
Abstract
Aims:
The aim of the present study was to investigate the biochemical properties of nitrosopersulfide (SSNO−), a key intermediate of the nitric oxide (NO)/sulfide cross talk.
Results:
We obtained corroborating evidence that SSNO− is indeed a major product of the reaction of S-nitrosothiols with hydrogen sulfide (H2S). It was found to be relatively stable (t1/2 ∼1 h at room temperature) in aqueous solution of physiological pH, stabilized by the presence of excess sulfide and resistant toward reduction by other thiols. Furthermore, we here show that SSNO− escapes the reducing power of the NADPH-driven biological reducing machineries, the thioredoxin and glutathione reductase systems. The slow decomposition of SSNO− produces inorganic polysulfide species, which effectively induce per/polysulfidation on glutathione or protein cysteine (Cys) residues. Our data also demonstrate that, in contrast to the transient activation by inorganic polysulfides, SSNO− induces long-term potentiation of TRPA1 (transient receptor potential ankyrin 1) channels, which may be due to its propensity to generate a slow flux of polysulfide in situ.
Innovation:
The characterized properties of SSNO− would seem to represent unique features in cell signaling by enabling sulfur and nitrogen trafficking within the reducing environment of the cytosol, with targeted release of both NO and polysulfide equivalents.
Conclusion:
SSNO− is a surprisingly stable bioactive product of the chemical interaction of S-nitrosothiol species and H2S that is resistant to reduction by the thioredoxin and glutathione systems. As well as generating NO, it releases inorganic polysulfides, enabling transfer of sulfane sulfur species to peptide/protein Cys residues. The sustained activation of TRPA1 channels by SSNO− is most likely linked to all these properties.
Introduction
Following the discoveries of nitric oxide (NO) and carbon monoxide (CO) as small endogenously produced signaling molecules, hydrogen sulfide (H2S) has emerged as the “new kid on the block” with an even wider ranging array of biological actions compared with the former two entities (1, 30, 33, 38 –40). Cooperative effects between NO and H2S were first observed by Kimura and colleagues, raising the possibility of a cross talk between their mediator functions (20). Several elegant studies since then contributed to accumulate further evidence for the reciprocal regulation of NO and sulfide biosynthetic pathways and the mutually cooperative nature of interaction at the level of cell signaling (23). Furthermore, as their biosynthetic machineries are often colocalized in a variety of cells and tissues, the potential for chemical interaction to produce secondary bioactive products has become an increasingly attractive field of investigation; those research activities will likely uncover many new layers of regulatory processes in redox biology in the near future. To conceptualize the chemical and functional interactions of NO- and H2S-derived reactive nitrogen species and reactive sulfur species with the long-studied reactive oxygen species, we recently introduced the concept of the “Reactive Species Interactome” (RSI) (10, 12). The RSI represents the biochemical sensing and adaptation system that enables individual cells (and entire organisms) to adjust their metabolic machinery to changes in demand or environmental conditions to stay “fit for purpose”. To this end, sulfur–nitrogen compounds likely play a fundamental role in enabling survival.
Innovation
Based on in vitro data, we propose that nitrosopersulfide (SSNO−) is an important chemical entity in the cross talk between nitric oxide (NO) and hydrogen sulfide (H2S)/persulfide signaling. It may act as a dual sulfane sulfur and NO trafficking molecule in biology, which can escape the cellular reducing power. By slowly releasing peptide/protein cysteine-persulfidating inorganic polysulfide species and NO distant to its site of generation, this S/N hybrid species may act as a mobile carrier of sulfane sulfur equivalents and NO to trigger sustained intracellular effects.
In their earlier studies, Whiteman et al. proposed the formation of thionitrous acid (HSNO), the smallest possible nitrosothiol, as a central intermediate in the chemical interactions of NO with sulfide species (43). Filipovic et al. later claimed that the reactions between S-nitrosothiols (RSNO) and sulfide give rise to the formation of HSNO; the authors proposed that HSNO is relatively stable (under physiological conditions the suggested half-life was 30 min) and acts as a source of NO+, NO•, and NO−, with the potential to support intracellular transnitrosation events (18). On the contrary, we and others argued that this compound is rather unstable [t1/2 ∼6s measured by Filipovic et al. themselves in Ref. (18)] and undergoes rapid isomerization, homolysis, and polymerization reactions, questioning its ability to serve as a central molecule in regulating intracellular signaling events (9, 26, 28). Moreover, accumulating evidence demonstrated that the direct chemical interactions between NO donors and H2S result in a network of cascading chemical reactions, with the generation of a plethora of bioactive molecules including nitrosopersulfide (SSNO−), sulfite (SO3
−), dinitrososulfite (SULFI/NO), and inorganic polysulfides (11). We demonstrated that SSNO− formation during the reactions of nitrosothiols with sulfide is a process in which in situ-generated inorganic polysulfides serve as autocatalysts.
Compounds in reactions 1–4 are depicted in their fully protonated states for the sake of simplicity, but they represent all acid/base derivative.
Cysteine (Cys)-per/polysulfide species were shown by a number of different research groups using different methods to be abundant in biological systems (2, 14, 15, 19, 21, 32, 45). Cys-polysulfides are in dynamic equilibrium with inorganic polysulfide species (6), which implies that their reactions with nitrosothiols could be a potential source of endogenous SSNO−. In addition, it was also shown to be generated in the chemical interactions of sulfide and persulfides with nitrite, representing another potential source of endogenous SSNO− (4, 8).
The early studies on SSNO− triggered lively debates on chemical grounds (4, 7, 22, 27, 41). For example, Wedmann et al. claimed that SSNO− was unstable and rapidly decomposed in the presence of thiols or cyanide. However, the kinetic measurements with different nucleophiles were performed in organic solvents or in mixtures of aqueous and organic solvents using bis(triphenylphosphine)iminium perthionitrite (PNP+SSNO−) as a reagent (which is inhomogeneous and contains substantial uncharacterized chemical entities, likely affecting its stability). Conversely, we and others provided robust evidence for the notion that SSNO− has considerable stability under physiological conditions and is resistant to thiols [such as dithiotreitol (DTT) or reduced glutathione (GSH)] and cyanide (4, 9, 11, 26, 28). These findings are corroborated by several experiments in the present contribution, and we propose that the main reason for these conflicting observations lies in methodological details of preparing and working with SSNO− solutions and misinterpretations in Ref. (42) by, for example, missing the 420 nm band being due to SSNO− [related to a solvatochromic effect, also addressed by Pluth and colleagues (4)], as well as erroneous assignment of the aqueous 314 ppm signal to SNO−, whereas it really pertains to SSNO− (27)
The central aim of the current study was to further investigate the chemical properties of SSNO− in relation to its potential biological actions. Importantly, we here demonstrate that contrary to other polysulfide species, SSNO− exhibits unexpected resistance toward the main NADPH-driven biological reducing machineries, the thioredoxin (TrxR) and the glutathione reductase (GR) systems. Furthermore, we now obtained mass spectrometric evidence that its slow decomposition gives rise to the formation of inorganic polysulfides, which in turn induce rapid per- and polysulfidation of peptide and protein Cys residues. Transient receptor potential ankyrin 1 (TRPA1) channels were previously proposed to be activated by inorganic polysulfides via persulfidation of their critical regulatory Cys residues (24). The cooperative actions of sulfide and NO on TRPA1 channel activation had been proposed earlier to be mediated via intermediate formation of nitroxyl (HNO) (16). On the contrary, Kimura et al. recently demonstrated that these effects are rather due to inorganic polysulfides arising as products of the chemical interaction between NO and sulfide (25). Here, we show that not only the rapidly generated inorganic polysulfides [that form concomitantly with SSNO− during the chemical reaction of nitrosothiols and sulfide (11)] can activate TRPA1 channels but also the slow release of these molecules upon SSNO− decomposition can sustain their activation status. Because contrary to polysulfides, SSNO− can escape the NADPH-driven reducing systems, we propose that its localized decomposition provides a slow in situ flux of sulfane sulfur species that sustains TRPA1 activation. These observations represent an example as to how the chemical interactions of reactive species in the NO and sulfide regulatory pathways can result in additional biological activities not entertained by either signaling entities alone. These observations make SSNO− a highly attractive candidate molecule for intracellular and possibly intercellular trafficking of NO and sulfane sulfur equivalents.
