
Abstract
Significance:
Senescence is an essential biological process that blocks tumorigenesis, limits tissue damage, and aids embryonic development. However, once senescent cells accumulate in tissues during aging, they promote the development of age-related diseases and limit health span. Thus, it is essential to expand the boundaries of our knowledge about the mechanisms responsible for controlling cellular senescence.
Recent Advances:
Cellular metabolism plays a significant role in the regulation of various signaling processes involved in cell senescence. In the past decade, our knowledge about the interplay between cell signaling, cell metabolism, and cellular senescence has significantly expanded.
Critical Issues:
In this study, we review metabolic pathways in senescent cells and the impact of these pathways on the response to DNA damage and the senescence-associated secretory phenotype.
Future Directions:
Future research should elucidate metabolic mechanisms that promote specific alterations in senescent cell phenotype, with a final goal of developing a new therapeutic strategy. Antioxid. Redox Signal. 34, 324–334.
Introduction
Hayflick and colleagues (33, 34) were the first to formally describe cellular senescence more than five decades ago. Later, cellular senescence was identified as a stable cell cycle arrest induced by damage or stress applied to proliferating cells (12). Induction of senescence limits tumorigenesis and tissue damage, and aids embryonic development (10). However, the accumulation of senescent cells decreases the regenerative potential of tissues and promotes tissue dysfunction and tumor development in a cell nonautonomous manner (11, 69, 84). Senescent cells produce multiple proinflammatory chemokines and cytokines, collectively named senescence-associated secretory phenotype (SASP), which strengthens senescence arrest by autocrine response and regulates immune surveillance of senescent cells (1, 18, 48, 49). Such secreted factors lead to the negative consequences of senescent cell accumulation, including tissue dysfunction, limited regeneration, and tumor development (68). Senescent cells express p16, a cyclin-dependent kinase (CDK) inhibitor that contributes to irreversible growth arrest in senescent cells (12). The elimination of p16-positive senescent cells extends mouse health span and life span, and limits the development of multiple age-related diseases in mouse models (5). Studies in mice demonstrated that p16-positive cells promote tumor progression and age-dependent changes, thus shortening a healthy life span. In contrast, the elimination of p16-positive cells reduces tumorigenesis and delays age-related deterioration of various organs without possible side effects (5). Senescence is an essential physiological process that comes with negative consequences during aging and disease once homeostasis of senescent cells is altered.
Senescence may result in response to multiple stress stimuli such as telomere dysfunction, oxidative stress, active oncogenes, and DNA damage (7, 15, 27, 28, 63, 72, 82, 98a) (Fig. 1). As a reaction to stress, the initiated DNA damage response (DDR) leads to activation of p53 and p16 (CDKN2A) pathways to initiate and sustain cell cycle arrest. DDR activates phosphorylation of different kinases such as ataxia-telangiectasia Rad3-related (ATR) and ataxia-telangiectasia mutated (ATM). Following activation, ATM and ATR phosphorylate checkpoint kinases CHK2 and CHK1, which then activates the p53 pathway and p21 (CDKN1A) expression (57).
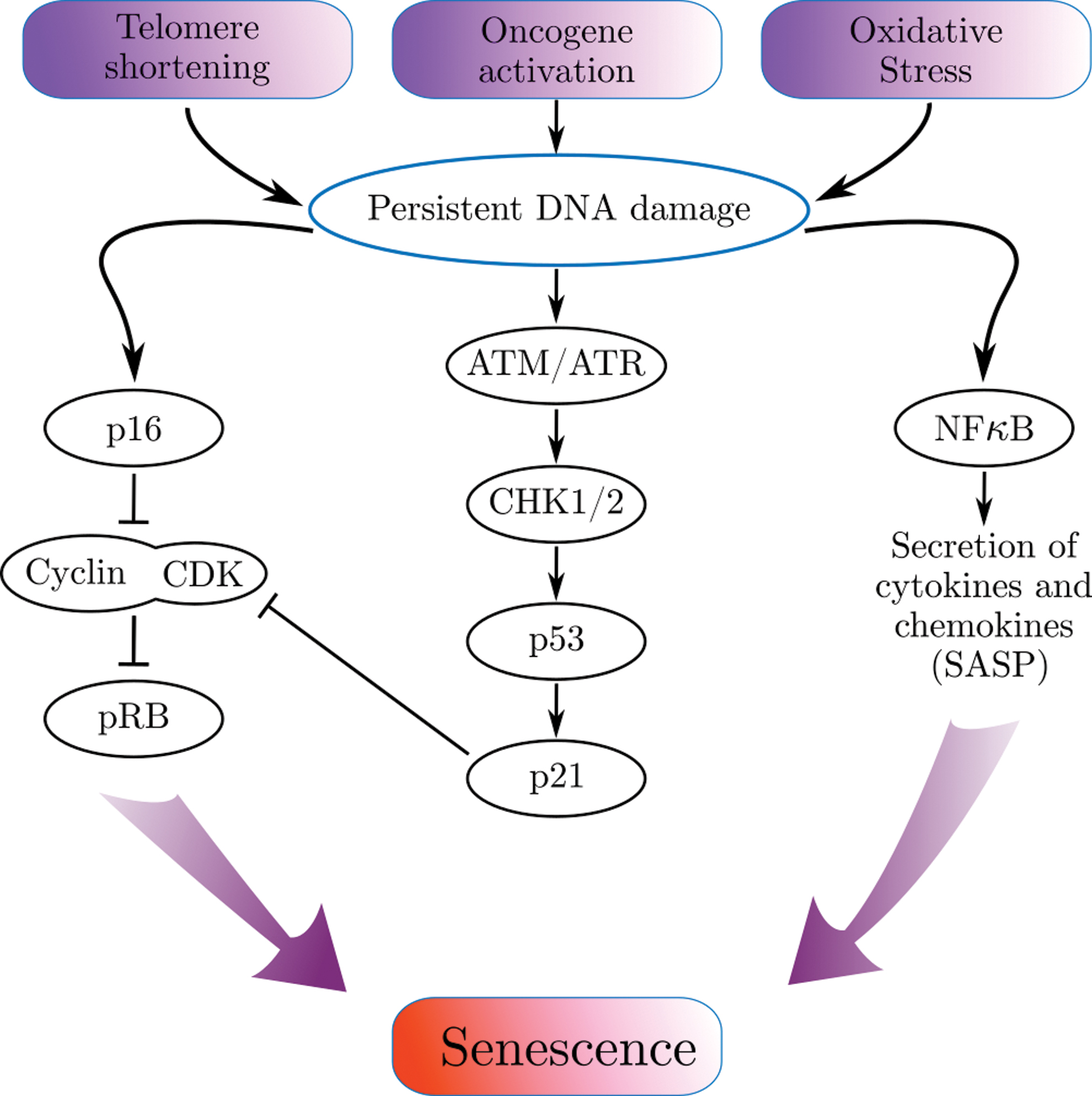
