
Abstract
Significance:
The cardiac side effects of hematological treatments are a major issue of the growing population of cancer survivors, often affecting patient survival even more than the tumor for which the treatment was initially prescribed. Among the most cardiotoxic drugs are anthracyclines (ANTs), highly potent antitumor agents, which still represent a mainstay in the treatment of hematological and solid tumors. Unfortunately, diagnosis, prevention, and treatment of cardiotoxicity are still unmet clinical needs, which call for a better understanding of the molecular mechanism behind the pathology.
Recent Advances:
This review article will discuss recent findings on the pathomechanisms underlying the cardiotoxicity of ANTs, spanning from DNA and mitochondrial damage to calcium homeostasis, autophagy, and apoptosis. Special emphasis will be given to the role of reactive oxygen species and their interplay with major signaling pathways.
Critical Issues:
Although new promising therapeutic targets and new drugs have started to be identified, their efficacy has been mainly proven in preclinical studies and requires clinical validation.
Future Directions:
Future studies are awaited to confirm the relevance of recently uncovered targets, as well as to identify new druggable pathways, in more clinically relevant models, including, for example, human induced pluripotent stem cell-derived cardiomyocytes.
Introduction
In the past 20 years, major advances in oncological treatments have significantly reduced cancer death rates but concomitantly highlighted cardiac disease as the leading cause of morbidity and premature mortality among cancer survivors. This is a critical issue, especially for patients who are often diagnosed at young age, such as lymphoma and leukemia patients, and are commonly treated with highly cardiotoxic drugs such as anthracyclines (ANTs). ANTs are potent anticancer agents that not only kill transformed cells but also significantly damage cardiomyocytes that, unable to regenerate, carry life-long alterations culminating in heart failure. ANT cardiotoxicity commonly occurs well after cancer remission and is generally irreversible and refractory to standard heart failure pharmacotherapy, leading to a poor prognosis (40, 140).
Early diagnosis of cardiotoxicity before overt cardiac deterioration occurs, that is, when the disease is still in a reversible and treatable phase, currently represents an unmet clinical need. Similarly, treatment options for patients diagnosed with cardiotoxicity are limited since small trials have shown only modest efficacy of standard heart failure treatments in patients with ANT cardiotoxicity. Antioxidants revealed to be equally ineffective, thus challenging the classical view in which reactive oxygen species (ROS) are the major culprits of the cardiac side effects of ANTs (127). In the past 10 years, the field of Cardio-Oncology has grown exponentially and has led to the awareness that the scenario of the molecular mechanisms behind ANT cardiotoxicity is more complex than previously anticipated and not just centered around the role of ROS (122).
The scope of this review article is to provide a comprehensive description of the major signaling pathways that are affected by ANTs within cardiomyocytes and whose deregulation has been causally linked to ANT cardiotoxicity. The role of ROS as signaling intermediates of ANT cardiotoxicity will be emphasized.
DNA Damage at the Interplay of ROS, Apoptosis, and Mitochondrial Dysfunction
The antineoplastic action of ANTs is linked to their ability to induce DNA damage, through both direct and indirect mechanisms, predominantly in proliferating cells in S and G2 phase. On one side, ANTs intercalate into DNA and form bulky adducts as well as cross-links that interfere with DNA replication and transcription. On the other side, ANTs can damage DNA by increasing the production of ROS, which in turn lead to oxidized nucleotides, base mismatches, point mutations, and DNA single-strand breaks. Last, ANTs interfere directly with DNA helicase activity and the ensuing DNA strand separation, as well as with type 2 topoisomerases (Top2) and DNA unwinding. The relevance of Top2 poisoning by ANTs recently emerged as a leading cause of cardiotoxicity, so that Top2 and the intertwined signaling pathways have been identified as promising therapeutic targets. However, the role of ROS has been reconsidered. Although initially considered as the triggering event of cardiotoxicity (the so-called ROS-driven hypothesis), oxidative stress is now recognized as a secondary event, associated with nuclear and mitochondrial DNA (mtDNA) damage (126).
The ROS-driven hypothesis
The susceptibility of cardiomyocytes to ROS-induced damage is related to their limited antioxidant resources. As a consequence of their prevalent oxidative metabolism, the antioxidant systems of cardiomyocytes are almost completely saturated at steady state (36) and any further source of ROS, such as ANTs, can exhaust their antioxidant capacity more rapidly than in any other cell type. ANTs are responsible for ROS generation since they are reductively activated to a semiquinone radical. This, in turn, undergoes redox cycling, thereby producing superoxide (O2−) and hydrogen peroxide (H2O2). A further mechanism of ANT-mediated ROS production involves iron. In the presence of iron, ANTs can form Fe3+-ANT complexes, which catalyze the Fenton reaction, where H2O2 is converted to various ROS, including the cytotoxic hydroxyl radicals (OH−) (22, 121) (Fig. 1). The ensuing massive ROS generation ultimately leads to cardiac cell death (17, 115).

Although ROS have long been recognized as pivotal mediators of ANT cardiotoxicity, the initial ROS-driven hypothesis has been challenged by a series of studies showing modest effects of ROS scavengers and iron chelators in preventing ANT cardiac side effects in chronic settings. Importantly, N-acetyl cysteine or combinations of antioxidants have been tested, with variable outcomes, but did not protect patients from ANT cardiotoxicity (114). Intriguingly, none has matched the effectiveness of dexrazoxane (ICRF-187 or DXR), a bisdioxopiperazine agent structurally related to ethylenediaminetetraacetic acid but with more potent chelating/antioxidant activities (118) (Table 1). This finding led to the hypothesis that the mechanism of action of DXR might be other than iron binding.
Relevant and Emerging Therapeutic Approaches to Counteract Anthracycline-Induced Cardiotoxicity
AMPK, adenosine monophosphate-activated protein kinase; AMPK-CA, AMPK constitutive activation; CPC, cardiac progenitor cell; CYP, cytochrome P450; EDTA, ethylenediaminetetraacetic acid; EGFR, epidermal growth factor receptor; LTCC, L-type Ca2+ channel; SIRT1, sirtuin 1; TLR, Toll-like receptor.
Yet, the contribution of redox cycling and primary ROS production to ANT cardiotoxicity is debated, and the most recent view is that the enhanced ROS generation is a secondary phenomenon resulting from mitochondrial dysfunction (48) or endoplasmic reticulum stress caused by accumulation of misfolded proteins (83).
Topoisomerase 2 poisoning and inhibition
In 2012, Zhang et al. showed that the production of ROS could also be secondary to the interaction of ANTs with the beta isoform of topoisomerase 2 (Top2) (143). DNA topoisomerases 2 are responsible for the catalysis of the transient breaking and adjoining of DNA double helix during DNA replication and transcription (18). As an intermediate of this process, a DNA-Top2 covalent complex, the so-called cleavage complex, is formed. ANTs act like Top2 poisons in stabilizing the complex, thereby preventing the religation of DNA and leading to the formation of DNA double-strand breaks, which are potent inducers of the DNA damage response (Fig. 2).