Results and Discussion
Corroborating evidence for the formation of SSNO− as a major product in the reaction of nitrosothiols with H2S and for its stability in the presence of thiols
SSNO− was first characterized in the 1980s by Seel and Wagner (35) in the form of its bis(triphenylphosphine)iminium salt (PNP+SSNO−). The chemical structure of the anion was determined by X-ray crystallography and its ultraviolet-visible (UV-Vis) spectrum was recorded in different organic solvents (35). The absorbance maxima of SSNO− in acetone and methanol were at 448 and 426 nm, respectively, with a calculated molar absorbance coefficient of 2800 M −1cm−1 in acetone. Previously, we reported the formation of a yellow product with a peak at λmax = 412 nm (denominated as the “412 species”), with a molar absorbance coefficient of 6000 M −1cm−1 that was generated in the reaction of RSNO with H2S in buffered aqueous solution. Based on high-resolution mass spectrometry data and a number of indirect experimental results (release of NO and sulfide under reducing conditions or sulfane sulfur equivalents under nonreducing conditions), we tentatively assigned this yellow product to SSNO− (9, 11). Our data suggested that SSNO− (the “412 species”) was relatively stable at room temperature (RT) and physiological pH and slowly decomposed (t1/2 ∼0.5–1 h, depending on conditions) with concomitant formation of NO and sulfane sulfur species. In addition, cell-based and in vivo data suggested that it could be one of the key bioactive intermediates of the chemical interaction between sulfide and NO that has a relatively long lifetime (on the order of tens of minutes rather than seconds). Notwithstanding these findings and Seel's earlier work, Filipovic and coworkers claimed that SSNO− was unstable at pH 7. Wedmann et al. also prepared crystalline SSNO− as its bis(triphenylphosphine)iminium salt (PNP+SSNO−) based on the procedure published by Seel and Wagner (42). After redissolving the solid material in organic solvents, the UV-Vis absorbance maxima of PNP+SSNO− in acetone and methanol were measured by them to be at 448 nm and 422 nm, respectively. They claimed that the molecule was highly unstable when the crystals were dissolved in aqueous solutions or water was added to organic solutes of PNP+SSNO−. Therefore, they suggested that the 412 nm absorbance observed by Cortese-Krott et al. “may only be attributed to the presence of the mixture of polysulfides/sulfur sols,” generated as by-products during NO–sulfide interactions. We prepared the same crystals following Seel and Wagner's procedure (see the Materials and Methods section); however, closer microscopic inspection and exploratory X-ray crystallography of the crystalline material suggested that it was highly inhomogeneous, with at least three clearly distinguishable morphologies, colors, and geometries (data not shown), indicative of different inclusion complexes. This had already been described in Seel et al.'s original article (35) and acknowledged by Wedmann et al. (42). Dissolution of this solid material in aqueous buffers resulted in the transient appearance of a 412 nm peak, followed by fast precipitation of a white solid (most likely colloidal sulfur), as observed when SSNO−/sulfide mixtures are acidified. However, when the crystals were first dissolved in dimethylsulfoxide (DMSO) followed by dropwise addition of an aqueous phosphate buffer, pH 7, containing 1 mM sulfide in addition, the characteristic 412 nm feature exhibited much greater stability with ∼20% decomposition over 5 min (Fig. 1).
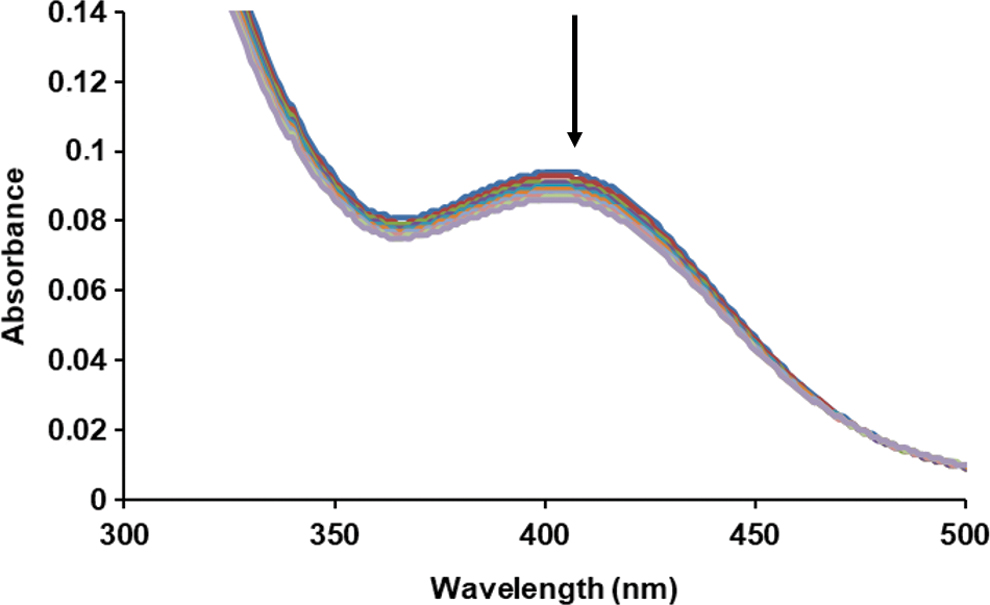
Curiously, Wedmann et al. also observed the appearance of a 420 nm peak with comparable stability when their crystals were dissolved in 300 mM phosphate buffer containing 1 mM sulfide [figure 7E in Ref. (42)]. However, they claimed that this could not possibly be due to SSNO− because the latter would rapidly react with sulfide to give HSNO; they concluded that it must therefore be due to inorganic polysulfides/sulfur sols. To shed further light onto this apparent paradox, we repeated their experiments using a different SSNO− preparation and reversing the order of solvent alterations. SSNO− (the 412 species) was prepared by the reaction of S-nitroso-N-acetyl-D,L-penicillamine (SNAP) with excess sulfide in Tris/HCl buffer, as described before (9, 11), and the aqueous mixture was then diluted 10-fold in methanol or acetone to investigate how its UV-Vis spectra in organic solvents compare to those measured after dissolving the PNP+SSNO− salt in similar solvents. Increasing the proportion of the organic solvent over water resulted in a bathochromic shift of the 412 nm feature, as described by Seel et al. and some of us earlier (9, 35). In a mixture of 90% acetone/10% Tris buffer, the absorbance maximum shifted to 439 nm, and in 90% MeOH/10% Tris buffer, it was at 426 nm (Fig. 2), very close to the ones observed by Wedmann et al., Seel et al., and us for the PNP+SSNO− salt. The close association between the observed absorbance maxima for the two distinct procedures/preparations, which apparently correlate very well with theoretically calculated solvatochromic effects using TD-DFT and QM/MM MD simulations (27), corroborate the notion that the observed characteristic UV-Vis bands in different solvents can both be attributed to SSNO−.