The inability to repair DNA damage leads to persistent DDR, upregulated p16 and p21 inhibit CDKs and restrained retinoblastoma protein (pRb) phosphorylation (16, 64, 65, 96). The hypophosphorylated pRb suppresses the transcription of genes responsible for the transition from phase G1 to S. It directly binds to the E2F transcription factors (on its transactivation domain) and blocks the transcription of E2F genes essential for cell cycle progression (46). Nuclear factor kappaB (NF-κB) is another pathway activated as a response to the DNA damage (38). Along with DNA repair and the activation of cell cycle checkpoints, it enables to preserve a normal cell function in cases of limited damage. In senescent cells, this signaling pathway is the major regulator of SASP (1, 14, 32, 75). The combination of signaling pathways of senescent cells orchestrates one of the main features of senescent cells—cell cycle arrest and non cell-autonomous effects. The impact of these pathways on the regulation of metabolism in senescent cells is understood in much less detail.
Despite the inability to proliferate, and dysregulation of several metabolic pathways, senescent cells are metabolically active (12). Moreover, cell metabolism is responsible for regulation of the DNA damage and DNA repair processes, by balancing the pool of nucleotides used for DNA repair and replication (90). Tricarboxylic acid (TCA) cycle, glycolysis, fatty acid (FA), and nucleotide synthesis are major metabolic pathways that are involved in the regulation of cell's homeostasis, along with regulation of the DNA repair machinery (90). DDR activation diminishes glutamine anaplerosis, while generating an expansion in nucleotide synthesis and anabolic glucose metabolism. Nutrients such as aspartate and glutamine are crucial for the de novo nucleotide synthesis. They serve as indicators for accessibility of the nucleotide pool and by that affect DNA replication and repair (90). Thus, metabolic alterations exhibit a significant influence on the DDR and therefore have a potential for impacting cell senescence.
In senescent cells, major regulators of senescence phenotype also attenuate metabolic pathways. For example, the p53 protein is one of the critical transcription factors that regulate cell proliferation and death (51). Evidences are accumulating regarding the ability of p53 to regulate cell metabolism and energy homeostasis. It was discovered that p53 balances amino acid metabolism and oxidative stress through regulation of oxidative phosphorylation (OXPHOS) and glycolysis, in addition to the autophagy pathway (92, 101). Translation of several factors involved in OXPHOS regulation such as fructose 2,6 bis phosphatase, TIGAR (TP53-induced glycolysis regulator), and synthesis of SCO2 (cytochrome c oxidase subunit) of complex IV of the electron transport chain (ETC) is governed by p53. In addition, p53 can influence energy metabolism indirectly by controlling the expression of glucose transporters (GLUTs), glutaminase 2 (GLS2), and fatty acid synthase (FAS). Moreover, studies of the NF-κB pathway during the past few years have underscored its central role in metabolic pathologies (87).
Multiple molecular effectors of stable cell cycle arrest and senescent cell secretory function have been established. In contrast, metabolic changes in senescent cells are not fully defined, and it is yet unknown whether metabolic changes are crucial for the senescent phenotype. There is an intricate interplay between pathways of DDR, senescence, and metabolism. This review discusses the unique metabolic features of senescent cells in light of central senescence pathways.
