
Abstract
Significance:
Despite their serious side effects, anthracyclines (ANTs) are the most prescribed chemotherapeutic drugs because of their strong efficacy in both solid and hematological tumors. A major limitation to ANTs clinical application is the severe cardiotoxicity observed both acutely and chronically. The mechanism underlying cardiac dysfunction under chemotherapy is mainly dependent on the generation of oxidative stress and systemic inflammation, both of which lead to progressive cardiomyopathy and heart failure.
Recent Advances:
Over the years, the iatrogenic ANTs-induced cardiotoxicity was believed to be simply given by iron metabolism and reactive oxygen species production; however, several experimental data indicate that ANTs may use alternative damaging mechanisms, such as topoisomerase 2β inhibition, inflammation, pyroptosis, immunometabolism, and autophagy.
Critical Issues:
In this review, we aimed at discussing ANTs-induced cardiac injury from different points of view, updating and focusing on oxidative stress and inflammation, since these pathways are not exclusive or independent from each other but they together importantly contribute to the complexity of ANTs-induced multifactorial cardiotoxicity.
Future Directions:
A deeper understanding of the mechanistic signaling leading to ANTs side effects could reveal crucial targeting molecules, thus representing strategic knowledge to promote better therapeutic efficacy and lower cardiotoxicity during clinical application.
Introduction
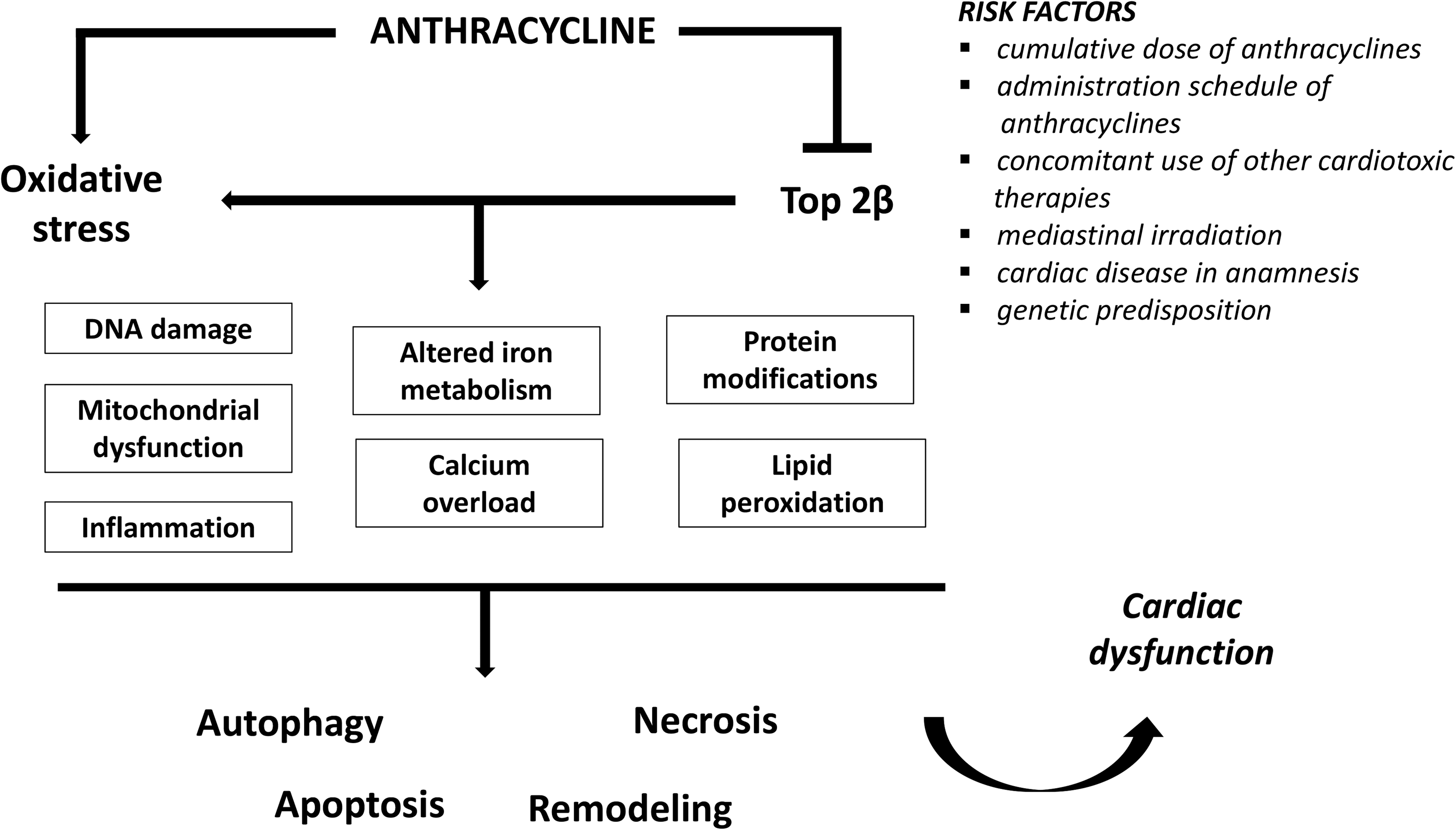
Introduced in the 1960s, anthracyclines (ANTs), primarily doxorubicin (DOX, also known as adriamycin), are among the most potent and prescribed chemotherapeutics for the treatment of hematological and solid tumors. Accordingly, ANTs remain a major component of the new-generation combination cancer therapy (112, 113, 152, 170). After the isolation of the ANT daunorubicin from Streptomyces peucetius (138) and of the ANT DOX from S. peucetius var. caesius (3), other ANTs have been developed and used in clinics. They include epirubicin, idarubicin (developed as a daunorubicin analog), and valrubicin (a semisynthetic analog of DOX). Unfortunately, the clinical use of ANTs is compromised by insidious cardiomyopathy and heart failure (HF). These are typically dose dependent and cumulative, and they often require modification, or even discontinuation, of potentially successful anticancer regimens, thus increasing morbidity and mortality (113, 152, 170). In DOX-based treatments, the incidence of congestive heart failure (CHF) corresponds to 5% in patients receiving a cumulative dose of 400 mg/m2, and it exponentially increases up to 48% with a cumulative dose of 700 mg/m2 (137). Subclinical events may occur in ∼30% of patients, even at doses of 180–240 mg/m2, up to 13 years after treatment (147). Also, lower DOX doses (i.e., 100 mg/m2) can be associated with cardiac dysfunction (15).
In addition to the total cumulative ANTs dose, several major risk factors for the development of ANTs-dependent cardiotoxicity should be considered. These include the administration schedule, the concomitant use of other cardiotoxic therapies such as trastuzumab, the humanized monoclonal antibody against HER2 (human epidermal growth factor receptor 2), or the anticancer paclitaxel (31), mediastinal irradiation, and cardiac diseases in anamnesis (8, 179). Cardiotoxic reactions to ANTs may be acute (i.e., during drug administration), subacute, or chronic (15). Acute cardiotoxicity mainly shows electrocardiographic changes and/or cardiac arrhythmias (38 and references therein). Despite the myocardial changes occurring during acute exposition to ANTs being resolved spontaneously, and not precluding further ANTs use, the clinically significant manifestations are related to both early- and late-onset events of chronic cardiotoxicity (15), characterized by structural changes of the human heart (77). The early-onset form occurs during the ANTs-based regime or within the first year after its completion (49), whereas the late-onset form can take place even several years (decades) after the chemotherapeutic treatment (15, 120).