As alluded to above, Filipovic and colleagues proposed that the 412 nm peak in the SNAP/sulfide reaction mixture was potentially due to inorganic polysulfides/sulfur sols since they could be reduced by DTT [figure 7J in Ref. (42)]. However, we demonstrated in our previous study that in contrast to polysulfides, the 412 nm species generated during the reaction of nitrosothiols with sulfide is fully resistant toward DTT and cyanide [figure S5 in Ref. (11)]. In fact, to eliminate the inorganic polysulfide species (which are formed as by-products in the reaction of nitrosothiols with sulfide) from SSNO− solutions, we here utilized this unique chemical property of the “412 species” the following way: SSNO− solutions were prepared by the reaction of 3 mM SNAP and 30 mM sulfide. This reaction mixture contains inorganic polysulfides (11), accounting for ∼50% of the initial SNAP concentration. Therefore, immediately after formation of the 412 nm peak the reaction mixture was passed through a Pierce Immobilized Reductant Column, containing beaded resin with immobilized sulfhydryl groups to selectively minimize the polysulfide content in that preparation. The solution was then bubbled with N2 for 5 min to eliminate the majority of reduced sulfide in the form of H2S from the mixture. Using this procedure, the “cleaned-up” SSNO− solutions contained <20% of the initially generated polysulfides.*
To also obtain kinetic evidence that the 412 nm species in aqueous buffer is responsible for the UV-Vis absorbance maximum at 439 nm measured in 90% acetone/10% aqueous buffer, this “cleaned-up” aqueous SSNO− solution was allowed to decompose on ice in the dark (the experiment was carried out on ice because in the absence of sulfide to stabilize SSNO− it is considerably less stable). Aliquots from this mixture were taken at distinct time points (at 20 min intervals), and a 10-fold dilution was performed either in 200 mM, pH 8.00, Tris/HCl buffer, or acetone. The recorded time-dependent decrease in the absorbance values of the 412 nm peak, which was measured after dilution with buffer, and that of the 439 nm peak, which was measured after dilution with acetone (i.e., in 90% acetone/10% aqueous buffer) was consistent with each other (Fig. 3). Using the previously estimated ɛ412 = 6000 M −1cm−1, the extinction coefficient at 439 nm in 90% acetone/10% aqueous buffer (ɛ439 = 4340 ± 202 M −1cm−1) was calculated as the average of the values that were measured at different time points. In light of the observed inhomogeneity of the crystalline material, this is reasonably close to the one originally reported by Seel et al. at 448 nm (ɛ448 = 2800 M −1cm−1).

In addition, we followed the decomposition of SSNO− at increasing concentrations of sulfide and GSH, at distinct concentrations of SSNO− and in solvents of different composition. The observed kinetic traces indicated that the reactions cannot be described by simple first-order rate equations and a detailed mechanistic study with product analyses would be required to obtain deeper mechanistic insight into the decomposition process of SSNO−. While this was beyond the aim of the current study, a number of conclusions can nevertheless be drawn from these analyses:
The kinetic traces depicted in Figure 4A and the measured half-lives of SSNO− decomposition from the data shown in Figure 4B demonstrated that sulfide concentrations up to ∼20 mM have a considerable stabilizing effect on SSNO− under the applied experimental conditions. Due to the fact that complete elimination of sulfide from the aqueous buffer solutions of SSNO− (where it is generated in the reactions of nitrosothiols with excess sulfide) is not possible, it is likely that the results shown in Figure 4A and B reflect conservative estimates of sulfide's true stabilizing effects. This could also partially account for the faster decomposition of PNP+SSNO− when the (inhomogeneous) crystalline material is dissolved in aqueous buffer systems, which contain no sulfide. In addition, Figure 4C and D shows that at a similar concentration range, also GSH stabilizes SSNO−. These observations, together with the fact that reactions of nitrosothiols with sulfide provide a mixture of reactive sulfur and nitrogen species (11), would suggest that the stabilizing effect of thiol compounds might possibly be due to capturing additional reactive species that facilitate SSNO− decomposition. An alternative explanation is that this effect is due to transition metal complexation, which has been demonstrated to contribute to the stabilizing effect of Cys and other thiols toward RSNO (17, 44). However, this effect is unlikely based on the observed similarities in the kinetics of SSNO− decomposition in the presence and absence of 200 μM of the metal chelator diethylenetriaminepentaacetic acid (DTPA) at both 0 or 10 mM added sulfide (Fig. 4B).
The results depicted in Figure 4A–D clearly indicate that in aqueous buffer systems at thiol concentrations up to ∼40 mM (thus well above the physiological range), the presence of either sulfide or GSH failed to accelerate the rate of SSNO− decomposition. However, at concentrations >40 mM, both thiols started to shorten the half-life of SSNO−, which may represent the emerging contribution of a bimolecular reaction between SSNO− and the added thiol to the kinetics of SSNO− decay. It should be noted that not fully eliminated polysulfides could have an effect on equilibrium (4) by shifting it to the right. However, if the bimolecular reaction between SSNO− and sulfide would be a major driving force of SSNO− decay, this phenomenon would only influence the decay kinetics by providing an intercept for a linear kobs (obtained from the exponential fits of the kinetic traces) versus sulfide concentration plot. Based on these observations, at physiologically relevant concentrations of thiols in aqueous systems, we propose that a bimolecular sulfur transfer reaction between sulfide or GSH with SSNO− can be excluded from mechanistic considerations regarding the fate of SSNO−. The decomposition kinetics of SSNO− is independent of the starting SSNO− concentration (Fig. 4E, F), suggesting that it is not a second-order process. This is in contrast to the disproportionation/conproportionation-driven decomposition of inorganic polysulfide species. Contrary to the observation by Filipovic et al. that SSNO− can be rapidly attacked by sulfides or other nucleophiles in pure aprotic solvents such as acetone or tetrahydrofuran [but not in mixtures with hydroxylic solvents, see Ref. (27)], the stability of SSNO− in our systems did not change substantially by changing the composition of the solvent system from 100% buffer to 60% buffer/40% acetone in the presence and absence of 20 mM GSH (Fig. 4G, H). These observations agree with our previous assumption that the relatively lower stabilities of PNP+SSNO− when it was dissolved in aqueous buffer systems or aprotic/protic solvent mixtures in the absence of sulfide were likely related to the inhomogeneity of the crystalline material (see Corroborating evidence for the formation of SSNO− as a major product in the reaction of nitrosothiols with H2S and for its stability in the presence of thiols section) and not to its self-decomposition and/or fast reactivity with sulfide or other thiols. To unambiguously reconcile these contradicting observations in mixed solvent systems, the preparation of a water-soluble perthionitrite salt will be necessary, which will allow to determine the contributions of inhomogeneity and solvent effects on the stability of SSNO−.

Thus, in refuting the claims by Wedmann et al., we conclude that the observed 412 peak is not due to inorganic polysulfides/sulfur sol because (i) it cannot be reduced by DTT, sulfide, glutathione, or cyanide, (ii) its decomposition kinetics are independent of the starting concentration, and (iii) in solutions in which preformed polysulfides as well as the residual sulfide was eliminated by immobilized -SH reductant and N2 bubbling, respectively, similar absorbance spectra were observed. Moreover, monitoring the decomposition of the 412 species by time-resolved UV-Vis spectrophotometry revealed that it has similar spectral characteristics to that of the previously reported PNP+SSNO− salt, with absorbance maxima at 448 nm and 426 nm in acetone and methanol, respectively. Based on all these observations, we argue that a major reaction product of SNAP and sulfide is indeed SSNO−, that its lifetime is relatively long at pH 7 and that it cannot be reduced by thiols.
SSNO− is also resistant toward the NADPH-driven cellular reducing systems
Motivated by the surprising observation that thiol compounds (DTT, Cys, or GSH) are unable to reduce SSNO− (11), we sought to investigate whether it might also escape the reducing power of the major NADPH-dependent cellular enzymatic machineries, that is, the thioredoxin and the GR systems. We previously demonstrated that thioredoxin reductase 1 (TrxR1) can use NADPH to reduce inorganic and protein per/polysulfides, in reactions that were further accelerated upon the addition of the TrxR1 substrate thioredoxin-related protein of 14 kDa (TRP14) (15). This observation was further corroborated here by following the enzymatic reactions using mass spectrometry-based detection of inorganic polysulfide species (Supplementary Fig. S1).
Furthermore, we recently obtained evidence in cells and in vivo that even the oxidized forms of protein per/polysulfides are substrates of the thioredoxin family of enzymes in a protein-specific manner (14). In addition, GR can use GSH and NADPH to catalytically reduce inorganic as well as protein per/polysulfides, in reactions that gain further catalytic power in the presence of glutaredoxin (Grx) (15). In light of these recent advances, it was particularly interesting to find that upon the addition of physiologically relevant concentrations of the Trx and GR systems in different enzyme compositions, we were unable to detect any effect on the slow “spontaneous” decomposition kinetics of SSNO− (Fig. 5). Initially, SSNO− was prepared by the reaction of 1 mM SNAP with 10 mM sulfide. Selective reduction of preformed inorganic polysulfides and sulfide removal was achieved by 500 μM DTT and N2 degassing, which was followed by sequential addition of the components of either the Trx or the GSH enzymatic systems (Fig. 5). Neither NADPH on its own nor in the presence of GR or TrxR1 or GSH+GR or TrxR1+TRP14 affected the slow decomposition of SSNO− in 200 mM Tris/HCl +100 μM DTPA buffer at pH 7.40 and RT.
