
Abstract
Significance:
Bioinformatics has brought important insights into the field of selenium research. The progress made in the development of computational tools in the last two decades, coordinated with growing genome resources, provided new opportunities to study selenoproteins. The present review discusses existing tools for selenoprotein gene finding and other bioinformatic approaches to study the biology of selenium.
Recent Advances:
The availability of complete selenoproteomes allowed assessing a global distribution of the use of selenocysteine (Sec) across the tree of life, as well as studying the evolution of selenoproteins and their biosynthetic pathway. Beyond gene identification and characterization, human genetic variants in selenoprotein genes were used to examine adaptations to selenium levels in diverse human populations and to estimate selective constraints against gene loss.
Critical Issues:
The synthesis of selenoproteins is essential for development in mice. In humans, several mutations in selenoprotein genes have been linked to rare congenital disorders. And yet, the mechanism of Sec insertion and the regulation of selenoprotein synthesis in mammalian cells are not completely understood.
Future Directions:
Omics technologies offer new possibilities to study selenoproteins and mechanisms of Sec incorporation in cells, tissues, and organisms.
Introduction
Selenium is an essential trace element present in selenoproteins in the form of the amino acid selenocysteine (Sec). The human genome encodes 25 selenoproteins (51), which play important roles in health and disease [reviewed in references (40, 54)]. Most selenoproteins are enzymes with Sec located at the active site and perform redox reactions in diverse cellular functions, including removal of hydroperoxides, reduction of thioredoxin, repair of oxidized methionines, and metabolism of thyroid hormones, among others.
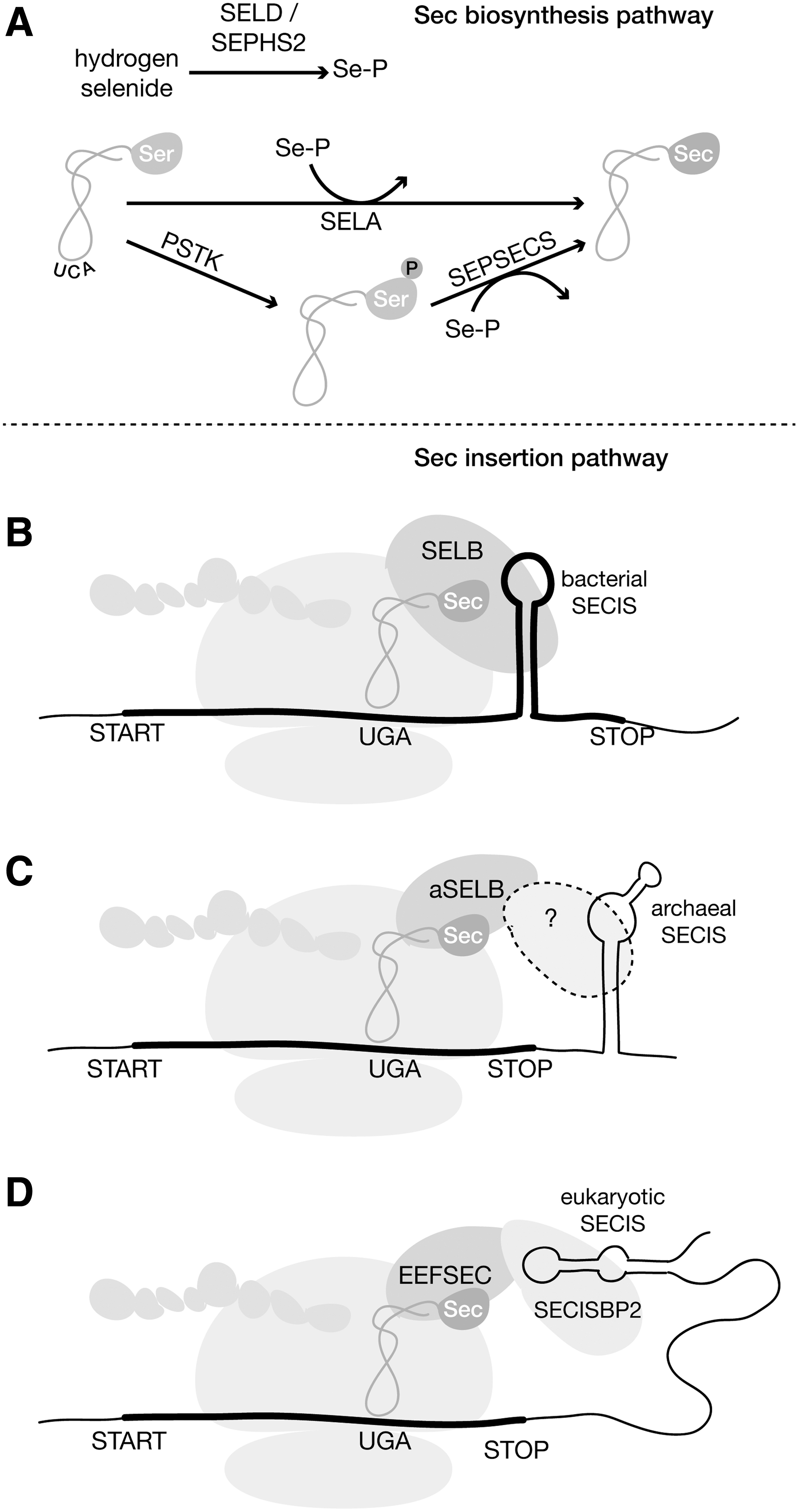
Sec is encoded by the opal stop codon UGA, which is recoded through the concerted action of a dedicated tRNA and protein machinery, triggered in selenoprotein mRNAs by the presence of specific RNA motifs. Selenoproteins are found scattered in eukaryotes, bacteria, and archaea, with important differences in the mechanisms of Sec insertion in the three domains (Fig. 1).
