proinflammatory factors and NOX activation in microglia drive the progression of oxidative stress and neuroinflammation in the brain.
Therapeutic Potential of Blocking NOX Enzymes in Neurodegenerative Diseases: Inhibitors of NOX
The failure of antioxidants and NSAIDs applied in clinical trials for treating neurodegenerative diseases has prompted the development of new therapeutic strategies. The focus has been turned to inhibiting the upstream oxidative stress and neuroinflammatory signaling pathways by inhibiting NOX enzymes—which in turn reduces superoxide production and overactivation of glial cells, thereby reducing the release of most proinflammatory factors (Fig. 4). Recent evidence suggests that NOX enzymes, especially NOX2, can be activated by noxious endogenous ligands, so-called danger-associated molecular patterns released by injured neurons, including μ-calpain, α-synuclein, and HMGB1, through an integrin CD11b-dependent pathway (13, 58, 76, 80, 94, 101). Furthermore, the inactivation of NOX2 abolishes the activation of microglia and neurodegeneration induced by these ligands (13, 58, 94, 101), indicating that blocking the activation of NOX enzymes appears to interrupt the vicious self-propelling cycle formed between chronically activated microglia and damaged neurons (Fig. 4). Thus, NOX enzymes have become promising therapeutic targets (107, 119), and NOX inhibitors are the most promising candidates for neurodegenerative diseases associated with oxidative stress and neuroinflammation (4).
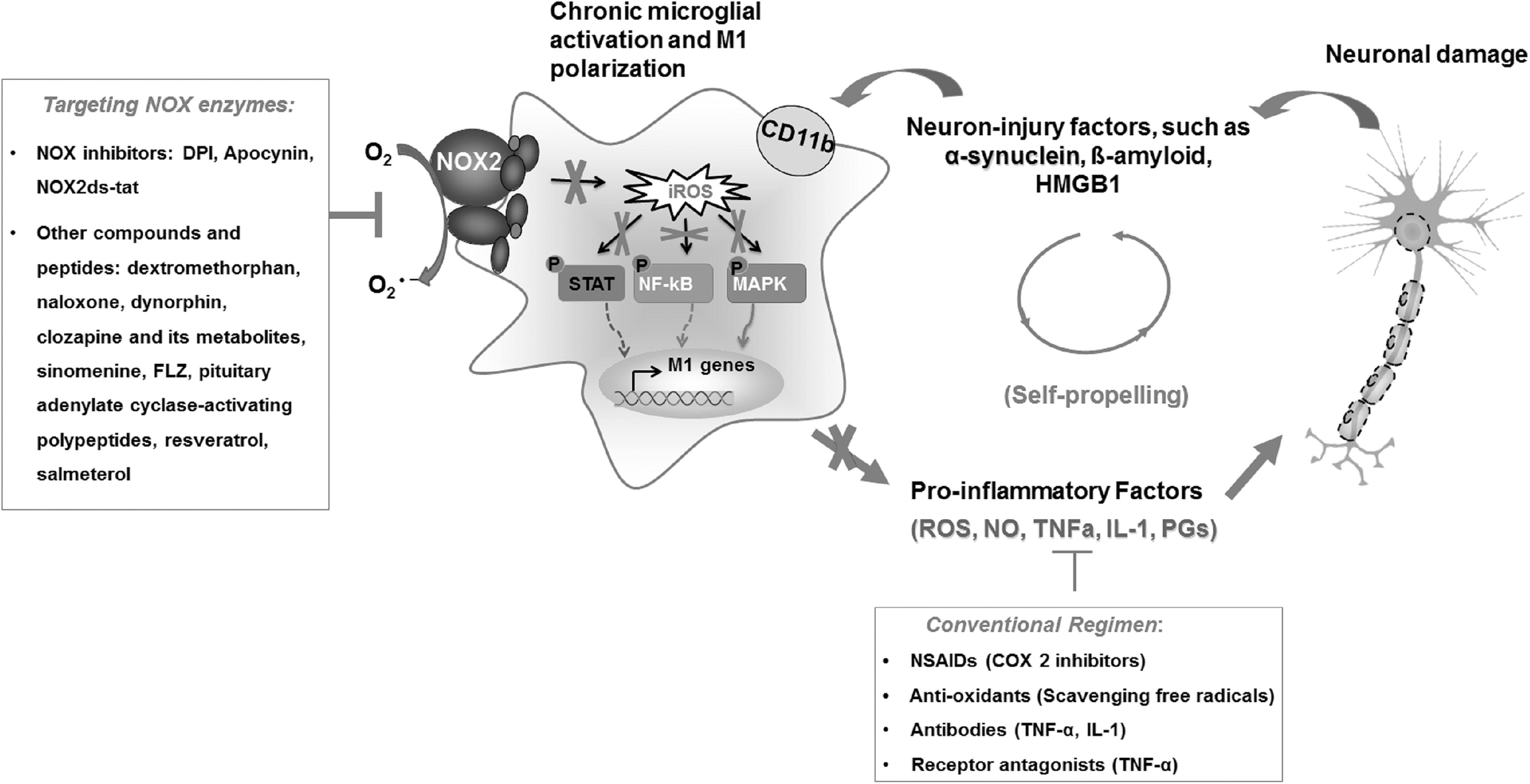
Currently, several compounds and small molecules have been developed as inhibitors of NOX enzymes. Some of these have been the focus of recent comprehensive review articles (4, 164) and are therefore only briefly mentioned here. DPI and apocynin are two of the most widely used inhibitors of NOXs, and display potent inhibitory capacities on activation of NOX enzymes. Other historical NOX inhibitors such as AEBSF or plumbagin have low potency for blocking the activity of NOX enzymes and are used less frequently (44). Due to several side effects of historical NOX inhibitors, novel small-molecule and peptide inhibitors have been developed. GKT136901 and GKT137831 are two structurally related compounds demonstrating preferential inhibition of NOX1, NOX4, and NOX5 with concentrations with 50% inhibition of activity (IC50) values in the three-digit nanomolar range (6, 136). Triazolopyrimidines, including