In this context, it is important to consider that patients show a significant variability in the susceptibility to ANTs. Although many patients tolerate therapeutic doses of ANTs without long-term complications, others show ANTs-dependent cardiotoxicity as early as after the first dose (12). In addition, some patients develop cardiotoxicity at a total cumulative dose of ANTs corresponding to 300 mg/m2 or even lower, whereas others have no significant cardiac alterations, despite the exposition to doses up to 1000 mg/m2 (154). Recently, a study estimated an overall incidence, corresponding to 9% of cardiotoxicity, defined as left ventricular ejection fraction below 50%, within the first year after completing ANTs-based chemotherapy. Moreover, another study showed, in childhood cancer survivors treated with both ANTs and radiotherapy, a high risk of cardiac events (the most common was CHF) at an early age, and a 12.5% risk of developing HF within 30 years after treatment (Table 1).
Anthracyclines and Therapeutic Use
The purpose of the present comprehensive overview is to provide an up-to-date insight into ANTs oxidative and inflammatory processes underlying the mechanistic bases of their related cardiotoxicity.
Impact of Oxidative Stress in ANTs-Dependent Cardiotoxicity
Physiologically, it is well known that, under aerobic condition, cells continuously react with reactive oxygen species (ROS), such as superoxide radical (O2 •−), hydrogen peroxide (H2O2), lipid and hydroxyl radical (OH•), and singlet oxygen (1O2), which derive from several metabolic reactions, and counteract their effects through a wide range of endogenous antioxidant enzymes (92, 139). Reactive nitrogen species (RNS) include radicals such as nitric oxide (NO•) and nitric dioxide (NO2 •), as well as nonradicals such as nitrous acid (HNO2) and dinitrogen tetroxide (N2O4) (149). Physiological levels of ROS and RNS play fundamental roles in the regulation of several cellular functions, whereas high and persistent ROS and RNS levels may directly or indirectly generate cardiac injury by inducing a dramatic imbalance between reactive species and antioxidant defense systems (91, 92, 103a, 139).
The endogenous antioxidant enzymes are necessary to preserve cellular integrity against oxidative stress-dependent damages and to repair structural oxidative deleterious modifications. In mammalian cells, glutathione (GSH)/glutathione peroxidase (GPX) and thioredoxin/thioredoxin reductase (TXNRD) systems, crucial enzymes belonging to the selenoproteins family, as well as Cu/Zn superoxide dismutase (SOD) and catalase (CAT), represent the principal endogenous antioxidant defense line involved in multiple redox-regulated signaling pathways (69, 89, 107, 109, 110, 144). Under pathological conditions, ROS/RNS can induce macromolecular damage due to their ability to react with specific amino acid residues present in the protein structure, inducing functional inactivation. They may also interact with deoxyribonucleic acid (DNA) and chromatin, thus causing mutations or double-stranded breaks. Other deleterious events induced by ROS/RNS include structural and functional mitochondrial alterations, disruption of the cellular calcium homeostasis, altered protein synthesis, and cell death (21 and references therein, 93, 110). This complex phenomenon is described as “oxidative and nitrosative damage,” and due to its implication and consequences in cellular and extracellular metabolic processes, it is overall known as “metabolic stress” (21, 44 and references therein, 110).
Today, it is widely accepted that metabolic stress is associated with diverse pathophysiological events, including cardiovascular diseases. The cardiac tissue is particularly vulnerable to ROS/RNS damage since the antioxidant resources, in terms of oxyradical-scavenging enzymes, such as SOD and CAT, or GPX, are lower than other tissues (e.g., liver) (84, 85, 149). In the heart, ROS/RNS overproduction is able to induce membrane lipid peroxidation with membrane and DNA damage, leading to activation of the cardiomyocyte death program, and to tissue replacement by connective, thus resulting in irreversible cardiac damage (149).
Because of the terminal differentiation and limited regenerative capacity of cardiomyocytes, they are highly vulnerable to long-term damage induced by ANTs. Cardiomyocytes' susceptibility to ANTs (in particular DOX) is under continuous intense investigation. It is considered a complex and multifactorial process, in which many factors contribute to induce irreversible cell damage (15, 97).
Several mechanisms have been proposed for explaining the reason of such an elevated cardiac susceptibility to ANTs toxic events. A first point is the relatively low myocardial array of endogenous antioxidants that characterize the heart. In addition, it should be considered that ANTs are more prone to be retained within cardiomyocytes (47), and they are able to downregulate the myocardial activity of key antioxidant enzymes (i.e., GPX), leading to a higher exposition of the heart to dangerous radicals (129). Accordingly, several evidences indicate that transgenic mice with cardiac overexpression of endogenous antioxidants such as CAT (51), GPX1 (162), mitochondrial SOD (167), and TXNRD1 (125) resulted in more protection, with respect to the control counterpart, to DOX-induced cardiac functional changes.
Other hypotheses regard the presence of large amounts of mitochondria in cardiomyocytes, and the unique contribution exerted by the NADH dehydrogenase-dependent pathway in cardiac mitochondria, generating semiquinones of ANTs and thus playing a crucial role in selective cardiotoxicity (129). An aspect that cannot be underestimated is the capacity of ANTs to selectively bind the phospholipid cardiolipin, localized in the inner mitochondrial membrane, that leads to mitochondrial accumulation of the drug (33). Through this interaction, ANTs can disrupt the electron transport chain by inhibiting complexes I and II, leading to additional ROS production. Via cardiolipin peroxidation, this can induce the release of mitochondrial factors, such as cytochrome c, that aggravates ANTs-induced injury (162).
Other mechanisms contributing to the ANT-induced cardiotoxicity are related to (i) the ANTs metabolism into a more hydrophilic and cardiotoxic compound that accumulates in cardiomyocytes, (ii) to ion dysregulation, and (iii) to the impaired expression of fundamental cardiac proteins, calcium homeostasis alteration and calcium overloading, mitochondrial structure and function alterations, activation of matrix metalloproteinase, interference with several pro-survival kinases, and alteration in beta-adrenergic receptor signaling (31, 58, 84, 88, 94, 102, 112, 128 –130, 142, 149, 175).
A peculiar aspect related to ANTs and their induced oxidative stress may be the occurrence of myocardial hibernation, observed in patients treated with chemotherapeutics (41). Several studies performed on animal models have demonstrated that myocardial hibernation occurs in the absence of detectable cardiomyocytes apoptosis (64, 106), also highlighting the emerging role of endothelial cell death (70) due to oxidative-mediated depletion of key angiogenic factors, including vascular endothelial growth factor that plays an adaptive role in accordance with the release of paracrine myocardial peptides (18).
Generation of toxic radicals
Despite the ongoing intense research, the complex mechanism responsible for ANTs side effects is not completely understood (15, 174). In particular, from a redox perspective, it is well known that ANTs generate ROS by redox cycling, iron complexation, chaotropic effects in mitochondria, and the consequent uncoupling of the electron transport chain (50, 59, 97, 155).