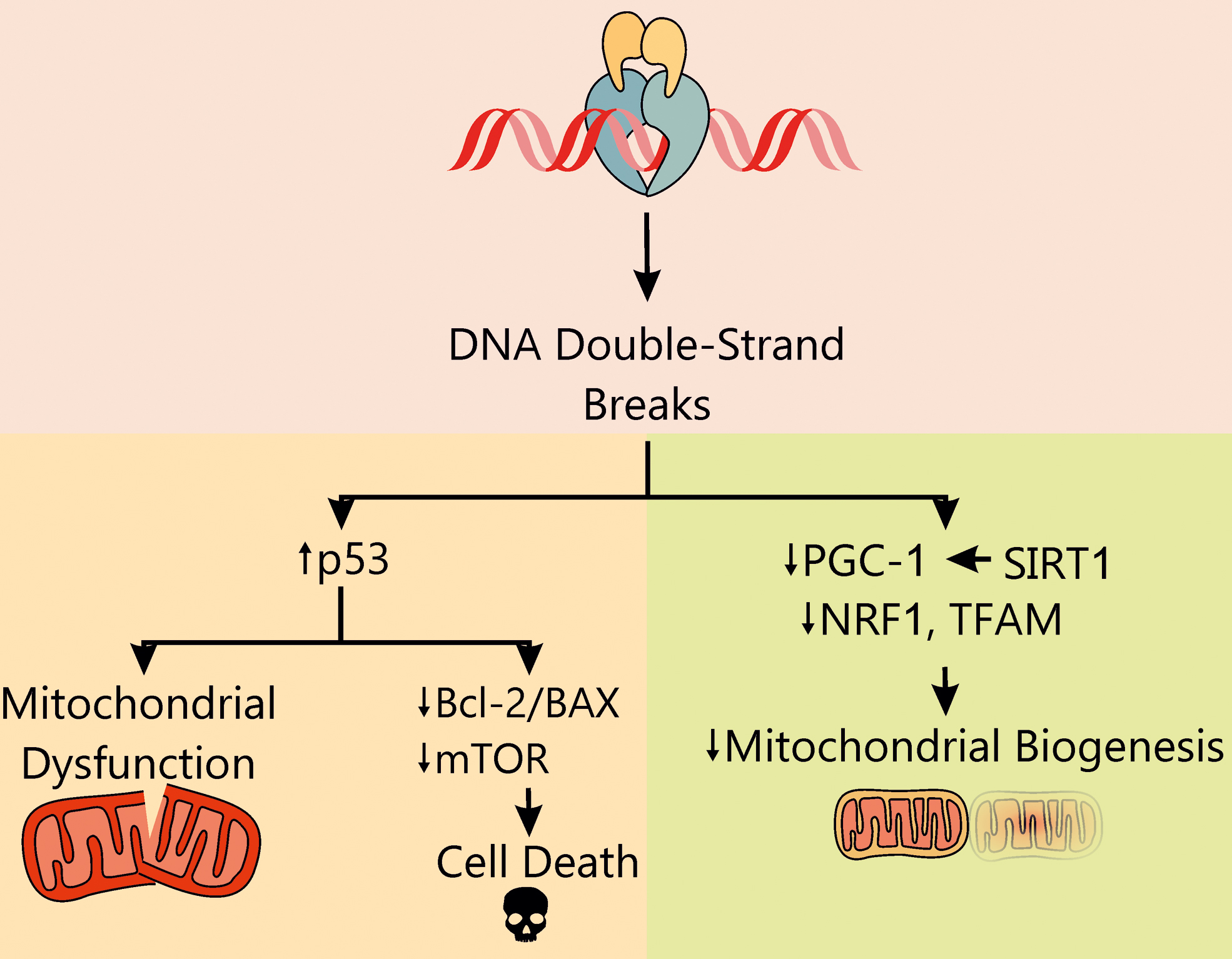
The DNA damage response to genotoxic stress is mainly regulated by three members of the phosphatidylinositol 3-kinase related kinase family: DNA-dependent protein kinase, ataxia-telangiectasia-mutated (ATM), and ATM and Rad3-related (ATR) (62, 66). The initial processing of double-strand DNA breaks, before repair, is mediated by the heterotrimeric Mre11-Rad50-Nbs1 (MRN) complex. Upon recognition of double-strand breaks by the MRN complex, ATM kinase is activated, leading to the phosphorylation of key regulators of cell cycle progression and DNA repair (78). However, if double-strand breaks are not properly repaired, DNA damage activates a set of apoptotic signaling routes, including the ATM/ATR-p53, as well as necroptosis (78, 102).
DNA damage-induced cell death is the key mechanism behind the anticancer action of ANTs and, at the same time, is responsible for their cardiotoxicity. Notably, the effects of ANTs in proliferating (tumor) and nonproliferating (cardiomyocytes) cells are mediated by two distinct isoforms of Top2 (Fig. 3). While the alpha isoform of Top2 (Top2-α) is overexpressed in proliferating cells in S and G2 phase, and it is almost undetectable in quiescent tissues, Top2-β is the unique isoform expressed by adult mammalian cardiomyocytes (12) and mature induced pluripotent stem cell (iPSC)-derived cardiomyocytes (4, 15). The interaction of doxorubicin (Doxo) with Top2-α generates a ternary Top2-Doxo-DNA cleavage complex that triggers tumor cell death. Similarly, the Top2-β-Doxo-DNA complex induces DNA double-strand breaks and activates a DNA damage response that is responsible for cardiomyocyte death. In cardiomyocytes, the activation of p53 stimulates DNA repair processes, but concomitantly represses genes involved in mitochondrial biogenesis (such as peroxisome proliferator-activated receptor-γ [PPARγ] coactivator [PGC]-1) and oxidative phosphorylation, impairs the ratio between beta-cell lymphoma 2 (Bcl2) and Bcl2-associated X (BAX), culminating in cardiac cell death (86, 143). Consistently, p53-knockout mice and adult mouse hearts expressing cardiac myocyte-restricted dominant-interfering p53 are partially protected against Doxo-induced cell death and myocardial dysfunction (147). The observed protection in these models likely stems from both inhibition of apoptosis and preserved mammalian target of rapamycin (mTOR) signaling, which may contribute to prevent cardiac mass reduction (147).
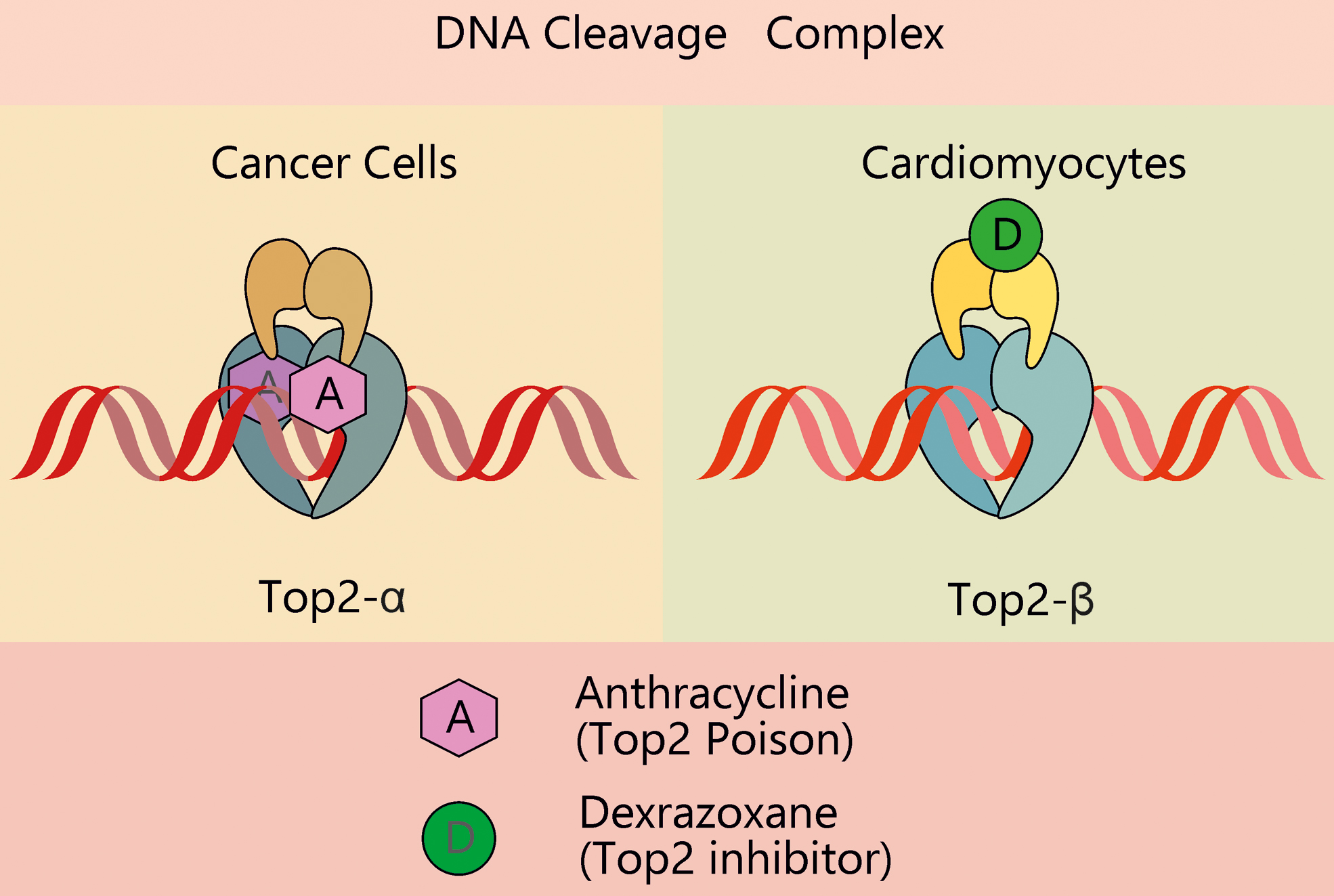
In further agreement with the role of Top2-β in ANT cardiotoxicity, murine embryonic fibroblasts (72) and cardiomyocytes (12) lacking Top2-β are protected from Doxo-induced damage. Of note, Top2-β appears to be a key player in the cardiotoxicity elicited by ANTs not only when they are used in monotherapy but also in the setting of combinatorial regimens, such as with monoclonal antibodies. For instance, both Doxo and trastuzumab alone induce Top2-β protein downregulation in cardiomyocytes, which is exacerbated in combined treatments. This further reduction in Top2-β activity leads to enhanced ROS production and apoptosis and might be one of the reasons of the aggravated cardiotoxicity of combination therapies compared with single agents (53). Of note, Atwal et al. showed that, in vitro, 10 μM Doxo (or higher) can interfere with the action of other drugs that act as Top2 poisons, such as etoposide, thus hindering the overall anticancer efficacy of combined treatments (4).