DDR and SASP
Different endogenous and exogenous factors can cause DNA damage. Endogenous genotoxic factors, for example, include by-products of normal cell metabolism, such as lipid peroxidation products, reactive nitrogen species, reactive oxygen species (ROS), and endogenous alkylating factors. Exogenous agents list among them ultraviolet radiation, ionizing radiation, and genotoxic compounds. Moreover, spontaneous reactions, such as hydrolysis, damage DNA. Those different types of DNA damage stimuli can induce the formation of double-strand breaks (DSBs) and single-strand breaks (SSBs) (21) (Fig. 2).

Cells have a developmental preserved pathway, the DDR pathway, which senses and repairs DNA damage. Activation of this pathway can arrest the cell cycle and prevent damaged DNA from entering into daughter cells. At the site of the DNA lesion, unique complexes sense double-strand and SSBs. Following detection of DNA breaks, they activate and recruit two protein kinases, ATR and ATM, respectively (57). Binding of both primary kinases to the damaged site results in immediate phosphorylation of the histone H2AX in cis. This step appears to be an essential for the activation of the DDR. To involve DDR components that operate remotely of the site of DNA lesion, the activity of local ATM and ATR required to be increased above a determined limit. When the threshold outpaces by DNA damage, the CHK2 is activated by ATM phosphorylation (97). Then, CHK2 passes easily throughout the nucleoplasm and transmits DDR signals in the nuclear space by phosphorylating its substrates (Fig. 2). Furthermore, once CHK1 is phosphorylated, mostly by ATR, it is not maintained in the nucleus at the DNA damaged cites, and diffuses (100) (Fig. 2). Ultimately, checkpoint requirement results from a number of upregulated pathways, for example, the cell-division cycle 25 (CDC25) and p53 phosphatases. As these phosphatases are necessary for proliferation, DNA damage, induced by CDC25 dysregulation, initiates accelerated cell cycle arrest. In comparison, its stabilization results in p53 upregulation after phosphorylation by DDR kinases. p53 also acts as a transcription factor that transactivates the expression of its target genes, including p21, which leads to a stable cell-cycle arrest. DDR activates several pathways responsible for the initiation and maintenance of cell cycle arrest. Moreover, it plays a crucial role in senescence induction.
Senescent cells undergo universal changes in gene expressions, including inflammatory cell secretion, growth factors, chemokines, matrix metalloproteases, extracellular matrices, and others, collectively named SASP. Those SASP components include among them interleukin (IL)-1β, IL-1α, IL-8, IL-6, CCL-2, GROα, GROβ, vascular endothelial growth factor (VEGF), transforming growth factor-beta (TGF-β), plasminogen activator, matrix metalloproteinase (MMP)-1, -3, and -10, and PAI-1. Notably, SASP is not just a consequence of senescence, but also serves as a senescence inducer. SASP contributes to tissue repair and to senescence recruitment in damaged cells. However, it can also lead to tissue dysfunction associated with age, and age-related diseases, among them cancer. Since SASP may cause both beneficial and harmful consequences, it may thus serve as a potent, dual-edged target for human disease pharmaceutical intervention (2, 74). The molecular connection and the interplay between SASP and DDR are well established (Fig. 3). SASP, unlike the rapid intracellular DDD signals, is a delayed and slow response. While primary DDR kinases such as ATM became active after several minutes, as a response to DNA damage, and the following DDR transcription reaction, other transcription factors such as p53 are detected in hours, SASP is increased over days, with associated factors such as IL-6, achieving highest secretion levels within 5–10 days after the initiation of DDR.
