
Abstract
Significance:
Sepsis is a health disaster. In sepsis, an initial, beneficial local immune response against infection evolves rapidly into a generalized, dysregulated response or a state of chaos, leading to multiple organ failure. Use of life-sustaining supportive therapies creates an unnatural condition, enabling the complex cascades of the sepsis response to develop in patients who would otherwise die. Multiple attempts to control sepsis at an early stage have been unsuccessful.
Recent Advances:
Major events in early sepsis include activation and binding of leukocytes and endothelial cells in the microcirculation, damage of the endothelial surface layer (ESL), and a decrease in the plasma concentration of the antioxidant enzyme, selenoprotein-P. These events induce an increase in intracellular redox potential and lymphocyte apoptosis, whereas apoptosis is delayed in monocytes and neutrophils. They also induce endothelial mitochondrial and cell damage.
Critical Issues:
Neutrophil production increases dramatically, and aggressive immature forms are released. Leukocyte cross talk with other leukocytes and with damaged endothelial cells amplifies the inflammatory response. The release of large quantities of reactive oxygen, halogen, and nitrogen species as a result of the leukocyte respiratory burst, endothelial mitochondrial damage, and ischemia/reperfusion processes, along with the marked decrease in selenoprotein-P concentrations, leads to peroxynitrite damage of the ESL, reducing flow and damaging the endothelial barrier.
Future Directions:
Endothelial barrier damage by activated leukocytes is a time-sensitive event in sepsis, occurring within hours and representing the first step toward organ failure and death. Reducing or stopping this event is necessary before irreversible damage occurs.

Introduction
Sepsis is a major health problem that requires specific and early diagnostic and treatment solutions. Sepsis has been recognized for many thousands of years, with descriptions dating back to 3000 BC in ancient Egypt (169).
Despite improvements in the understanding of the pathogenesis of sepsis and in therapeutic management, sepsis remains a silent public health disaster, with 337 cases occurring per 100,000 inhabitants every year in the United States, associated with a 30%–50% mortality rate (237). Moreover, patients who survive sepsis frequently have reduced long-term quality of life (133, 228). Sepsis accounts for more than $20 billion (5.2%) of total U.S. hospital costs (133, 276). The risk of a worldwide major infectious pandemic leading to multiple patients with sepsis is a persistent threat, as illustrated by the recent coronavirus disease 2019 (COVID-19) pandemic (60). More severe pandemics may occur in the future, of natural origin or as a result of use of biological weapons, as has already happened since medieval times, if not earlier (12, 62, 123, 173).
Severe noninfective events, such as extensive burns or irradiation, multiple trauma, some intoxications, acute pancreatitis, and major surgery, induce an acute innate response that shares similarities with sepsis (35, 185, 316).
Sepsis is not a disease but a complex syndrome resulting from an overwhelming dysregulated innate host response to invasive infection (59, 133, 187, 241, 276). According to the third international consensus definition for sepsis, an infection with a mild innate response is no longer called sepsis, and what was previously called “severe sepsis” is now defined as “sepsis” (169, 276). Despite some progress in early management, sepsis remains a major health problem worldwide; early detection is crucial to optimize treatment, but can be difficult (11, 48, 187, 199, 215, 222, 315).
Numerous phase III studies performed over the last few decades have failed to demonstrate improved outcomes of patients with sepsis with the interventions studied, most probably because of the great complexity of the pathophysiology of sepsis (14, 90, 208). Most of these interventions, including antioxidant approaches, were aimed at blocking one or more pathogenetic cascades (90, 153, 208, 226), and it has been suggested more recently that early therapeutic interventions should rather target the microcirculation or mitochondrial dysfunction (196).
The objective of this article, written in two parts, is to provide a clearer understanding of the early events of sepsis in terms of redox potential and free radical damage, to propose a new approach for early diagnosis and treatment, which will be covered in a separate part B article.
Here we focus on the oxidative consequences of the simultaneous activation and binding of activated neutrophils to endothelial cells, which induces the release of reactive oxygen, halogen, and nitrogen species (ROHNS), especially peroxynitrite, compounds that are effective against pathogens but deleterious for the endothelium. As a consequence of their hyperactivation, the intracellular redox potential of leukocytes, and especially neutrophils, increases and immature forms are released by the bone marrow. Excessive release of ROHNS overwhelms plasma antioxidant defenses, especially that provided by selenoprotein P, which protects against peroxynitrite-mediated oxidation, and, to a lesser extent, by plasma glutathione peroxidase (GPX3). The ROHNS damage the inner surface of the endothelial wall, especially the endothelial surface layer (ESL), and also the endothelial cells, especially the mitochondria.
These events lead to disseminated shock-induced endotheliopathy (SHINE), the first step to multiple organ failure (MOF) and ultimately death.
Duality of the Innate Response
Beneficial against infection
The innate response is an ancient mechanism adapted from plants to mammals to counteract the exponential growth of pathogens (195). The innate reaction at the site of an infection must occur rapidly to control the infection locally. Toll-like receptors (TLRs) are activated by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (191, 258). Phagocytosis, which involves phagocytic cells such as neutrophils (also called polymorphonuclear cells), is at the crossroad of the innate response with all the cascades implicated in sepsis (108, 150, 154, 165, 192, 203). This process releases ROHNS, which contribute to the killing of microorganisms (113, 160).
In sepsis, the innate response changes from a beneficial localized response against infection into a dysregulated and generalized response, leading to organ dysfunction and failure, especially circulatory, and to death (108, 191, 263, 281). The mortality rates associated with sepsis remain high, despite improvements in management, in particular more widespread, adequate early resuscitation, and improved supportive care in the intensive care unit (ICU) (24, 191, 241, 276, 280). The formal 3-h and 6-h bundles have recently been combined into a 1-h bundle, reinforcing the need for an efficient early intervention at the very early phase of sepsis (170).
Detrimental effects and cascade signaling in the early phase of sepsis
The availability of improved supportive care enables patients with sepsis to survive beyond their natural body functions. In this “unnatural” state, the innate response may be inadequate and deleterious (241, 314) (Fig. 1). Thus, the different forms of supportive care in this context could be likened to survival attractors (320) (Fig. 1). In the absence of such supportive care, most patients die (148). At this stage of sepsis, the dysregulated innate and host response is mixed, with complex pro- and anti-inflammatory reactions (61, 212).
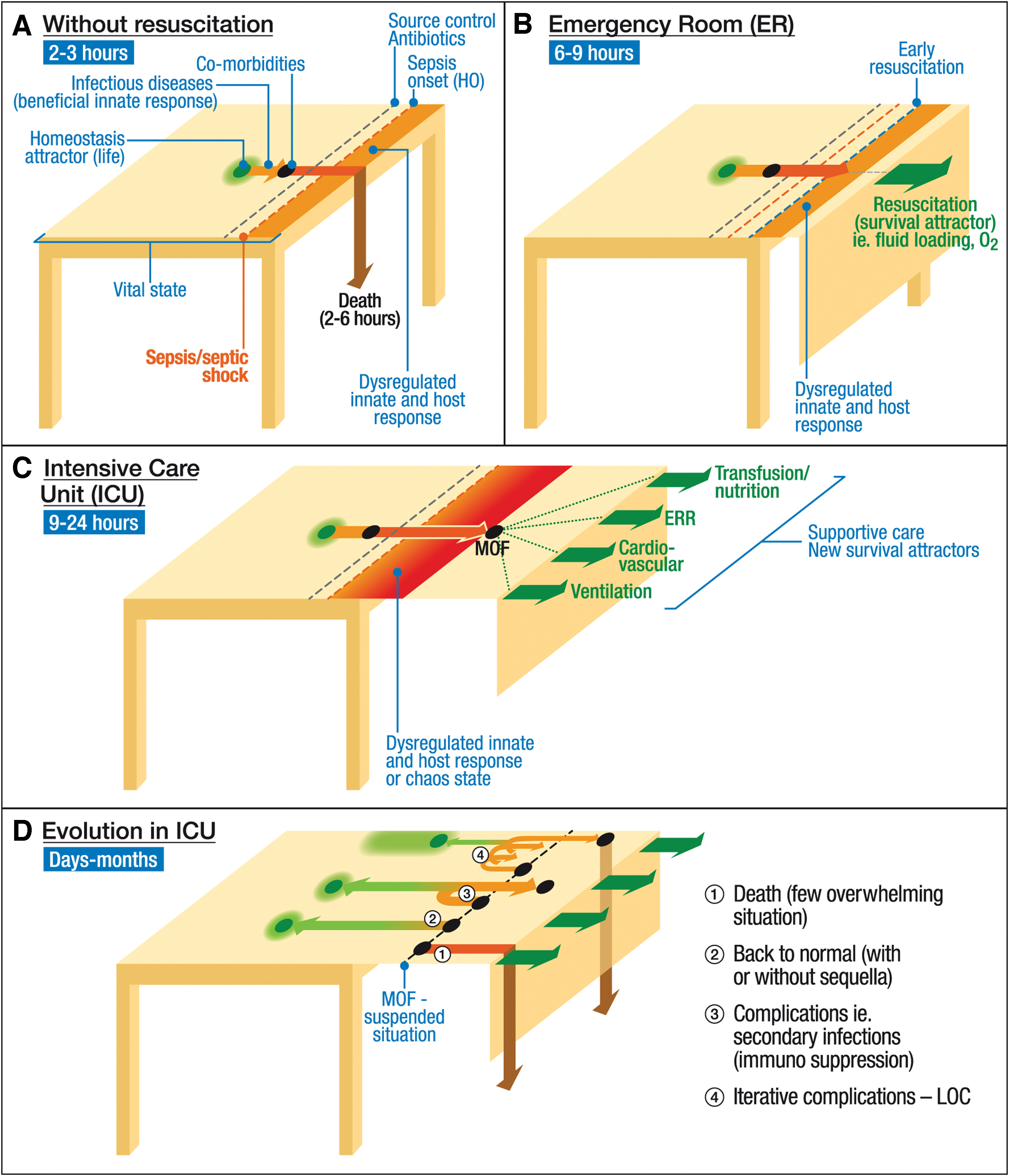
In such a situation, natural coping mechanisms may no longer be adequate, resulting in a state of chaos (3, 8, 321). Although the chaos theory is still debated, it is nevertheless widely acknowledged that the host response involves many cascades, with positive loops and complex feedback pathways and interactions (212).