VAS2870 and VAS3947, have also emerged as promising inhibitors of NOX activity (4). ML171 (also known as 2-acetylphenothiazine) has been identified as a potent inhibitor of NOX enzymes, with IC50 values of 130–250 nM for NOX1 and 3–5 μM for NOX2–4 (60). NOXA1ds and NOX2ds-tat are rationally designed peptide inhibitors for NOX1 and NOX2, respectively, preventing the assembly of active NOX complexes (4, 134). Celastrol, one of several bioactive compounds extracted from the medicinal plant Tripterygium wilfordii, has also been shown to block NOX1, NOX2, NOX4, and NOX5 activities in neutrophils or NOX-overexpressing HEK293T and CHO cells, with concentration–response curves exhibiting higher Hill's coefficients and lower IC50 values for NOX1 and NOX2 compared with NOX4 and NOX5 (90).
Neuroprotection of NOX inhibitors in AD
AD is one of the most common neurodegenerative diseases of the CNS, characterized by progressive cognitive dysfunction, memory impairment, and decreased cognitive function (112). A typical pathological feature of AD is the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles in the brain (112). The original evidence linking NOX enzymes to AD comes from the analysis of the postmortem brain tissue of AD patients, in which activated NOX2 and increased membrane translocation of p47phox and p67phox are observed in cerebral cortices (148), suggesting chronic activation of NOX enzymes during the process of neurodegeneration. Consistently, multiple mouse models of AD also display high expressions and activity of NOX isoforms in the brain compared with wild-type controls (141, 180). For example, Russo et al. observed enhanced expression of NOX2, as well as the p47phox and p67phox subunits, in the cerebellum of the TgCRND8 mouse AD model (141). Although the exact mechanisms remain unclear, the activation of NOX enzymes in AD might be related to the accumulation of Aβ since Aβ oligomers induce the membrane translocation of the subunit p47phox and thereby NOX2 activation in microglial cells (85, 180). The elevated expression and activity of neuronal NOX2 and NOX4 have also been observed in Aβ-treated neuronal cell cultures (29, 85). DPI, apocynin, and VAS2870, three inhibitors of NOX enzymes, are capable of blocking Aβ-induced ROS generation, glutathione depletion, and mitochondrial depolarization in both neurons and glial cells (2, 121), further confirming the activation of NOX enzymes in response to Aβ.