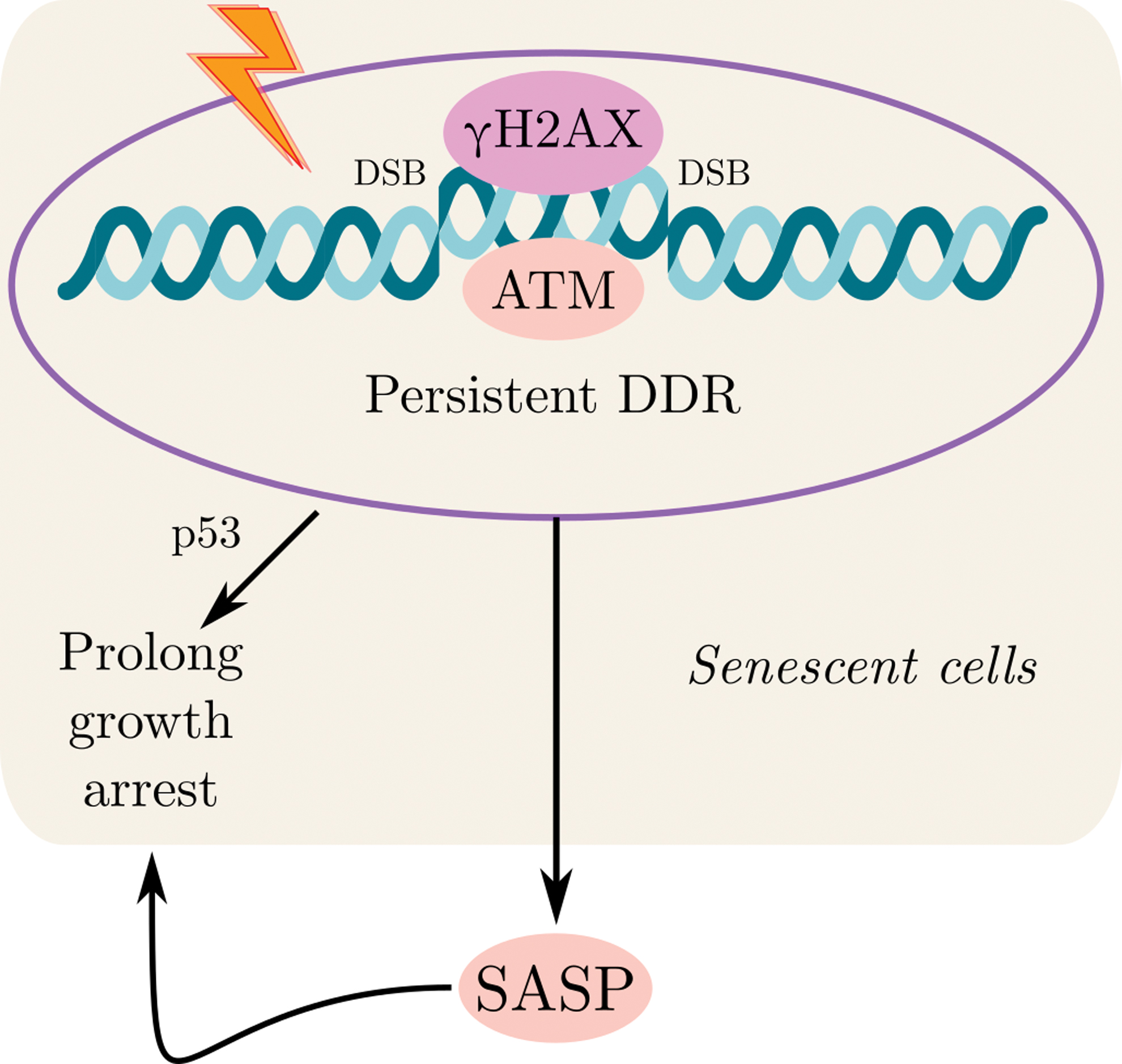
There are several transcription factors that are known as regulators of SASP. The inflammation-associated transcription factor NF-κB is a key regulator of the SASP (79). The expression of several SASP factors, including IL-8 and IL-6, relays on the p65 (NF-κB subunit) activation, and its reinforcement to the chromatin (14). Several studies also demonstrated that interplay between activated ATM and the NF-κB essential modulator (NEMO) can directly stimulate the activation of NF-κB signaling. NEMO serves as a regulatory subunit of the inhibitor of NF-κB signaling, the IκB kinase (IKK) complex. Following the ATM/NEMO complex exports into the cytoplasm, DDR activation occurs. In the cytoplasm, this complex binds to IKKα/β and activates it. This activation is followed by phosphorylation of IκB proteins and the initiation of NF-κB signaling. CCAAT-enhancer-binding protein (C/EBPβ) is also one of the transcription factors known as regulator of inflammatory process. In cooperation with NF-κB, C/EBPβ also contributes to SASP induction (49) (Fig. 4). Moreover, in senescent cells, PARP1 [poly(ADP-ribose) polymerase 1] is an essential sensor of DNA damage, DDR regulator, and also plays a significant role in the activation of NF-κB (67).