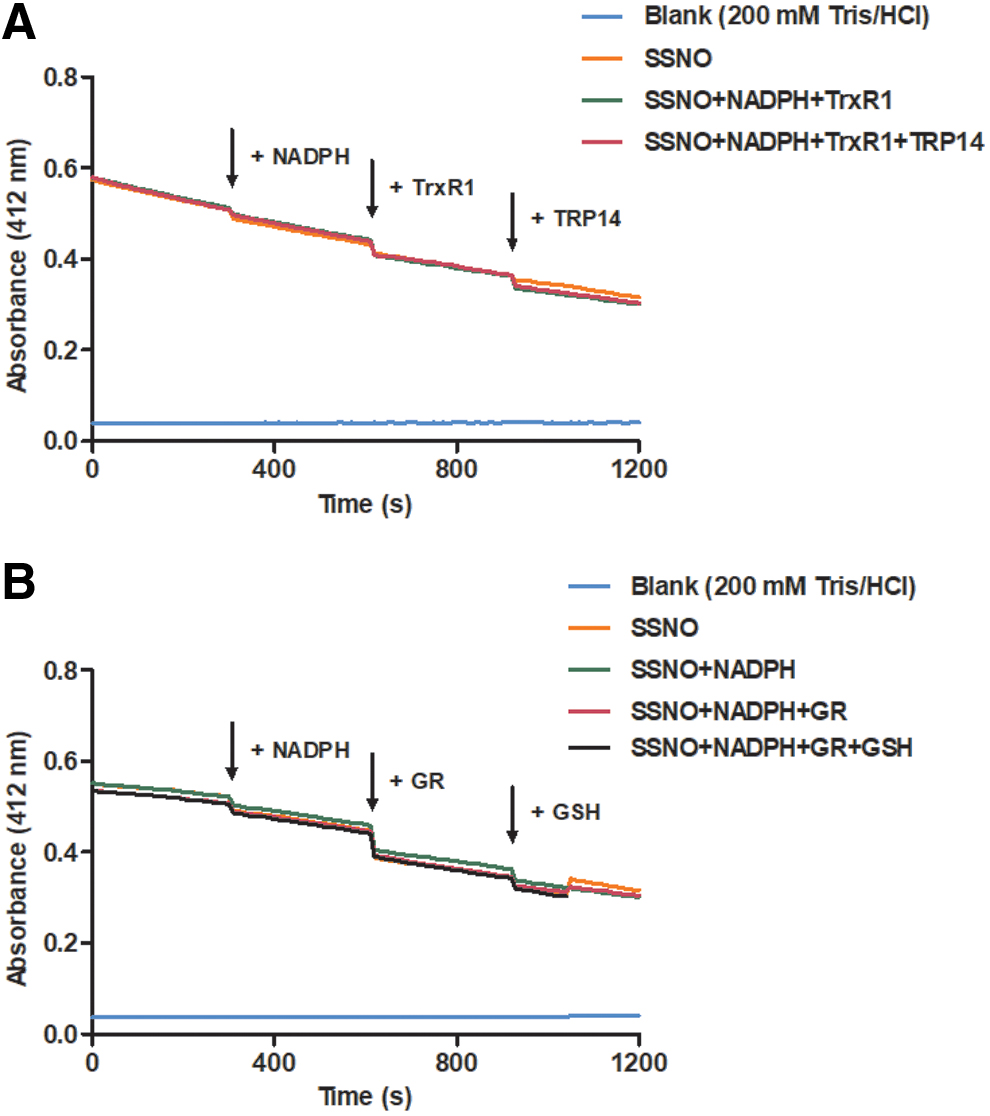
Thus, to the best of our knowledge, SSNO− to date represents the only investigated sulfane sulfur carrying compound that is capable of escaping the reducing power of these extremely efficient enzymatic systems. Olabe and colleagues estimated the pK a for HSSNO to be ∼5 (27), which means that at physiological pH the majority of it would be deprotonated. This could contribute to the inherent insensitivity of this molecule toward thiol-mediated reduction by hindering the nucleophilic attack of thiolates and strengthening the S-N bond. This observation suggests that SSNO− might function as a sulfane sulfur trafficking molecule, generating polysulfides in situ upon its slow decomposition, while being resistant toward the intracellular NADPH-driven reducing machineries (basically working as a slow endogenous sulfane sulfur donor). Inorganic polysulfides were, however, reduced by the Trx system in the presence of SSNO−. Based on repeated (n = 5) quantification by cold cyanolysis and the methylene blue method [as in Ref. (11)] after the formation of SSNO− was completed upon mixing 1 mM SNAP and 10 mM sulfide, the reaction mixture contained 560 ± 190 μM sulfane sulfur equivalents and 9 ± 3 mM sulfide besides 300 ± 25 μM SSNO−. Incubation of these mixtures with 200 nM TrxR1, 650 μM NADPH ±2 μM TRP14 for 10 min at RT, protected from light, followed by degassing with N2 for an additional 5 min at RT in the dark, reduced the sulfane sulfur equivalents to 253 ± 93 μM and the residual sulfide to 111 ± 20 μM. (Note that the enzymatic reduction was likely much more efficient because the residual sulfane sulfur equivalents also contain inorganic polysulfides that were eliminated from SSNO− during the incubation with cyanide in the cold cyanolysis measurements.)
These observations indicated that the Trx system was functional and that selective reduction of polysulfides can be achieved while preserving the integrity of SSNO−. This selective reactivity of the Trx system also has methodological implications because it indicated that it can be used to enzymatically diminish inorganic polysulfide contents of freshly made SSNO− solutions, which can be followed by the elimination of the Trx family proteins using size-exclusion chromatography (ultrafiltration) to avoid reduction of subsequent SSNO−-derived polysulfides (for more details, see the Materials and Methods section).
Inorganic polysulfides were identified as sulfane-sulfur-carrying products upon decomposition of SSNO−
The formation of SSNO− in the reaction of nitrosothiols and sulfide is accompanied by autocatalyzed formation of inorganic polysulfides. SSNO− appears to be sufficiently stable to be of physiological significance but slowly decomposes to generate sulfane sulfur species. In our previous study, we used cold cyanolysis and chloroform extraction-based methods to show that stoichiometric amounts of sulfane sulfur equivalents are released upon the decay of SSNO− (11). However, the chemical nature of these slowly generated sulfane sulfur species has not been investigated. Here, we used liquid chromatography with tandem mass spectrometry (LC-MS/MS) to identify what inorganic polysulfides are generated in this system. Following the preparation of SSNO− by the reaction of SNAP with excess sulfide and enzymatic purification with 2 mM NADPH and 200 nM TrxR1, reaction mixtures were incubated at RT in the dark for 2 h. Twenty-five microliters aliquots of these mixtures were subjected to LC-MS/MS analyses after alkylation with 25 mM β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) (final concentration, 30 min, RT). Inorganic polysulfides were detected by selected reaction monitoring [as in Ref. (6), for seleted reaction monitoring (SRM) parameters, see the supporting information] with chain lengths up to seven sulfurs (see S1–S7 chromatograms in Fig. 6). In this context, we caution that the actual speciation of the different chain length inorganic polysulfides shown in Figure 6 may differ substantially from that in real biological systems, due to the sensitivity of their distribution to the composition of the chemical environment as well as the experimental parameters used for detection. For example, we demonstrated that the nature and concentration of the alkylating agent that is used for their stabilization as well as the time of alkylation can substantially perturb polysulfide speciation equilibria (6). Thus, while the results of the experiment depicted in Figure 6 provide qualitative information only, it unequivocally demonstrates that inorganic polysulfides are eventual products of the SNAP–sulfide interaction and SSNO− decomposition. To obtain quantitative data on how much of the sulfane sulfur equivalents can be accounted for as inorganic polysulfides upon the formation and decomposition of SSNO−, isotopically labeled derivatives of all polysulfide species would have to be prepared, independently quantified and used as internal standards, which was well beyond the scope of the present study.