Numerous unsuccessful attempts, including some large multicenter randomized phase III studies, have targeted single-cascade interventions, suggesting that multicascade interventions may be more effective (90, 133, 208, 318). Because of lack of an early effective treatment for sepsis and improved general intensive care management, most of the associated mortality is currently related to the effects of immunodepression (76, 133, 317).
One of the reasons for the numerous failed attempts to demonstrate the effectiveness of immunomodulatory therapies against sepsis may be the lack of an animal model that can adequately reflect sepsis and septic shock in critically ill patients (9, 89, 211). One of the important differences between animal models and patients may lie in serum proteins rather than in intrinsic cellular differences (327). Another reason for the lack of success is the complex cascade of interactions involved in the early phase of sepsis, which varies over time and with illness severity (90, 133, 237, 318).
At the extracellular level, these processes include activation of TLRs by PAMPs and DAMPs, cytokine storm (145, 207), coagulation activation (87, 95, 166, 198, 256, 285), formation of microparticles (236), and activation of the complement system (325), proteases, heat shock proteins, neuromediators, and the neuroendocrine axis (13). In addition, at the macrocirculation level, cross talk is observed between organs. One example of this cross talk is the effects of a decrease in perfusion of the small intestine, which induces ischemia/reperfusion injury and leads to a second hit by bacteria and compounds such as endotoxin, further enhancing the inflammatory response (110, 111, 255, 335).
Microcirculatory Dysfunction As an Early Event and SHINE
Importance of leukocyte binding to the endothelium
In the early phase of sepsis, interactions and binding between leukocytes, and more specifically neutrophils, and the endothelium are a major event at the level of the microcirculation (6, 11, 79, 94, 129, 133, 137, 140, 142, 150, 154, 165, 168, 192, 203, 209, 225, 236, 256, 268, 279, 338). Initially limited to the site of infection, this process becomes generalized and dysregulated in sepsis and is involved in many of the cascades cited earlier (Fig. 2). Before developing this concept further, we first describe the physiological functioning of the microcirculation (Fig. 2, upper panel).
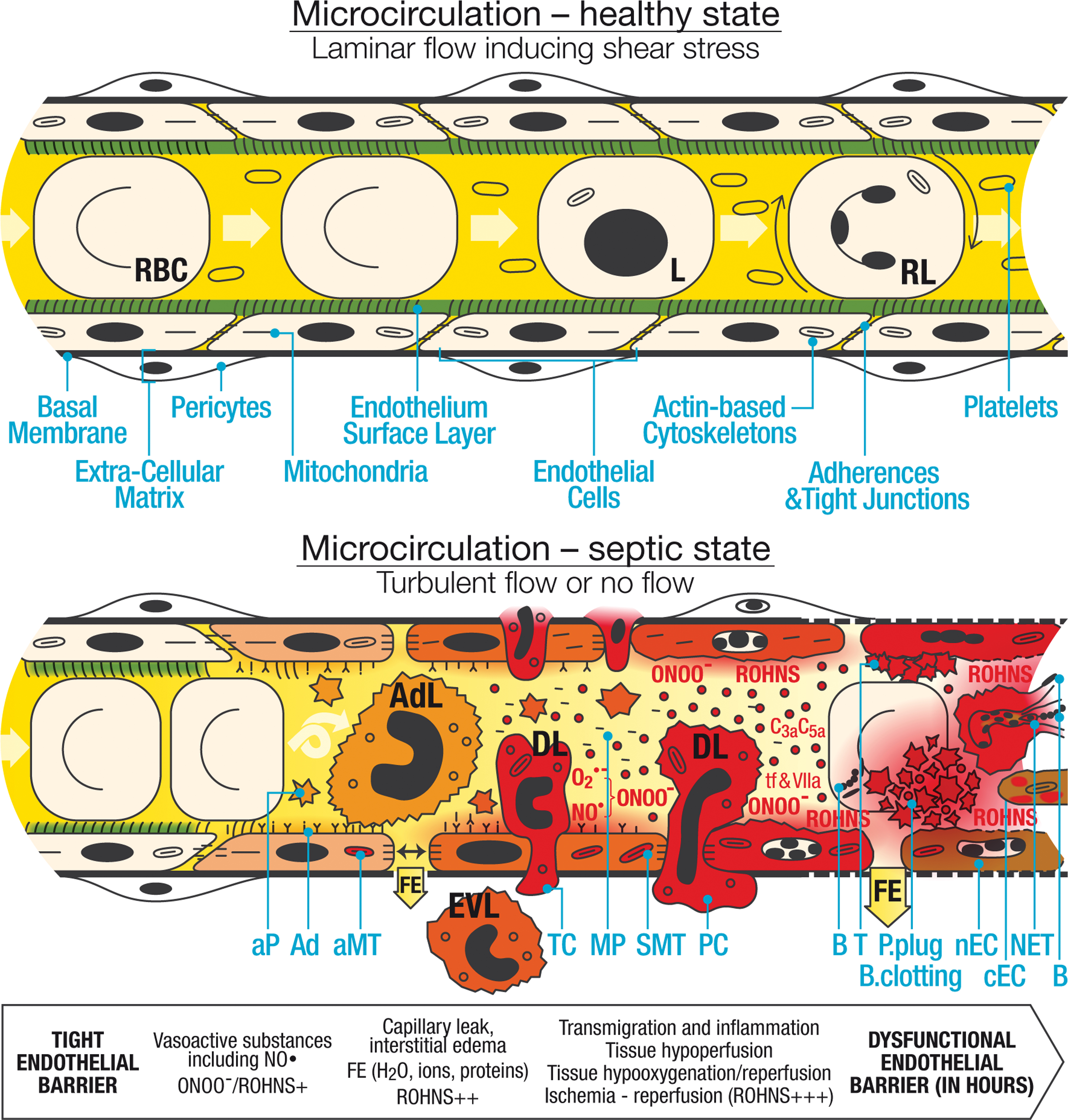
Physiological functioning of the microcirculation
The endothelium, which should be considered an organ, plays a pivotal role in the microcirculation (4, 5, 209). It forms a monocellular layer, the surface area of which is about 4000–7000 m2 (4 –6, 304), and acts as a border between a liquid medium—the plasma—and a solid structure—the basal membrane (6, 210). Physiologically, the endothelium is coated by glycoproteins, forming the glycocalyx (translated from the Greek meaning sugar coat) and, more broadly, the ESL (65, 230, 231). The endothelium has a wide range of physiological functions and can be viewed as an input/output system (5).