Manipulating NOX activity induces potent neuroprotection in both in vitro and in vivo AD models. The suppression of NOX activity by KHG26693 (a new thiazole amine derivative), cilostazol (an antiplatelet agent), and resveratrol (a nonflavonoid polyphenol naturally found in red wine and grapes) significantly reduces Aβ-induced oxidative stress and neurotoxicity in cortical neurons or SHSY5Y cells (25, 123a). In a coculture system containing microglia and human neuroblastoma cells that overexpress wild-type or mutated APP, inhibition of microglial NOX enzyme activity by DPI also exhibits potent neuroprotection (130). Furthermore, pharmacological or genetic inactivation of NOX2 has been shown to reduce the destructive properties of Aβ plaques and delay disease progression in AD mouse models due to decreased ROS production and neuroinflammation (154, 155). In the APP mouse AD model, the membrane-permeable NOX2 inhibitor NOX2ds-tat attenuates neuronal oxidative stress, cerebrovascular dysfunction, and behavioral deficits (125). Chronic administration of apocynin in human amyloid precursor protein (751) (SL) AD mice also attenuates Aβ plaque size (106). The neuroprotective effects elicited by blocking NOX enzymes might be related to inhibition of oxidative stress and microglia-mediated neuroinflammation. Qin et al. found that inhibiting that activity of NOX enzymes with DPI prevents neurotoxicity mediated by activated microglia in neuron–glial mixed cultures (131). The administration of apocynin in an animal model of AD (aged APP-overexpressing Tg2576 mice) markedly reduces oxidative stress, neuroinflammation, and Aβ-related pathological processes (65, 107). Apocynin is known to protect neurons by switching the activation form of microglia from “proinflammatory” M1 to the “anti-inflammatory” M2 in response to intracerebroventricular injection of LPS or Aβ (36). In addition, ibuprofen, a widely used NSAID, is suggested to inhibit NOX isoforms in an enantiomer-specific manner, leading to a dramatic reduction in oxidative damage and Aβ deposition in aged R1.40 mice (176). Recently, Nortley et al. demonstrated that Aβ accumulation can result in decreased cerebral blood flow, a vascular risk factor for AD, in both humans and AD mouse models by inducing pericyte contraction via NOX4-derived ROS and endothelin-1 (123). Interestingly, NOX4 inhibition by GKT137831 abolishes Aβ-evoked ROS production and capillary constriction (123). These studies suggest that blocking NOX isoforms, especially NOX2 and NOX4, could be a promising therapeutic strategy for AD.
Neuroprotective effects of NOX inhibitors in PD
PD, the second most prevalent neurodegenerative disorder, affects 1%–2% of the elderly population (92). PD is clinically associated with some motor symptoms, including resting tremor, rigidity, bradykinesia, and late postural instability (50). Although the pathogenesis of PD remains unclear, NOX-derived oxidative stress is thought to be a crucial factor (17). Several studies have revealed variations in the presence of NOX isoforms, such as NOX1, NOX2, and NOX4, in the substantia nigra of patients with PD and in mouse PD models (33, 178, 189). Many studies using in vitro and in vivo PD models have demonstrated a correlation between the inhibition of NOX2 activity overactivation and dopaminergic neuroprotection (54, 177). In primary mesencephalic cultures, dopaminergic neurodegeneration induced by diverse classical PD-related factors, including MPP+, 6-OHDA, LPS, and formylmethionyl-leucyl-phenyl-alanine, nanometer-size diesel particles, angiotensin II, and the pesticides rotenone and paraquat, is significantly attenuated by DPI, apocynin, or genetic NOX2 deletion (12, 55, 59, 133, 138, 139, 179). In addition to NOX2, NOX1 inhibition displays potent dopaminergic neuroprotection. Blocking NOX1 by GKT137831 prevents 6-OHDA-induced death of N27 cells, a dopaminergic cell line (35).
Inhibitors of NOX enzymes also display potent neuroprotective effects in vivo. Pretreatment with apocynin in the drinking water or by systemic injection significantly reduces dopaminergic neuron loss, α-synuclein aggregation, and/or motor deficits in mouse PD models induced by paraquat and maneb (98), LPS (146, 147), or MPTP (126). DPI is a nonspecific inhibitor of NOX2 with high toxicity (3, 172). Recent studies demonstrated that DPI at an ultralow dose (10 ng/kg/day, subcutaneously infused for 2 weeks) successfully protects dopaminergic neurons in two inflammation-related mouse models after postadministration even when 30% of nigral dopaminergic neurons were already lost (172). The therapeutic potential of DPI has been further verified in a subchronic MPTP mouse PD model, and parallel experiments in NOX2-deficient mice indicated that the target of ultralow dose DPI is NOX2 (172). More importantly, DPI at this ultralow dose does not produce gross toxicities in mice and does not affect the function of peripheral immune cells in both mice and humans (172). These findings suggest that DPI at ultralow doses could be considered a candidate for future clinical trials.