This review discusses the bioinformatic approaches used to address the challenges of selenoprotein annotation in genomes, the most relevant computational tools and resources in selenium genomics, and the current knowledge in the evolution and distribution of selenoproteins across the tree of life.
Preface: the discovery and characterization of Sec
Following the identification of selenium as an integral constituent of some proteins in 1973 (3, 27, 86, 96), a series of studies elucidated the molecular and genetic basis for the synthesis of the newly discovered selenoproteins. It was established that selenium was present in selenoproteins in the form of Sec (22, 49), and that the Sec residue was encoded by an in-frame TGA codon (UGA in mRNA) (45, 112). A Sec-inserting tRNA (tRNA-Sec) was identified as the SelC gene in Escherichia coli (56) and animals (55). Thus, Sec was recognized as an expansion to the genetic code, through which the nonsense codon UGA is recoded as a sense codon to specify Sec insertion. The letter “U” was added into the one letter amino acid code to designate Sec (9).
The sufficient Sec recoding signal was identified as a cis-acting RNA secondary structure present in selenoprotein mRNAs, which was termed Sec-insertion sequence (SECIS) (8, 98, 113) (Fig. 2). Besides tRNA-Sec, additional trans-acting protein factors required for Sec insertion were then identified, including a Sec-specific elongation factor (EEFSEC) and an SECIS-binding protein (SECISBP2), revealing lineage-specific differences between the bacterial, archaeal, and eukaryotic Sec systems (54). Sec is the most widespread expansion to the genetic code in nature, and UGA is the only codon with ambiguous meaning in all domains of life. For this reason, selenoproteins are commonly mispredicted by standard gene finding programs, and misannotated in genome annotation projects and protein databases.

Selenoprotein Gene Annotation
Selenoproteins are usually mispredicted in nonmodel organisms because standard gene annotation programs consider UGA only as stop signal, whereas correct identification of Sec-UGA codons requires additional curation steps. Thus, selenoprotein sequences are commonly truncated in protein databases. Typical misannotations include Sec-UGA considered stop, the Sec-UGA-containing exon being skipped, or coding sequence (CDS) starting downstream of Sec-UGA (Fig. 3D). Errors in selenoprotein annotation are propagated as protein sequences from databases are used to annotate new genomes.