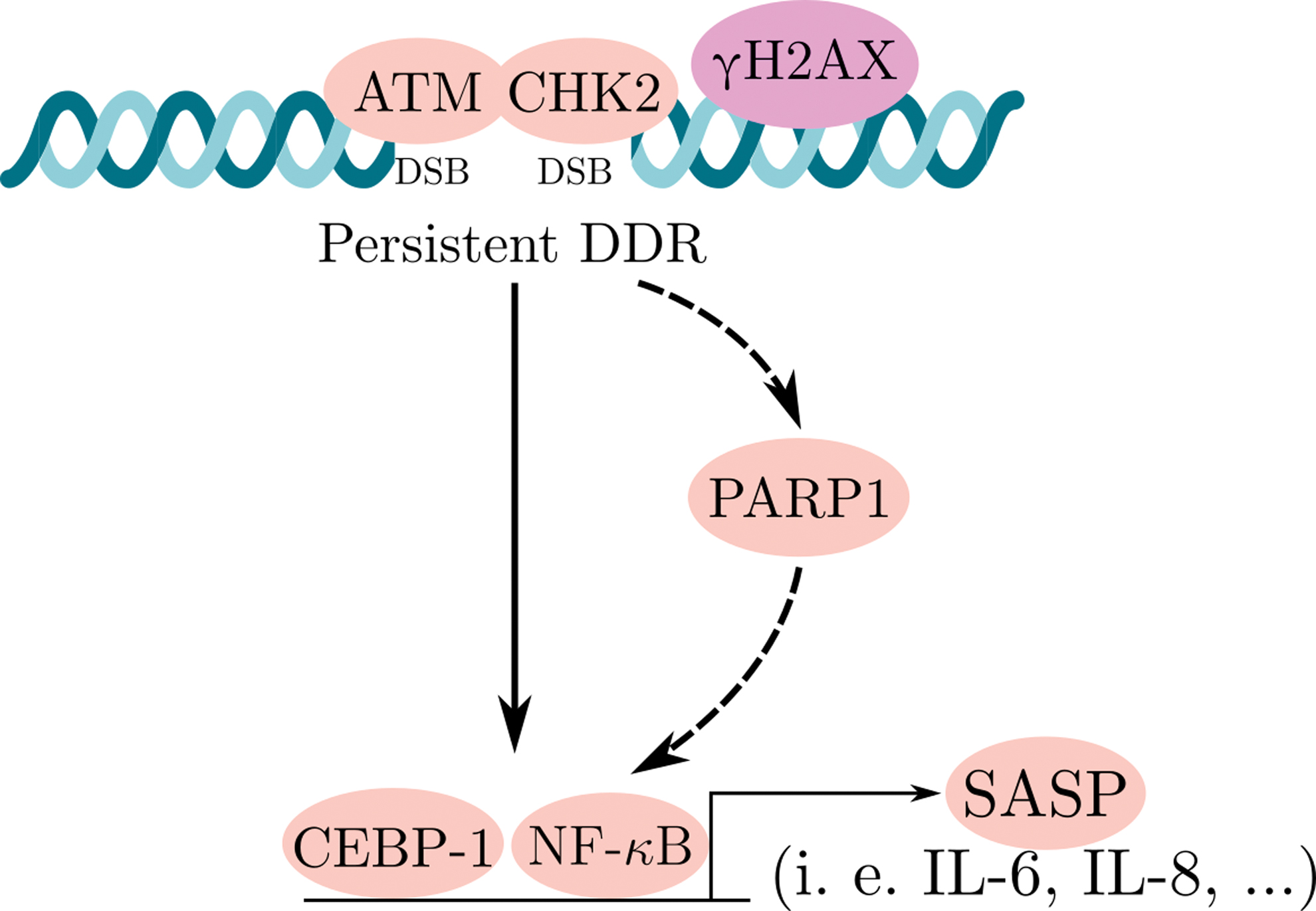
Remarkably, different transcription factors involved in DDR and SASP regulation are also known as cellular metabolism activators. Understanding the relationship between those pathways may lead to a discovery of novel mechanisms involved in SASP regulation in senescent cells.
Cellular Metabolism and Its Major Pathways
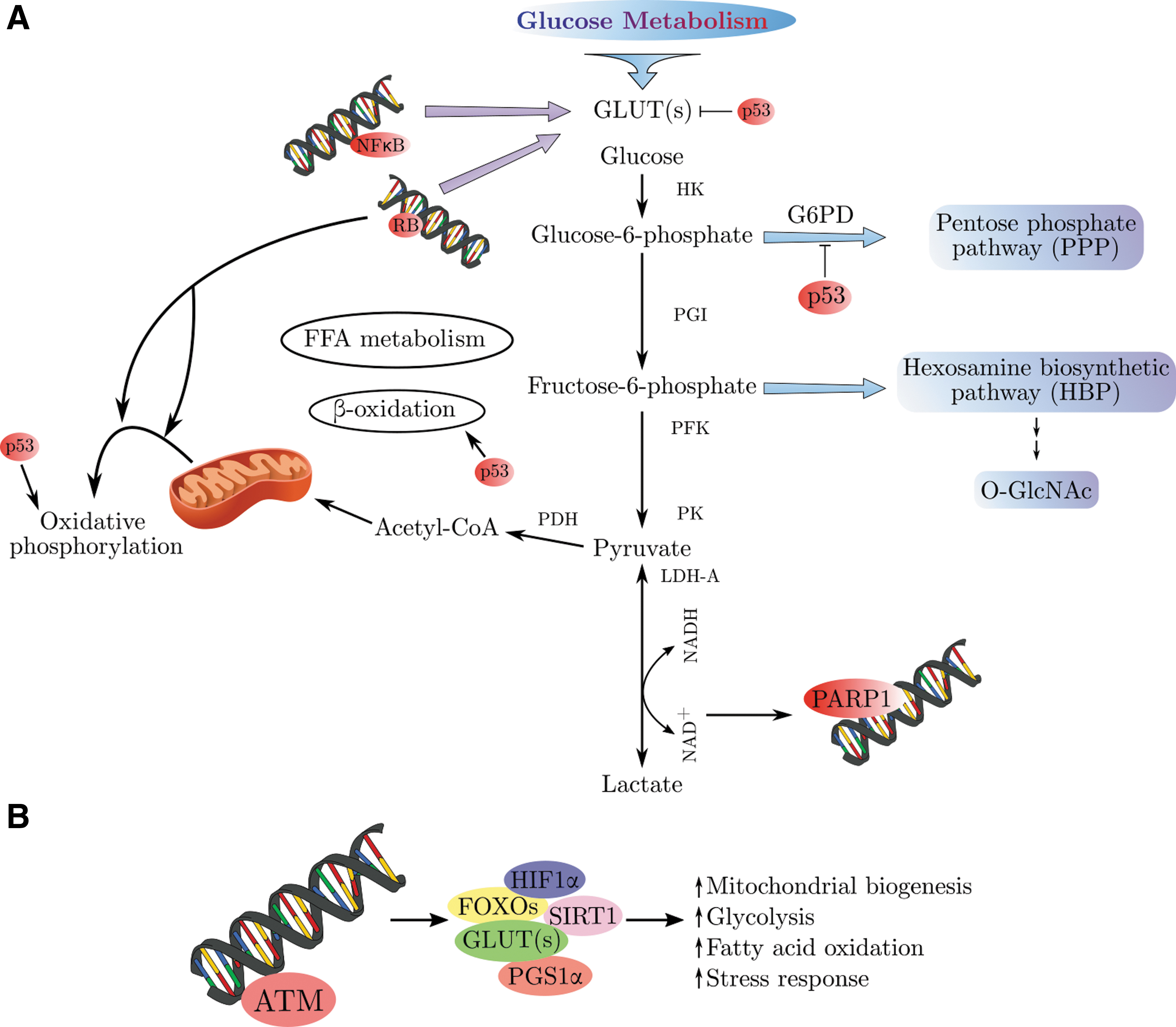
Cellular metabolism is a complicated network that consists of numerous chemical reactions. It affects all cellular processes and plays a vital role in biology. Although cellular metabolism is often discussed with regard to individual pathways, organism survival ultimately relies on the combination of all metabolic pathways. In addition, no significant metabolic pathway works entirely alone other than glycolysis. For instance, the pentose phosphate pathway (PPP) regularly depends on glucose-6-phosphate (G6P), glycolysis intermediate to move forward, and without input of nicotinamide adenine dinucleotide phosphate (NADPH) and adenosine 5-triphosphate (ATP) from two or more metabolic pathways, lipid synthesis cannot proceed. Among many other examples, the key argument is that biochemical processes cannot be easily understood individually. It is important to mention that nowadays our understanding of metabolic pathway architecture and their physiological significance has dramatically increased (Fig. 5).

Glycolysis
Glucose is one of the main sources of energy for most tissues. The metabolism of glucose begins with a 10-step reaction in the cytoplasm known as glycolysis or the glycolytic pathway. At the end of this reaction, one molecule of glucose is converted into two pyruvate molecules. In the first step, glucose is transported across the plasma membrane by GLUTs according to the concentration gradient. Hexokinase (HK), the first glycolytic enzyme, converts glucose to G6P. Following this reaction, G6P will generally enter the glycolysis pathway to produce NADH, ATP, and pyruvate, or the PPP. PPP's main and significant role is synthesis of nucleic acids for RNA and DNA. Moreover, PPP produces NADPH for the synthesis of lipids and maintaining of the