Given the selective expression of the beta isoform in cardiomyocytes, the development of Top2-α-specific drugs may provide a valuable means to reduce cardiotoxicity, while maintaining the anticancer efficacy of the drug. This is a key aspect to be considered in the development of cardioprotective agents against ANT toxicity. On these grounds, the increased risk of secondary malignancies in patients treated with DXR has limited its application as cardioprotective agent in the past, although its use in children and adolescents has been recently reconsidered (98). Mechanistically, the cardioprotective action of DXR has been suggested to be independent of its antioxidant properties, rather it has been ascribed to its ability to compete with ATP binding on Top2-β, thereby producing a configuration change that prevents the formation of a complex with ANTs (72). In accord with this view, the DXR analogue JR-311 shows significant cardioprotective effects, which depend on Top2-β modulation, while being independent of its iron-chelating properties (10). Despite its cardioprotective function, caution is still used while prescribing DXR, and this drug is nowadays not recommended for children aged 0–18 years who are expected to receive a cumulative dose of Doxo (or other ANT) lower than 300 mg/m2.
Considering the isoform selectivity and opposing effects of Top2 poisoning versus inhibition, it is reasonable to assume that all patients receiving ANTs would benefit from the development of new therapeutic regimens including Top2-α-specific poisons and Top2-β-specific inhibitors.
Rac1 as a master regulator of DNA damage and ROS production
As Top2 emerged as a good pharmacological target to prevent ANT cardiotoxicity, new Top2 regulators have started to be investigated. Among these, Rac1 has been shown to contribute to Doxo-mediated cardiomyocyte death [for a complete review, see Ref. (38)], in both ROS-dependent and ROS-independent manners (73) (Fig. 4). Rac1 is a small guanosine triphosphate-bound protein, belonging to the Rho family, a group of molecular switches and signal transducers that transmit extracellular stimuli from the inner plasma membrane to intracellular signaling pathways. Rac1 is involved in a variety of cellular processes, such as cell–cell and cell–substrate adhesion, polarization, migration, invasion, proliferation, transcription, vesicle formation, and apoptosis, which depend on its spatiotemporal distribution (94). Several studies have identified the presence of a nuclear pool of Rac1 and have unraveled the mechanisms underlying its nuclear localization (94), which include DNA damage itself (41). This is in agreement with the finding that oxidized DNA bases can act as Rac1 guanine nucleotide exchange factors (35). In the nucleus, Rac1 was recently found to be associated with Top2 (108), where it is responsible for the activation of a DNA damage-ATM-p53-apoptosis pathway (139).

Pharmacological inhibition of Rac1 signaling with NSC23766 interferes with Doxo genotoxicity by preventing the formation of the cleavage complex, thereby reducing the generation of double-strand breaks and the activation of the DNA damage response (46). Of note, the effect of NSC23766 can be further improved by the lipid-lowering drug lovastatin (46). Inhibition of the HMG-CoA reductase by statins causes the depletion of the cellular pool of isoprene precursors, which are essential for the C-terminal prenylation, and the subsequent membrane localization, of Rho proteins (26). Thus, prevention of Rac1 prenylation by statins promotes Rac1 translocation to the nucleus, suggesting that the therapeutic effect of statins might be linked to the upregulation of the nuclear pool of Rac1 versus the cytosolic counterpart. Nevertheless, the mechanism by which nuclear Rac1 regulates Top2 activity is still unclear and deserves further experimental efforts. It was shown that in acute (73) and subacute (39) models of ANT cardiotoxicity, cardiac Rac1 knockout confers cardioprotection by means of ROS-independent as well as ROS-dependent mechanisms. In fact, Rac1 contributes to the assembly and activation of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (42), and cardiomyocyte-specific deletion of Rac1 was shown to impair Doxo-induced NADPH oxidase activation, ROS production, DNA fragmentation, cardiomyocyte apoptosis, and finally improve cardiac function. Consistently, mice with impaired Nox2 NADPH oxidase complex are less sensitive to ANTs (145).
Overall, statins interfere with two of the main mechanisms underlying ANTs cardiotoxicity, that is, ROS production and Top2-mediated DNA damage. Their beneficial effect in models of ANT cardiotoxicity and their favorable tolerability make them promising candidates for the prevention and/or treatment of ANT cardiotoxicity, as confirmed in small-scale clinical studies (1). More importantly, statins may sensitize certain tumors to chemotherapy, thereby enhancing the anticancer efficiency of ANT regimens and at the same time protecting the heart (25).
mtDNA and mitochondrial homeostasis
Another interesting aspect related to Top2 poisoning by ANTs is that both Top2-α and Top2-β isoforms were found in mitochondria (70, 141). Top2-β is likely the most relevant isoform in cardiac mitochondria (141). In cardiomyocytes, ANT-mediated Top2-β poisoning might affect not only nuclear DNA but also mtDNA, further contributing to increased ROS production and cytotoxicity. Accordingly, ANT-related chronic heart failure is characterized by imbalanced mitochondrial mass and reduced nuclear expression of genes pivotal to mitochondrial biogenesis and homeostasis (54). These include the PGC-1α and β, nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM). Accordingly, the protective effect exerted by the histone deacetylase and PGC-1α activator, Sirtuin 1 (SIRT1), against ANT cardiotoxicity (11) is likely due, at least in part, to the regulation of mitochondrial biogenesis.
In addition, ANTs can induce mitochondrial damage through the uncoupling of the electron transport chain, disruption of mitochondrial membrane potential, and generation of ROS, especially in conjunction with mitochondrial iron metabolism (32). However, the relative contribution of direct Top2 poisoning and of secondary oxidation of nucleotides by ROS on mtDNA depletion has yet to be clarified.
Recently, Jean et al. (51) showed that mice treated with a modified form of Doxo, which exclusively targets mitochondria but not nuclei, are resistant to ANT cardiotoxicity, downsizing the contribution of mtDNA damage to the disease. In contrast, Khiati et al. (56) showed that mice lacking mitochondrial Top1 are hypersensitive to ANTs and display severe mitochondrial damage. Doxo-induced mitochondrial defects, including mitochondrial cristae disorganization and impaired production of respiratory chain proteins, ultimately result in decreased O2 consumption, increased ROS production, and cardiac damage leading to lethality.