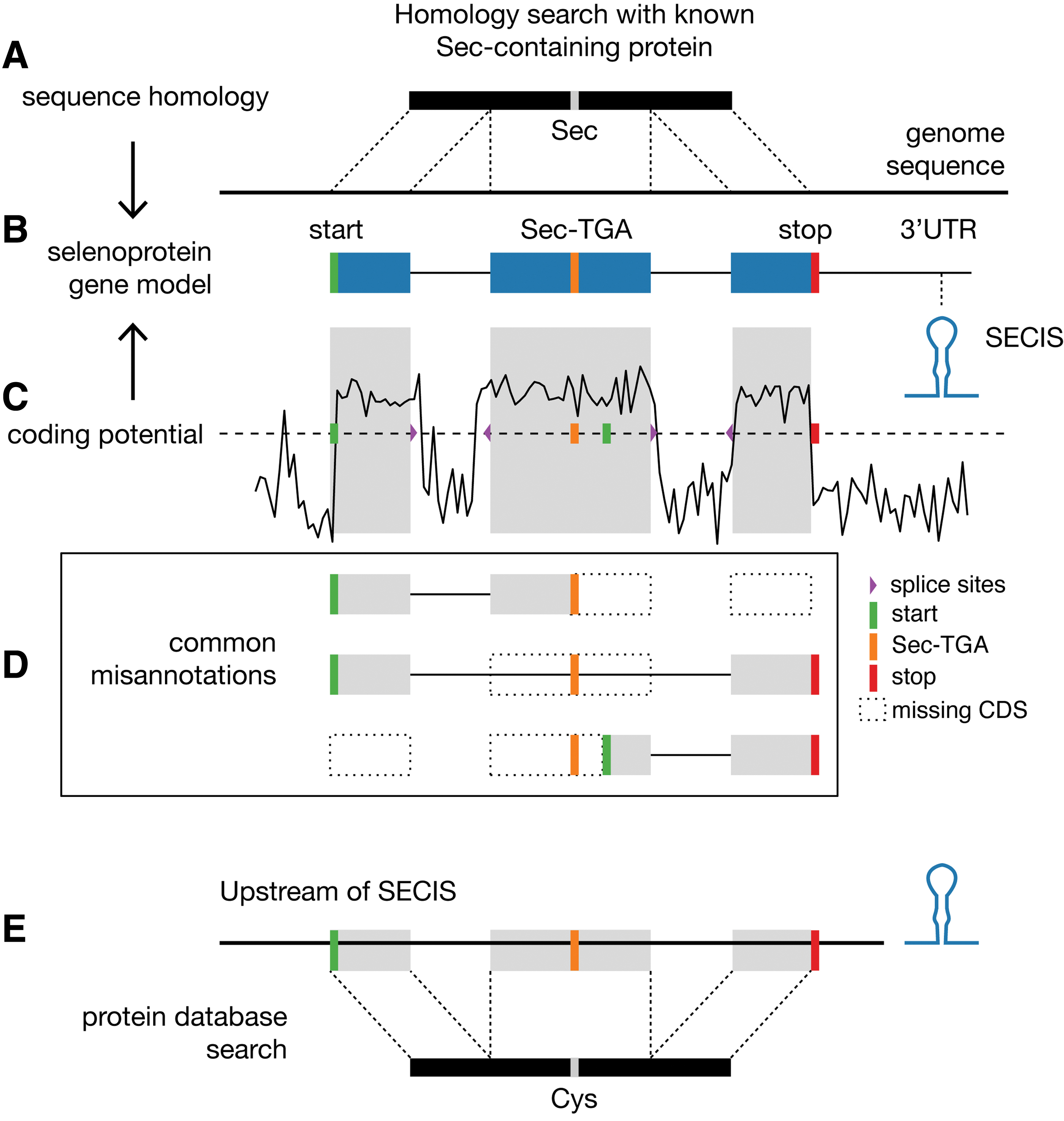
The main features to support the correct identification and annotation of selenoprotein candidates are (i) the presence of a properly located SECIS; (ii) identification of selenoprotein homologues (Sec/Sec alignment); (iii) identification of cysteine (Cys)-containing homologues (Sec/Cys alignment); and (iv) sequence signatures of protein coding potential both at the 5′ and 3′ of UGA. Finding selenoproteins encoded in genomic sequences can be divided into two conceptually different problems: finding known selenoproteins and finding novel selenoproteins.
Gene finding for known selenoproteins
Selenoprotein genes can be identified by their homology to sequences of previously characterized selenoproteins (Fig. 3A). Briefly, a “query” Sec-containing amino acid sequence is used to scan a “target” DNA or RNA sequence assembly (e.g., genome, transcriptome, or metagenome) using Tblastn (2) or analogous approaches. Tblastn translates the nucleotide sequence into all six frames and finds high-scoring regions of amino acid sequence similarity between query and target. Hits that align the Sec residue in the query with an in-frame TGA in the target are potential indicators of a selenoprotein gene. Typically, SECIS elements are then searched at an appropriate distance from candidates (SECIS prediction is summarized below). Homology searches allow identifying not only selenoproteins but also additional standard genes in the same protein families. Typically, these homologues carry Cys in place of Sec at the active site, so we refer to them as Cys-homologues.