intracellular redox homeostasis. Under sufficient oxygen conditions, pyruvate from glycolysis can enter the mitochondria, and there become fully oxidized to produce more ATP. Under limited oxygen conditions, however, pyruvate will prefer to be distributed as lactate. This makes glycolysis the main source for ATP production. Glycolysis also generates two molecules of ATP and reduces two NAD+ molecules, which both serve as metabolic energy supply for driving other biological processes (56).
The TCA cycle
The TCA cycle is the heart of energy metabolism. It serves as a main core for macromolecule synthesis and redox balance. TCA cycle occurs in the mitochondrial matrix and consists of series of biochemical reactions, which allow utilizing fuel sources and providing energy and macromolecules, as well as maintaining the redox balance of cells under aerobic conditions (4). While glycolysis provides only two ATPs, the two major by-products of the TCA cycle, flavin adenine dinucleotide (FADH2) and NADH, allow the production of additional 34 ATPs through OXPHOS in the ETC. Generally, the entry point into the TCA cycle is through glucose-derived pyruvate or FAs, after conversion to acetyl coenzyme A (acetyl-CoA). However, various other factors can serve as substrates for the TCA cycle, such as glutamine, following conversion to α-ketoglutarate, and the deaminated amino acids such as alanine, cysteine, glycine, serine, and threonine, following conversion to pyruvate. In addition, the TCA cycle, like glycolysis, can be used to export biosynthetic intermediates to other parts of the cell through a process called cataplerosis. There are several pathological processes with aberrant TCA cycle function. For instance, deficiency of fumarase (FH), one of the TCA cycle enzymes, is linked to genetic diseases (78). Furthermore, some of TCA cycle enzymes are dysregulated in obesity, for example, citrate synthase. It was demonstrated that citrate synthase has reduced activity in obese mice (19). In addition, several neurodegenerative disorders such as Alzheimer's disease are correlated with diminished activity of the α-ketoglutarate dehydrogenase complex (KGDHC) (30).
