
Abstract
Significance:
Senescence is a cellular state induced by internal or external stimuli, which result in cell cycle arrest, morphological changes, and dysfunctions in mitochondrial and lysosomal functionality as well as the senescence-associated secretory phenotype. Senescent cells accumulate in tissues in physiological and pathological conditions such as development, tissue repair, aging, and cancer.
Recent Advances:
Growing evidences indicate that senescent cells in vivo are a heterogeneous cell population due to different cell-autonomous activated pathways and distinct microenvironmental contexts.
Critical Issues:
In this review, we discuss the different contexts where senescence assumes a key role with beneficial or harmful outcomes. The heterogeneous nature of senescence pushes toward resolution of the specific molecular profile and secretome to typify senescent cells in physiological and pathological contexts.
Future Directions:
Future research will enable exploring the heterogeneity of the senescent population to precisely map the progression of cells through senescent trajectories and study the impact of the therapeutic advantage of senolytic drugs for translational strategies toward supporting the health span. Antioxid. Redox Signal. 34, 294–307.
Introducing Cellular Senescence
Cellular senescence is a cell state defined as irreversible cell cycle arrest triggered by a plethora of stresses induced by external or internal insults, including genotoxic agents, irradiation, oxidative stress, mitochondrial dysfunctions, and oncogene activation (9, 16, 20, 34, 111). While cellular senescence was originally described in cells cultured in vitro (60), raising the question of whether the observation was an artifact of the experimental system, several studies later demonstrated that senescent cells accumulate in tissues of living organisms in physiological and pathological states, like development, tissue repair, aging, and cancer (62, 89). Senescent cells exhibit specific properties, including intracellular signals and extracellular secreted molecules, distinguishing them from quiescent or terminally differentiated cells (55, 92). Several cellular processes are associated with senescence, such as DNA damage, cell cycle arrest, senescence-associated secretory phenotype (SASP), morphological and membrane composition changes, and mitochondrial and lysosomal dysfunctions. One of the first molecular features associated with senescence is telomere shortening that occurs as a result of DNA end-replication. Telomere erosion during proliferation activates the DNA damage response (DDR), causing cell cycle arrest (52). In addition to DNA damage accumulation at telomeres, other agents such as radiation, chemotherapeutics, and oxidative agents can cause senescence by inducing irreparable DNA double-strand breaks (DSBs) (99). DSBs promote the recruitment and binding of ATM kinase to the DNA damage sites, driving phosphorylation of histone H2AX that orchestrates the assembly of DNA repair complexes (11). Persistence of the DDR induces p53 phosphorylation that triggers a transcriptional cascade, which ends with cell cycle arrest. Oncogene expression might induce senescence, referred as oncogene-induced senescence (OIS), which is associated with DNA damage or hyper-replication (40). Indeed, it has been demonstrated that in senescent cells, cytoplasmic chromatin fragments (CCFs) leave nuclei after the loss of nuclear envelope integrity (66) and activate the innate immunity cytosolic DNA-sensing cGAS-STING pathway, leading to both short-term and chronic inflammation, the former to eliminate damaged cells and the latter in association with cancer (42). Recently, it has been recognized that the involvement of dysfunctional mitochondria coupled with the downregulation of mitochondrial oxidative phosphorylation genes is an upstream signaling event that prompts CCF formation and hence the SASP (113). Irreparable DNA damage and oncogenic activation can induce senescence or apoptosis, depending on DNA damage intensity. While highly damaged cells are eliminated by apoptosis, senescence activates several prosurvival factors to counteract apoptosis (27).
In senescent cells, the expression levels of CDK2 inhibitor p21 Waf1 and CDK4/6 inhibitor p16 INK4a increase, resulting in persistent activation of the RB family of proteins, inhibition of E2F transactivation, and cell cycle arrest (15). The persistence of inhibition of cell cycle genes is associated with epigenetic silencing as trimethylation of histone 3 (H3) in lysine 9 (K9), a transcriptionally repressive heterochromatin mark of E2F target genes (104). However, senescence-associated cell cycle arrest might not be necessarily terminal (15). It has been described as light senescence, where p16 INK4a expression levels are high, and deep senescence, where heterochromatization levels are increased and are responsible for maintenance of the senescent status (77). Recent studies have shown that cells released by senescence have returned to the cell cycle with increased clonogenic growth compared with identical populations equally exposed to chemotherapy, but only those that had never been senescent (80). However, whether this escape from the senescent state takes place only in incompletely senescent cells or also in those cells that acquired a fully senescent phenotype is still poorly understood.