In blood, neutrophils account for 50%–70% of all circulating leukocytes. They are normally produced at a rate of 1011 per day and stored in the bone marrow, with only a small fraction (1%–2% in mice) released each day (165, 268). Under physiological conditions, neutrophils have a short half-life of 7–12 h in the circulation, where they are quiescent, and 1–2 days in tissues, because of spontaneous apoptosis (192, 219, 268). During the spontaneous apoptosis of neutrophils, both mitochondria- and death receptor-mediated apoptotic signalings are activated (219). This spontaneous apoptosis is inhibited by neutrophil activation as developed later. Rolling leukocytes can be observed and red blood cells (RBCs) are flexible. RBCs and leukocytes, including neutrophils, move evenly with platelets that are not activated (Fig. 2, upper panel).
In health, blood flow within the microcirculation is laminar, applying shear stress evenly on the surface of the endothelial cells. The ESL, which includes the glycocalyx, covers all endothelial cells and is highly fragile (56, 100, 229, 232) (Fig. 2, upper panel). It is closely regulated and differs, especially in thickness, from one organ to another (e.g., in the brain, the glycocalyx is thicker in hippocampal than in cortical microvessels) (56, 272, 308). With a thickness ranging from 0.5 to 1 μm, it forms a step between blood cells and the endothelium (229, 232).
The ESL is divided into a thin, cell-attached glycocalyx (about 70 nm) that includes proteins, glycolipids, glycoproteins, and proteoglycans, and a layer of linked molecules comprising (i) adsorbed plasma proteins, (ii) a soluble component, for example, extracellular superoxide dismutase (SOD), derived from endothelial cells, (iii) large linear hyaluronic acid interacting with cell surface glycoprotein CD44, and (iv) covalently bound glycosaminoglycan side chains (e.g., heparin sulfate and chondroitin sulfate), which are negatively charged (56, 100, 272, 308). In equilibrium with the flowing plasma, the ESL generates an exclusion zone for cellular blood components, and contains about 700 mL of adult plasma. The ESL acts to reduce the filtered protein concentration, reduces the effective oncotic pressure across the endothelium, and protects the endothelium against ROHNS (56, 231, 272). It plays an important role in normal vascular homeostasis, including flow resistance, transmission of shear stress, regulation of blood flow, oxygen transport, capillary barrier function including the glomerular filtration barrier, vascular permeability, creation of an anti-inflammatory and anticoagulation phenotype, inhibition of leukocyte adhesion, and angiogenesis (56, 100, 229, 232, 272). The ESL is thus intrinsically linked to vascular functionality (272). Tight junctions and adherent junctions, sealing the paracellular space between cells, join the endothelial cells and, with the ESL, form a double barrier that limits macromolecule transit (56). These components are linked to the actin-based cytoskeleton. The basal membrane is coated with pericytes, forming a layer. In this healthy state, mitochondrial activity is normal and the intracellular redox potential is low.
In endothelial cells, as in all cells, the antioxidant selenoenzymes are ubiquitously present, playing an important role in redox signaling, protection against ROHNS, and selenium (Se) metabolism in tissues and in the plasma (44, 53, 104, 159, 178, 288). Every selenoprotein contains only one atom of Se per molecule; with the exception of selenoprotein-P (see later) (104). In all selenoproteins, the Se is always included in the 21st amino acid, selenocysteine (50, 53, 104, 223).
By contrast with other essential trace elements, such as iron (Fe), copper, or zinc, Se is not present in ionic form, or is present in small molecules in plasma, most probably because of the oxidant properties linked to the particular electronic nature of Se (27, 53, 130, 261, 289). Se is required at the active site of all the antioxidant selenoenzymes of the 25 known selenoprotein genes in humans, as a consequence of the particular electronic properties of Se (70, 158).
Intracellular selenoproteins, which include GPXs, thioredoxin reductases (TXNRDs), and iodothyronine deiodinases, (44, 103, 104, 205), have crucial antioxidant functions, controlling the intracellular redox potential. These proteins therefore affect, in a complex manner, many intracellular metabolic processes (44, 104, 282, 284). The ambivalent electronic properties of Se account for its complex and highly energy-consuming intracellular metabolism (see separate part B article) (50, 53, 70, 130). Se can also replace sulfur in the amino acid selenomethionine, thus forming a storage pool of Se, which could be released in case of protein catabolism, but is not considered to have biological properties (53).
Se and Fe are both required for antioxidant defense and metabolism in eukaryotic cells, but also in almost all pathogens (105, 162, 172, 283, 286). Physiologically, plasma Fe, which is at physiologic pH in ferric state (Fe3+), is mainly complexed to transferrin. Each protein has two iron binding sites (218). Nontransferrin-bound iron (NTBI) comprises Fe3+ citrate, acetate, and Fe loosely bound to albumin (286, 287). Cells use transferrin receptors to acquire Fe.
The biologically active form of Se is transported in the plasma from the liver to the tissues within the nine selenocysteine aminoacids of the C terminal part of selenoprotein P, which has bifunctional activity (53, 104, 126, 249, 261). Selenoprotein P binds to specific receptors (apolipoprotein E receptor-2—ApoER2) (53, 126, 260) or megalin (53, 157, 260).
During anaerobic exercise, selenoprotein P also binds to heparin sulfate proteoglycan, favored by the low pH, and has antioxidant enzymatic activity through the single selenocysteine of the N terminal part of the protein (42, 52, 53, 85, 132, 193, 220).
Selenoprotein P accounts for about 60% of plasma Se with marked homogeneity between mammals, especially in terms of the N terminal portion (118, 121, 127, 176, 201). It has a short half-life of about 4 h in rats (53). It is glycosylated and belongs to the heparin binding proteins (307). In physiological states, selenoprotein P has the crucial function of transporting Se and regulating the delivery of biologically active Se from the liver to the tissues (53, 261).
The second plasma selenoprotein, GPX3, contains about 30% of plasma Se, and is synthesized in the kidney; it is dependent on selenoprotein P intake (128, 262). GPX3 does not seem to participate in Se transport (53). GPX3 antioxidant function in plasma is unlikely in physiological conditions because of the low plasma glutathione (GSH) concentration (31, 115). Its possible antioxidant function during sepsis is discussed later. The remaining plasma Se is included in proteins, notably albumin, within the nonbiologically active aminoacid, selenomethionine (53).
Microcirculatory Dysfunction and Endothelial Damage During Sepsis
Large increase in neutrophil activity, redox potential, and release of immature neutrophils
During sepsis, neutrophil production dramatically increases to 1012 per day, consisting of mature and immature forms, even very immature forms, to compensate recruitment and margination as the first line of cellular defense (190, 247, 268, 294). Neutrophil differentiation includes the myeloblast, promyelocyte, myelocyte, meta-myelocyte, band, and segmented (mature) neutrophil stages (165). This includes band cells characterized by a curved but not lobular nucleus (77, 165, 185, 247, 273). The immature forms may be more aggressive, increasing oxidative stress (185, 190, 294, 311). During the early phase of sepsis, neutrophils also have an increased life span in the circulation, from less than a day to up to 5 days. This is related to delayed apoptosis and further enhances the innate response (76, 192, 219, 268).
In neutrophil cell signaling in sepsis, the recognition of PAMPs (e.g., lipopolysaccharide [LPS], bacterial deoxyribonucleic acid [DNA], …) and DAMPs (e.g., heat-shock proteins, uric acid, …), and other signaling molecules, results in stimulation of neutrophils and endothelial cells by kinase activation, inducing the expression of transcriptional factors, such as nuclear factor-kappa B (NF-κB), and other factors of the rel family, central to the acute inflammatory response (1). Secretory neutrophil vesicles can rapidly transport their contents to the cell surface and can kill pathogens by phagocytosis and degranulation of neutrophil extracellular traps (NETs) (154, 247). The ability of neutrophils to kill pathogens is immediate, nonspecific, and does not depend on previous exposure (247).
It is important to specify that the redox potential modifies function of all cells, from bacteria to eukaryotic cells (15, 117, 177, 239). The redox potential depends notably on the redox environment, the intracellular production of reactive oxygen species (ROS) by the mitochondria (16, 277), and the peroxide-associated enzymes linked to cellular redox couples (49). An increase in the degree of oxidation of the intracellular redox potential modifies cell function from starvation to multiplication or activation (activation of transcription factors, protective enzymes, increase in Ca2+), to preapoptosis, and finally to cell death by apoptosis, necrosis, or ferroptosis (49, 106, 112, 114, 143) (Fig. 3).