Gene finding for novel selenoproteins
The strategies used for identification of novel selenoproteins (i.e., those without any Sec-containing homologue) rely on the identification of SECIS elements. These cis-acting elements differ among the domains of life in location (3′ untranslated region [UTR] in eukaryotes, archaea; flanking Sec-UGA in bacteria) (Fig. 1), as well as RNA structure and identity (Fig. 2), so that every domain requires dedicated adjustments of the general approach. Briefly, a DNA or an RNA sequence assembly is scanned to find SECIS candidates with dedicated methods (detailed later). The corresponding region of each SECIS is analyzed for the occurrence of TGA-containing open reading frames (ORFs), and then evaluated in terms of their likelihood to be coding for a selenoprotein. In the field of gene prediction, two main principles exist to assess the “coding potential” of a sequence: (i) ab initio gene finding and (ii) homology-based matching.
In ab initio gene prediction (Fig. 3C), coding regions are distinguished from noncoding regions by virtue of intrinsic sequence features, such as nucleotide composition, codon usage, and triplet periodicity. The conserved patterns of the splice junctions are also used to build complete gene models, including intron/exon structures. Ab initio prediction is used for de novo identification of selenoproteins, searching for such protein coding signature extending past a TGA codon with a predicted SECIS at the appropriate location (15, 46).
In homology-based matching, nucleotide sequences nearby SECIS elements are translated in six frames and aligned to a database of proteins. This procedure is analogous to the detection of known selenoprotein outlined above, in which a TGA is aligned to Sec residue of an annotated protein. For novel selenoproteins, however, matches between a TGA codon and a Cys residue of an annotated protein are considered. The assumption is that Cys-homologues already exist annotated in protein databases for selenoproteins, which are not discovered yet. Since almost all selenoproteins have Cys-homologues, this strategy allows discovering novel selenoproteins.
Sec machinery factors
The biosynthesis and cotranslational insertion of Sec require the action of a dedicated genetic machinery. While essential for human and other vertebrates, Sec is not ubiquitous to life. The identification of the Sec machinery in the genomes of different species has been used to perform large-scale surveys of the distribution of Sec usage across the tree of life (55, 73, 79, 82, 88). Besides the cis-acting SECIS element, the Sec pathway in eukaryotes requires six obligate trans-acting factors: tRNA-Sec (Sec-tRNA[Ser]Sec) charged with Sec (100), selenophosphate synthetase SEPHS2 (99),
The biosynthesis of Sec occurs on its cognate tRNA, whose anticodon UCA is complementary to UGA. tRNA-Sec is initially aminoacylated with serine (Ser) by seryl-tRNA synthetase, which provides the backbone for Sec synthesis. In bacteria, the pyridoxal phosphate-dependent protein Sec synthase (SELA) converts Ser to Sec. In eukaryotes and archaea, a two-step reaction occurs, in which Ser is first phosphorylated by the kinase PSTK, producing phosphoserine, which is then converted to Sec by the Sec synthase SEPSECS (100) (phylogenetically distinct from bacterial SELA). The active selenium donor selenophosphate is synthesized from selenide by selenophosphate synthetase SEPHS2 (SELD in prokaryotes) (34, 64).
Cotranslational incorporation of Sec is promoted by the SECIS secondary structure and requires the Sec-specific elongation factor EEFSEC in eukaryotes and SELB in prokaryotes. In bacteria, SELB also functions as an SECIS-binding protein, whereas, in eukaryotes, SECISBP2 binds the SECIS (EEFSEC has no SECIS-binding activity). The Sec pathway in archaea is analogous to eukaryotes, but notably there is no SECISBP2 and its function is still not assigned in this lineage (84). Identification of Sec machinery in all domains is typically performed by homology matching. Specialized tools have been developed for this task, described later.
The Rise of Selenium Genomics
Selenium genomics of eukaryotes
The investigation of selenoprotein genes in genomes was kick-started by the development of programs to detect SECIS elements in sequences. The first computational tool for eukaryotic SECIS finding was SECISearch (53). The program was used to predict SECIS elements in transcript sequences, which were then analyzed to find corresponding TGA-containing ORFs with the selenoprotein-coding potential. An analogous approach was carried out (57). These two studies led to the first three novel selenoproteins identified through computational means, and Sec incorporation into these proteins was confirmed by labeling the cells with radioactive selenium. These three proteins are currently known as MSRB1, SELENOT, and SELENON (33).
The first complete selenoproteome was characterized 2 years later in the then newly sequenced genome of Drosophila melanogaster (15, 75). One study used an ab initio strategy, in which TGA-containing ORFs near predicted SECIS elements were assessed for coding potential using the program geneid (37). Geneid was modified specifically for this task to allow in-frame TGA in ORFs. Another study used a similar approach focusing on the prediction of SECIS elements followed by the identification of selenoprotein ORFs (75). This resulted in the identification of three selenoproteins in D. melanogaster, two of which were novel.