The senescence process is characterized by profound epigenetic changes that modulate gene expression. In particular, DNA methylation plays an important role in senescence with specific CpG site methylation changes, and DNA methylated regions are usually enriched with specific methylated histone residues, including H3 K27 and K9 trimethylation (trimethyl histone H3 lysine 27 [H3K27me3]; trimethyl histone H3 lysine 9 [H3K9me3]). Indeed, H3K27me3 and H3K9me3 enrichment contributes to the formation of senescence-associated heterochromatin foci (SAHFs) (25). A recent study that combines chromatin immunoprecipitation (ChIP)-seq and genome-wide chromosome conformation capture (3C)-based technologies such as Hi-C approaches reveals the role of condensin in orchestrating the three-dimensional (3D) reorganization of the genome and the transcriptional outcome. In particular, the authors show a switch between heterochromatic and euchromatic regions, which occurs both in OIS and in replicative senescence. The data highlight the role of condensin in activating the SASP and the p53/p21 pathway, possibly establishing specific 3D genomic contacts (67). Besides intrinsic changes, senescent cells also secrete several factors that influence neighboring cells (Fig. 1). The SASP comprises interleukins (ILs), chemokines, growth factors, secreted proteases, and extracellular matrix components (31). The SASP can induce senescence in a paracrine manner in neighboring cells (3) and leads to reinforcement of cell cycle arrest by cytokines such as IL-6 or IL-8 (73). SASP proteins promote the recruitment of immune cells, including macrophages and natural killer (NK) cells, which in turn are responsible for the clearance of senescent cells (82, 107). Thus, transient and regulated induction of the SASP prevents tumor formation (56). Conversely, prolonged and persistent SASP can have detrimental effects in promoting cancer (38). In addition to secreted proteins, some senescent cells release small extracellular vesicles that, through delivery of microRNAs (miRNAs), proteins, and other nucleic acids such as CCFs to recipient cells, exert an impact on the tissue microenvironment (108) with proproliferative and proinflammatory outcomes (5). A recent study allows the generation of a comprehensive SASP atlas, including both exosomal and soluble protein content that has proven useful to identify SASP components or candidate biomarkers for senescence, aging, and related diseases. These data suggest that core SASP factors are enriched among plasma biomarkers of aging in humans (14).

At the morphological level, senescent cells present diverse alterations such as irregular cell body, cytoskeleton rearrangements, and changes in nuclear lamins such as lamin B1. Lamin B1 is a major component of nuclear lamins and together with lamin A/C sets the structure and function of the nucleus. Lamins are essential organizers of genome topology, allowing a spatial distribution of chromatin inside the nucleus and eventually modulating gene transcription (116). Indeed, the nuclear lamina plays an important role in the structural organization and function of the nucleus. The role of lamin B1 in coordinating DNA repair mechanisms has been previously described (103) as well as its role in the formation of SAHFs and in gene regulation during senescence (97). The plasma membrane changes its composition and presents upregulation of caveolin-1, DeP1, B2MG, and an oxide form of vimentin that represent markers of senescent cells (63). Senescent cells also exhibit an increased mitochondrial mass, which lose their functionality as a consequence of decreased membrane potential (87). The senescent state is characterized by upregulation of lysosomal proteins and activation of lysosomal enzymes such as senescence-associated (SA) beta galactosidase (SA-β-Gal), which constitutes one of the first cellular markers employed to identify senescent cells (41). During senescence, lysosomes increase in number and size, and this becomes evident by increased cytoplasmic granularity observed microscopically (91). Among lysosome proteins, lipofuscin has been described to accumulate in senescent cells, resulting as an established hallmark of senescence (44). Recent work has demonstrated that impaired lysosomal function leads quiescent cells progressively into a deep quiescent and senescence-like irreversibly arrested state. Conversely, increasing lysosomal function gradually pushes cells into shallower quiescence by lowering oxidative stress (51), suggesting that quiescence is positioned as an intermediate path between proliferation and senescence in aging contexts.