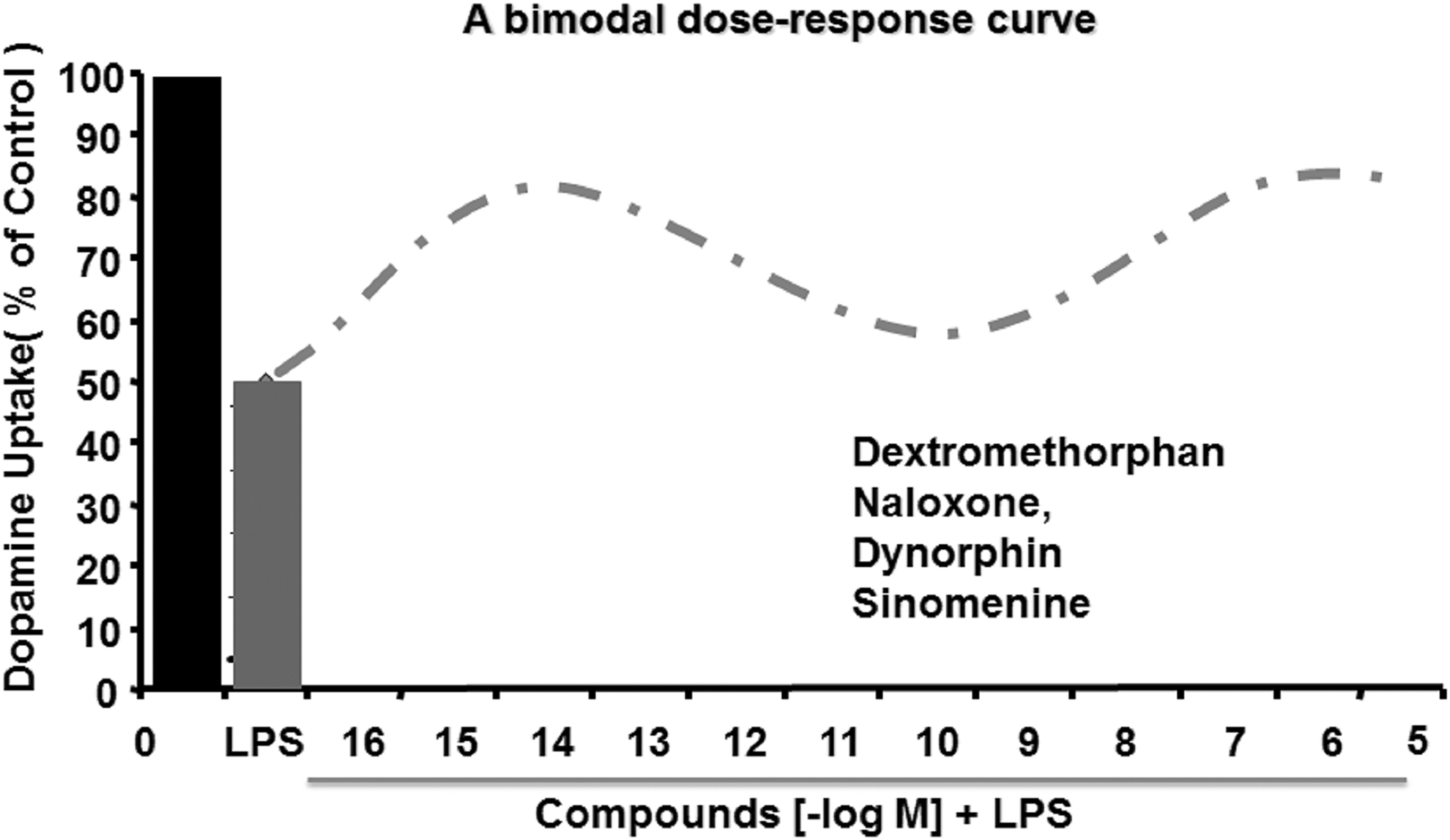
In addition to classic NOX inhibitors, a variety of compounds, such as taurine (27, 77), dextromethorphan (102), naloxone (174), dynorphin (173), clozapine, and its metabolites (84, 91), sinomenine (129), the squamosamide derivative FLZ (8), pituitary adenylate cyclase-activating polypeptides (184), transforming growth factor-β (127), resveratrol (193), and salmeterol (128), have been reported to protect dopaminergic neurons through the inhibition of NOX2 and related superoxide production (Fig. 5). Moreover, the neuroprotective effects of these compounds are not observed in NOX2-deficient primary neuron–glial cultures (57, 196). For instance, taurine, a major intracellular free β-amino acid in mammalian tissues, significantly attenuates paraquat- and maneb-induced ROS production, 4-HNE content and p47phox membrane translocation, as well as dopaminergic neuron loss in vitro and in vivo (27). Dextromethorphan, a commonly used antitussive agent, and its metabolite 3-hydroxymorphinan (3-HM) significantly inhibit NOX2 activation and superoxide production, as well as dopaminergic neurodegeneration in mesencephalic neuron–glial cultures and in C57BL/6J mice treated with LPS or MPTP (102, 195, 196). Interestingly, such neuroprotection is not observed in neuron–glial cultures and in mice deficient in NOX2 (102, 195, 196), indicating that NOX2 is a crucial mediator of the neuroprotective properties of dextromethorphan and 3-HM. While evaluating the dose-related neuroprotective efficacies of the abovementioned compounds, we made a fortuitous discovery that some of these compounds display bimodal dose–response curves for their neuroprotective effects in neuroinflammation-induced dopaminergic neuron loss in vitro. Specifically, dextromethorphan (102), naloxone (174), dynorphin (173), and sinomenine (129) are effective at submicromolar (10−7–10−5 M) and subpicomolar (10−14–10−13 M) concentrations, but the intermediate concentrations (10−11–10−10 M) of these compounds are less effective (Fig. 6). A similar submicromolar-protecting efficiency of DPI has also been observed in primary midbrain neuron–glial cultures (170). Recently, fucoidan, a sulfated fucosylated polymer from brown algae, has been shown to suppress dopaminergic neurodegeneration through the inhibition of NOX1-triggered oxidative stress in the 6-OHDA-lesioned rat PD model (194), indicating that blocking NOX1 can also exert potent neuroprotection.