FA β-oxidation
The β-oxidation FA pathway is an evolutionarily well-preserved mechanism of metabolizing the FAs in the mitochondria to produce acetyl-CoA and ATP. β-oxidation is generally more complicated than glycolysis. FA molecules must first be conjugated to carnitine in the cytoplasm, and then delivered to mitochondria via a rate-limiting enzyme called carnitine palmitoyltransferase I (CPTI) before breakdown (89). Moreover, β-oxidation is also less efficient than glycolysis, at least in terms of oxygen consumption. For example, 23 molecules of O2 are consumed in the process of breaking down 1 palmitate molecule (yielding 129 ATP molecules), while only 6 O2 molecules are needed in the complete breakdown of one molecule of glucose (yielding 38 ATP molecules). Finally, the breakdown of FAs can cause significant oxidative stress to cells, in particular, for those not adequately equipped to handle an acute rise in ROS production. This is because FA breakdown delivers electrons to the ETC from NADH and FADH2, thereby yielding superoxide as a by-product (28). Importantly, very-long-chain FAs >22 carbons and branched FAs are initially oxidized in peroxisomes rather than mitochondria (29). Finally, this ends in the production of octanoyl-CoA, which is then delivered to the mitochondria for further breakdown. A distinct difference between β-oxidation in mitochondria and that in peroxisomes is that the latter does not involve ATP synthesis. Instead, NADH-carried electrons are transferred to O2 to produce H2O2, ultimately yielding H2O and O2 by the enzyme catalase (3).
Lipid metabolism
Lipids are a complex family of biochemical molecules—the major structural elements of biological membranes—including FA, triglycerides, sterols, phospholipids, and glycolipids. Lipid synthesis is an extremely regulated and controlled molecular program coordinated by the sterol regulatory element-binding proteins (SREBPs) (83). Following activation by the cellular nutrient status and upstream signaling pathways, it controls the expression of enzymes implicated in FA and cholesterol synthesis and uptake. In mammals, FASN is an important enzyme that is responsible for catalyzation of production of endogenous long-chain FA. FASN converts dietary carbohydrates to long-chain saturated FAs through NADPH, malonyl-CoA, and acetyl-CoA.
Glycolysis and DNA Damage : Contribution to Senescence
Glucose is one of the major carbon sources for most organisms, and it serves as an energy source and precursor for generating building blocks in the cell. Normal cells utilize glucose via glycolysis to obtain the final product pyruvate. Glycolysis terminates with the entrance of pyruvate into the TCA cycle and the mitochondrion in the presence of oxygen. During glycolysis, free energy is released and utilized to generate the high-energy compounds, such as ATP and NADH. Under oxygen deprivation, pyruvate is converted to lactate, which is pumped out of the cell.
In senescent cells, lactate production and glucose consumption are elevated during replicative aging (9, 37, 104). The glycolysis is elevated in senescence as a result of various triggers, including oncogene-induced senescence (59), genotoxic stress-induced senescence, and replicative senescence (22, 37, 53, 85). Elevated glycolysis was accompanied by increased extracellular acidification, resulting from increased production of lactate (37). In vivo experiments in a murine lymphoma model, where senescence was induced by chemotherapy, demonstrated that blocking the glucose usage resulted in tumor regression and improved patient outcomes (22). This positive effect was mediated, at least partially, by eliminating senescent cells from tumor and their SASP. These studies together indicate a functional connection between senescent phenotype and a highly glycolytic condition.
The cellular response to DNA damage is strongly involved in the processes that lead to alterations in glucose metabolism. One of the pathways activated in senescent cells as a response to persistent DNA damage is NF-κB. Studies from different groups (43, 44, 94) demonstrated that elevated glycolysis increases O-GlcNAcylation of IKKβ that finally activates the NF-κB pathway, as a response to DNA damage. O-GlcNAcylated protein levels increased during aging, and an increase in IKKβ protein O-GlcNAcylation enhances NF-κB activity during aging (6). NF-κB is one of the main regulators and activators of proinflammatory alterations in tissues during aging (1, 14, 52, 70). Such changes may promote glycolysis as cytokines are well-established stimulators of glycolytic activity (8, 86, 93, 98). Thus, the interplay between glycolysis and proinflammatory changes during senescence may be crucial for SASP development.
p16 protein accumulates with aging and inhibits complex of cyclin D with the CDK4 or CDK6, thus suppressing the phosphorylation of retinoblastoma (RB) tumor suppressor protein. RB acts as a transcriptional modifier and activates several metabolic pathways, including increased protein turnover and secretion, and enhanced glucose uptake and OXPHOS (35). Taken together, these data demonstrate the involvement of persistent DNA damage activation in glycolysis regulation, thus pointing at a direct influence of essential senescence pathways on metabolic state.