Although multiple senescent inducers and physiological and pathological senescence-related processes have been identified (Fig. 2), to date there is no single reliable hallmark for senescent cells, thus measurements of multiple markers are required to identify a cellular senescence status (55, 101).

Cellular Senescence: The Beauty and The Beast
Senescence in adulthood and in aging
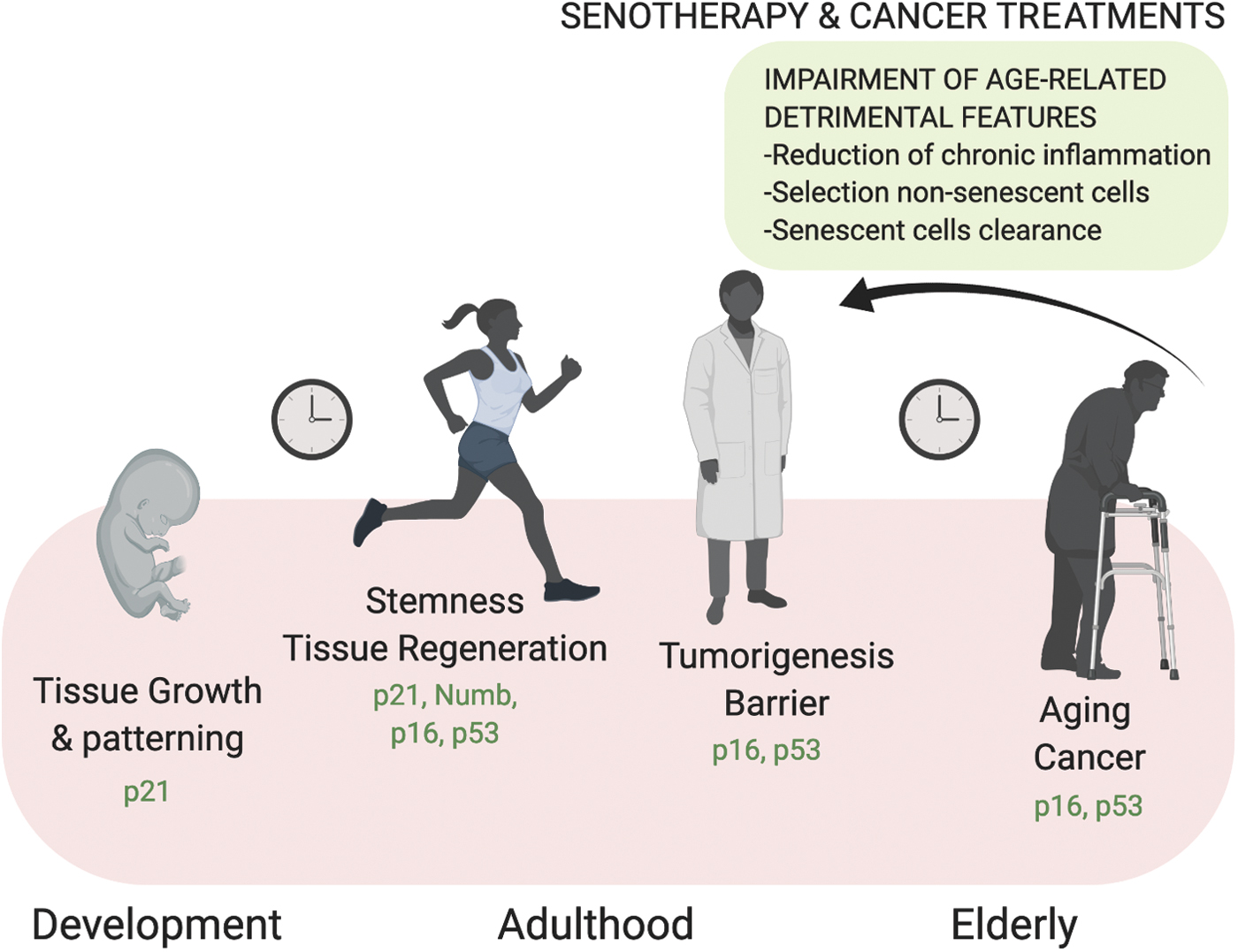
Although a correlation between cellular senescence and organismal aging is well defined, senescence and aging are distinguishable processes (92). Indeed, cellular senescence occurs at different stages of our life, from embryogenesis until adulthood and in the elderly. According to the antagonistic pleiotropy theory of aging, senescence exerts both beneficial and detrimental effects in organisms (22). In fact, senescence functions as a crucial barrier to tumorigenesis (29), promotes tissue repair (39), and constitutes a programmed mechanism that regulates organism development (82, 107). However, at the same time, senescence contributes to a variety of age-related pathologies, including cancer, as well as to reduced tissue renewal and functions (111). These apparently opposite features of senescence coexist and gain increased significance during the life span (Fig. 3).