Parameters for Tandem Mass Spectrometry Detection of the β-(4-Hydroxyphenyl)Ethyl Iodoacetamide-Labeled Inorganic Polysulfide Species
SSNO− decomposition products induce slow and sustained polysulfidation of GSH
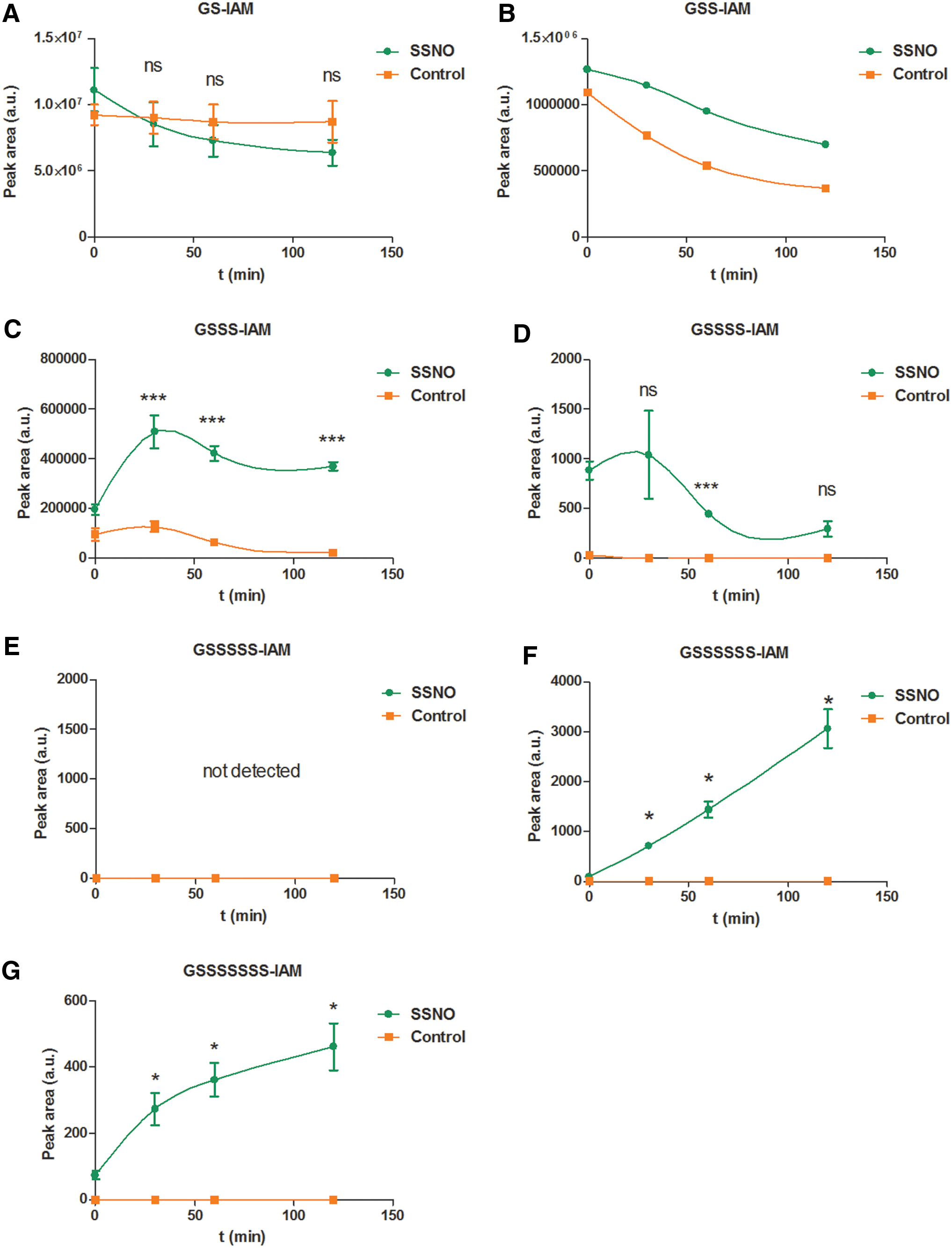
To experimentally address whether SSNO− indeed induces polysulfidation of Cys residues, we first investigated this using GSH. SSNO− working solutions were prepared immediately before start of the kinetic experiments by the following procedure: SSNO− was generated by reacting 1 mM SNAP with 10 mM Na2S in 200 mM Tris/HCl +100 μM DTPA buffer at pH 7.40. As stated above, besides SSNO− (∼300 μM), the reaction mixture also contained ∼500 μM sulfane sulfur equivalents and ∼9 mM sulfide. To eliminate the majority of the polysulfide content from this SSNO− stock, we utilized the polysulfide-selective reducing potential of NADPH/TrxR1 (for experimental details, see the Materials and Methods section and the “SSNO− is also resistant toward the NADPH-driven cellular reducing systems” section). A control solution containing similar amounts of total polysulfides and sulfide (made by mixing freshly made sodium disulfide (Na2S2) in water and sulfide solutions in 200 mM Tris/HCl, 100 μM DTPA buffer at pH 7.40) was subjected to the same enzymatic reduction. TrxR1 was removed from the “cleaned-up” SSNO− working solutions and from the controls by ultrafiltration. The reactions were started by mixing these solutions (final concentration of SSNO− was 87 ± 16 μM) or controls (using a similar dilution factor as for the SSNO− working solutions) with 1 mM GSH in 200 mM Tris/HCl +100 μM DTPA buffer at pH = 7.40 (the mixing time was considered as the 0 min time point of the kinetic runs); reactions were let to proceed at RT in the dark. At 0, 30, 60, and 120 min, 50 μL aliquots were sampled from the incubation mixtures, and the reactions quenched by alkylation with 100 mM IAM (allowing for a minimum of 30 min alkylation at RT). Time-resolved formation and decomposition of the alkylated GS(S)n–IAM (n = 1–7) species were detected by LC-MS/MS analysis. As shown in Figure 7, the reactions with SSNO− induced significantly more glutathione per- and polysulfidation than the control samples at all time points (polysulfide formation in the controls, in particular the short-chain derivatives, was likely due to incomplete elimination of polysulfur species). An interesting observation in this system was the fact that the concentrations of longer chain glutathione polysulfides (S6 and S7) steadily increased in the SSNO− incubated with GSH, a behavior not observed in the controls. While polysulfide speciation may not reflect the true distribution of these products in the incubator [as discussed previously (6)], these experiments nevertheless demonstrate a sustained polysulfidation potential of SSNO− using a biologically relevant thiol target (GSH) over a timescale similar to that of its decomposition. This observation together with the fact that under these conditions a direct reaction between SSNO− and GSH was excluded on kinetic grounds (Fig. 4C, D) suggests that the time resolved per/polysulfidation of GSH shown in Figure 7 was induced by SSNO− released inorganic polysulfides.
SSNO− decomposition products can induce sustained polysulfidation of thiol proteins
Next, SSNO−-induced persulfidation was studied using the protein model, human serum albumin (HSA). We previously demonstrated that the free sulfhydryl group of the only Cys residue that is not engaged in intramolecular structural disulfide bonds, Cys34, is prone to polysulfidation by inorganic polysulfides, which can be detected by our Protein Persulfide Detection Protocol (ProPerDP) method (15). This system can therefore be used to study the potential polysulfidating effects of SSNO− or its decomposition products on protein thiol targets. To this end, SSNO− and control mixtures were prepared and purified from preformed polysulfides as described for the GSH experiments above, before reaction with HSA (containing reduced Cys34, see the Materials and Methods section) at RT, protected from light to give a reaction mixture of 48 ± 9 μM SSNO− (or control solution at a similar dilution factor) with 7.5 μM HSA. Aliquots from each reaction mixture were taken at 0, 20, 4,0 and 60 min, followed by the ProPerDP assay to detect protein persulfide formation (13). The persulfidated fraction of HSA was detected following separation on nonreducing 12% polyacrylamide gels, followed by Western blotting and visualization as described in Ref. (15) (for the detailed protocol, see the Materials and Methods section). As shown in Figure 8, increasing amounts of human serum albumin persulfide (HSA-SSH) were detected on a similar timescale as SSNO− is expected to decompose under these conditions. No HSA-SSH was detected in the control samples, suggesting that the selective abolishment of in situ-generated inorganic polysulfide by-products by the NADPH–TrxR1 system was effective. [Note that this method is less sensitive compared with the mass spectrometry-based detection of polysulfide species shown in Fig. 7 and that HSA-polysulfidation by inorganic polysulfides represents a true equilibrium, where an excess of polysulfides is needed to fully polysulfidate Cys34 (15).] Figure 8 indicated that the kinetics of HSA-SSH generation upon SSNO− treatment is indeed consistent with the kinetics of SSNO− decay under these conditions, corroborating that the delayed persulfidating effects are caused by its decomposition products rather than SSNO− itself (the concentration of which was highest at the beginning).