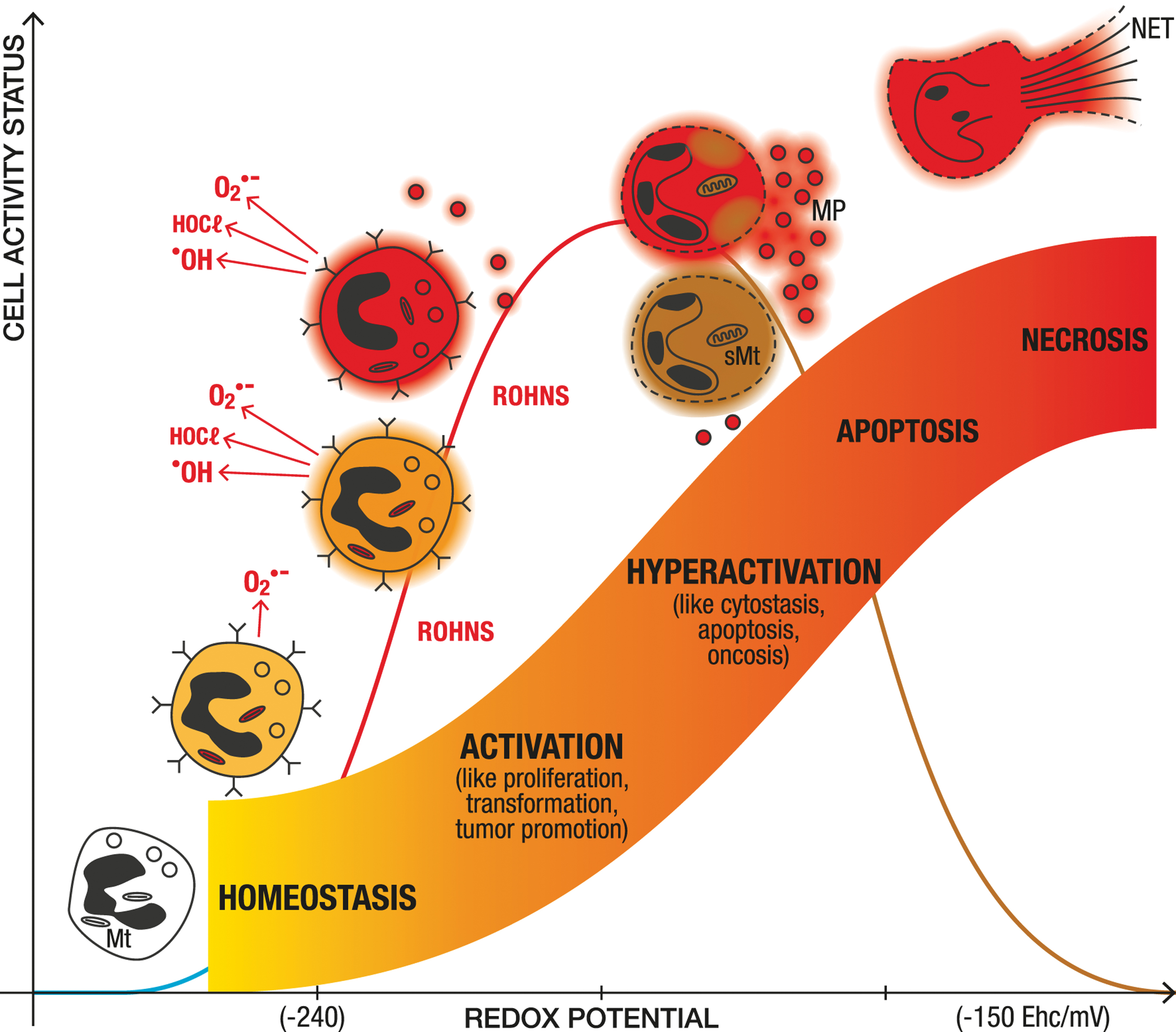

Main Reactive Oxygen, Halogen, and Nitrogen Species Within Cells and in Plasma Near the Endothelial Vascular Membrane: Radical Species (One-Electron Oxidant)
See Balligand (23), Halliwell (113, 114), Martinez-Cayuela (188), Sies (271), Vanasco et al. (312), Winterbourn et al. (334), and notably Halliwell (113) and Winterbourn et al. (334). The most reactive species are in bold. Respiratory burst is part of the immediate innate response of sepsis (113, 334). O2 uptake by neutrophils can increase from 20- to 100-fold (141, 238, 264, 265, 333). ROHNS can be produced within the phagosome or secreted into the blood especially near the endothelial vascular membrane (46, 297).
iNOS increases NO• synthesis by a factor of up to 1000. It has numerous effects in sepsis: vasodilation, migration, and activation of leukocytes (151, 296). Hereditary deficiency of the NOX system leads to chronic granulomatous disease. Patients have persistent and multiple infections, especially by Staphylococcus aureus, Pseudomonas, and Aspergillus, and chronic inflammatory lesions that may be due to feedback disruption (113). This is also observed in knockout NOX mice (114). Anoxia also decreases phagocyte killing (113).
cNOS, constitutive (or endothelial) nitric oxide synthase; COX, cyclooxygenase; DNA, deoxyribonucleic acid; Fe-S, iron/sulfur; H2O2, hydrogen peroxide; iNOS, inducible nitric oxide synthase; I/R, ischemia/reperfusion; LPS, lipopolysaccharide; MPO, myeloperoxidase; NO•, nitric oxide; NOX, NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase); NOx, nitrite+nitrate; PUFA, polyunsaturated fatty acid; ROHNS, reactive oxygen, halogen, and nitrogen species; XO, xanthine oxidase.
Main Reactive Oxygen, Halogen, and Nitrogen Species Within Cells and in Plasma Near the Endothelial Vascular Membrane: Nonradical Species (Two-Electron Oxidant)
See Balligand (23), Halliwell (113, 114), Martinez-Cayuela (188), Sies (271), Vanasco et al. (312), Winterbourn et al. (334), and notably Halliwell (113) and Winterbourn et al. (334). The more reactive species are in bold. ROHNS are an important part of the killing of pathogens, but also are a major factor in endothelial damage leading to multiple organ failure (113, 297). ROHNS can be within the phagosome or secreted into the blood, especially near the endothelial vascular membrane (46, 297). They can increase very rapidly within 30 min in in vitro experiments (141).
Peroxynitrite (ONO2 −), produced by the reaction of NO• with O2 •−, rapidly oxidizes sulfhydryl groups and thioethers, and induces nitration and hydroxylation of aromatic compounds, including tyrosine, tryptophan, and guanine. Peroxynitrite is required for effective antimicrobial defense. Mice that are double knockout for gp91 NOX and iNOS are nonviable to endogenous bacteria, whereas simple knockout mice are less sensitive to infection (270). On the contrary, ONO2 − is most probably the more dangerous oxidative agent leading to endothelial damage, our key topic (297). Its formation is assumed to be spatially associated with O2 •− near the plasma endothelial membrane. In acute inflammatory conditions, the ESL and glycocalyx are the first line of the endothelium/blood interface. They are extremely sensitive to oxidative stress, such as peroxynitrite, and a first step toward endothelial dysfunction (138, 186, 272, 278). Microcirculatory disorders and endothelial mitochondrial damage are considered to be linked to oxidative and nitrosative stress (196). The pathogenic role of peroxynitrite has been considered to lead to multiple organ failure (297).
CO2, carbon dioxide; ESL, endothelial surface layer; HOCl, hypochlorous acid; ICU, intensive care unit; iNOS, inducible nitric oxide synthase; NET, neutrophil extracellular traps; SCN, thiocyanate.
However, the response to an increased oxidation state varies from cell to cell (134). Immune cells, especially lymphocytes, have increased apoptosis, whereas apoptosis is delayed in monocytes/macrophages and neutrophils, which may be related to the particular properties of their mitochondria (76, 247, 285, 309, 326). The increase in intracellular superoxide anion (O2 •−) concentration, due to extra- and intracellular production, and the increase in redox state are involved in neutrophil priming before complete activation (74, 180, 247, 293, 298) (Table 1). The increase in redox potential increases neutrophil activation by membrane perturbation (293), activating the arachidonic acid cascade (171), Janus kinase (JAK) activation as reported by Brigelius-Flohe, especially with effects on thiol residues (43, 45, 57, 149, 293), redox activation of NF-κB (293), nitration of protein tyrosine by peroxynitrite (ONOO−) (57, 293), and mitochondrial one-electron way blockage (41).