Neuroprotective effects of NOX inhibitors in ALS
ALS is a chronic progressive motor neuron disorder characterized by the degeneration of motor neurons in the brain and spinal cord (18). The pathological basis of ALS remains unknown, but many studies have shown that the neuropathology of ALS is accompanied by neuroinflammation and oxidative stress. Increased ROS and oxidative stress markers, such as MDA, have been observed in the blood and spinal cord of ALS patients and in the SOD1G93A mouse model (15, 175). The activation of NOX enzymes, especially NOX2, might contribute to oxidative stress in ALS since elevated NOX2 expression and activity were detected in the affected regions of the spinal cord in ALS patients (145) and in mouse ALS models (17, 145, 155). Increased levels of oxidative stress markers in the serum and NOX2 expression in cytoplasmic inclusion bodies containing mutant SOD1 and a number of other proteins are a pathological hallmark of familial ALS. ALS-associated SOD1 mutations, such as L8QSOD1 and G93ASOD1, stimulate NOX2 activation to produce superoxide through coupling with Rac1, a small GTPase that is essential for NOX2 activation (68). Furthermore, studies in SOD1G93A transgenic ALS mice show that the deletion of NOX2 and, to a lesser extent, NOX1 can significantly slow disease progression and improve the survival of ALS mice (14, 68, 136, 177). Marrali et al. also found that lower NOX2 activity significantly increases the survival time of ALS patients (113). Apocynin administration can retard the progression of the disease and prolong survival in the SOD1G93A transgenic ALS mouse model (14, 68, 136, 177). Seredenina et al. treated SOD1G93A mice with the broad-spectrum NOX inhibitors perphenazine and thioridazine, and found that both compounds significantly decrease superoxide levels in the spinal cord of mice (145). Thioridazine has been further shown to induce an immediate and temporary enhancement of motor performance in SOD1G93A mice (145). Therefore, blocking the activation of NOX enzymes appears to be a promising therapy for slowing down the progression of ALS (155).