The tumor-suppressive function of senescence relies on the postmitotic nature of senescent cells, which has been shown to act as a barrier against tumor progression in several tissues, including the colon, lung, lymphoma B cells, and pancreas (13, 18, 23, 28, 57). The studies led by Serrano et al. demonstrated that cellular senescence is activated in response to oncogenic stimuli, such as Ras expression, becoming part of the anticancer response (100). p53 and p16INK4a activation is required for OIS as a safe mechanism to limit cellular transformation and cancer development (47). Senescence acts as an efficient anticancer barrier against lymphoma formation in an established transgenic mouse model of Burkitt's lymphoma resulting from telomere attrition (46). The authors provided evidence that short telomere-associated senescence retains potent tumor-suppressive function despite abrogation of the apoptotic program, highlighting that the pathway of downstream short telomeres includes both p53 and p16INK4a (46). Accordingly, the vast majority of human cancer cells accumulate mutations in the p53-p21Waf1 and/or the p16INK4a -RB1 axis (2, 85, 102, 112). Recently, the use of a new p16INK4a reporter cell line recapitulating endogenous transcriptional activity led to the identification of a novel mechanism of p16INK4a transcriptional regulation via a cis-regulatory element adjacent to the ARF promoter, adding knowledge to how the locus is regulated in human cells (115). Parallel studies have focused on the causal link between DDR activation and OIS. In particular, DDR and senescence have been both detected in precancerous tissues, while progression to carcinoma is associated with decreased scores for both activation of the DSB checkpoint and senescence (13). In addition, it has been revealed that oncogene activation generates augmented DNA hyper-replication in vivo, eventually leading to stalling of DNA replication forks, which in turn cause the formation of DSBs and senescence (40). Together with genomic instability, DSBs activate p53 that coordinates apoptosis and senescence to remove harmful cells and exerts a tumor suppressor function. Therefore, two key features of cancer, genomic instability and high incidence of p53 mutations, converge toward DNA damage accumulation (59) (Fig. 4).

Despite the senescence-mediated anticancer barrier being imposed mostly by a cell-autonomous arrest of cell proliferation, senescence can also display detrimental features through cell-nonautonomous mechanisms. Senescent cells secrete multiple factors, including chemokines, cytokines, ILs, growth factors, angiogenic factors, and matrix metalloproteinases (30), giving rise to an SASP that mediates many pathophysiological processes in neighboring cells, with different outcomes depending on the biological context (83, 107). This paracrine effect, revealed by direct and transwell-mediated coculture experiments together with transcriptome and proteomic analyses, has been shown to be p16INK4a
Several studies provide evidence that the accumulation of senescent cells in different tissues occurs during the organismal life span (20), but whether senescent cells are directly involved in age-related phenotypes is of relatively recent demonstration. Several murine models of accelerated senescence and aging have been utilized to study these processes. The mouse models that recapitulate premature aging disorders might be classified according to the types of genetic manipulations and whether they impair the integrity of nuclear DNA and the repair machinery, mitochondrial DNA, telomere attrition, or the nuclear architecture (49). For instance, mice defective for the mitotic spindle assembly checkpoint (i.e., BubR1 mice) display a shortened life span, recapitulating several age-related defects such as sarcopenia, cardiac abnormalities, cataract, and lordokyphosis (8). To study the effect of senescent cell removal, BubR1 mice were genetically modified to obtain BubR1/INK4-ATTAC (apoptosis through targeted activation of caspase), in which p16INK4a-positive cells are eliminated by apoptosis through administration of the synthetic compound AP20187 that induces dimerization of a membrane-bound myristoylated FK506-binding protein–caspase 8 (FKBP–Casp8) fusion protein (10). The authors demonstrated that senescent cells accumulate in different tissues, including the eye, skeletal muscle, and adipose tissue. Their removal not only delays the onset but also attenuates the progression of all described age-related phenotypes, providing the first evidence of a causal link between senescent cell accumulation and aging-related decreased tissue fitness (10). The same group extends the analysis on physiological aging, further demonstrating a negative impact of p16INK4a-positive senescent cells on the life span (7). Accordingly, a targeted approach to delete senescent cells in the elderly might constitute a possible resource to prolong both health span and life span.