A major advance in the field was the characterization of the complete human selenoproteome (51). This study exploited the sequence conservation of SECIS elements in orthologous selenoprotein genes between human, rat, and mouse, to improve the specificity of SECIS genomic searches. Twenty-five human selenoproteins were identified, seven of which were novel.
Later studies also used comparative approaches to discover additional selenoproteins in other organisms. For example, the selenoprotein U (SELENOU), found in fish and present as Cys-homologue in mammals, was found by comparing TGA-containing ORF predictions from the puffer fish Fugu and human (16). The limitations of the comparative approaches, which require Sec- or Cys-containing sequence homologues, were highlighted by the ab initio discovery of selenoprotein J (SELENOJ) in another puffer fish, which has no homologue in mammals (14). Since then, many more selenoproteomes have been described using computational approaches (5, 47, 48, 60, 61, 71, 93, 111).
Selenium genomics of prokaryotes
Naturally, analogous computational approaches were applied to bacteria and archaea, accounting for the differences in the domain-specific SECIS elements (Fig. 1) (50). The bacterial SECIS (bSECIS) is located immediately downstream of the UGA, within the CDS (7), and its function is constrained by the stem-loop structure and the distance to the UGA codon (21). The conserved structural features among known bSECIS elements were used to build a consensus model, which allowed developing the program bSECISearch to identify selenoproteins in bacterial genomes (105). The program scans a nucleotide sequence and examines the occurrence of potential bSECIS downstream of each UGA triplet. SECIS-independent approaches based on homology matching with Cys-containing proteins have also been applied to bacterial genomes and environmental samples, yielding many novel selenoproteins (104, 106).
More recently, a novel selenoprotein named DUF466, with a predicted C-terminal Sec residue, was identified in Helicobacter pylori (25), and analysis on bacterial genomes with tRNA-Sec but without known selenoproteins revealed putative Sec-containing “redox-active disulfide 2” genes in Brachyspira bacteria (88). As of today, more than 50 bacterial selenoproteins are known (103). It is estimated that only 20%–25% of bacteria use Sec, with a scattered distribution across different phyla (73, 79).
Archaeal selenoprotein genes possess, unlike bacteria, an SECIS element located in the 3′ UTR (98). A SECIS-based approach was developed to identify selenoproteins in archaeal genomes (52). The number of archaeal genomes that contain selenoproteins is limited. Until recently, they were confined to Methanococcales and Methanopyrales (85). Then, analyses of uncultivated metagenomes from the newly discovered Asgard archaea revealed selenoproteins in Lokiarchaeota (70) and Thorarchaeota (59).
Computational Tools for Selenoprotein Gene Prediction
The fast pace of genome sequencing in the last decade has rendered impractical the manual identification of selenoproteins. Thanks to the following computational tools dedicated to selenium genomics, a large number of genomes can be analyzed nowadays with little or no manual intervention.
Selenoprofiles (68) is a homology-based pipeline for prediction of known selenoproteins in genomic sequences. The program is also suited for finding Cys-containing homologues, as well as the protein factors of the Sec machinery. The program comes with a default set of manually curated profiles for all known selenoproteins and machinery protein factors, and can be extended to annotate standard genes (89). Selenoprofiles is available for download.
SECISearch3 (67, 69) is currently the most efficient and widely used method for identification of eukaryotic SECIS elements. SECISearch3 constitutes an improvement of the original SECISearch (53) through the incorporation of covariance models, improving speed and accuracy of RNA motif finding. The program is also a component of Seblastian (67, 69), an SECIS-dependent pipeline for the identification of eukaryotic selenoproteins. Seblastian analyzes the regions upstream of SECIS elements to find selenoprotein genes by homology. Seblastian can be used either to identify known selenoproteins (matching ORFs to Sec-containing annotated homologues) or to predict novel selenoproteins (matching to Cys-homologues). Both SECISearch3 and Seblastian are available online as web servers. Then, structural analysis of eukaryotic SECIS can be performed with a dedicated tool, SECISaln (18).
For finding bacterial SECIS elements, the method bSECISearch (105) is used. This program, available online, scans nucleotide sequences and returns potential bSECIS elements as well as their host ORF. bSECIS elements are identified by matching a predefined structural pattern and several sequence constraints.
SelGenAmic (46) is an ab initio gene predictor specifically developed for selenoprotein genes. The program is based on geneid (37) and uses the coding potential and splice sites to build gene models that include an in-frame TGA codon. Coupled with SECIS prediction, the program has been used to identify selenoproteins in various metazoans (47, 48).