SSNO− can induce sustained activation of TRPA1 ion channels
We next investigated whether SSNO−, the major product of NO and sulfide interaction, might also account for the previously observed cooperative action of NO and sulfide species on TRPA1 channel activity (16). In these experiments, SSNO− was prepared using a different S-nitrosothiol precursor, S-nitroso-L-glutathione (GSNO), to avoid the possible TRPA1 antagonistic effects of N-acetyl-DL-penicillamine generated as a by-product of the SNAP/sulfide interaction (36).
The SSNO− preparation generated from GSNO/sulfide was subjected to enzymatic reduction of concomitantly generated inorganic polysulfides, followed by N2 bubbling to remove the majority of the remaining sulfide (as for the other SSNO− preparations and described in the Materials and Methods section). SSNO− and a control sample (a solution of 9 mM Na2S + 0.5 M Na2S2, which was subjected to the same treatment) were allowed to decompose at RT in the dark. Aliquots from the incubated solutions were taken at 0, 30, 60, and 120 min, diluted to a final concentration of 10 μM SSNO− (using a similar dilution factor for the control samples) and added to TRPA1-expressing Chinese hamster ovary (CHO) cells previously stained with the Ca2+ ion-specific fluorescent dye Fluo-4. The intracellular fluorescence signal generated was immediately analyzed by flow cytometry. TRPA1-deficient cells were also treated and analyzed in the same manner, but they did not reveal any intracellular fluorescence above baseline upon any treatment, which corroborated that the detected Ca2+ influx can indeed be attributed to the activation of TRPA1 ion channels in this cell line (Supplementary Fig. S2). A parallel batch of cells was reacted with the selective TRPA1 receptor agonist mustard oil [100 μM in extracellular solution (ECS)] as positive control. The obtained average fluorescence signal in these positive control samples was set as 100%, and the fluorescence intensity in the presence of SSNO− or control solutions was expressed as % of this value.
Figure 9 shows that treatment of cells with 10 μM SSNO− resulted in a constant TRPA1 activation over 2 h corroborating that most likely due to the slow but continuous release of inorganic polysulfides, SSNO− can induce a sustained activation of these ion channels. On the contrary, a statistically significant (p = 0.0136) decrease in Ca2+ influx was detected in cells treated with the control solutions, which is in line with a transient activation of TRPA1 by the relatively unstable residual inorganic polysulfides (due to noncomplete TrxR1-mediated reduction and/or remaining sulfide).
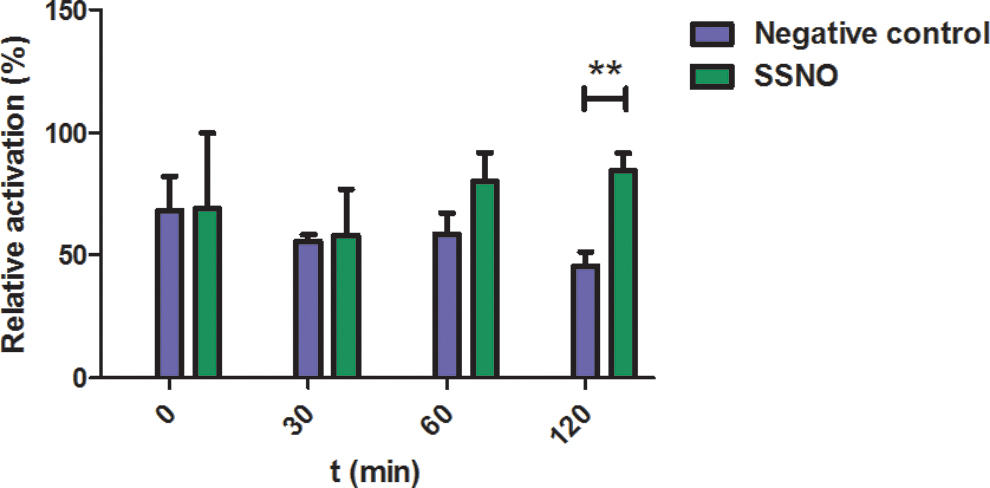
Taken together, our data suggest that SSNO− indeed plays a role in the cooperative actions of NO and sulfide on TRPA1 channel activation. In addition, our data agree with the proposal by Kimura and coworkers that the immediate activation of TRPA1 channels by reaction mixtures of nitrosothiols and sulfide is likely due to preformed polysulfide species (31). However, preformed polysulfide species would only elicit a transient activation of TRPA1 channels, whereas SSNO− induces sustained TRPA1 activation by providing a slow flux of inorganic polysulfide species.
Conclusions
In the present study, we obtained corroborating evidence for the fact that SSNO−, a major product of the chemical reactions between sulfide and NO species, is indeed relatively stable in aqueous solutions of physiological pH. Furthermore, we confirm that it is stabilized by the presence of excess sulfide and fully resistant toward reduction by thiols. Unexpectedly, we also find that SSNO− escapes the reducing power of the NADPH-driven biological reduction machineries. We believe that these properties have important biological implications. To the best of our knowledge, SSNO− is the first biologically relevant sulfane-sulfur-carrying molecule that cannot be effectively reduced by thiols or by the TrxR and/or GR systems. Furthermore, we here show that the slow decomposition of SSNO− liberates inorganic polysulfides, which efficiently convert glutathione and protein Cys thiols into their corresponding per- and polysulfide derivatives. The potential biological implication of these reactions was demonstrated exemplary by the activation of TRPA1 channels. Specifically, we demonstrated that in contrast to the transient activation of these channels by polysulfides, SSNO− induces long-term potentiation of TRPA1 channels by generating a slow in situ flux of inorganic polysulfides. Notwithstanding the use of simple in vitro model systems to demonstrate biological relevance, these properties of SSNO− would seem to offer unique opportunities for cell signaling by enabling sulfur and nitrogen trafficking within the reducing environment of the cytosol, with targeted release of both NO and polysulfide equivalents.
Materials and Methods
Reagents, SSNO− preparation
Na2S2 was kindly provided by Dojindo Laboratories (Munich, Germany and Kumamoto, Japan). Recombinant rat TrxR1 and human TRP14 were kindly provided by Prof. Elias Arnér. GR was from Sigma–Aldrich. All other reagents were purchased at the highest purity available from Sigma–Aldrich, Cayman Chemicals, VWR Chemicals, Santa Cruz Biotechnology, or Thermo Fisher Scientific.
Sulfide stock solutions were prepared in water as previously described (33) and diluted further in the indicated buffer immediately before use. Although all precautions were undertaken to avoid the formation of oxidation products (including polysulfides) in these stock solutions, their presence (at least in trace amounts) cannot be prevented completely under aerobic conditions. Polysulfide stock solutions were prepared either in ultrapure water or in the indicated buffer by dissolving technical potassium polysulfide (K2Sx, a mixture of sulfide and polysulfides of different chain lengths), Na2S2, sodium trisulfide (Na2S3), or sodium tetrasulfide (Na2S4) salts. All solutions were freshly prepared immediately before use in experiments.
10 mM aqueous stock solutions of SNAP or GSNO were freshly prepared by dissolving crystalline material in 200 mM tris(hydroxymethyl)aminomethane (Tris) buffer, pH 7.4, and diluted further immediately before use or divided into aliquots and kept at −80°C until use. Stock solutions of SSNO− were prepared by reacting 1 mM SNAP or GSNO with 10 mM Na2S in 200 mM Tris (pH 7.4).