Extracellular formation of ROHNS, such as hydrogen peroxide (H2O2) and ONOO−, can induce neutrophil priming (293). Intracellular myeloperoxidase (MPO) activation is associated with decreased GSH concentration by hypochlorous acid (HOCl), resulting in irreversible protein thiols and in GSH oxidation (57, 74) (Table 2), whereas H2O2 detoxification by GPXs leads to reversible glutathione disulfide (GSSG) (57). Irreversible GSH oxidation by HOCl decreases the antioxidant defense and may contribute to increased redox potential (57). Following phagocytosis and MPO lipid peroxidation, an increase in protein carbonyl is observed in the neutrophil cytosol (330).
However, additional enhanced redox potential by increased intracellular H2O2 concentration contributes to reduce the inflammatory phenotype (337). The initial stage of neutrophil apoptosis is linked to changes in the expression of genes encoding for the redox pathway, including GSH-, thioredoxin- and heme metabolism (247). Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX2) and MPO are considered key players in postphagocytotic events, such as activation of cell death and NETs (Fig. 4). This is in accordance with the central impact of redox potential on cell function (49, 106, 112) (Fig. 3).
When comparing the redox status in resting and oxidative burst conditions of the human leukemia cell line-60 (HL-60), a cell line from an acute promyeolocytic leukemia patient, to that of neutrophils, these cells share similarities (74, 97). However, they also differ. For example, in resting and oxidative burst states, recycling of peroxiredoxins, thiol peroxidases with a key role in antioxidant defense and redox signaling, may be deficient in neutrophils compared with HL-60 cells (74).
Neutrophils are also implicated in many of the numerous extracellular cascades in the early phase of sepsis, such as the cytokine storm, coagulation activation, microparticle release, and complement activation, among others (90, 133, 237, 318). Another important cellular cross talk is the interaction of neutrophils with other immune cells, such as activation of natural killer (NK) cells, leading to (219) a self-amplifying loop of inflammation with dendritic cells, while inhibiting the proliferation of T lymphocytes (154, 192). Neutrophil/macrophage interactions are important in both the initiation and the resolution phases of sepsis, especially in the process of phagocytosis-induced cell death of neutrophils by macrophages as underlined by Kennedy and DeLeo (146, 192, 285). Finally, neutrophils interact with platelets, in particular, through microparticles displaying the platelet activating factor (PAF) (192). Generation of NETs, which is an effective mechanism for bacterial trapping, is an additional deleterious neutrophil/endothelium interaction (78). Together, these mechanisms result in activation and binding of neutrophils and endothelial cells (6, 11, 79, 94, 129, 133, 137, 140, 142, 150, 154, 165, 168, 192, 203, 209, 225, 236, 256, 268, 279, 338).
Hyperactivation and interaction between endothelium and leukocytes, and ROHNS formation
In sepsis, the endothelium receives information from mediators of inflammation, including cytokines, growth factors, chemokines, complement and coagulation factors, ROHNS (including oxidized lipid), and circulating cells (leukocytes, especially neutrophils, but also RBCs and platelets). Endothelial cells in return alter the vasomotor tone, plasticity, and fluidity (230, 231), and interact with circulating cells, especially neutrophils. In addition, sepsis-induced microcirculatory alterations are characterized by heterogeneous abnormalities in blood flow and pathological shunt, in which the different degree of expression of inducible nitric oxide synthase (iNOS) and nitric oxide (NO•) production in different organ beds plays an important role in the ischemia/reperfusion process (72, 73, 139, 209, 305).
Dramatic increase in ROHNS production
ROHNS play a major role in the early phase of sepsis (Tables 1 and 2) (Figs. 2 and 4) (23, 113, 114, 188, 271, 334). A hyperoxidation redox state in sepsis was first reported as a major part of the innate response in 1990 by Novelli and colleagues and confirmed later by others (96, 202, 204). This hyperoxidation can be related to three main mechanisms (5, 92, 204, 217, 250): (i) the respiratory burst from hyperactivated leukocytes and concomitant activation of the endothelium, which is probably the most important early event (81, 113, 119, 136, 152, 180, 192, 204, 300, 329); (ii) the onset of ischemia/reperfusion (204, 250); and (iii) blockage of the mitochondrial respiratory chain (39, 40, 98, 119, 274).
In the early phase of sepsis, neutrophil activation induces an abrupt increase in oxygen consumption, termed the respiratory burst (247, 264, 334). ROHNS are produced by activated leukocytes (especially O2 •− and halogen species) and by endothelial cells (especially NO•). Neutrophils or macrophages via NOX2 and MPO represent most of the protein oxidation and lipid-derived free radicals in the very early 6-h phase following LPS instillation (243, 253). (Figs. 2 and 4) (Tables 1 and 2). ROHNS are ambivalent molecules that are major antimicrobial agents, and are also toxic to endothelial cells (23, 113, 114, 188, 227, 236, 270, 271, 312, 331, 334). Among the ROHNS, ONOO− should be highlighted as it can cross membranes, and lead to the formation of extremely reactive species. ONOO− can be split into hydroxyl (•OH) and nitrogen dioxide (NO2•) radicals when protonated or to NO2• and carbonate radical (CO3•) when reacting with carbon dioxide (CO2) (40, 80, 113, 141, 235, 297, 334). Superoxide anion (O2 •−) rapidly dismutates H2O2, activating MPO or producing •OH via Fe++ - or copper (Cu+)-catalyzed Fenton/Haber-Weiss reactions (144, 244, 296, 332). In addition, ONOO− enhances the release of NETs, which further increase the oxidative stress (183) (Figs. 2 and 3).
Adhesion of activated leukocytes to the endothelium is important for ROHNS production in plasma at the edge of the vascular luminal surface in the microcirculation (122). The simultaneous production of O2 •− and NO• at this location leads rapidly, in less than 30 min in vitro, to the formation of ONOO− (46, 141, 297). Oxygen consumption by hyperactivated leukocytes increases by a factor of 20%–100%, leading to a marked release of O2 •− in phagosomes and in the plasma (141, 238, 265, 266, 333).
Release of NO• by endothelial cells increases from a picomolar range by constitutive (or endothelial) NOS (cNOS) to a nanomolar range from iNOS (151, 296). The gradient of NO• concentration, decreasing exponentially with distance from the cell, favors a spatially increased concentration of ONOO− near the endothelial vascular membrane (46, 245, 290, 297). This process targets the ESL, endothelial cells, especially their mitochondria, and multiple other targets (46, 138, 186, 196, 272, 278, 297) (Tables 1 and 2) (Figs. 2 and 4). HOCl, initially synthesized in the phagosome via MPO, can be released or synthesized in the plasma, despite being buffered by hypothiocyanite (22, 116, 141, 333) (Table 2 and Fig. 4). In addition, multiple positive redox loops increase ROHNS production in the acute phase of sepsis, an unnatural, dysregulated, or chaotic immune response, made possible by supportive care (122, 151, 296) (Fig. 1).
In the early phase of sepsis, there is also a complex alteration of the microcirculation, with closing and reopening of small vessels and opening of shunts leading to ischemia/reperfusion and NOS activation. Activated macrophages and ischemia/reperfusion processes through xanthine oxidase (XO) and cyclooxygenase participate in the plasma increase in O2 •− and H2O2 concentrations (120, 224, 242, 339) (Table 1), as do NOX2, mitochondria, and NO• by iNOS (69, 109, 115, 227, 243). These processes are an important part of ROHNS production and interact with neutrophil activation (109, 180).
Finally, in the early phase of sepsis, inside the endothelial cells, and later within tissues, mitochondrial dysfunction—by reversible blocking by NO• of complex I and IV and later irreversible blocking of complex I by ONOO−—increases the production of O2 •− (40, 213, 235). In turn, mitochondrial isoforms of NOS further increase NO• synthesis within cells (101, 235). NO• also reacts with heme to form nitrite (NO2 −) and nitrate (NO3 −) (151) and may react with O2 •− to form ONOO− within the mitochondria (235) (Tables 1 and 2). Damage to mitochondria is discussed later as part of endothelial cell dysfunction.