Thus, over the past 30 years, one of the commonly accepted explanations of ANTs-dependent cardiotoxicity is oxidative stress, probably the most widely studied mechanism for DOX-mediated iatrogenic cardiotoxicity. Oxidative stress can be first generated by the chemical structure of DOX because of its intrinsic propensity to generate ROS during the drug metabolism (84). The quinone group of the tetracyclic quinone-hydroquinone moiety of DOX, as well as of other ANTs, undergoes a one-electron reduction, which forms a semiquinone radical (81). This quickly regenerates the parent quinone by reducing molecular oxygen to superoxide anion (O2 −). The dismutation of the latter generates H2O2, which can elevate other potentially harmful ROS and RNS (81). This induces a continuous redox cycling of the quinone moiety, with consequent oxidative stress generation and deleterious effects for cardiomyocytes.
Despite quinone species being among the most important redox cyclers with a pivotal role in many biological mechanisms, futile quinone redox cycling can lead to a deleterious ROS cascade that generates toxic effects (132). In particular, the futile redox cycling, with NADH dehydrogenase considered as the most likely site of ANTs reduction, represents one of the most important mechanisms responsible for the compromised therapeutic utility of ANTs due to dose-dependent cardiotoxicity (52). In fact, within the mitochondria, ANTs semiquinone radicals strongly react with molecular oxygen, presumably to produce O2 −, to complete the redox cycle (Fig. 1). Moreover, a preferential accumulation of selective adducts in cardiac mitochondrial DNA (mtDNA) after DOX exposition have been reported in experimental animal models.
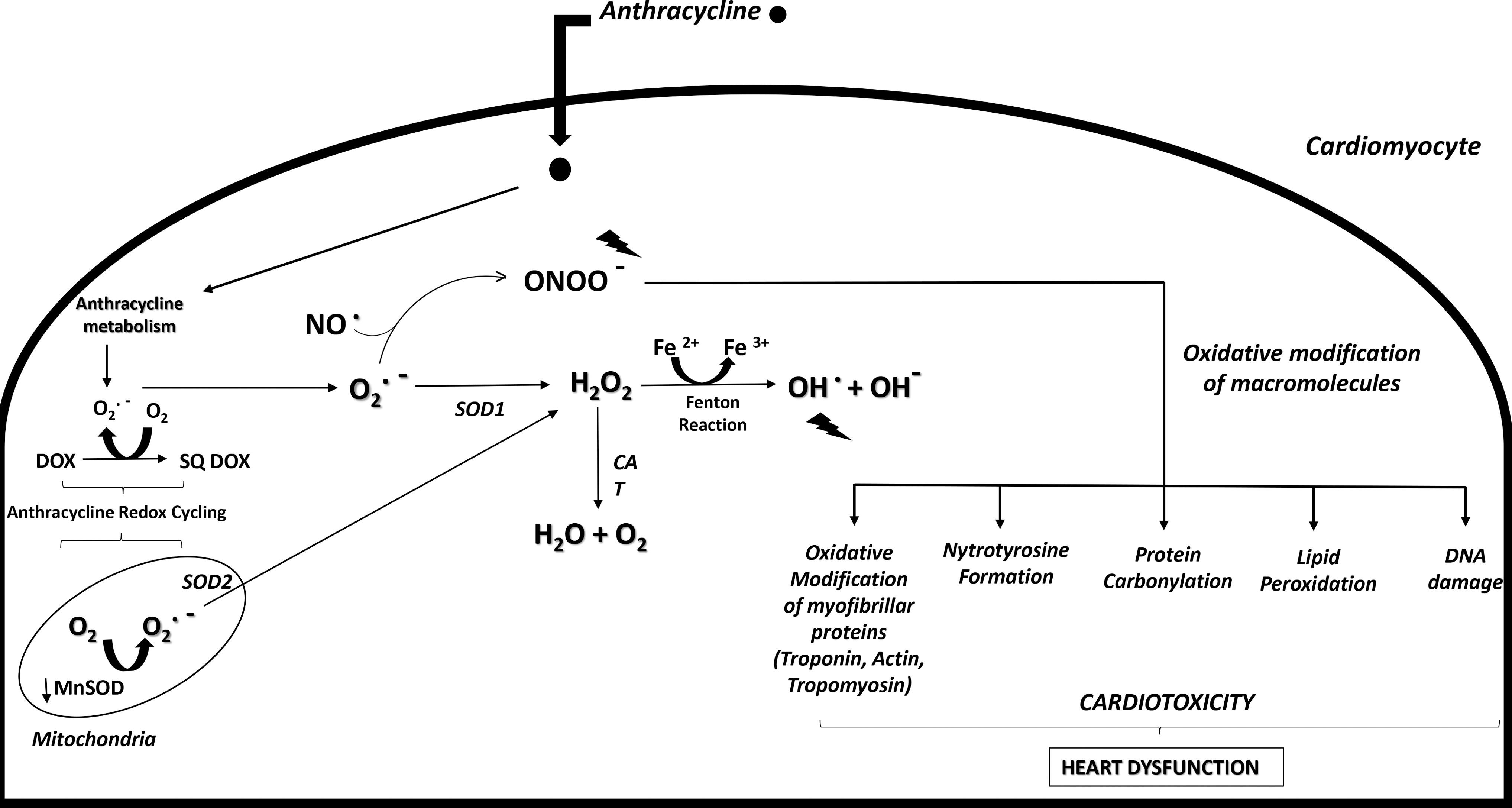
The circular and covalently closed nature of mtDNA facilitates the DOX intercalation with consequently mtDNA alterations, superoxide generation, and respiratory chain dysfunction (58, 129). Further, during ANTs metabolism, two-electron reduction of the carbonyl group in the side-chain of the drug generates a secondary alcohol metabolite. Because of its more polar nature, compared with its parent ANTs, these alcohol metabolites show reduced cardiac clearance and the propensity to form a long-lived toxic ANTs reservoir (81). In particular, doxorubicinol, formed after a two-electron reduction of the side-chain carbonyl group of DOX, is known to interact with thiol groups of proteins, exacerbating cell damage and disturbing Ca2+ release from sarcoplasmic reticulum vesicles (90, 97). Thus, the accumulation of the alcohol metabolite in the heart contributes to the time-dependent component of DOX cardiotoxicity, through a mechanism that could involve perturbations of Ca2+ homeostasis, with consequences on the contractile function (97).
In this scenario, an excessive ROS and RNS generation occurring during ANTs metabolism overcomes the endogenous capacity of antioxidant-producing enzymes, such as peroxidase, and SOD, including the mitochondrial enzymes, NOSs, NAD(P)H oxidases, and CAT (38, 149 and references therein). This consequently leads to damages on DNA, RNA, proteins, and membrane lipids, with typical redox modifications of macromolecules, including nitrotyrosine formation, protein carbonylation, and lipid peroxidation (33).