ANT-Induced Impairment of Calcium Handling
The failure of antioxidants to protect against ANT cardiotoxicity led to the view that ROS likely represent signaling intermediates of Doxo-induced damage, but not the triggering event of ANT cardiotoxicity. Increasing evidence highlights the impairment of cellular and mitochondrial Ca2+ signaling, rather than the induction of oxidative stress, as one of the primary pathological events.
Cytosolic Ca2+ impairment
Isolated cardiac myocytes perfused with Doxo show increased Ca2+ sparks occurrence and decreased Ca2+ transients (104). Of note, the Doxo metabolite, doxorubicinol, has been shown to target calsequestrin type 2, thus increasing cytoplasmic Ca2+ concentration and favoring arrhythmic conditions (1) (Fig. 5). Doxo also inhibits the transcription of the sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) and therefore reduces Ca2+ uptake in the sarcoplasmic reticulum (SR) (2). Moreover, Doxo and its metabolite doxorubicinol modify ryanodine receptor (RyR) and SERCA2a activity by binding to the proteins via thiol oxidation (37) and induce calcium/calmodulin-dependent protein kinase-II (CaMKII)-dependent Ca2+ leakage from the SR (104, 124). Overall, the effects on RyR, SERCA2a, and phospholamban (PLN), mediated by the activation of protein kinase A (PKA) and CaMKII, result in transient intracellular Ca2+ alterations. These, in turn, lead to a temporary depression of Ca2+ transients that precede structural defects and overt cardiac dysfunction in models of cardiotoxicity induced by chronic low-dose Doxo (69). Although it is known that ROS activate both PKA and CaMKII (8, 141), the two main regulators of the excitation-contraction coupling, the contribution of β-adrenergic signaling in the setting of ANT cardiotoxicity has been postulated but still requires experimental confirmation (69).
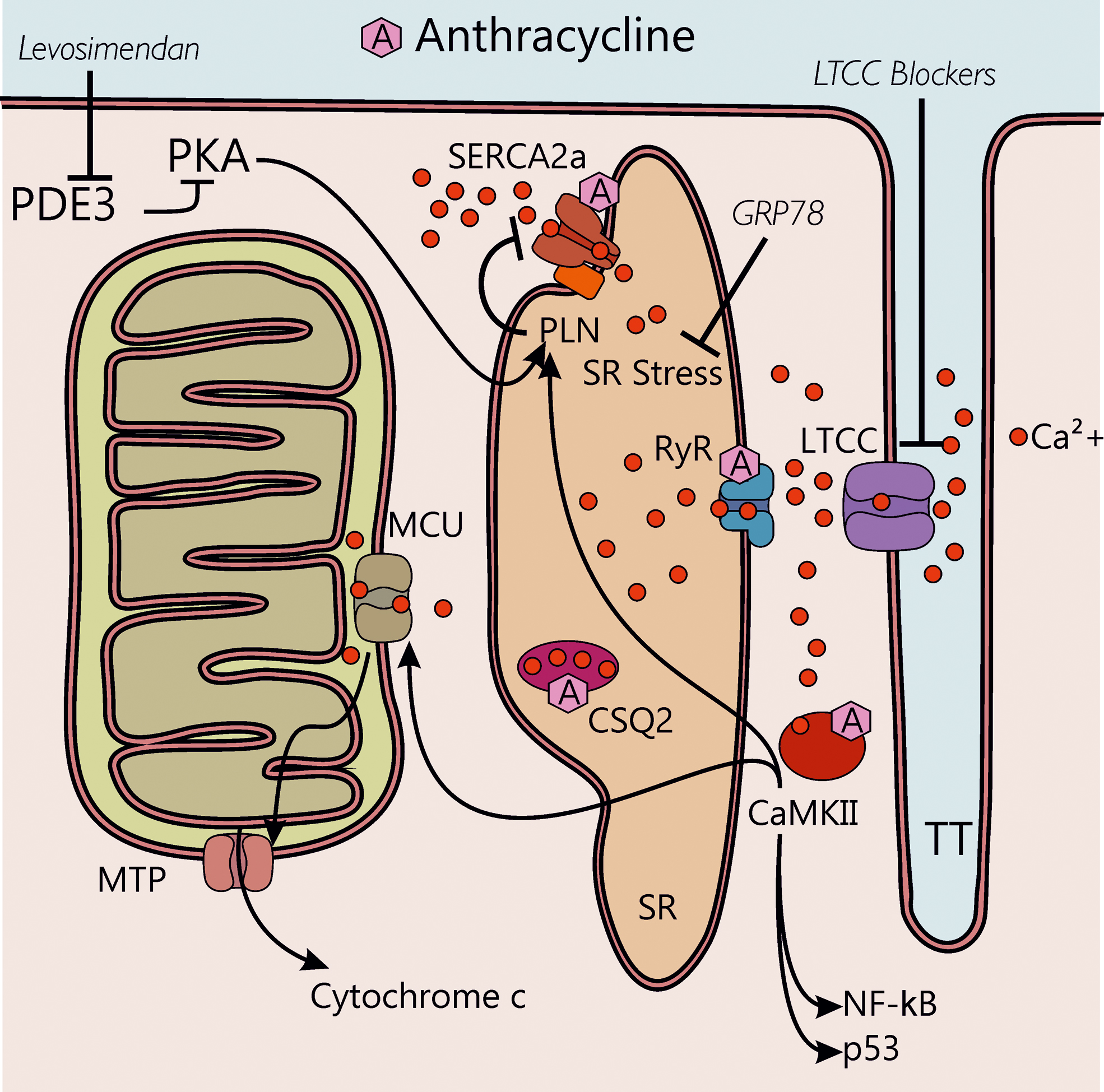
The ensuing disruption of sarcoplasmic Ca2+ handling machinery leads to intracellular Ca2+ overload and, finally, to sarcomeric disarray and myofibril deterioration, which is further aggravated by Doxo-activated Ca2+-dependent calpain proteases (68).
Mitochondrial Ca2+ impairment
Besides excessive accumulation of Ca2+ in the cytoplasm, impaired Ca2+ fluxes have been reported in mitochondria in response to Doxo treatment (19). Ca2+ enters mitochondria through the mitochondrial calcium uniporter and, above a physiological threshold, mitochondrial Ca2+ triggers the opening of the mitochondrial permeability transition pore (MTP), resulting in dissipation of transmembrane potential, mitochondrial swelling, and increased permeability of the outer membrane to apoptotic factors, such as cytochrome c (144). In turn, in the cytosol, cytochrome c forms a complex with the adaptor protein apoptosis protease activator protein-1 (Apaf-1) and caspase-9, the so-called apoptosome, which finally activates the apoptotic cascade (144).
Emerging therapeutic strategies to rescue Ca2+ homeostasis
As the relevance of SR Ca2+ leakage in ANT cardiotoxicity emerged, new therapeutic approaches have been explored, including cardiac gene therapy with the GRP78 chaperone, which is responsible for amelioration of SR stress (124). Intriguingly, adeno-associated virus-mediated GRP78 overexpression partly protects cardiomyocytes from Doxo-induced cell death by modulating Ca2+/CAMKII-dependent pathways and preventing p53 accumulation (124). This study suggests that SR stress is a valuable therapeutic target in the context of Doxo-induced, and possibly other, cardiac disease.
Further highlighting the central role of CaMKII in this process, it has been recently demonstrated that pharmacological or genetic blockade of L-type Ca2+ channel (LTCC) attenuates Doxo-induced cardiomyocyte apoptosis by suppressing intracellular Ca2+ accumulation and the ensuing activation of CaMKII-nuclear factor-kappa B (NF-κB) pathway (49). Hence, LTCC blockers might be potential therapeutic agents against ANT-induced cardiomyopathy.
Recently, Efentakis et al. (23) showed that targeting both impaired cytosolic Ca2+ homeostasis and mitochondrial alterations is a promising strategy. A single-dose of levosimendan, an inotropic vasodilator (92) that is clinically used for the treatment of decompensated heart failure (24), prevented subchronic Doxo-induced cardiotoxicity, as indicated by preserved left ventricle systolic function and structure, by acting on cAMP/PKA and PLN pathway (23). Intriguingly, levosimendan and its active metabolite OR-1896 act as PDE3 inhibitors, with whom they share structural similarity (90). Of note, besides counteracting cytoplasmic Ca2+ accumulation, levosimendan displays anti-inflammatory and antioxidant activities, sensitizes Troponin C to Ca2+ (inotropic effect), and activates the ATP-sensitive K+ channel (24), events that all converge into a cardioprotective effect.