Other radicals, oxidants, and secondary species are produced, such as singlet oxygen 1O2, alkyl peroxynitrites RONOO−, alkyl radicals RO• in reaction with lipids (especially unsaturated), and many other reactive molecules as detailed by Halliwell (33, 68, 88, 113, 160, 216, 226, 243, 312) (Tables 1 and 2).
Decrease in Plasma Antioxidant Defense by Selenoenzymes
In parallel to the increase in ROHNS production, there is increased nonspecific adhesion of selenoprotein P to the endothelium via its heparin binding domain (7, 18, 52, 124, 157), especially to the ESL, which contains heparin sulfates (17, 18). This binding is favored by acidic pH (42, 52, 53, 132); it is also sensitive to ionic strength (18).
In addition, multiple mechanisms support a downregulation in the synthesis and excretion of selenoprotein P by the liver during the acute phase of sepsis, including (i) negative regulation by inflammatory cytokines, especially interleukin (IL)-1β (83, 201); (ii) hypoxic conditions favoring the synthesis of phospholipid hydroperoxide glutathione peroxidase (GPX4) instead of selenoprotein P (30); (iii) decreased selenoprotein P excretion and selenoprotein P messenger ribonucleic acid (mRNA) in an LPS mouse model (240); (iv) decreased hepatic transcription and protein content of factors required for Se insertion into selenoproteins (269); and finally, (v) liver mitochondrial dysfunction in sepsis may impair selenoprotein synthesis, which is highly energy requiring, especially for synthesis of selenoprotein P, which contains 10 selenocysteine residues (39, 41, 119, 136, 184, 226, 243, 246, 313). This counterregulation of selenoprotein P synthesis and excretion will be further developed in a separate part B article.
As a result of increased binding, downregulated liver excretion, and fluid extravasation, plasma Se and selenoprotein P concentrations decrease by up to 75% in a few hours in mice, rat, and sheep septic models. The more severe the model, the more rapid is this decrease (182, 240, 252, 291, 324) (Fig. 5). In a resuscitated peritonitis sheep model, a halving of plasma Se concentration was observed 4 h after the onset of peritonitis. On admission to the ICU, a profound decrease in plasma Se and selenoprotein P concentrations is observed in patients with sepsis and septic shock (32, 91, 93, 131, 251).
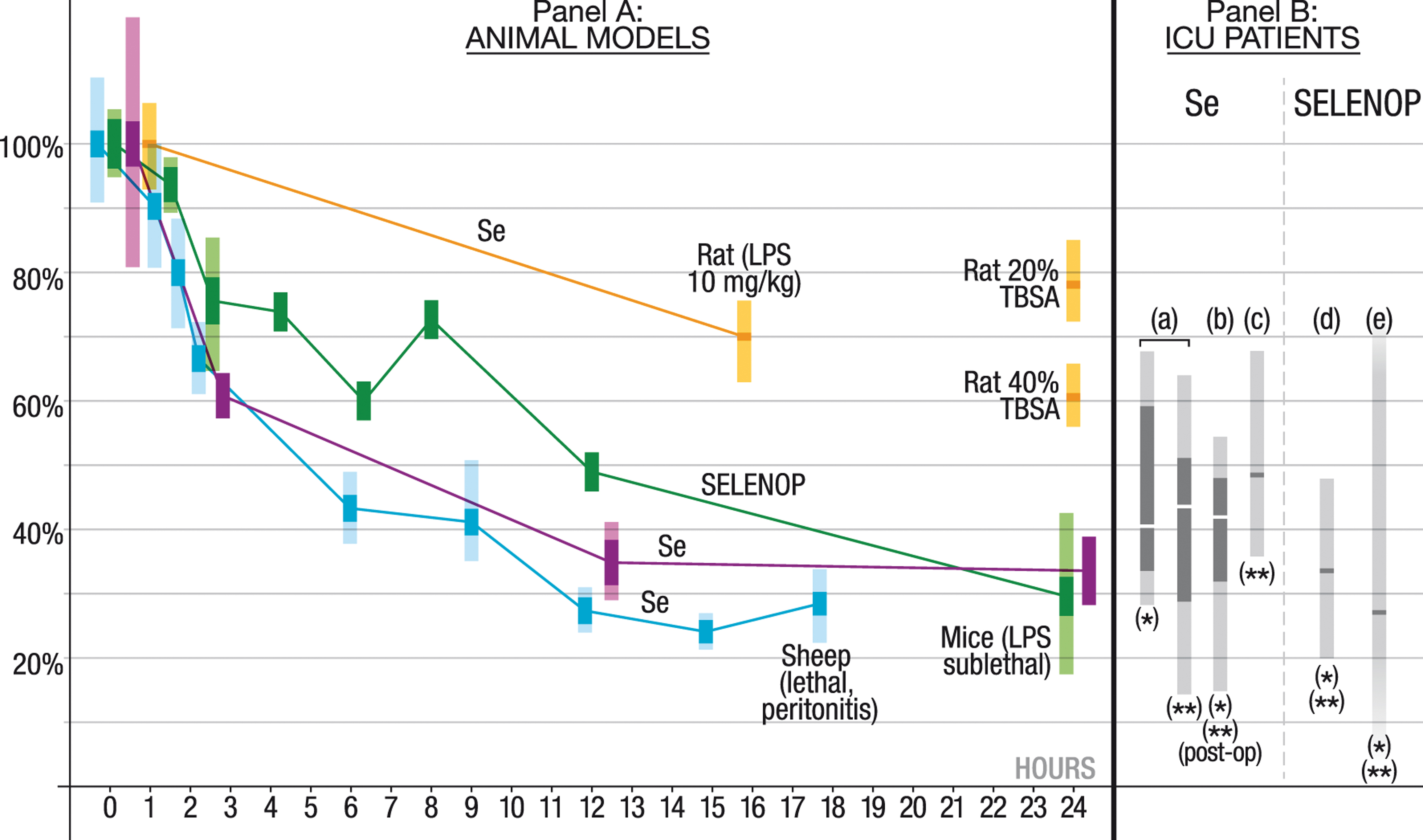
In the early phase of sepsis, this marked decrease in plasma selenoprotein P concentration limits the availability of Se to pathogens, but also decreases plasma antioxidant defense, especially at the ESL level; the N-terminal part of selenoprotein P has high antioxidant enzymatic activity (53, 248, 299), especially against ONOO− (19, 20, 42, 51, 54, 201, 288, 299, 303).
Later, GPX3, whose synthesis depends on selenoprotein P, may exert its antioxidant action after specific binding to basement membranes or the extracellular matrix (ECM). According to the double barrier hypothesis, this effect may play an important role in limiting macromolecule transit and fluid extravasation (55, 56, 254) (Fig. 2).
Involvement of Iron in ROHNS Production
A parallel could be made between altered Se and Fe metabolism during sepsis. As previously indicated, bacteria, with few exceptions, require Fe for growth and proliferation (162, 200, 286). However, in the presence of O2 •− and H2O2, Fe catalyzes Fenton/Haber-Weiss reactions leading to the production of the extremely reactive •OH (Table 1) (144, 332). Most of the body's Fe content (4–5 g) is incorporated into hemoglobin (Hb) (2.5 g) (162, 221); the remainder is in other hemoproteins (such as myoglobin [Mb]), iron/sulfur (Fe-S), and Fe binding and storage proteins (218). Within the RBC, Hb is protected against O2 •− by antioxidant enzymes, such as SOD, and against H2O2 by catalase (CAT) and intracellular cytosolic glutathione peroxidase (GPX1) (28, 99). Auto-oxidation of Hb, or oxidation by plasma nitrite, may release H2O2 or diffusible protein radicals (28, 147, 175).
Nevertheless, in the acute phase of sepsis, the main impact of ROHNS, especially ONOO−, on RBCs is oxidation of their membrane and their cytoplasmic proteins, including the cytoskeleton (175, 206) (Table 2). This oxidation decreases RBC deformability, increases RBC aggregation (Fig. 2) (28, 206, 292), increases blood viscosity, and alters tissue oxygenation (28, 292). In addition, oxidized RBCs may induce ROHNS formation by macrophages and neutrophils as part of multiple positive loops (206). Macrophages also play a pivotal role in erythrophagocytosis (162). Inflammatory anemia occurs at a later stage of sepsis (28, 37, 221, 295), but anemia resulting from previous Fe deficiency, blood loss, hemodilution, or frequent blood sampling may alter tissue oxygenation (37, 221).