Inorganic polysulfides are present in sulfide stock solutions as contaminants and produced in situ during SSNO− preparation or decomposition. Their selective reduction (where indicated) was carried out either using a Pierce™ Immobilized Reductant Column (supported with 6% cross-linked beaded agarose and activated by a thiol-based reducing agent to enable solid-phase reduction of peptide and protein disulfide bonds) based on the manufacturer's instructions (
Preparation of crystalline PNP+SSNO− and UV-Vis spectra of PNP+SSNO− in phosphate buffer in the presence of sulfide
Preparation of crystalline PNP+SSNO− was carried out in a three-step procedure following literature protocols (29, 34, 35). Briefly, first, bis(triphenylphosphine)iminium chloride (PNP+Cl−) was made treating PPh3 in 1,1,2,2-tetrachloroethane with one equivalent of chlorine gas at −20°C and subsequent boiling of the resulting slurry in the presence of hydroxylamine hydrochlorine (34). PNP+Cl− was isolated by adding the reaction mixture to ethyl acetate. Subsequent recrystallization from hot water afforded pure PNP+Cl− as a white crystalline solid. PNP+Cl− was then converted to the nitrite salt by treating its warm concentrated aqueous solution with saturated NaNO2 solution (29). The solid formed was filtered, dried, and recrystallized using Et2O/acetone. For the preparation of PNP+SSNO−, the white needles of the nitrite salt (PNP+NO2 −; 2.5 g, 4.28 mmol) were reacted with elementary sulfur (S8; 0.274 g, 8.56 mmol) in predried, freshly distilled, and nitrogen-saturated acetone (70 mL) under an atmosphere of purified N2 using standard Schlenk techniques in the dark (35). After overnight reaction, the dark red reaction mixture was filtered under N2 and chilled to −78°C. Carefully, absolute Et2O (stored over sodium and saturated with N2 before use) was layered on top of the filtrate in a Schlenk vial and left to warm up to RT in the dark. After 3 days, the mother liquor was decanted under N2 from the orange-red crystalline solid formed. The solid was rinsed with three portions of dry Et2O (3–4 mL) and dried under vacuum (yield: 0.487 g, 18%). UV-Vis spectra were recorded using a Perkin Elmer Lambda 2S UV-VIS spectrophotometer using quartz cuvettes with 1 cm path lengths.
UV-Vis spectra of the reaction products of SNAP and sulfide in solvent mixtures
SSNO− stock solutions were prepared by reacting 3 mM SNAP with 30 mM Na2S in 200 mM Tris/HCl (pH 7.4) (9). Following the selective reduction of in situ-generated polysulfides by a reductant column (see Reagents, SSNO− preparation section), the solutions were activated by 10 mM DTT and sulfide removal by degassing with N2 for an additional 5 min at RT. The SSNO− stock solution was allowed to decompose at RT, protected from light. Aliquots from this incubation solution were taken at 0, 20, 40, and 60 min and 10 × dilutions were performed at each time point both in 200 mM Tris/HCl (pH 7.4) and in acetone. UV-Vis spectra of these diluted solutions were taken using an Agilent Cary 8454 diode-array spectrophotometer with quartz cuvettes of 1.00 cm path length, and concentrations of SSNO− at each time point were calculated using the measured absorbance and previously determined ɛ values (which were found to be 6000 M −1cm−1 in aqueous media). The ɛ = 4340 ± 202 M −1cm−1 value for SSNO− in 90% acetone 10% 200 mM Tris (pH 7.4) was calculated from the obtained spectra at 439 nm by using the calculated concentrations of the decomposing SSNO− solution at different time points in buffered media before dilution with acetone.
Effects of H2S, glutathione, SSNO− concentrations, and solvent composition on the decomposition kinetics of SSNO−
SSNO − stock solutions were prepared by mixing 6 mM SNAP with 60 mM Na2S in 100 mM Tris/HCl (pH 7.4). This was followed by the reduction of in situ-generated polysulfides by 100 nM TrxR1 and 1 mM NADPH for 15 min at RT and sulfide removal by degassing with N2 for an additional 10 min. Concentration of SSNO− stock solution was calculated using measured absorbance at 412 nm and previously determined ɛ value of 6000 M −1 cm−1 in aqueous media (11).
Na2S solutions were prepared as described above and mixed with SSNO− in 1:1 ratio immediately before measurement. DTPA (when added in Fig. 4B) was introduced to the reaction mixtures at this time point.
GSH stock solutions were prepared by dissolving crystalline material in 200 mM Tris/HCl (pH 7.40). The pH of GSH stock solutions was set to 7.4 before further dilution with buffer or buffer/acetone mixtures. GSH in buffer solutions was mixed with SSNO− in 1:1 ratio immediately before measurement and acetone-containing solutions with or without GSH were added to SSNO− immediately before measurement in a 1:1 ratio.
Decomposition of SSNO− was followed at 412 nm using Tecan Spark 10 M plate reader in 96-well microplate format.
Studying the reduction of SSNO− toward the NADPH-driven cellular reducing systems
To study the effects of the NAPDH–TrxR1–Trx/TRP14 or NAPDH–GR–GSH enzymatic systems on the decomposition of SSNO−, an aqueous mixture of 1 mM SNAP and 10 mM sulfide was prepared in 200 mM Tris/HCl buffer, pH 7.40. Selective removal of in situ-generated inorganic polysulfides was achieved by reacting this mixture with 500 μM DTT at +4°C, protected from light for 15 min, then the solution was degassed with N2 at +4°C, protected from light for an additional 15 min, to eliminate sulfide excess. After polysulfide and sulfide removal, 250 μL of this solution was pipetted into wells, and then each components of the enzymatic system were separately added to the solution. All kinetic experiments were carried out in Tris buffer (200 mM Tris/HCl and 100 μM DTPA, pH 7.40) at RT, in 96-well microtiter plates using a BioTek Powerwave XS plate reader. The presence of the enzyme systems on the decomposition of SSNO− was studied by following the absorbance changes at 412 nm, which is the λmax of SSNO−. Timescale and concentration conditions were optimized according to preliminary experiments by monitoring the reaction at 340 nm, which is the λmax of NADPH. The path length correction mode of the plate reader was used to record proper absorbance values (l = 1 cm) for further calculations.
LC/MS measurements of inorganic polysulfides
Enzymatic reduction of inorganic polysulfides
To study timescale and efficiency of the enzymatic reduction of inorganic polysulfides, 100 μM Na2S2 was incubated with 250 μM NADPH and 100 nM TrxR1, or 250 μM NADPH, 100 nM TrxR1, and 2 μM TRP14 at RT, protected from light. Twenty-five microliters of these reaction mixtures were taken at 0, 5, 10, 15, 30, and 60 min and incubated with 25 μL of 50 mM HPE-IAM at RT for 30 min. Then, 5 μL of 50% trichloroacetic acid (TCA) was added to the samples, followed by vigorous vortexing and centrifuging at 3000 g for 5 min. Samples were subjected to LC/MS analysis and the corresponding S1–S7 – HPE-IAM adducts were detected. The system consisted of a Thermo Ultimate 3000 HPLC equipped with binary pump, cooled autosampler, and UV-Vis detector connected to a Thermo LTQ-XL linear ion trap mass spectrometer. After HPE-IAM labeling, 20 μL of the acidified samples was injected on a Phenomenex Kinetex C18 2.6 μm 50 × 2.1 mm column for separation using eluents H2O/0.1%FA (A) and MeOH/0.1% FA (B) and the following profile: Initial composition of 5% B was increased linearly to 90% B in 15 min, followed by a 2 min decrease to 5% B and a 3 min equilibration before the next injection. MS/MS detection was used to quantify the species, unless stated otherwise, in which case only the parent ions were detected in single ion monitoring mode. In all cases, an ESI ion source was used in positive mode employing a spray voltage of 4 KV, capillary voltage of 14 V, and a capillary temperature of 300°C. The detected SRM parameters using collision-induced dissociation (CID) at a normalized energy of 35% are shown in Table 1, for SIM mode only, the parent masses were selected without further CID fragmentation. Isolation windows for all selected ions were set to ±0.5 m/z.