As increased intracellular Ca2+ levels emerged as key triggers of ROS production, which, in turn, further disrupts intracellular and mitochondrial Ca2+ homeostasis, new therapeutic tools aimed at preventing or restoring Ca2+ balance may prove effective against ANT-induce toxicity.
The Controversial Role of Autophagy in ANT-Induced Cardiotoxicity
The finding that antioxidants and iron chelators fail to prevent ANT cardiotoxicity suggested that additional mechanisms may be involved and, among these, autophagy recently emerged to play a key role (55). However, there is still controversy regarding its beneficial versus maladaptive role in ANT-induced cardiotoxicity (111).
Adenosine monophosphate-activated protein kinase/mTOR signaling pathway
Autophagy is the major cellular recycling process, essential for maintaining cellular homeostasis (113). The initiation of autophagy is under the control of autophagy-related genes (ATG)1, also known as activated unc-51-like autophagy activating kinase 1 (Ulk-1), which phosphorylates Beclin-1 (5, 85), thus triggering the formation of autophagosomes. The activity of Ulk-1 is under the control of adenosine monophosphate-activated protein kinase (AMPK) and mTOR signaling pathways. AMPK and mTOR promote and inhibit autophagy, respectively, by finely regulating the kinase activity of Ulk-1. Consistently, ANTs have been found to interfere with mTOR and AMPK regulatory pathways leading to autophagy dysregulation.
ANTs inhibit autophagy
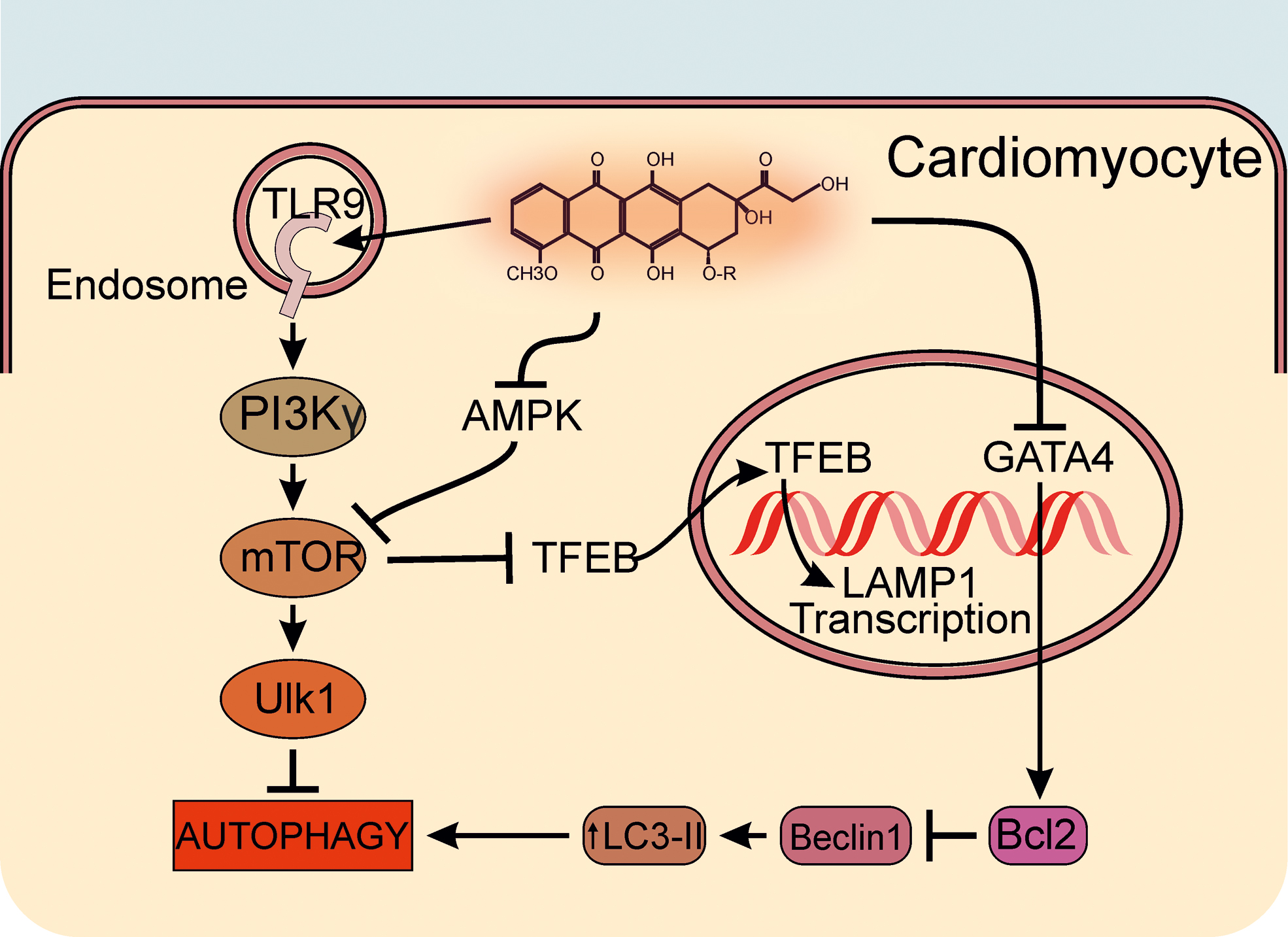
ANTs activate mTOR that, in turn, induces the inhibitory phosphorylation of Ulk-1 at Ser757, thereby preventing Ulk-1-mediated autophagosome initiation (132). However, ANTs inhibit the phosphorylation of AMPK, an upstream positive regulator of autophagy initiation, further contributing to the block of the autophagic process. Interestingly, Li et al. (65) recently identified another kinase, which is involved in autophagy regulation, and whose activity is critically affected by ANTs. The authors show that, in cardiomyocytes, Doxo engages phosphatidylinositol 3-kinase (PI3K)γ signaling, converging on autophagy inhibition and maladaptive metabolic reprogramming. In particular, Doxo activates PI3Kγ/Akt signaling downstream of Toll-like receptor (TLR) 9, leading to increased phosphorylation of mTOR and of its downstream target Ulk-1 and, in turn, to autophagy inhibition. Accordingly, genetic or pharmacological blockade of PI3Kγ reactivates autophagy by releasing the inhibitory brake of mTOR and protects the heart against ANT-induced cardiotoxicity (65). Notably, the enhanced autophagy triggered by PI3Kγ inhibition protects cardiomyocytes against multiple Doxo-induced toxic effects, such as DNA damage, Ca2+ dysregulation, and ROS production. Indeed, in cardiomyocytes, Doxo treatment results in ROS accumulation, which is further enhanced by the addition of the autophagy blocker BafA1, while being reduced by PI3Kγ inhibition. Of note, BafA1 completely abrogated the protective effect of PI3Kγ blockade, thus corroborating the view that PI3Kγ inhibition protects against Doxo-induced oxidative stress and cardiotoxicity via activation of autophagy (65).
Another recent study (131) confirms that Doxo impairs the autophagic flux and the transcription of ATG by increasing the expression and phosphorylation of mTOR. Activated mTOR, indeed, retains in the cytoplasm the transcriptional regulator transcription factor EB (TFEB), which normally drives the expression of autophagy and lysosomal genes, such as LAMP1 (57, 109, 110, 132, 148). The transcriptional activity of TFEB is inhibited by Doxo-induced phosphorylation of mTOR, whereas the impaired nuclear localization of TFEB causes the decline of lysosomal biogenesis and function (77, 82), resulting in increased levels of Beclin-1, LC3-II, and P62. In line with these findings, inhibition of mTOR activity with Torin-1 in H9c2 cardiomyoblasts restores the nuclear localization of TFEB and autophagy, preventing Doxo-induced apoptosis and improving cell viability (6).