Secmarker (88) is a program for the identification of tRNA-Sec in genomes, built upon realizing that existing tools for generic tRNA finding in genomes performed badly for tRNA-Sec. This gene is a good marker for the presence of selenoproteins in a genome. Therefore, Secmarker can be used to quickly scan thousands of genomes and predict which ones might encode for selenoproteins. The program is available online as web server and for download.
Selenoprotein databases
Due to the historic unreliability of broadly used bioinformatic resources for selenium research, a number of specialized databases have been created for selenoproteins. SelenoDB was built to provide manually curated selenoproteomes, which include gene, protein, and SECIS sequences for a number of model organisms (13). Its second release expanded the database with automatic annotations by Selenoprofiles to include a larger number of genomes, mostly vertebrates (81). Then, the database dbTEU is a collection of proteins from both prokaryotes and eukaryotes, which are related to trace elements, including selenoproteins (108). Next, recode is a database for genes that use a nonstandard translation through recoding events, such as Sec and other types of stop codon redefinition (6). Finally, recent efforts were made to reannotate selenoproteins in the NCBI Reference Sequence database (RefSeq) for prokaryotes (39) and vertebrates (80).
Distribution of Selenoproteins Across the Tree of Life
The rise of sequencing technologies brought great biodiversity at the disposal of scientists in the form of nucleotide sequences. Many researchers analyzed sequences throughout the tree of life to profile the taxonomic distribution of selenoproteins and other forms of selenium utilization. There is compelling evidence to support that the Sec trait evolved only once, for example, prokaryotes and eukaryotes use analogous Sec biosynthesis and insertion pathways, and some selenoproteins are shared among the three domains. The Sec trait was then lost in many lineages independently resulting in a scattered distribution of selenoproteins in nature (72, 73, 109).
Eukaryotes
The distribution of selenoproteins in eukaryotes outlines a highly dynamic evolutionary history (Fig. 4). Many selenoprotein families are shared between single-cell eukaryotes and vertebrates, notably selenophosphate synthase (SEPHS2), glutathione peroxidases (GPXs), thioredoxin reductases, and methionine-R-sulfoxide reductase, among others (54), indicating an early origin for most eukaryotic selenoproteins (32). Some of them subsequently replaced Sec by Cys, or were lost altogether, in different lineages (61). The number of selenoproteins varies greatly between organisms: the largest selenoproteome described to date corresponds to the harmful pelagophyte alga Aureococcus anophagefferens, with 59 selenoproteins (35, 36), while the nematode Caenorhabditis elegans encodes only a single selenoprotein (93), and many other eukaryotes lack selenoproteins completely (73).

Selenoproteins are widespread among metazoans. The richest selenoproteomes among animals are found in vertebrates and other deuterostomes [such as lancelet (48) and echinoderms]. The reconstructed ancestral vertebrate selenoproteome consists of 28 genes, whose evolutionary history was thoroughly described (71). The 25 human selenoproteins are tightly conserved across mammals (mouse has 24, with GPX6 containing Cys instead of Sec). Fish genomes encode up to 38 selenoprotein genes, product of multiple lineage-specific gene duplications (71). Three selenoprotein families are vertebrate-specific: SELENOI (EPT1 or SelI), SELENOV (SelV), and SELENOE (Fep15, found only in fish). Interestingly, SELENOI is involved in neural development (42) and appeared to be one of the most important human selenoproteins, based on selective constraints against loss-of-function (LoF) variants in the general population (87).
The patterns of selenoprotein evolution in vertebrates have been studied to investigate the evolutionary forces that shaped them. Two environmental factors have been proposed to play important roles in the evolution of selenoproteins: availability of selenium and aquatic environment. The two effects may be intertwined, as selenium is abundant in water but scarce in land; however, an alternative explanation is that selenoproteins are more beneficial in aquatic than terrestrial environments because of different exposures to oxidants. Several observations support the role of these factors. Aquatic organisms typically encode for larger sets of selenoproteins than terrestrial ones (5, 61).
Further insights come from the analysis of SELENOP, the only human selenoprotein with multiple Sec residues. SELENOP is a secreted plasma protein that distributes selenium throughout the body, prioritizing tissues with high requirements for this element (11). SELENOP functions as a quantitative marker of selenium utilization at various evolutionary scales: its concentration in plasma reflects selenium availability in mouse and human (11, 41), and its number of encoded Sec residues correlates with the selenoproteome size across genomes of vertebrates (63) and other animals (5). Notably, SELENOP has fewer Sec residues in mammals compared with teleost fishes (63), again supporting the role of aquatic environment in shaping selenoprotein evolution. Indeed, recent evolutionary analyses showed that the SELENOP Sec content is under selection in fish, but in contrast it declined in a neutral manner in mammals (90). In the same study, terrestrial vertebrates showed relaxed evolutionary constraints throughout the whole set of genes that use or regulate selenium, when compared with fish (90).