Oxidative stress also targets ion channels, as in the case of K+ channels. This affects membrane currents of the ion, causing action potential propagation abnormality and cardiac arrhythmias. It also inhibits calcium sensor proteins in the excitation
Iron contribution to the oxidative stress
As stated earlier, ANTs may also promote ROS generation, chelating free iron and forming iron–ANTs complexes. In turn, iron undergoes redox cycling and triggers oxygen radicals, including the high-toxic hydroxyl radical (OH•) by Fenton and Haber-Weiss reactions, with consequent alteration of iron homeostasis (33, 152). The iron complex could cause lipid peroxidation through its interactions with the negatively charged membranes (97). Compared with other radicals (such as O2 •− or H2O2), OH•, generated by ANTs–iron interaction, shows a higher reactivity with any oxidizable compounds, thus inducing lipid peroxidation, nucleic acids mutations, and protein modifications (Fig. 1) (129). Notably, some evidences indicate that the toxicity of the combination of iron and some ANTs (in particular DOX) seems not to be a simple additive effect. For example, ANTs–Fe complexes can induce oxidative stress by involving two different mechanisms, depending on the presence or absence of a reducing system, that can be represented by NADH cytochrome P450 reductase or the thiols of cysteine or GSH (129). In either case, the end-effect converges on OH• production.
Octavia et al. reviewed the complexity of iron-dependent ANTs cardiotoxicity, proposing that DOX effects on iron metabolism are not mediated by DOX–iron interactions, but rather via proteins that sequester and bind intracellular iron (97). This corroborates the presence of a very complex regulatory mechanism that overcomes the simple concept of “iron-ANT complex” formation in the pathogenesis of DOX-induced cardiotoxicity.
Relevance of topoisomerase 2 in oxidative stress
Mechanistic insights of ANTs cardiotoxicity are now focusing on topoisomerase 2β (Top2β). This is one of the two types of Top2, and a target of these anticancer drugs (42, 152). By forming complexes with Top2α, ANTs induce DNA breaks and prevent DNA and RNA synthesis (140). The consequent ternary Top2-DOX-DNA cleavage complex induces cell death (71). Top2α is predominantly found in proliferating cells in which it reaches a peak in the G2/M phase of the cell cycle. It is overexpressed in tumors but absent in quiescent cells. Thus, it is considered a marker of cell proliferation (153, 172). In contrast, Top2β is present in all quiescent cells, including cardiomyocytes (38). It is generally assumed that one of the major mechanisms of ANTs cardiotoxicity is via inhibition of Top2β, the only Top2 present in the heart. This can largely contribute to the development of ANTs-dependent cardiotoxicity. Its inhibition is responsible for double-stranded breaks in DNA and for cardiomyocyte death (153, 172). This ANTs-mediated mechanism is required for the p53 activation and its dependent apoptotic pathway (Fig. 2) (118, 127, 172).

Lyu and colleagues showed that mouse embryonic fibroblasts lacking Top2β are protected from DOX-induced cytotoxicity, whereas Zhang et al. (172) demonstrated that the cardiomyocyte-specific deletion of Top2β (Top2β−/−) is able to protect mice from the DOX-dependent progressive cardiomyopathy. Moreover, authors reported that, in the presence of Top2β, DOX decreases the transcription of key genes involved in mitochondrial regulation, inducing perturbation in mitochondrial biogenesis and function, and ROS formation (172). In particular, authors found that, compared with the wild type mice (Top2β+/+), Top2β−/− mice show, after DOX exposure, a remarkable reduction of ROS levels in the heart. This DOX-dependent effect was due to a significant reduction in transcripts of crucial antioxidant enzymes (i.e., SOD, peroxiredoxin, and TXNRD), in Top2β+/+ mice, with respect to the mice with cardiac depletion of Top2β, thus suggesting that DOX-induced ROS generation is a Top2β-dependent process (Fig. 2) (42, 152, 172).
Mechanistically, the mitochondrial dysfunction observed after DOX administration in Top2β+/+ mice was secondary to the suppression of peroxisome proliferator–activated receptor-γ coactivator 1-α (PGC-1α) and peroxisome proliferator–activated receptor-γ coactivator 1-β (PGC-1β) transcription (172). These genes are fundamentally involved in the regulation of several mitochondrial functions (61), and the double-knockout in mice is associated with severe HF derived by structural and functional dramatic mitochondrial alterations, which result in immediate postnatal death (151, 153). Since PGC-1α is one of the key regulators of SOD (101, 131) in the presence of Top2β, DOX can increase ROS generation through SOD downregulation (Fig. 2).
The clinical implication of the observations just cited could refer to the evaluation of Top2β expression in cardiomyocytes, whose low levels are associated with a decreased susceptibility to cardiotoxic events, ANTs-dependent cardiotoxicity, and the monitoring of Top2β in peripheral blood, for evaluating the individual susceptibility to ANTs-induced cardiotoxicity (151). Moreover, drugs that specifically target Top2α, instead of Top2β, could be associated to a reduced cardiotoxicity (172), based on the assumption that Top2β is not required for tumoricidal activity of ANTs and that their anticancer activity should be preserved, sparing the heart at the same time (153).
This corroborates the assumption that the classical “ROS hypothesis,” by which ANTs induce cardiotoxicity through redox cycling and ROS generation, should not be considered the single and/or the principal event in ANTs-dependent cardiotoxicity. The role of oxidative stress, as a result of the ROS/RNS overproduction in ANTs-related cardiotoxicity, is absolutely undisputed (141). Indeed, despite several studies (78, 79) indicating that antioxidant drugs can prevent and/or mitigate ANTs-dependent cardiac dysfunction (149, 171), none of these molecules was clinically approved as cardioprotective adjuvants during ANTs regimens. This presumably depends on the necessity to fine-tune the redox balance since ROS also play important physiological roles (114). Notably, ROS are considered promoters of innate defenses (23a, 145) and this may explain why in many cases chronic treatments with antioxidants failed in preventing cardiotoxicity. For this reason, research is searching for alternative pathways.
Several evidences demonstrated that neither antioxidants nor some iron chelators can provide therapeutic benefits in preclinical models and clinical trials, failing in preventing cardiac toxicity caused by ANTs (152, 172). Likewise, since most cardiomyocytes are terminally differentiated, the molecular mechanism of ANTs-induced cardiotoxicity might not be related only to its anticancer effects due to Top2β inhibition.
Among iron chelators tested so far, only dexrazoxane appears promising in preventing or reducing the cardiac injury induced by ANTs (65). This molecule is the only cardioprotective agent against ANTs cardiotoxicity approved by the Food and Drug Administration due its proven efficacy in cancer patients receiving ANTs chemotherapy (160). It was initially believed that dexrazoxane mediates cardioprotection via its metal-chelating activity. In particular, it was traditionally postulated that it undergoes hydrolysis for displacing iron from ANTs–iron complexes, or chelating free or loosely bound cellular iron, thus preventing the formation of toxic reactive oxygen radicals within cardiomyocytes (129). However, other iron chelators, such as deferasirox (40), do not show cardioprotective effects, despite their efficient iron chelating capability and rapid intracellular distribution (129 and references therein), suggesting that dexrazoxane exerts its effects by additional protective mechanisms.