In contrast, Zhu et al. report that mice with cardiomyocyte-specific and constitutively active mTOR are protected against ANT-induced acute cardiac dysfunction (147). In this work, the authors observed that Doxo treatment in vivo decreases mTOR activity, as evidenced by decreased mTOR phosphorylation, inducing cardiomyocyte death and mass reduction, whereas the activation of the mTOR pathway effectively retains normal cardiac mass and function.
ANTs induce autophagy
In line with the study of Zhu et al., other studies have reported that ANTs increase autophagy, leading to maladaptive cardiac remodeling. A first study of 2009 by Lu et al. (71) demonstrated that ANTs induce an upregulation of the autophagy-related marker Beclin-1 and autophagic vacuole formation. In agreement with this study, Kobayashi et al. (58) showed that Doxo markedly increases LC3-II protein expression, leading to cardiomyocyte death via inhibition of antiapoptotic protein B cell lymphoma 2 (Bcl2), a negative regulator of Beclin-1. Conversely, Bcl2 is directly upregulated by the transcription factor GATA4 (61), which appears to participate to autophagy regulation (58) also by binding to and blocking Beclin-1 (93). Of note, the preservation of GATA4 activity attenuates Doxo cardiotoxicity by inhibiting autophagy through modulation of the expression of Bcl2 and ATG.
Li et al. (64) further investigated the role of Beclin-1 in ANT cardiotoxicity in vivo using Beclin-1 heterozygous-deficient mice (Beclin-1+/−) since Beclin-1 knockout is embryonic lethal. Beclin-1+/− hearts emerged to be protected against ANT-induced damage. Moreover, Beclin-1 inhibition limits the initiation of autophagy, thus preventing the accumulation of damaged cellular components within autophagosomes and reducing cardiomyocyte apoptosis (58, 64). Consistently, later studies confirmed the activation of autophagy as a primary cause of cardiomyocyte programmed cell death in response to ANTs and suggested inhibition of autophagy as a valuable tool to significantly improve cardiac function and prevent ANT-induced cardiotoxicity (71, 135, 142).
Although autophagy has been revealed as an important mechanism underlying ANT-induced cardiotoxicity, whether ANTs induce or inhibit autophagy (Fig. 6), and if autophagy is beneficial or harmful in this context is still a matter of debate. Future studies are awaited to clarify the scenario.
Mitophagy as a Key Mechanism of ANT-Induced Cardiotoxicity
Mitochondria, as principal integrators of energy metabolism, are particularly important for maintaining cellular ATP levels to sustain myocardial contractile activity and cardiomyocyte survival. Growing evidence demonstrates that structural damage and dysfunction of these organelles are implicated in the development of cardiovascular diseases, including Doxo-induced cardiomyopathy (28). Of note, Doxo preferentially accumulates inside mitochondria: as a cationic drug, it binds to cardiolipin, an anionic-charged phospholipid located in the inner mitochondrial membrane leading to mitochondrial engulfment and toxicity (31). A selective form of mitochondrial autophagy, namely mitophagy, plays a pivotal role in the regulation of mitochondrial homeostasis, by regulating and eliminating dysfunctional organelles (59). In cardiomyocytes, mitophagy occurs via two pathways: (i) the PTEN-induced kinase 1 (PINK1)/Parkin (E3 ubiquitin ligase) pathway and (ii) BH3-only protein Bcl2-like 19 kDa-interacting protein 3 (BNIP3)/BNIP3-like protein Nix, an effector of apoptosis pathway (105). In the following paragraphs, we will describe how these two pathways are affected by ANTs.
BNIP3 and BNIP3L/Nix
Both BNIP3 and BNIP3L/Nix act on the outer mitochondrial membrane as direct mitophagy receptors, as they have an LC3-II recognition motif, thereby allowing autophagosomal engulfment of mitochondria. ANTs lead to severe necrosis in cardiomyocytes, promoting upregulation and translocation of BNIP3 to the mitochondrial membrane, which, in turn, triggers the opening of the permeability transition pore and the increase in ROS production (Fig. 7) (20). Conversely, inhibition of BNIP3, with either shRNA against BNIP3 or through the expression of a mutant BNIP3 that fails to target mitochondria, rescues ANT-induced defects of the mitochondrial respiratory chain in vitro in cultured cardiomyocytes. Furthermore, mitochondria morphology and cardiac function are preserved in BNIP3-knockout mice (BNIP3−/−) treated with ANTs, confirming a maladaptive role of BNIP3 in ANT cardiotoxicity (20). Additionally, BNIP3 activation in cardiomyocytes treated with ANTs is associated to loss of nuclear Hmgb1 and release of classical markers of necrosis (i.e., lactate dehydrogenase and cardiac troponin T), supporting a primary role of BNIP3 in necrosis (20).

New compelling evidence establishes that NF-κB signaling, which transcriptionally silences BNIP3 activation under basal conditions, is dramatically reduced in cardiac myocytes treated with Doxo (19). This, in turn, promotes BNIP3 gene activation, mitochondrial injury including calcium influx, permeability transition pore opening, and necrotic cell death. Interestingly, restoring NF-κB signaling suppresses BNIP3 expression, mitochondrial perturbations, and necrotic cell death, thus suggesting a new potential therapeutic approach for cancer patients treated with Doxo.
PINK1/Parkin pathway
However, PINK1 accumulates and recruits Parkin to the outer membrane of depolarized mitochondria, mediating the ubiquitination of mitochondrial proteins and their recognition by p62 and LC3, consequently leading to the initiation of mitochondrial autophagy (105).
In Doxo-induced cardiomyopathy, mitophagy is critically involved in the elimination of vacuolated and dysfunctional mitochondria (47). Hull et al. (47) demonstrated that PINK1/Parkin-dependent mitophagy is suppressed early after ANT treatment (2–8 days post-treatment), but is restored at a later stage, when PINK1 and Parkin are significantly upregulated. Interestingly, the mitophagy inhibitor peptide, Mdivi-1, prevents Doxo-induced cardiotoxicity, indicating that excessive mitophagy contributes to Doxo cardiotoxicity (29).
Nevertheless, the molecular mechanisms by which ANTs inhibit mitophagy have still to be uncovered. A recent study provides novel and compelling evidence that overexpression of SESN2, a member of the sestrins family, protects cardiomyocytes against Doxo-induced cardiomyopathy via the improvement of mitochondrial function and rescue of mitophagy (129). Of note, the expression of SESN2 is significantly reduced following Doxo stimulation, in vitro and in vivo. Mechanistically, SESN2 interacts with Parkin and p62 and promotes Parkin recruitment to mitochondria, subsequently activating mitophagy. These results establish SESN2 as a new player in the regulation of mitochondrial function and provide a potential therapeutic approach to prevent Doxo-induced cardiomyopathy.
Interestingly, mitophagy appears to be coregulated with mitochondrial biogenesis, through a shared signaling pathway involving nuclear translocation of NRFs and a functional interaction between PGC-1α and Parkin (137). Doxo inhibits the expression of PGC-1α and its downstream targets, NRF1 and TFAM, thus reducing the expression of mitochondrial proteins. Inhibition of mitophagy by mdivi-1 attenuates Doxo-mediated activation of the PINK1/Parkin pathway and rescues mitochondrial biogenesis by preserving the expression of PGC-1α (137).
Association Between Apoptosis and Autophagic Dysregulation
The present section focuses on the interplay between a naturally programmed cell death process, apoptosis, and the cellular recycling process of autophagy. Autophagy and apoptosis have a key role in maintaining cellular homeostasis and respond to similar types of signals and stress, including increased intracellular Ca2+ concentration and ROS accumulation.