Neuroprotective effects of NOX inhibitors in multiple sclerosis and other neurodegenerative diseases
Multiple sclerosis (MS), an autoimmune demyelinating disease in the CNS, is characterized by the loss of motor and sensory function (115). The myelin loss is mainly caused by oligodendrocyte dysfunction, and microglia- and astrocyte-mediated inflammatory responses further aggravate the demyelination (115). Although the exact cause of this disease has not been clearly identified, oxidative stress and activation of NOX enzymes are thought to contribute to the damage to the myelin sheaths (167). Genome-wide microarrays and immunohistochemistry analyses reveal upregulated expression of various NOX subunits, including gp91phox, p22phox, p47phox, NOX1, and NOXO1, in initial MS lesions compared with control white matter (48). A closed relationship has been found between the formation of NOX-dependent oxygen radicals in peripheral blood leukocytes and disease severity in patients with MS (117). Furthermore, persistent activation of microglia and NOX enzymes has been shown to drive cognitive and synaptic hippocampal dysfunction during the remission phase of experimental MS in mice (42). Moreover, the administration of apocynin orally once daily in mice with experimental autoimmune encephalomyelitis (EAE), a model of MS, has been found to mitigate the clinical symptoms of EAE mice, and this mitigation is accompanied by suppression of demyelination, reduced infiltration by encephalitogenic immune cells, decreased ROS production, and inhibition of blood–brain barrier (BBB) disruption (32). In a myelin oligodendrocyte glycoprotein peptide-induced EAE mouse model, NOX inhibition by melatonin and n-acetylserotonin, two tryptophan metabolites, mitigates clinical scores, CD4+ T-cell infiltration, elevated p67phox expression, the loss of mature oligodendrocytes, demyelination, and axonal injury in the white matter when administered before or after symptom onset (175a).
HD is a hereditary neurodegenerative disorder caused by a CAG trinucleotide expansion in the huntingtin (HTT) gene and characterized by the presence of progressive chorea, psychiatric symptoms, and cognitive decline (190). In a mouse striatal cell culture model of HD, mutant HD cells are more susceptible to toxicity induced by chlorpyrifos (CPF), a commonly used organophosphate insecticide, compared with wild-type cells (46). CPF-induced cytotoxicity of mutant HD cells is significantly attenuated by apocynin (46). Apocynin has also been reported to display neuroprotective effect in an in vivo model of HD. Using a rat model of HD induced by intrastriatal injection of quinolinic acid (QUIN), Maldonado et al. reported that apocynin administration 30 min before and 1 h after QUIN injection decreased NOX activity and superoxide production, which are associated with mitigated circling behavior and histological damage in the striatum (109).
n-Hexane is a widely used organic solvent, and can induce central-peripheral neuropathy in rodents and humans (67). We recently reported that exposure to 2,5-hexanedione, the active metabolite of n-hexane, results in damage to the dopaminergic system through αMβ2-NOX2 axis-mediated microglial activation (198). Inhibition of NOX enzymes, especially NOX2, by apocynin significantly attenuates 2,5-hexanedione-induced neurotoxicity (198), suggesting that pharmacological inhibition of NOX enzymes may also have beneficial effects against environmental toxin-induced neuropathy.