In the acute phase of sepsis, the circulating ionic form of Fe is decreased by hepatocyte expression of hepcidin, which (i) increases H-ferritin (Fe3+ storage in macrophage); (ii) downregulates Fe efflux; (iii) increases secretion of ceruloplasmin, a copper ferroxidase; and (iv) decreases transferrin concentration (162, 218). Lactoferrin also chaperones Fe in neutrophils (162, 218). This occurs in less than 3–6 h in sepsis models (162, 221), inducing hypoferremia and low NTBI (306).
In septic patients admitted to the ICU, low serum Fe, transferrin concentration, and transferrin saturation, and high ferritin concentrations are observed (37, 221). In sepsis models, hepcidin agonists have been shown to decrease mortality, and intravenous Fe administration increased mortality (218, 286, 287, 295). In septic ICU patients, a higher concentration of catalytic Fe (similar to NTBI) and a lower concentration of hepcidin have been associated with higher mortality rates (163). On the contrary, small quantities of Fe2+ can serve as a catalyst in the phagolysosome for Fenton/Haber-Weiss reactions (340). In neutrophils, lactoferrin may also enhance such a reaction (26). Catalytic Fe may contribute to positive inflammatory loops (197).
Cell-free Hb or Mb is released in the plasma during hemolysis, hemorrhage, and rhabdomyolysis (cell-free Mb), the latter being implicated in renal toxicity (267). These compounds induce the expression of (i) acute-phase proteins hemopexin and haptoglobin, which bind to heme and Hb; and (ii) heme oxygenase-1 (HO-1), which catabolizes hemopexin-bound heme by the hepatocytes (84, 161, 162). However, this clearance mechanism can be overwhelmed (84). During sepsis, hemolysis has been suspected to be the main source of catalytic Fe, free heme, and free Hb, which in turn further increase hemolysis (84, 161, 163). These products increase the severity of sepsis in mice (161, 163, 194, 267) and also have the characteristics of a “danger signal,” amplifying the innate response. Nevertheless, their importance in oxidative stress during the acute phase of sepsis is debated (84). Hemolysis is certainly an important mechanism in acute malaria and this may also be the case in infections by pathogens secreting hemolysins, such as Staphylococcus species (spp.), Stretococcus spp., and Clostridium spp., or in case of disseminated intravascular coagulation (84, 200).
Damage of the First Barrier, the ESL
Damage of the ESL plays a major role in sepsis pathophysiology, modifying hematocrit, flow resistance, permeability, coagulation, leukocyte adhesion, and the ability to bind a variety of proteins, including selenoprotein P, to heparin sulfate (17, 18, 56, 100, 138, 245, 308). The damage induces positive amplification loops, through (i) destruction of the glycosaminoglycan chain and unsulfated hyaluronic acid and release of hyaluronic acid fragments (82, 107, 272); (ii) cleavage of heparan sulfate from proteoglycans, themselves cleaved from endothelial cells (308); and (iii) loss of the heparan sulfate-bound extracellular SOD—directly or through activation of heparanase—that further increases oxidative stress (272).
As illustrated in Figure 2 (lower panel) and Figure 4, in the early phase of sepsis, ROHNS are central players in the destruction of the ESL, which is extremely sensitive to free radicals and oxidants (11, 29, 38, 82, 107, 186, 210, 245, 272) (Tables 1 and 2). ONOO− and HOCl play an important role in ESL damage and enzyme activation (38, 272) (Fig. 4). These oxidant molecules activate matrix metalloproteinase, a zinc-dependent endopeptidase, and heparanase (272). The damage to the ESL, the first line barrier of the blood/tissue interface, further exposes endothelial cells (Fig. 2). Adhesion molecules are unmasked (272). Following rupture of the tight junctions, according to the double barrier hypothesis, this leads to fluid extravasation and protein-rich leakage (Fig. 2) (11, 56, 165, 272).
In animal models and human volunteers, LPS administration results in reduction in the depth of the ESL and glycocalyx shedding within hours, with a similar kinetic to that of the decrease in selenoprotein P concentrations (56, 82, 182, 240, 291, 308, 324). This effect may be initiated within minutes during the ischemia/reperfusion process (245) and coincided with vascular dysfunction in a rat LPS study (186). As a consequence, numerous glycocalyx degradation products are under investigation as early sepsis markers (56, 308).
Damage may vary from one organ's circulation to another. As an illustration, the hippocampal region, which physiologically has a thicker glycocalyx than the cortex, seems to be especially susceptible to damage, altering the blood/brain barrier and making it one of the first organs to suffer in sepsis, engendering long-term sequelae (56). Similarly, early glycocalyx damage on the thicker glycocalyx present in the pulmonary vasculature compared with that in the systemic vasculature may be an important first step toward acute respiratory distress syndrome (56).
Damage to Endothelial Cells with Loss of the Second Barrier and Mitochondrial Dysfunction
During sepsis, there is a change in endothelial properties from a noncoagulant, nonadherent, vasodilator phenotype to a procoagulant, adherent, vasoconstrictor phenotype (6, 137, 139, 164, 167, 209, 301, 329). Under the expression of adhesins (P- and E-selectins; vascular cell adhesion molecule [VCAM]; intercellular adhesion molecule) (322), slow rolling of leukocytes is observed, leading to leukocyte arrest and adhesion to the endothelial surface, leukocyte crawling, and diapedesis. Diapedesis may be of trans-endothelial cells or para-endothelial cells. Extravascular leukocytes are observed beyond the ECM. Phagocytosis can be observed with intracellular bacteria. Degranulation by leukocytes can also contribute to killing of bacteria.
Breakdown of the second endothelial barrier, after ESL degradation, is due to physical endothelial disruption with breakdown of the actin cytoskeleton, and rupture of the tight junction and the basal membrane, resulting in opening of intercellular gaps. There is increased microvascular permeability with fluid extravasation and retraction of endothelial cells.
Flow is reduced, decreasing the shear stress. The endothelial cell membrane becomes unstable with formation of microparticles, which originate from endothelial cells and also platelets and leukocytes. These particles are mainly procoagulant, providing an increased area of tissue factor exposure and increasing activation of factor VIIa. They induce the expression of enzymes related to inflammation and ROHNS, increase cytokines and exposure of adhesion molecules, and participate in the immunosuppression (5, 6, 11, 17, 36, 58, 142, 164, 179, 226, 234).
Abnormal coagulation leads to activated platelet aggregation, fibrin deposition, and thrombus (e.g., exposure of tissue factor, activation of the complement system C3a, C5a, and b, and von Willebrand factor…), which induce ischemia/reperfusion and ROHNS formation (O2 •−) through intermittent flow. There is a formation of platelet plugs, including microparticles, which trap bacteria. Blood clotting and thrombi are also observed, favored by impaired anticoagulant pathways and fibrinolysis. NET—DNA structures released with adherent bactericidal proteins, such as MPO, participates in the trapping and killing of bacteria and in endothelial damage (78).
ROHNS and especially ONOO− have a specific toxicity toward endothelial cell mitochondria (Fig. 4, lower part). (41). This disruption of the mitochondrial respiratory chain is probably due to the structural analogy between mitochondria and bacteria, as mitochondria evolved from captured bacteria (75). These damaged mitochondria swell, which increases ROHNS formation (O2 •−) (2, 25, 39, 41, 67, 119, 136, 233, 246, 274, 275, 323, 336) (Fig. 4, lower part). Together with the endothelial wall damage, mitochondrial damage in endothelial cells and later in tissues, especially the liver, is recognized as a key factor in the pathophysiology of the early phase of sepsis (39, 41, 46, 119, 136, 184, 196, 226, 243, 246, 313). ROHNS damage aerobic and anaerobic microorganisms that are killed by exposure to oxygen, but also eukaryotic cells, especially mitochondria (75).
Mitochondria in eukaryotic cells are protected against oxygen toxicity by a low PO2 tension of around 0.5 mmHg. This allows eukaryotic cells to use oxygen for energy production (i.e., the mitochondrial respiratory chain) and metabolic transformation (i.e., by cytochrome P450). Mitochondria have a key role in energy production, supplying 80%–90% of the adenosine triphosphate (ATP) required by mammalian cells (115, 243). They are the source of most cellular free radical production in physiological conditions (277). However, they are also organelles that are sensitive to oxidative damage. The mitochondrial damage checkpoint induces apoptosis, as demonstrated in aging and cancer (277). In addition, ferroptosis or pyroptotic cell death was recently proposed as a mechanism in ischemia/reperfusion and sepsis (143), involving lipoperoxidation membrane damage that may evolve with oxidation of the inner membrane space of mitochondria in case of ferroptosis, and caspase 11 activation for pyroptosis. These effects seem to be related to overwhelming antioxidant defense by the cell membrane and mitochondrial membrane selenoenzyme GPX4, requiring reduced GSH (64, 143, 156). Acute mitochondrial damage induces a decrease in ATP production, threatening cell viability (46, 243) (Fig. 4, lower part).