Autophagy promotes apoptosis
Apoptosis is regulated by Bcl2 and p53 genes, which are closely associated with autophagy. In general, autophagy restrains apoptosis, while apoptosis-associated caspase activation shuts off the autophagic process (76). However, in particular cases, such as following Doxo stimulation, autophagy is associated with high levels of apoptotic cell death. Autophagy is dramatically increased in Doxo-treated cardiomyocytes and associates with increased cleavage of caspase 3 and PARP, which indicate apoptosis initiation (58). Similarly, in human umbilical vein endothelial cells. Doxo induces autophagy early after treatment (since 3 h), while the level of apoptosis as well as lysosomal membrane permeabilization (LMP) and mitochondrial outer membrane permeabilization (MOMP) increase at later stages (12 h) (52). This finding implies that Doxo-induced autophagy and the ensuing autophagy-dependent LMP are upstream events, followed by distal events including MOMP and cytochrome c release, which finally triggers caspase-dependent apoptosis. Consistently, inhibition of Doxo-induced autophagy, through the autophagy inhibitor 3-MA or Beclin-1 knockdown, results in significant attenuation of cell death (52, 58). Conversely, activation of autophagy by rapamycin or Beclin-1 expression exacerbate apoptosis in H9C2 cells and in mice hearts treated with Doxo (117, 134).
In addition, Doxo-induced cell death correlates with depletion of GATA4, a transcription factor essential for cardiomyocyte growth and survival (89). GATA4 is both sufficient and necessary to inhibit autophagy by controlling the gene expression of Bcl2, a survival factor with anti-autophagy activity (93). Concurrently, GATA4 reduction induces an upregulation of ATG genes through p53, leading to the excessive activation of autophagic flux, which ultimately contributes to Doxo-induced cardiomyocyte death.
Autophagy inhibits apoptosis
The above-presented findings are challenged by the observation that activation of autophagy, by stimuli such as AMPK activation by glucose-depletion or mTOR inhibition by rapamycin, results in enhanced cell viability and reduced apoptosis following Doxo treatment in H9C2 cardiomyoblasts and mice overexpressing GFP-LC3 (117). In this study, the induction of autophagy by rapamycin reduces the activity of cleaved caspase-3, thus supporting an inhibitory role of autophagy on apoptosis, and indicating a therapeutic potential of rapamycin against Doxo-induced myocardial damage (117).
Further confirming the beneficial role of autophagy against apoptosis, it has been shown that autophagy inhibition, by decreasing the expression of AMPK or knocking out AMPKα1 in mice (AMPKα1−/−), increases the sensitivity to Doxo-induced apoptosis, while restoration of autophagy by adenovirus-mediated AMPK constitutive activation or 5-aminoimidazole-4-carboxamide ribonucleoside significantly limits the iatrogenic effects of Doxo (130). Consistently, genetic deletion of AMPKα1 (AMPKα1−/−) enhances Doxo-related reduction of p53 phosphorylation, establishing a critical role of AMPK in Doxo-induced p53-dependent DNA damage and apoptosis (130).
Similarly, Doxo inhibits autophagy by activating E2F1/mTORC1 pathway and further induces apoptosis by activating E2F1/AMPKα2 pathway in starved H9C2 cardiomyoblasts (33). The same result was observed in a mouse model of Doxo-induced cardiotoxicity, in both myocardial ischemic and nonischemic conditions, in which resveratrol was able to repress E2F1, thereby activating autophagy and reducing apoptosis (33).
Finally, p53, that is mostly studied for its role as a transcription factor, displays a key role in cell death and autophagy regulation (13, 119). Accumulation of cytosolic p53 can directly activate BAX to permeabilize mitochondria, thereby inducing caspase activation and apoptosis (13). p53 also functions as an endogenous repressor of autophagy (80). An additional function of cytosolic p53 has been discovered in Doxo-treated cardiomyocytes (44), in that it inhibits mitophagy by blocking Parkin translocation to damaged mitochondria (Fig. 7). The inhibition of Parkin-mediated mitophagy compromises mitochondrial function, resulting in aging-related and Doxo-mediated decrease in cardiac contractility. In keeping with this mechanism, p53-deficient mice and Parkin-overexpressing cardiomyocytes show a less severe decline of mitochondrial integrity and cell death after treatment with Doxo (44).
Inflammation-Mediated Cell Death in ANT Cardiotoxicity
Inflammation appears to be significantly involved in Doxo-induced cardiotoxicity (81), as acute and chronic myocarditis occur in a dose-dependent manner in patients treated with Doxo (63). Doxo triggers the inflammatory signaling cascades by promoting the release of different cytokines, including interleukin (IL)-1 and tumor necrosis factor (TNF)-α, in combination with the activation of various signaling pathways, such as the NF-κB, p38-MAPK, and the autophagy pathways (97, 107).
NF-κB is a master regulator of the inflammatory cascade and is maintained in the cytoplasm in an inactive form by the inhibitor of κB protein (IKK). Upon Doxo treatment, inhibition of autophagy and the ensuing p62 overexpression induce IKK signaling downstream of TLRs, thereby engaging NF-κB and triggering the inflammatory response. Conversely, when autophagy is upregulated, p62 is reduced and blocks the NF-κB signaling pathway (146). Therefore, upregulation of autophagy by AMPK/mTOR signaling pathway results in an anti-inflammatory effect, with a reduction of inflammatory cytokine production (97).
In addition, Doxo induces the expression of TLRs, which contribute to cardiac damage in virtue of their role in the activation of proinflammatory NF-κB (96). TLRs are a class of 13 members normally expressed in leukocytes in response to microbial pathogens or dead/damaged cell structures, but recent studies uncover their implication in Doxo-mediated cellular injury and related inflammation (101). Among them, TLR2 and TLR4 are also found in cardiomyocytes and are implicated in ANT-induced cardiotoxicity. TLR2 regulates cytokine release, such as TNF-α, IL-6, and IL-1β, through NF-κB activation, as NF-κB DNA binding activity is increased in wild-type mice but reduced in TLR2−/− mice after Doxo administration (87). Of note, genetic ablation and pharmacological inhibition of TLR2 in mice and in cardiomyocytes, respectively, treated with Doxo result in cardioprotection, with reduced release of cytokines and enhanced mice survival (87), as well as prevention of cardiomyocyte death, cardiac dysfunction, and inflammation (74, 106, 136).
On the contrary, the role of TLR4 in ANT cardiotoxicity is still controversial. On one side, it controls cardiac inflammation and appears to be involved in Doxo-induced cardiomyopathy through the inhibition of the transcription factor GATA4 since cardiac inflammation is decreased in TLR4-knockout mice (TLR4−/−) (99). On the other side, blockage of TLR4 with neutralizing antibodies results in an enhanced inflammation and inhibition of autophagy (74). New compelling evidence about TLR4 implication in Doxo-induced cardiac inflammation has been shown by Tavakoli Dargani and Singla (120). They demonstrate the implication of TLR4 in a new form of programmed cell death characterized by proinflammation, termed pyroptosis. In this study, Doxo activates TLR4-pyrin domain containing-3 (NLRP3) and caspase-1, leading to inflammasome formation and pyroptotic cell death in H9c2 cells (120). Moreover, Doxo stimulates the release of cytokines and inflammatory markers, such as ILs (IL-1β, IL-6), TNF-α, and NF-κB (34), which are associated to cardiac dysfunction and apoptosis. Interestingly, the use of embryonic stem cell-derived exosomes (ES-Exos) inhibits TLR4 and NLRP3 inflammasome markers and inflammasome-induced pyroptosis in mice under Doxo treatment, by inducing the release of anti-inflammatory cytokines, such as IL-10 (116, 120). Moreover, ES-Exos-transplanted mice display improved cardiac function, suggesting that the protective effects of ES-Exos could be used as a future therapeutic option to treat Doxo-induced cardiac dysfunction (116, 120).
Emerging Role of microRNAs in ANT Cardiotoxicity
Recently, microRNAs (miRNAs or miRs), a class of endogenous 22-nucleotide noncoding RNAs, emerged as pivotal modulators of target genes in a variety of heart disease conditions, such as myocardial infarction and hypertensive heart disease (14), as well as Doxo-induced myocardial injury. Of note, several studies have shown increased levels of circulating miRNAs, including miR-21, miR-34a, miR-146a, and miR-532-3p in response to Doxo treatment (125).
Doxo treatment significantly increases miR-21 expression in both mouse heart tissue and H9C2 cells, whereas Doxo-induced apoptosis results attenuated by miR-21 overexpression in cardiomyocytes through the modulation of the antiproliferative factor, B cell translocation gene 2 (Btg2) (123).