Potential drawbacks of current NOX inhibitors in clinical translation
Although most of the abovementioned inhibitors of NOX enzymes display potent neuroprotective potency in neurodegenerative disease, they have significant drawbacks that cannot be ignored. The first is nonspecificity. For most inhibitors, no study has shown inhibition across all NOX isoforms in assay systems appropriate to assess direct NOX activity or the relative potency of various NOX isoforms (155). Consistently, none of these inhibitors mentioned above, including DPI, apocynin, AEBSF, GKT136901, GKT137831, VAS2870, VAS3947, and ML171, are isoform specific or display selectivity for NOX2 oxidase over other NOX enzymes (4, 6, 47, 52, 136). Moreover, DPI acts as a general flavoprotein inhibitor, and therefore also inhibits iNOS, NADH dehydrogenase, xanthine oxidase, thioredoxin reductase, cytochrome p450 reductase, NADH-ubiquinone oxidoreductase, and proteins of the mitochondrial electron transport chain (119, 170). In addition, some of these inhibitors may not directly inhibit NOX activation, but may inhibit pathway activity upstream or downstream of NOX enzymes. For instance, apocynin needs to be metabolized to become a NOX inhibitor and is not supposed to act in a given biological process (119). Furthermore, apocynin has been proven to be an indirect inhibitor of NOX enzymes, probably via an ROS scavenging mechanism and myeloperoxidase or Rho kinase pathway-dependent manner (73, 144, 181). GKT136901 potently scavenges peroxynitrite, an RNS created from the reaction of superoxide with nitric oxide (143). AEBSF is also recognized to irreversibly inhibit serine proteases (44). One excellent study performed by Augsburger et al. (7a) recently provided a detailed analysis of several small-molecule inhibitors of NOXs. They found that except for DPI, all tested molecules, including apocynin, diapocynin, ebselen, GKT136901, and VAS2870, displayed interesting pharmacological characteristics, but did not meet the criteria for a bona fide NOX inhibitor. Moreover, VAS2870 is cytotoxic at low concentrations and presents an unspecific redox mode of action.
The second concern is toxicity. Studies have revealed the benefits of using apocynin and DPI as inhibitors of NOX enzymes in alleviating symptoms in animal models (119, 162). However, there are notable exceptions, including an ALS mouse model and the Tg19959 mouse model of AD, where apocynin is inactive (155) or deleterious (155), respectively. The third is the lack of detailed information for in vivo use. Many of these inhibitors of NOX enzymes do not have suitable pharmacokinetic properties for in vivo studies. For example, celastrol displays low water solubility, reduced oral bioavailability, a narrow window of dosage, and side effects that limit its clinical application (90). The newly developed peptide inhibitors, including NOXA1ds and NOX2ds-tat, might also be excluded as clinical drugs since they are unlikely to be orally active or display a suitable pharmacokinetic profile in vivo (47, 162). Moreover, other potential side effects of inhibiting NOX enzymes must be considered. Completely blocking NOX enzyme activity, especially NOX2, may severely compromise patients' immunological function, resulting in CGD-like symptoms. For this aspect, ultralow dose acting compounds, such as DPI and dextromethorphan, might offer good options because they attenuate only the induced activity, without affecting the basal activity of NOX2 in microglia. Moreover, NOX2 activity in peripheral immune cells is not affected by ultralow dose DPI (102, 172, 195, 196). In addition, DPI at ultralow doses fails to inhibit other cytochrome enzymes and displays high specificity toward NOX2 (170).
Conclusions
Oxidative stress and neuroinflammation are two common features shared by neurodegenerative diseases. Dysregulated activation of NOX enzymes is identified as a major regulator of both oxidative stress and inflammatory responses in these diseases. Administration of NOX enzyme inhibitors attenuates neurodegeneration in various models of neurodegenerative diseases. This novel class of therapeutic candidates seems to be more efficacious than most of the conventional antioxidants and NSAIDs (107, 164, 166), providing a novel direction for combating neurodegenerative diseases. However, in view of the limitations of NOX inhibitors, novel isoform-selective NOX inhibitors with improved efficacy, specificity, and BBB permeability and favorable toxicity and bioavailability should be developed for clinical therapeutics. GSK2795039 is the first identified NOX2 inhibitor through competition for the NADPH binding site of NOX2 with orally available, nontoxicity and CNS-permeability properties (75, 155). Although GSK2795039 displays potent neuroprotective effects in vitro, in vivo studies are still lacking (155, 185). Further research on GSK2795039 should be conducted. Furthermore, ultralow dose acting compounds, such as DPI and dextromethorphan, are worth pursuing in the future because of their improved safety profile and high specificity. We look forward to an exciting future with increasingly more studies focusing on NOXs and neurodegenerative diseases, as well as novel NOX inhibitors, with the specificity, safety, and suitability for clinical applications to treat neurodegenerative diseases.
Footnotes
Funding Information
This work was supported by National Natural Sciences Foundation of China (81973087, 81703264), National Natural Sciences Foundation of LiaoNing Province (2019-MS-077), LiaoNing Revitalization Talents Program (XLYC1907026; XLYC1808031), LiaoNing BaiQianWan Talents Program (No. [2017]90), Program for LiaoNing Innovative Talents in University (LR2016008), “QiZhen” talent project of Dalian medical university (No. 201122).