It is likely that iron chelating-dependent ROS formation does not represent the predominant mechanism of action of dexrazoxane. Being a catalytic inhibitor of Top2 (71), the cardioprotective effects of dexrazoxane could be due to its interaction with Top2β (136). It has been observed that dexrazoxane antagonizes DOX-induced DNA damage through its ability to induce changes in the Top2β configuration, preventing its interface with ANTs, thereby avoiding the formation of Top2-DNA complexes (60, 71). This has been confirmed by experimental findings indicating that dexrazoxane derivatives lacking activity on Top2β showed no cardioprotection in models of ANTs-induced cardiotoxicity (46, 75).
Inflammation in ANTs-Induced Cardiotoxicity
As previously mentioned, except for dexrazoxane, both ROS scavenging and iron chelators failed to limit chemotherapy-induced cardiotoxicity (129, 147), and this is contrary to the general assumption that the iatrogenic ANTs-induced cardiotoxicity was mainly linked to altered iron metabolism and ROS production. It was found that two DOX analogues, 5-iminodaunorubicin and zorubicin (ZRN), are relatively noncardiotoxic, even if ZRN forms ROS as efficiently as DOX (5, 6). This indicates that DOX may use other mechanisms for inducing cardiotoxicity. Moreover, several anti-inflammatory agents showed important cardioprotective actions, suggesting that inflammation has a significant role in chemotherapy-dependent cardiomyopathy (14). On these bases, it is possible that a strong connection exists between oxidative stress, inflammation, and ANTs-induced cardiac dysfunction. Oxidative stress itself is able to activate a systemic inflammation by triggering cytokine expression, leukocyte chemotaxis, and complement activation that, in turn, induce cell damage (20). Even if ROS contribute to DOX systemic inflammation by increasing the levels of crucial inflammatory receptors, such as Toll-like receptors (TLRs) (22), the possibility exists that chemotherapy can directly induce systemic toxicity (157). It is generally accepted that DOX treatment is responsible for a severe systemic inflammation that involves many organs, with cardiac toxicity as a major adverse effect. It is also known that DOX administration evokes an increase of proinflammatory cytokines levels, whose inhibition was found to mitigate the resulting tissue damage (176).
These observations prompted researches aimed at finding novel pathways involved in ROS-independent mechanisms underlying ANTs-dependent side effects, and at designing new agents that are able to counteract cardiac injury. The modulation of the mechanisms controlling DNA damage responses, gene expression, cardiomyocytes pro-survival cascades, and energetic stress is emerging as a crucial actor in cardiotoxicity. In particular, the role of systemic and cardiac inflammation is under growing attention (30). As reviewed by Salazar-Mendiguchía et al. (115), the possibility exists that oxidative stress and inflammation are not exclusive or independent of each other, since both of them are shown to contribute to chemotherapy-induced multifactorial cardiotoxicity.
Toll-like receptors
An increasing body of experimental data point to innate immunity and to activation of inflammatory pathways as key factors linked to cardiovascular disease and, in particular, to HF (100, 169). The failing heart shows increased levels of inflammatory molecules and an altered expression of innate immune genes, suggesting that inflammation, together with the innate immune system, may represent an early response of cardiomyocytes to injury (74, 169). Within the innate immune responses TLRs play a crucial role in drug-related cardiomyopathy (96). Activation of their intracellular signaling pathways leads to nuclear localization of pro-inflammatory transcription factors with the production of cytokines (96). This is believed to play a central role also in DOX-dependent cardiotoxicity (84).
TRLs belong to the germline-encoded pattern recognition receptors that are able to activate the cardiac innate immune response, triggering homeostatic and reparative responses (169). They are differentially expressed in endothelial cells, smooth muscle cells, and cardiomyocytes (87). TLRs are made up of an extracellular domain, a transmembrane, and an intracellular domain that is characteristic of type I transmembrane glycoproteins (98). Depending on their subcellular localization, TRLs can be classified in two groups: one including those expressed on the plasma membrane (TRL-1, TLR-2, TLR-4, TLR-5, TLR-6, and TLR-11) and the other including those located in endosomes (TLR-3, TLR-7, TLR-8, and TLR-9) (19).
These receptors recognize both exogenous pathogen-associated molecular patterns and endogenous stress signals, namely damage-associated molecular patterns (DAMPs), that are able to influence a progressive myocardial remodeling and a cardiac functional decline that leads to dilated cardiomyopathy (DCM) and HF (11, 146). Downstream pathways of TLRs can be divided into myeloid differentiation factor 88 (Myd88)-dependent pathway and Myd88-independent pathway. All TRLs use Myd88 cascades, except for TLR-3 that activates the TIR domain-containing adaptor protein inducing interferon (IFN)-β-mediated transcription factor (TRIF) (169). TLR-4 activates both Myd88-dependent and -independent signaling (169). Lastly, these pathways stimulate the production of pro-inflammatory cytokines and IFNs via the induction of nuclear factor-κB (NF-κB) and IFN regulatory factors (Fig. 3) (169).
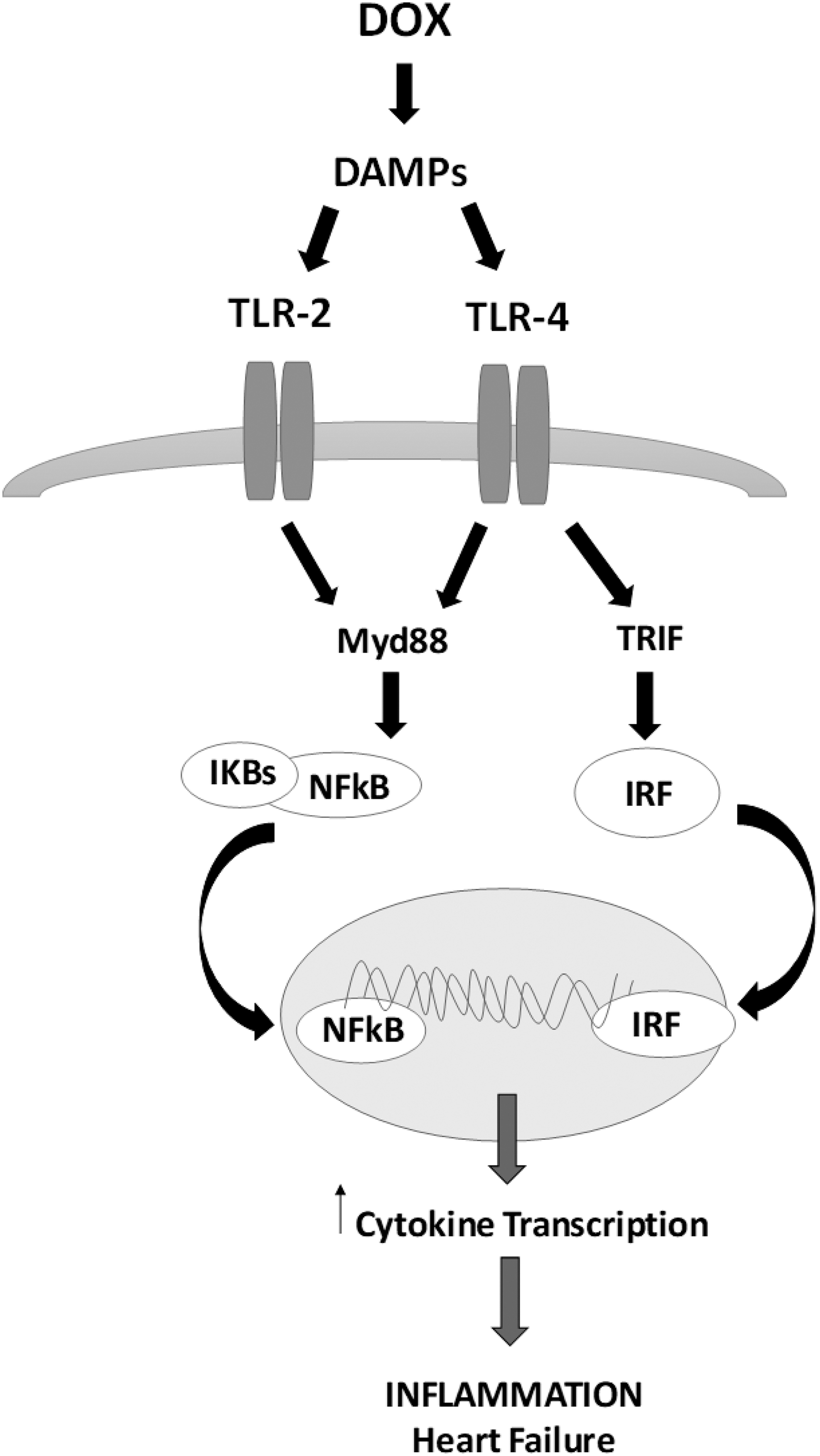
A large body of experimental data describe that activation of myocardial TLRs by exogenous ligands (27, 96), such as oxidative stress (26), plays a crucial role in cardiac dysfunction and in ventricular remodeling after ischemia (126). Since TLRs are engaged during different forms of cardiac damage, also related to chemotherapy (74, 143), it was proposed that activation of innate immune mediated by myocardial TLRs is important in HF. Based on these observations, it is not surprising that TLRs, highly expressed in cardiac cells (96), have been recently linked to DOX-induced HF and cardiotoxicity.