Regarding the role of miR-146a, Tong et al. showed that in a model of acute Doxo cardiotoxicity, miR-146a overexpression further aggravates Doxo-mediated myocardial apoptosis, by targeting a key component of neuregulin-1-ErbB signaling, ErbB4. The increased expression of miR-146a drives ErbB4 downregulation and leads to cardiomyocyte death, whereas inhibition of miR-146a or overexpression of ErBb4 could prevent and improve cardiomyocyte survival (123). However, Pan et al. recently proved a protective role of miR146-a in a different model of Doxo cardiotoxicity characterized by a low-dose Doxo treatment leading to late-onset cardiomyopathy (91). In this study, the expression of miR-146 is significantly reduced upon Doxo treatment, leading to cardiomyocytes death and, conversely, to autophagy inhibition. Of note, these pathological events are reversed by overexpression of miR-146a, which plays an anti-apoptotic role through the p53 pathway. In particular, by targeting TATA-binding protein-associated factor 9b, a coactivator and stabilizer of p53, miR-146 downregulates the expression of p53, thereby attenuating apoptosis and restoring autophagy in cardiomyocytes (91). New compelling evidence about the implication of miR-146a-5p in Doxo-induced cardiotoxicity has been recently provided by the work of Milano et al. (79). The authors demonstrate that intravenous administration of human cardiac progenitor cells (CPCs) exosomes, highly enriched in miR-146-a5p, attenuates Doxo/trastuzumab-mediated oxidative stress in cardiomyocytes. Hence, specific silencing of miR-146-a5p results in enhanced Doxo-induced cell death (79).
In addition, recent findings demonstrate that miR-34a levels are increased and trigger senescence and apoptotic mechanisms involving p16INK4a and p53 in rat CPCs exposed to Doxo (87). Mesenchymal stem cell (MSC)-based therapies have been reported to modulate Doxo-induced cellular senescence in H9C2 cells via inhibition of miR-34a (133). The mechanism underlying the anti-senescence function of MSCs involves the miR34a-SIRT1-p53 axis, where the inhibition of miR-34a leads to a reduction of p53 and activation of SIRT1, which elongates telomere length and increases telomerase activity (133).
Another miR, miR-532-3p, has been recently shown to have a proapoptotic role, inducing mitochondrial fission and apoptosis in Doxo-treated cardiomyocytes. Mechanistically, these effects are explained by a negative regulation of the expression of apoptosis repressor with caspase recruitment domain (ARC) (128).
In addition, Doxo induces miRNA-320a overexpression, which leads to cardiac dysfunction and endothelial injury by dysregulating the vascular endothelial growth factor signaling pathway (137). Accordingly, miR-320a-knockdown mice have attenuated damage of endothelial cells as well as reduced cardiac dysfunction in response to Doxo.
However, a number of miRNAs, such as the miR-30 family, are downregulated by ANTs. Of note, the miR-30 family has a protective role against Doxo-induced cardiotoxicity by targeting multiple members of the β-adrenergic pathway, such as β1AR, β2AR, and Gαi-2, as well as members of the mitochondrial apoptotic pathway, such as the proapoptotic gene BNIP3L (100).
Overall, the differential and altered expression of miRNAs in response to ANT treatment highlights the possibility of exploiting miRNAs not only as therapeutic targets but also as diagnostic biomarkers, as in the case of miR-216b, that it is upregulated in the first stage of Doxo-induced cardiotoxicity and may act as an early marker of cardiac damage (60).
Future Perspectives for the Treatment of ANT-Induced Cardiotoxicity
A basic requirement for clinically relevant cardioprotective strategies is the ability to preserve the efficacy of the anticancer treatment. Even more interesting are pharmacological approaches, which potentially provide a dual beneficial effect, the so-called ability to “kill two birds with one stone” (106), where the cardioprotective agent not only prevents the cardiac side effect of chemotherapy but also enhances its antitumor action.
Among this class of dual drugs are CYP1 inhibitors, derived from Visnagin, which have been recently shown to be promising therapeutic agents against Doxo-induced cardiotoxicity (3). CYP1 belongs to a family of highly conserved monooxygenases that are usually responsible for the metabolism of toxic compounds, including polycyclic aromatic hydrocarbons that share structural similarity with Doxo (112).
On one side, CYP1 plays an important role in the metabolism of polyunsaturated fatty acids (21), such as arachidonic acid metabolites, which have been previously implicated in the pathogenesis of Doxo cardiotoxicity (75). Moreover, CYP1, in particular the CYP1B1 isoform that is overexpressed in a wide range of tumor types, including breast cancer (84), plays a role in estrogen-mediated tumor formation (9, 67). These observations suggest that CYP1 inhibition may simultaneously confer cardioprotection, while enhancing the antitumor effect of Doxo, and make CYP1 inhibitors appealing candidates for future clinical trials.
Besides CYP1, neuregulin, an agonist of the receptor tyrosine kinases belonging to the epidermal growth factor receptor (EGFR) family, has elicited interest as a potential therapeutic against ANT cardiotoxicity. EGFR family includes EGFR/HER1/ErbB1, ErbB2/HER2 (that has no ligand), ErbB3/HER3, and ErbB4/HER4, and among these, ErbB2 and ErbB4 are expressed in differentiated cardiomyocytes. Evidence for the involvement of the ErbB family in ANT cardiotoxicity emerged from the increased risk of cardiotoxicity in patients receiving concurrent Doxo and trastuzumab (103), a monoclonal antibody targeting ErbB2 receptor used to treat HER2-postive breast and advanced stomach cancer. Moreover, in mouse models of cardiotoxicity, ANTs downregulate cardiac expression of ErbB4, potentially by increasing the levels of miR-146a (43). However, cardiac-specific overexpression of ErbB2 in mice is protective against cardiac damage (16), including that elicited by ANTs, likely through the upregulation of antioxidant mechanisms (7).
Neuregulin-1 acts as a primary ligand to initiate ErbB signaling and plays a critical role in cardiomyocyte development, homeostasis, and disease (88). Although administration of neuregulin-1 reduces ANT-induced cardiomyocyte apoptosis via PI3K/Akt (27), its use as a cardioprotectant is limited by potential pro-neoplastic effects (45). Ongoing work is focusing on the development of an engineered neuregulin, displaying reduced neoplastic potential, but preserved cardioprotective function, in mice (50). Neuregulin-mediated cardioprotection thus appears to be a promising approach for patients undergoing treatment with ANTs and deserves further studies and clinical validation.
Conclusions
The exponential growth of the field of Cardio-Oncology of the last decade has greatly expanded our knowledge of the molecular basis of chemotherapy-related cardiotoxicity. The emerging view is that cardiotoxic anticancer drugs, such as ANTs, affect intertwined signaling pathways, which are key to the survival not only of cancer cells but also of cardiomyocytes. Despite ROS are no longer considered the main determinant of cardiotoxicity, they are crucial signaling intermediates that intervene in the response of cardiomyocytes to cardiotoxic insults. The current hope is that the signaling pathways that have just emerged to be targeted by ANTs could be therapeutically manipulated to prevent and/or treat cardiotoxicity. While some pharmacological approaches have already been tested in preclinical models, the way toward the clinical application is still arduous and requires conclusive validation of the most promising targets in models that faithfully reflect the fundamental biology or cardiotoxic responses of the human myocardium. These include for instance human iPSC-derived cardiomyocytes, which are just emerging as a powerful tool for drug discovery, and the realization of precision medicine in Cardio-Oncology (30, 95).
Footnotes
Acknowledgment
We gratefully acknowledge Giulia Prono for figure layout.
Author Disclosure Statement
Prof. A.G. and E.H. are co-founders and board members of Kither Biotech, a startup biotech focused on the development of PI3K inhibitors.
Funding Information
This work was supported by grants from Progetto d'Ateneo-Compagnia di San Paolo (PICANCAR to A.G.), Fondazione Cariplo (GR 2017-0800 to V.S.), Ricerca Sanitaria Finalizzata (GR-2013-02355449 fellowship to V.S.), and Leducq Foundation (09CVD01 to E.H.).