Blockage of the mitochondrial respiratory chain induces an increase in O2 •−, but also in ONOO−, a decrease in energy production and membrane potential, and mitochondrial damage inducing apoptosis (39, 243, 246, 313) (Fig. 4, lower part). Mitochondrial ROS production, especially in the lung, is particularly enhanced when sepsis appears on a background of chronic ethanol abuse (47). Mitochondrial impairment is more likely to participate in functional endoplasmic reticulum (ER) stress, as shown in the LPS cellular model, inducing a proapoptotic state (155). The ER is a structure that is highly sensitive to increased cellular redox state and is involved in sepsis lung damage (86, 155). Tumor necrosis factor-alpha (TNF-α) increases ROS production via mitochondria, especially in tumor cells (67). In human endothelial cells, TNF-α induced rapid mitochondrial ROS production at the ubisemiquinone site; the ceramide-dependent signaling pathway was implicated (67). H2O2 is involved in NF-κB upregulation of iNOS, creating a positive loop in endothelial cells (243). NO• may also activate the ER (86).
The increased redox potential in the endothelium is associated with a decrease in reduced GSH. This reduction is partially related to mitochondrial dysfunction, which increases the oxidative stress and activates cell apoptosis (11, 135, 136, 142, 243, 331). Endothelial cell metabolism is markedly modified, with resistance to insulin (302, 310). Microparticles are released from the damaged and activated endothelial membrane, as well as from other cells, and play an important role in vascular redox signaling and endothelial dysfunction (189, 236, 319). All these phenomena increase platelet as well as leukocyte adhesion to endothelial cells, which in turn increases the endothelial dysfunction, as already discussed (Figs. 2 and 4).
At a later stage, which was achieved in 2–4 h in a murine cecal ligation and puncture (CLP)-septic model, endothelial cells directly cause apoptosis by a caspase-dependent mechanism (102), or necrosis with the release of DAMPs, particularly mitochondrial DAMPs (192, 328). This alteration of the endothelium is linked to the hyperactivation of neutrophils (72, 133, 137, 165, 192) (Figs. 2–4).
Sepsis is a time-sensitive condition ideally requiring prevention of irreversible endothelial damage (66, 170). It is also an unnatural situation, made possible by supportive care, leading to a dysregulated innate response or a state of chaos (Fig. 1).
The ubiquitous intracellular antioxidant selenoenzymes, GPX1, GPX4, and TXNRDs (cytosolic TXNRD1 and mitochondrial TXNRD2), play a key role in the antioxidant defense of endothelial cells. If their antioxidant capacity is exceeded, there is increased redox potential, complex modifications of cell functioning, and cell damage (10, 42, 44, 117, 152, 178). In the acute phase of sepsis, intake of a biologically active form of Se by endothelial cells is reduced as a result of the previously mentioned downregulation of liver selenoprotein P synthesis. In addition, even if a biologically active form of Se is available, synthesis of selenoenzymes is an energy-intense process, and energy production may be impaired in situations of mitochondrial dysfunction (50, 53, 70, 130). Moreover, excess oxidative stress and hypoxia may reduce selenoenzyme synthesis (30, 214, 259). Finally, endothelial apoptosis and necrosis are observed as well as circulating endothelial cells. All these phenomena contribute to the dysfunction of the endothelial barrier. This dysfunction takes hours to occur in CLP models, leading to SHINE (6, 11, 79, 94, 129, 133, 137, 140, 142, 150, 154, 165, 168, 192, 203, 209, 225, 236, 256, 268, 279, 338).
Consequences and Deleterious Effects of ROHNS in the Early Phase of Sepsis
Endothelial damage (SHINE) is the first step to MOF and death (63, 72) (Fig. 1) and occurs within hours of the start of sepsis (5, 69, 71, 94, 125, 154, 165, 181, 192, 219, 256). As previously highlighted, the marked decrease in plasma Se and selenoproteins, specifically selenoprotein P, is an indirect argument in favor of a major deleterious effect of ROHNS (91, 93, 131) (Fig. 5). SHINE is a first step to sepsis-related organ dysfunction, characterized by a profound modification of tissue cell metabolism with a low degree of apoptosis, despite major organ dysfunction or failure requiring organ support in the ICU to survive. It is also associated with secondary immunosuppression that begins simultaneously with hyperinflammation (dysregulated innate and host response) (4, 6, 21, 34, 63, 133, 142, 209, 275) (Fig. 1). Organ dysfunction is related to the severity of the early endothelial damage, coupled with immunosuppression due to immune cell apoptosis (4, 63, 72, 94, 142, 174, 209, 210, 236, 243), and, in surviving patients, these two factors are most probably responsible for the long-term consequences of sepsis (133, 228).
Considering the importance of the increased redox potential and activity of leukocytes, and especially neutrophils, to ROHNS production and endothelial damage, a therapeutic intervention would conceptually need to be very potent and active at multiple sites to be effective. One target of such an intervention could be to reduce the hyperactivity of leukocytes, especially neutrophils, while protecting the endothelium. Such an approach could potentially be achieved using a combination of oxidant and antioxidant selenocompounds and might enable a major part of the complexity and heterogeneity of sepsis to be bypassed. We will develop this possibility further in a separate part B article.
Conclusion
In summary, endothelial damage occurs rapidly in the first few hours following the onset of sepsis, largely due to the extensive interaction between activated leukocytes and the endothelium. Dysfunction of the ESL, especially as a result of ONOO− and HOCl toxicity, is the first step of this damage. The role of mitochondrial dysfunction, especially as a result of ONOO− toxicity, should also be stressed. Concomitantly, there is a major decrease in plasma concentrations of selenoprotein P, a protein with antioxidant enzymatic functions, especially against ONOO−. In this early phase of sepsis, which can be considered inducing SHINE, ROHNS play a major role. Consequently, the hyperactivated leukocytes with myelemia and release of immature cell forms could be considered an acute, transient, and benign hematological tumor.
With development of supportive care, patients can survive in a state of dysregulated and overwhelming host response. This response involves many overlapping cascades, with positive loops and interaction of pro- and anti-inflammatory processes (dysregulated innate and host response). The result could even be likened to a state of chaos, an unstable condition falling between a stable situation controlled by homeostasis and a stable situation provided by supportive care, including the treatment of opportunistic infections.
Together, sepsis must be considered a time-sensitive acute condition requiring early identification and efficient treatment before endothelial damage becomes established and immunosuppression occurs. Ideally, such a treatment would reduce leukocyte hyperactivity and overwhelming ROHNS production by neutrophils, while protecting the endothelium.
Footnotes
Authors' Contributions
X.F.: conceived the presented review; X.F., P.V.A., and J.-C.P. contributed to the writing of the article. P.V.A. especially participated and validated the biochemical data. J.-C.P. had a particular implication in the plan of the review and participated and validated the clinical implication.
Acknowledgments
We thank Prof. Anne Marie Roussel, emeritus Professor in Biochemistry at Grenoble University, for her thorough rereading of this article and valuable advice; and Prof. Jean Neve, emertitus Professor at the Université Libre de Bruxelles, for his valuable advice on oxidative stress and septic shock. We also thank Ms Evelaine Guilbert for her help in the realization of the graphic illustrations.
Author Disclosure Statement
Dr. Xavier Forceville started a very small start-up in 2005 (Sérénité-Forceville). In 2015, Pharm. V Cotereau volunteer manager of Sérénité-Forceville, former vice president of the French pharmacist association, filed a new patent for the treatment of sepsis entitled: “Kit for treating sepsis and/or any systemic (SIRS) or damaging cellular hyper-inflammation.” Its reference numbers are as follows: PCT number PCT/FR2016/051569, European patent application 16742342.5, U.S. patent application Attorney Docket Number: 0727–1267. I declare that I am the main shareholder of this company. One of my brothers is also a shareholder. All other authors declare no competing financial interests.
Funding Information
There is no funding information to declare.