Toll-like receptor-2
TLR-2 expression and activation are upregulated not only in experimental models and humans affected by HF (9, 71) but also in DOX-induced HF and cardiotoxicity. This suggests that these receptors play a major role in these diseases (169). TLR-2 is involved in endothelial cell inflammation (168). Its activation may induce ROS production and cardiac injury (72). The contribution of TLR-2 to cardiomyocytes' oxidative stress, and the subsequent cardiac alterations have been described (26). It was found that TLR-2 ablation is able to rescue the cardiac damage induced by DOX (96), whereas its stimulation generates 1O2 and activates the NF-κB, with resulting inflammatory effects (26).
A pioneer study performed by Nozaki et al. (96) revealed that, in DOX-treated animals, TLR-2 ablation preserves the cardiac function. It improves the survival rate of KO-mice by reducing proinflammatory cytokines production, caspase-3 activation, and TUNEL-positive nuclei in the myocardium (72). This suggested, for the first time, the importance of TLR-2 in chemotherapy-induced heart dysfunctions. Subsequently, Ma et al. showed that TLR-2 inhibition induces a strong protective effect, by attenuating cardiomyopathy with amelioration of cardiac performance and fibrosis, and a decreased inflammation (72).
Many studies demonstrated that TLR-2 stimulation induces chronic inflammation, and tissue injury, by a mixed immune response (T-helper 1, T-helper 2, T-helper 17, regulatory T cell). TLR-2 inhibition reduces cardiac fibrosis markers, such as transforming growth factor-β1 (83), and the inflammation and fibrosis promoter, interleukin (IL)-17A (68). These data are of remarkable interest since cardiac fibrosis is a major detrimental factor in cardiac dysfunction and HF also under DOX treatment.
During DOX administration, among the DAMPs able to activate TLR-2, an interesting endogenous ligand is represented by the high mobility group box 1 protein (HMGB1). It is a cytokine secreted by activated macrophages and monocytes in response to infection, injury, and inflammation (72). Ma et al. (72) suggested a positive feedback loop involving TLR-2 and HMGB1. They showed that TRL-2 inhibition suppresses HMGB1 expression, thus suggesting that HMGB1 may be involved in DOX-induced cardiomyopathy through TLR-2 activation (72).
Toll-like receptor-4
Also, TLR-4 plays a crucial role in cardiovascular pathologies. It is the most expressed cardiac TLR in myocardial inflammation, ischemia/reperfusion (I/R) injury, HF, and hypertension (67, 74, 166). TLR-4 upregulation is believed to drive the systemic inflammation activated by DOX (157). In DCM and HF it is upregulated, similar to TLR-2. However, if, under DOX exposure, TLR-4 is inhibited, the cardiac inflammation and the related functional alterations are amplified (72). This has been suggested to be linked to a decreased autophagy (72).
It is known that TLR-4 activates TH1 immune response and this produces an acute inflammation. At the same time, a T-helper 2/regulatory T cell-dependent inflammation, strictly linked to both cardiac fibrosis and dysfunction, is induced when TLR-4 is inhibited (72). These data describe TLR-4 as a beneficial receptor in the scenario of DOX cardiotoxicity, suggesting that inhibition of TLR-2 without affecting TLR-4 activity may represent a possible therapeutic strategy.
Despite these observations, other studies contradict the protective role proposed for TRL-4. By using a TLR-4 KO animal model, it was found that loss of this receptor is associated with a mitigation of DOX-dependent cardiotoxicity (108). These opposite results may depend on various factors, including the different timing of TLR-4 inhibition. In fact, by using KO mice, Riad et al. preventively inhibited TLR-4 (108), whereas Ma et al. performed a therapeutic antagonism after DOX treatment (72). The possibility exists that a preventive blockage of TLR-4 attenuates DOX inflammation and the consequent cardiac injury, whereas TLR-4 antagonism after DOX administration may interfere with its ability to promote the resolution of cardiac inflammation and fibrosis (165). Notably, in the same study, Riad et al. (108) found that the absence of TLR-4 reduces the oxidative stress produced by DOX, thus suggesting that this receptor is activated by ROS and this contributes to oxidative stress, as also demonstrated in I/R oxidative injury (124).
TLR-4 is also critically involved in cardiac infiltration by activated lymphocytes, monocytes, and macrophages. In fact, TLR-4 KO mice show a mitigation of this DOX-dependent phenomenon (108). The absence of TLR-4 also prevents DOX-related downregulation of GATA-binding protein 4, an important transcription factor that is essential for the function of the adult heart (108).
TLR-4 is importantly involved in innate immune responses against bacterial infection (157). Wang et al. (157) were the first to postulate that, under DOXO treatment, TLR-4-mediated systemic inflammation could be triggered by the entry of endotoxins in the circulation. It was proposed that a major function of TLR-4 is to recognize bacterial components (103). Endotoxins belonging to Gram-negative bacteria are an important TRL-4 ligand (103). Gram-negative bacteria resident in the intestine are the primary source of body endotoxins (133). They are physiologically confined by the healthy barrier of the intestinal mucosa and this prevents their translocation into the circulation (157).