Protostomes, the sister clade of deuterostomes (which includes vertebrates), encompass a huge diversity of invertebrate animal life-forms. Insects and nematodes encode minimal selenoproteomes: D. melanogaster has three selenoproteins, and C. elegans only one. Some noninsect arthropods encode larger selenoproteomes (Fig. 4), but no systematic study has been reported for these groups yet. All known selenoprotein-less animals belong to arthropods or nematodes. Within arthropods, Sec was lost independently in several insect orders (17, 62, 66), and in at least two genera of mites (arachnids) (88). Some parasitic plant nematodes are also devoid of selenoproteins (78), while other nematodes have minimal selenoproteomes (93) (Fig. 4).
An extensive study describing the evolution of SELENOP in Metazoa, including diverse invertebrate lineages, was recently reported (5). SELENOP is present throughout Metazoa, but it was lost in tunicates, crustaceans, platyhelminthes, most nematodes, and most insects. The Sec content appeared to be particularly high in some Lophotrochozoa (lineage including Annelida, Nemertea, and molluscs): SELENOP of pacific oyster Magallana gigas has 46 Sec-UGAs; in freshwater mussel Elliptio complanata, it has 132 Sec-UGAs (5). The remarkable diversity of invertebrates is particularly interesting for the study of the evolution of selenoproteins and can provide valuable insights into the biology of selenium in animal life.
Selenoproteins were recently discovered in fungi, a lineage that was previously believed to be devoid of these proteins (72). At least nine genomes from diverse early-branching fungal phyla appear to encode selenoproteins and the Sec machinery. The absence of Sec in most fungal genomes is the product of multiple independent Sec extinction events (Fig. 4). Notably, Sec was lost at the root of Dikarya, which comprises Ascomycota and Basidiomycota, and led to the absence of selenoproteins in Saccharomyces cerevisiae and other yeasts. No selenoproteins have been identified in land plants so far (73), whereas algae use Sec and are often selenoprotein rich (4, 58, 61, 77).
Protists are a paraphyletic group of unicellular eukaryotes representing massive and largely unexplored biodiversity. The distribution of selenoproteins is highly scattered throughout protist genomes (73, 88) including many selenoprotein-less species, as well as the largest selenoproteomes known (4, 36) (Fig. 4).
Bacteria
Analysis of fully sequenced genomes indicates that selenoproteins are used by 20%–25% of bacteria, displaying a widely scattered distribution across different lineages (73, 79, 109). Around 50 selenoprotein families have been identified so far in bacteria (103), only a few of which are also present in eukaryotes: selenophosphate synthetase (SelD), GPX, deiodinase-like, methionine-S-sulfoxide reductase (MsrA), alkyl hydroperoxide reductase C (AhpC) and radical SAM domain proteins (RSAM). Most bacterial selenoproteins were predicted using bioinformatics in genomes or metagenomes derived from environmental sequencing projects (19, 52, 104 –106).
Despite the fact that the majority of bacteria (∼75%) do not use Sec, most of the clades contain members with selenoproteins. Selenoprotein-rich bacteria were identified in three phyla: Deltaproteobacteria, Clostridia, and Synergistetes (79), with the largest selenoproteome to date identified in Syntrophobacter fumaroxidans (Deltaproteobacteria) with 39 selenoproteins (107). Selenoprotein-containing bacteria encompass a great diversity, but most of them are obligate or facultative anaerobic (83, 92). Many of the characterized selenoproteins are involved in energy metabolism, as well as other processes such as redox cycling and Sec synthesis itself (83, 92). However, most bacterial selenoproteins were identified through bioinformatic analysis and their functions could only be inferred by sequence homology. Additional studies will be necessary to better understand the biology of selenium in prokaryotes.
In addition, it is very likely that many selenoproteins have not been identified yet, and more efficient and accurate tools will be necessary to analyze the overwhelming number of genomes and metagenomes being sequenced. Recently, it was discovered that some bacteria use non-UGA codons for Sec incorporation (76), in a remarkable example of flexibility of the genetic code. tRNA-Sec genes were identified that recognize the stop codons UAG and UAA, and 10 other sense codons. Accordingly, selenoprotein genes that contain the corresponding non-UGA codon in the Sec position were identified, and Sec insertion by those tRNAs was confirmed experimentally.