Mitochondrial Activity and DNA Damage During Senescence
Mitochondrion is an important cell organelle, which produces metabolites and ATP, through the TCA cycle and ETC (26). TCA cycle produces reducing equivalents of FADH2 and NADH, which deliver their electrons to the ETC, utilizing oxygen for respiration. Moreover, mitochondria serve as the main source for ROS within most mammalian cells (102, 103). Mitochondrial activity plays an essential role in senescence progression, and mitochondrial dysfunction is one of the senescence hallmarks and aging drivers (42, 47). Changes in mitochondrial OXPHOS are primarily associated with early stages of cellular senescence (50). As a result of mitochondrial dysfunction, the production of ROS is elevated in senescence cells, resulting in enhanced DNA damage and activation of DDR (20, 54, 66). The intracellular factors released by the dysfunctional mitochondria, mainly mitochondrial DNA fragments and ROS, are identified by NOD-like receptors (NLR), initiating inflammasome assembly and triggering of proinflammatory cytokines IL-1β and IL-8, which are known as SASP components (2, 47, 79). Elevated ROS levels are associated with damage- or oncogene-induced senescence, or replicative senescence (17, 39, 55). ROS induced demethylation in the promoter region of p16, leading to the upregulation of its levels and induction of senescence, providing a link between ROS and p16 (80). Moreover, long-term activation of p21 during senescence induces mitochondrial dysfunction and production of ROS (73). There is close cross talk between the induction of ROS and senescence pathways.
Senescent cells exhibit significant changes in metabolites related to mitochondrial metabolism (91). Low NAD+/NADH ratio is known as a significant inducer of cellular senescence (29). NAD+ consumption is increased due to chronic DNA damage, mitochondrial damage, and impaired functioning of the ETC during senescence. However, the intracellular NAD+ pool is decreased in senescent cells (41). This elicits PARP1 activity as a response to DNA damage (13a, 61). PARP1 is responsible for mediating multiple DDR pathways and can impair mitochondrial activity (13a, 81). PARP1 utilizes ADP(ribosyl)ate and other related proteins as a substrate for NAD+. The accelerated accumulation of poly (ADP-ribose) (PAR) at the damage sites regulates the site's accessibility through a variety of specific chromatin modifiers and determines the selection of the DNA repair pathway. Targeting PARP1/NAD+ signaling attenuates the induction of senescence (99). In addition to its effects on senescence induction, the NAD+ signaling axis might promote the proinflammatory SASP by enhancing glycolysis and mitochondrial respiration (62).
Besides, its impact on NAD+ senescence pathways affects TCA cycle metabolites. FH is an important enzyme essential for catalyzing the fumarate-to-malate transformation reaction during the TCA cycle. FH translocates to the nucleus upon DNA damage and participates in repairing DSBs in an enzymatic activity-dependent manner (36, 40). Recent studies demonstrated the effect of FH on expression levels of p53, p21, p16, and ROS production (24).
There are several transcriptional regulators impacting both cell metabolism and DNA repair processes. For example, the forkhead box O (FOXO) proteins such as FOXO3a, hypoxia-inducible factor 1α (HIF1α), and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), and several sirtuin family members, are known to coordinate multiple mitochondrial processes by targeting cell metabolism and triggering cell (Fig. 2) senescence (25, 45, 95). The interplay between DNA repair proteins and nuclear sirtuins provides a potential link between DDR and regulation of cell metabolism (Fig. 6).
Lipid Metabolism and DDR
The organelle membrane is mainly consisting of lipids. Synthesis of lipids has a major contribution to the metabolic state of the cells, and it changes significantly during aging (58). Changes in lipid synthesis lead to an increase in membrane lipid contents, and accompanied by augmented expression of lipogenic enzymes, such as ATP citrate lyase, FAS, and acetyl-CoA carboxylase (71). Lipids are integrated in the production of sphingolipids (SLs). SL metabolites, together with sphingosine, sphingosine 1-phosphate (S1P), and ceramide, are bioactive lipids playing pivotal roles in cell survival, growth, and death (31, 60). SLs are key components of cell membranes and are involved in many cellular processes, including cell differentiation, apoptosis, growth arrest, and senescence (88). At the heart of the SL, pathways are ceramides. ATM and p53 activation results in elevation of ceramide levels (13). This elevation can cause cell cycle arrest at the G2 phase, through induction of p21 (76), or at G0/G1 due to loss of pRb resulting in ceramide elevation (13). Further to the elevation of ceramides, the FAS pathway provides long-chain FAs utilizing the FASN enzyme to combine acetyl-CoA supplied from glycolysis, with malonyl-CoA (23). For example, inhibition of FASN reduces the expression of the key SASP factors ILs (e.g., IL-6, IL-1α, and IL-1β), and suppresses the secretion of small extracellular vesicles (23) Therefore, several essential lipid metabolites undergo multiple changes in senescent cells as a response to DDR.
Conclusions
Cellular senescence is a driver of various age-related and metabolic diseases. Here, we have highlighted how persistent DDR in senescent cells can alter the metabolic state. We suggest that exploiting the metabolic variations between senescent cells and nonsenescent cells holds the potential to become a useful approach to reduce the detrimental effects of senescence and provide a feasible strategy for targeting diseases associated with accumulation of senescent cells.
Footnotes
Acknowledgment
V.K. is an incumbent of The Georg F. Duckwitz Professorial Chair.
Funding Information
V.K. was supported by grants from the Israel Science Foundation (634-15) and Sagol Institute for Longevity Research.