However, it was observed in patients that DOX is able to destroy this barrier (82). Epithelial cells are particularly vulnerable to chemotherapy, since they are constantly under self-renewal. DOX-treated patients suffer from gastrointestinal problems that lead to a leakage of endotoxins into the circulation. This exacerbates intestinal inflammation and triggers systemic inflammation (157 and references therein). It was found that DOX produces effects that are typical of endotoxin-induced inflammation, such as ROS production, tumor necrosis factor (TNF) activation, and expression of proinflammatory cytokines (32, 39, 53). DOX-dependent damage is aggravated by intestinal resident bacteria. In fact, these bacteria can produce additional pro-inflammatory agents, such as nucleic acids or polysaccharides, and this contributes to the detrimental effects of DOX via TLR-2/TLR-9–mediated pathways. Accordingly, when removing gut microbiota, DOX inflammation can be rescued (157).
Toll-like receptor-3
Little is known about the role of TLR-3 in DOX-induced cardiotoxicity. Many studies describe a protective activity of this receptor in cardiac pathologies, especially in myocardial infarction (156). With respect to other TLRs, stimulated TLR-3 is able to increase circulating IFN-γ-induced protein 10, which is known to be protective (34). TLR-3 activation recruits a TRIF pathway and this contributes toward reducing cell death, apoptosis, and macrophage infiltration; toward enhancing autophagy; and toward inhibiting the expression of inflammation factors (29). It was observed in a DOX-treated model that mRNA, protein, and serum expression of TLR-3 are downregulated, contrarily to TLR-2 (63). This was associated to cardiac dysfunction. Of note, the imbalanced TLR-2–TLR-3 equilibrium observed in DOX-induced HF suggests a predictive role in cardiotoxicity (63).
Immunometabolism modulation
Additional data supporting the role of the immune system and inflammation in ANTs-induced cardiotoxicity come from a recent field of research interested in the multifaceted interactions existing between the metabolic and the immune systems. This is called “immunometabolism,” a term proposed in 2011 (76) starting from the assumption that both the innate and the adaptive immune cells are able to control and modulate metabolism (73 and references therein).
It was found that acute and chronic treatments with DOX produce a defective immunometabolism. This, by modulating the activity of crucial immune-sensitive enzymes, in particular cyclooxygenase (COX) and lipoxygenases (LOXs), results in cardiac damage (45). It is well known that COX-2 elicits cardioprotection via prostacyclins production (97), and that LOXs finely regulate cardiac immune metabolism by producing resolvins, immune responsive bioactive mediators involved in the resolution of the cardiac infarction-derived inflammation (36). Both COX and LOXs use essential fatty acid, which is enriched in the spleen, to biosynthesize autocoids (45). This is counteracted by DOX via their irreversible dysregulation (45). The consequent impairment of immunometabolism altered cardiac homeostasis in terms of inflammation
It is known that P450 is involved in arachidonic acid (AA) metabolism, thus influencing cardiovascular physiology and pathophysiology (24). The metabolites produced by the action of this enzyme on AA, such as EETs and HETEs, are able to mediate cardiac physiological events, including anti-inflammation, cell proliferation, vasodilation, and cardioprotection (4, 99, 135). Very recently, a role of cytochrome P450 (CYP2J2), strongly expressed in human cardiomyocytes, was suggested in relation with DOX-induced cardiotoxicity (5). Surprisingly, the DOX metabolite, 7-deoxydoxorubicin aglycone, is supposed to bind to CYP2J2 and this results in inhibition of AA metabolism and an alteration of the ratio of its protective derivatives, EETs and HETEs (5). As a consequence, it is presumed that DOX treatment significantly modulates cardiac expression of P450 and EH enzymes, thus causing an imbalance in protective and toxic signaling in the heart (1).
Cell Death Modulation in ANTs-Induced Cardiotoxicity
Apoptosis
Apoptosis is a finely regulated homeostatic mechanism of cell death that is not associated to inflammation (55). ANTs, particularly DOX, are able to evoke an immunogenic form of apoptosis (2). Several evidences describe TLR-4 as a strategic link between oxidative stress, inflammation, and apoptosis in DOX cardiotoxicity (84). It is known that, both in vivo and in vitro, DOX is able to evoke an upregulation of apoptotic markers, such as Bax (108, 134). This effect is abolished in TLR-4-deficient animals, suggesting a pivotal role of this receptor. TLR-4 absence is associated with a reduction of cardiac TUNEL-positive apoptotic cells and to an increase of the anti-apoptotic protein Bcl-2 (56, 108). In the experimental model used by Krysko et al. (55), an intraperitoneal injection of DOX induces a sterile inflammation typical of apoptosis that recruits neutrophils producing pro-inflammatory mediators, such as IL-6. The hypothesis is that the apoptotic cells produced by DOX, mainly monocytes/macrophages, may represent a DAMPs source that is able to stimulate TLRs, resulting in inflammation. A pivotal role is mainly played by TLR-2, which initiates the DOX-related inflammation in response to the apoptotic cell (55).
Pyroptosis and NLR family pyrin domain containing 3
To date, even if DOX-induced cardiotoxicity is considered to be related to cardiomyocyte apoptosis, other forms of cell death have been recently considered (165). This is the case of pyroptosis, a programmed cell death, different from apoptosis since it is characterized by proinflammation (13, 66). The phenomenon is regulated mostly by caspase-1, which produces active IL-1β, IL-18, and Gasdermin D (13). They, in turn, are activated by pyroptotic inflammasome sensors, such as the NLR family pyrin domain containing 3 (NLRP3), involved in different cardiovascular pathologies (100). Several evidences highlight the role of the pro-inflammatory IL-1β in ANTs-associated side effects. This cytokine increases in plasma under DOX treatment (176), and this points to a role of NLRP3. It was found that both DOX and Daunorubicin strongly activated NLRP3 (119). This activates caspase-1, with the subsequent production of IL-1β (119).
DOX-induced release of IL-1β in the circulation also stimulates the production of additional inflammatory cytokines and chemokines, such as IL-6, TNFα, IL-6, GCSF, and CXCL10/IP-10 (119). Recently, Meng et al. showed that DOX is able to increase cardiomyocytes pyroptosis through an increase of NLRP3/caspase-1 activity, resulting in cardiac injury and dysfunction (80). They propose that DOX modulates transcription factors, such as the promoter TINCR, to increase the expression of NLPR3 and cell death (80). Since NLRP3 inhibitors were able to block DOX-induced cardiac damage (80), it is conceivable that pyroptosis contributes to this side effect of the drug. Very recently, the cardioregulatory protein chromogranin A has been found to mitigate DOX-induced pyroptosis, in rat and mouse models, by reducing both NLRP3 expression and myocardial and peripheral inflammatory markers (111).
Autophagy
Autophagy represents a catabolic process that eliminates cellular components (122). Several metabolic stresses, such as ROS, hypoxia, and energy deficiency, are able to activate autophagy-related genes and lysosomal proteolytic mechanisms that, in turn, regulate autophagy (95). This process takes place by five distinct steps, namely initiation, nucleation, expansion, maturation, and degradation (25). Each of them depends on corresponding regulators (161). A major regulatory signal of autophagy is represented by adenosine monophosphate-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) (43).