Archaea
Selenoproteins in archaea show a very narrow distribution, observed so far only in three phyla (84). Nine selenoprotein families are known, the majority of which are involved in methanogenesis (84, 92). The selenoprotein synthesis pathway in archaea resembles the eukaryotic system, rather than the bacterial one (92). The recent discovery of selenoproteins in the newly described Asgard lineage, the closest archaeal relatives to eukaryotes (102), brought key insights into the evolution of Sec insertion (70). Asgard archaea have selenoproteins with SECIS elements that resemble eukaryotes, rather than other archaea (methanogens), indicating that the eukaryotic SECIS motif predated the origin of eukaryotes. A key feature of this motif, the kink-turn core, was also found in genomes of methanogens: it forms part of the archaeal SECIS of a single selenoprotein gene, vhuD, leading to speculation that vhuD is the progenitor of eukaryotic SECIS elements. A long-standing question regarding the selenoprotein synthesis pathway in archaea still remains elusive: the identity of the archaeal SECIS-binding protein is unknown (84).
Beyond Gene Prediction
Functional studies: Ribo-seq
The synthesis of selenoproteins is essential for mammals and its disruption causes lethality in mice [reviewed in ref. (23)]. Mutations in selenoproteins or in genes required for their synthesis result in various diseases in humans [reviewed in refs. (28, 30, 91)]. Selenoprotein synthesis has been extensively studied, and all the required and sufficient factors for Sec insertion in eukaryotes are known (38). Yet, many aspects of the mechanism for Sec incorporation are still unclear, including the roles of the various factors, its efficiency, and regulation (44). Ribosome profiling (Ribo-seq) has emerged as an important tool for studying the mechanism of selenoprotein synthesis (26). This relatively new technique is based on high-throughput sequencing of ribosome-protected fragments (RPFs), ∼30-nucleotide long reads roughly corresponding to the sequence covered by a single ribosome. The analysis of RPFs reveals which regions of the transcriptome are under active translation. Quantification of RPFs provides a measure of ribosome abundance with codon resolution.
Ribo-seq experiments are useful to study many aspects of translation. Particularly, for selenoproteins, it allows to quantify UGA readthrough as a proxy for Sec incorporation efficiency. Translation of selenoproteins in mouse liver has been used to study the effects of dietary selenium (43, 94), the role of Secisbp2 on UGA recoding and selenoprotein mRNA stability (29), and the effects of pathogenic missense mutations in Secisbp2 mouse models (110). One particularly interesting example of UGA recoding is the gene SelenoP, which contains two SECIS elements and multiple UGA codons. Ribo-seq was applied to study the role of each of the two SECIS in mouse (74), and more recently, to study the translation of the 46 UGA-containing SelenoP of pacific oyster (5).
Population genetics
The study of genetic variation in selenoprotein genes from human populations is a powerful tool to study human adaptation, selection, and disease. Recently, the population dynamics of variants in selenoproteins and selenium-related genes was analyzed to address the response of different populations to variable selenium levels in the soil (97).
More recently, selenoprotein genes were analyzed to assess the frequency of LoF variants in populations, yielding estimated levels of tolerance to the loss of these genes in human (87). These were compared with knockout phenotypes in mice. While good correspondence was found between human and mouse, notable differences were found in some genes, such as difference in tolerance of GPx4, in which mice were more sensitive, strikingly, iodothyronine deiodinases seemed to not tolerate LoF variants in humans, whereas their deletion in mice produced only a mild phenotype (31).
Conclusions
Twenty years on, bioinformatics has been pushing forward the field of selenium research. Since the main genetic features of selenoproteins were established, scientists found in selenoproteins intriguing questions that could be addressed by sequence analysis. Selenoproteins were an early application of bioinformatics, before the human genome was published. The prediction of selenoproteins in nucleotide sequences is still challenging, mainly due to the growing number of genomes and transcriptomes available. Automated programs alleviated this, and now allow carrying out large-scale surveys, which provided a much-detailed map of Sec usage across the tree of life.
Selenoprotein research also benefited from new sequencing techniques, such as ribosome profiling, for it is now possible to estimate the efficiency of Sec incorporation in vivo. Sequencing of the genomes and exomes of human populations, which is now in the range of hundreds of thousands of individuals, as well as sequencing of diseased patients, promises new insights into the molecular mechanisms of selenoproteins and the role of selenium in health and disease.
Funding Information
Supported by the National Institutes of Health grants to V.N.G.