Detection of inorganic polysulfides upon the decomposition of SSNO−
To study the formation of inorganic polysulfides during SSNO− decomposition, a mixture of 1 mM SNAP and 10 mM sulfide was prepared and enzymatic purification was performed by incubation with 2 mM NADPH and 200 nM TrxR1, protected from light. Twenty-five microliters aliquots from this reaction mixture were taken at the indicated time points and reacted with 25 μL of 50 mM HPE-IAM as described in the Materials and Methods section. Protein removal was achieved by TCA precipitation, and then, samples were centrifuged and subjected to LC-MS/MS analysis as described previously.
SSNO−-induced sustained polysulfidation on GSH
To study the per- and polysulfidation effects of SSNO− on GSH, 1 mM GSH was incubated with enzymatically purified (10 min of incubation with 200 nM TrxR1 + 2 mM NADPH, followed by 5 min degassing with N2) 87 ± 16 μM SSNO− at RT, protected from light. Aliquots were taken from the reaction mixture at 0, 30, 60, and 120 min and alkylated with 100 mM IAM for 30 min at RT. Samples were then subjected to ultra-performance liquid chromatography with tandem mass spectrometry (UPLC-MS/MS) analysis or kept at −25°C until analysis. Separation was performed on a 1.6 μm diameter Modus 100 × 2.2 mm Aqua UPLC column (Chromatography Direct). Glutathione per/polysulfur species were detected by a Waters Xevo TQ–S triple quadrupole detector and identified in multiple reaction monitoring mode as described previously (37). As a control, a solution containing 500 μM Na2S2 and 9 mM Na2S was prepared, purified, and reacted with 1 mM GSH in parallel with the SNAP–sulfide reaction mixture. Samples from this control reaction were taken similarly at 0, 30, 60, and 120 min, alkylated with 100 mM IAM for 30 min at RT, and then subjected to UPLC-MS/MS analysis or kept at −25°C until analysis.
Time-resolved formation of HSA-SSH upon SSNO− treatment
The reduction of Cysteine-34 of HSA was achieved by incubation with 5 mM DTT in 200 mM Tris/HCl/100 μM DTPA buffer at pH 7.40, at +4°C for at least 30 min (3). Excess DTT was removed by desalting with Zeba Desalting Spin Columns (0.5 mL, 7KMWCO). Protein quantification was performed by the Bradford assay, followed by the treatment of 0.5 mg/mL of pre-reduced HSA with enzymatically (10 min of incubation with 650 μM NADPH and 200 nM TrxR1) purified SSNO− and control solutions. Aliquots were taken from the reaction mixtures at 0, 20, 40, and 60 min, samples were desalted using Zeba Desalting Spin Columns (0.5 mL, 7K MWCO), and HSA-SSH was detected by the ProPerDP method (15). Briefly, desalted protein solutions were alkylated with 1 mM iodoacetyl-biotin (IAB) (1 h at RT, protected from light). IAB excess was removed by gel filtration and by ultrafiltration using Amicon microconcentrators (using AMICON ULTRA 0.5 mL–30 kDa cutoff, Ultracel-30 regenerated cellulose membrane, or 0.5 mL sample volume; Merck Millipore). Biotinylated proteins were pulled down by streptavidin-coated magnetic beads (Sigma) according to the following procedure: after incubation on a rocking platform at RT for 30 min, magnetic beads were separated from the solution with a magnetic particle separator (Dynal MPC-M). Then, supernatant was placed in a clean tube, and the beads were washed with Tris-buffered saline containing 0.05% Tween 20 (TBST) three times, to eliminate nonspecific adhesion. Beads were then resuspended and incubated with 5 mM tris(2-carboxyethyl)phosphine (30 min, RT) while shaking, followed by magnetic separation. Beads were finally boiled at 100°C for 3 min in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer to elute all bound materials. The samples were then analyzed by SDS-PAGE followed by Western blot analyses (see Western blotting against HSA section). Protein concentrations were monitored throughout the process by the Bradford assay (Bio-Rad) and adjusted to the same concentration of HSA in each sample.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
Equal amounts of total protein were incubated with the streptavidin-coated beads in the case of each given gel and blot. Samples containing ≈50 ng protein were run on nonreducing 12% polyacrylamide gels at 120 V for at least 90 min. Gel bands were visualized following Western blotting, followed by 5-bromo-4-chloro-3-indolyl phosphate–nitro blue tetrazolium (BCIP-NBT; Merck) staining and imaged on Syngene G:Box gel documentation system.
Western blotting against HSA
Following SDS-PAGE, plasma samples were transferred to polyvinylidene difluoride membranes (130 V, 90 min), which were then blocked overnight in 3% bovine serum albumin (BSA) solution at 4°C. Membranes were subsequently incubated for 1 h in anti-albumin antibody (Sigma A0433), diluted 1:10,000 in 3% BSA. Subsequently, the membranes were washed three times with TBST buffer and incubated for 1 h in alkaline phosphatase-conjugated secondary antibody (Sigma A3687), diluted 1:10,000 in 3% BSA. Membranes were washed again three times in Tris-buffered saline, 0.1% Tween 20 (TBST) buffer, and bands were visualized by BCIP-NBT staining. Quantitative analysis of the bands was performed by using the ImageJ software (NIH).
Measurement of Ca2+ influx in TRPA1-expressing CHO cells in response to SSNO− by flow cytometry
TRPA1-expressing CHO cells were kindly provided by Prof. Zoltán Sándor and maintained in culture as described previously (5). For cell harvest, the culture medium [500 mL Dulbecco's-modified Eagle's medium (DMEM), 50 mL fetal BSA, 10 mL L-glutamine (200 mM), 10 mL MEM nonessential amino acid solution, 500 μL penicillin and streptomycin] was gently removed from the cells and trypsin solution (250 μL, 0.1% in phosphate-buffered saline) was applied for 5 min. For each sample, ∼104 TRPA1-expressing CHO cells were resuspended in 100 μL cell culture medium. Cells were incubated with Fluo-4 AM (Invitrogen, 0.4, 1 μg/μL in DMSO) for 30 min, at 37°C. Four hundred microliters of ECS was then added [400 μL, containing (in mM): NaCl, 160; KCl, 2.5; CaCl2, 1; MgCl2, 2; HEPES, 10; glucose, 10; pH 7.3] to reach a final volume of 500 μL. Then, the appropriate volumes of SSNO− and control solutions were taken from the reaction mixtures, diluted to 500 μL in ECS to reach a final 10 μM concentration of SSNO−, added to the cell suspensions, and analyzed by flow cytometry immediately. Fluo-4 AM was excited by 488 nm laser; fluorescence was detected at 504 nm. Mean green fluorescence of the samples was compared with base fluorescence of dye-loaded control cells. As positive control, cell groups were reacted with the selective TRPA1 receptor agonist mustard oil (100 μM in ECS) and analyzed by flow cytometry.
Data analysis
Plotting and statistical analysis were performed using GraphPad Prism 5.03 (GraphPad Software, Inc.). Measured values were compared using Student's t-test. To determine tendencies, deviation of the slopes from zero from linear regression analysis was tested. Relevant significances are denoted on the graphs depending on the obtained p values in the following way: ≥0.05ns, 0.01 to 0.05*, 0.001 to 0.01**, ≤0.001***.
Footnotes
Acknowledgments
We thank the NIHR Southampton Center for Biomedical Research and the University of Southampton Medical School for support with the mass spectrometry experiments and Dr. Mateusz Pitak at the EPSRC National Crystallography Service & Southampton Diffraction Center of the University of Southampton for assistance with X-ray diffraction studies of the different components of the [PNP+SSNO−] crystals. We are indebted to Prof. Elias Arnér for providing the thioredoxin family proteins, Prof. Miriam Cortese-Krott for stimulating discussions in the early part of the project, and the unknown reviewers of this article for their insightful criticism. Noémi Balog, Éva Sághy, Maya Payrits, Gábor Pozsgai, and Éva Szőke are acknowledged for their supports to different aspects of the experimental work discussed above.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This work was supported by the 2019 Hungarian Thematic Excellence Program (TUDFO/51757/2019-ITM), by the Hungarian National Research, Development and Innovation Office under grants number KH_126766 and K_129286, by EFOP 3.6.1-16.2016.00004, EFOP-3.6.3-VEKOP-16-2017-00009, OTKA PD 112171 grants, and by the ÚNKP-16-1 and ÚNKP-17-1 New National Excellence Program of the Ministry of Human Capacities.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.