It has been postulated that DOX can induce cardiac damage by affecting autophagy (158), although conflicting results have been observed. A dual role of autophagy in DOX-dependent cardiotoxicity has been postulated (161). Some studies propose that DOX upregulates autophagy and this is an adverse actor in cardiotoxicity (54). In contrast, other studies strengthen autophagy activity as a pro-survival for cardiomyocytes (54). In this context, Kobayashi et al. (54) found that DOX induces cardiomyocyte death by increasing, via inhibition of Beclin-2, the expression of the Light Chain 3, a regulator of autophagosome biogenesis. In addition, it was observed that DOX upregulates Beclin-1 by increasing autophagy and cardiomyocytes apoptosis (164).
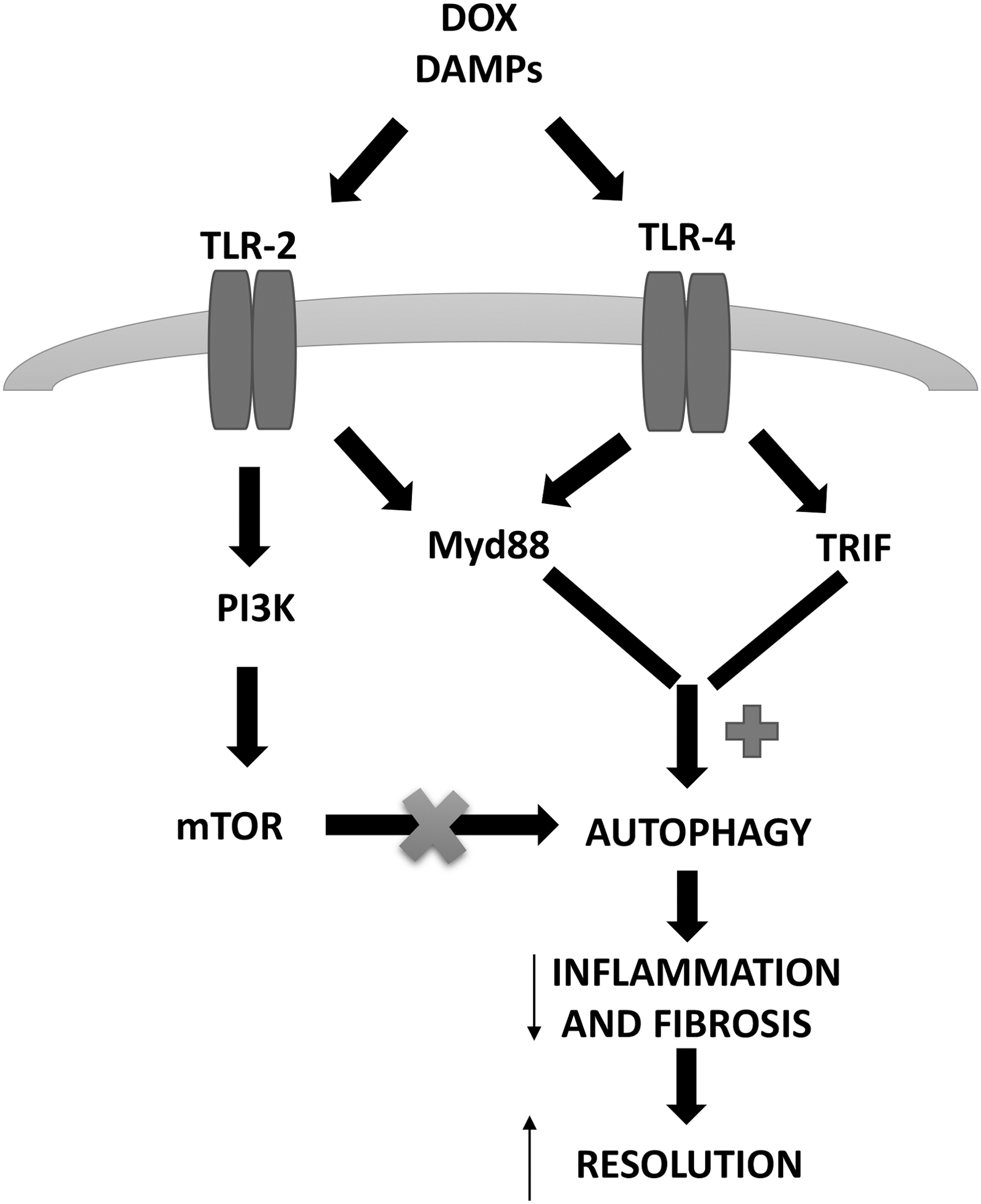
On the other hand, increased autophagy was proposed to mitigate DOX-induced cardiotoxicity by activating the AMPK/mTOR/ULK1 signaling (62) and by reducing the extent of the p53/mTOR/p62 pathway (48). Based on the available data, a definitive conclusion is prevented since different animal models, experimental conditions, and experimental reagents and doses have been used in the available studies (161). Interestingly, several researches suggest that the activation of autophagy before DOX administration can protect from cardiac toxicity. A role is suggested for TLR-4-activated autophagy. TLR-4 inactivation increases cardiac damage and dysfunction induced by DOX, because of an increased inflammation and a decreased autophagy (72). TLR-4-related autophagy is mediated by TRIF- and MyD88-p38 kinase-dependent pathways (164) and is strictly required for chronic inflammation and fibrosis resolution in damaged tissues (165). When TLR-4 is inhibited, the two downstream pathways are blocked, and this leads to a complete suppression of autophagy in cardiomyocytes (72). On the other hand, a biphasic regulation of autophagy is proposed for TLR-2. Its activation may induce autophagy through the activation of P38 kinase, but it may inhibit the process through the PI3K-Akt-mTOR pathway (117). Anyway, autophagy represents an alternative mechanism for cell survival under stress stimuli that induce cardiac remodeling, such as DOX-derived detrimental effects (23, 35), and targeting distinctly inflammatory mediators, such as TLR-2 and/or TLR-4 may represent a possible way to go for therapeutic or preventive strategies (Fig. 4) (105).
Conclusions
Even if ANTs are related to severe cardiac dysfunction and toxicity, they remain among the most potent and prescribed chemotherapeutics that are used in both hematological and solid tumors. Unfortunately, together with their therapeutic efficacy, these drugs induce oxidative stress and inflammatory responses that are responsible for the detrimental process observed in the heart that limits their clinical application.
It is out of doubt that a deeper understanding of ANTs-related molecular and genetic disorders could represent key knowledge to promote better therapeutic efficacy and lower cardiotoxicity (Fig. 5).
Overall, it is important to consider that the study of the side effects of chemotherapy and of their mechanistic basis is studded with several limitations that complicate the translational potentiality of this field. Notably, it is not straightforward to exactly reproduce a tumor animal model considering the wide range of cancer-related modifications depending on metabolic or immunologic adaptations and on animal species and strain. In addition, it should not be underestimated that in tumor-bearing animals ANTs are mostly administered intraperitoneally, quite different from the intravenous application used in clinical practice, with a subsequent “first-pass” hepatic metabolism that modifies the drug availability. A great limit also comes by the conflicting results obtained when using different experimental models and experimental reagents/doses.
Footnotes
Funding Information
This work was partially supported by “Fondazione Umberto Veronesi” Post-Doctoral Fellowship (2019) to Teresa Pasqua and by Doctorate School in “Life Science and Technology,” University of Calabria.