Recently, several studies have focused on the use of pharmacologically active small compounds, known as senolytics, to deplete senescent cells as a promising approach to counteract the detrimental features associated with aging. In particular, senolytics can trigger apoptosis by inhibiting prosurvival pathways, promoting senescent cell natural clearance, or altering SASP and its detrimental consequences, thus highlighting their potential use in clinical trials (86). For instance, systemic administration of ABT263 (a specific inhibitor of the antiapoptotic proteins BCL-2 and BCL-xL) depletes senescent cells by inducing apoptosis, leading to rejuvenation of the hematopoietic compartment in aged mice (26).
In a recent study, p16-3MR transgenic mice were employed. This model takes advantage of the use of the trimodal reporter protein (3MR) technology (a fusion protein consisting of Renilla luciferase, red fluorescent protein, and herpes simplex virus thymidine kinase that converts the pro-drug ganciclovir into a toxic guanosine analogue) to identify, track, and selectively kill p16INK4a-positive senescent cells in vivo. Both the INK/ATTAC and the p16-3MR mouse models have been used to investigate the causal role of senescent cells in osteoarthritis. Administration of AP20187 and UBX0101 senolytic drugs is effective in eliminating senescent cells, attenuating articular cartilage degeneration, and creating a proregenerative environment (26). The p16-3MR animal model has been used to monitor the role of hematopoietic senescent cells during organismal aging. The study demonstrates that senolytic treatment of aged mice or mice that underwent irradiation (both conditions that trigger cellular senescence and age-associated decline in tissue fitness) rejuvenates the phenotype of hematopoietic and muscle stem cells (MuSCs) (26). Similar findings of therapeutic benefits of eliminating senescent cells were shown in other contexts, such as post-traumatic osteoarthritis and neurodegenerative diseases (19, 68).
Noteworthy, the accumulation of p16INK4a-positive cells in astrocytes and microglia has been shown to mediate degenerative processes in cortical and hippocampal neurons, thereby affecting cognitive functions (19). In particular, to unravel the role of senescence accretion in neurodegenerative disorders, the authors employed the transgenic mouse line MAPTP301SPS19, which expresses the mutant human tau specifically in neurons, crossed with the INK/ATTAC strain. This allows the demonstration that senescent cells promote insoluble tau aggregates and reduce the dentate gyrus area, driving neurodegenerative disease. AP20187 administration counteracts all age-associated morphological features and improves cognitive functions, assessed by short-term memory tests (19).
All the mouse models that exhibit age-related features serve as useful tools for development of therapeutic treatments that target senescence cells to improve physiological aging and age-related diseases, otherwise known as senotherapy (55). Through a screening of compounds aimed at finding a broad spectrum of senolytic agents, cardiac glycosides (CGs) have been identified recently, which can be combined with anticancer therapies to improve the elimination of tumor cells and to counteract senescent cell accumulation (58). In the context in which chemotherapy and radiotherapy lead to senescence induction as one of the side effects, CG follow-up might provide additional benefits that are elimination of senescent cells induced by anticancer treatments and removal of age-associated senescent cells, resulting in a reduction of chronic inflammation (58). However, whether the main effects of these pharmacological treatments are cell autonomous, that is, rescue of function of individual cells that underwent senescence, or nonautonomous, selection of nonsenescent cells in tissues in live organisms is still unclear.
One limitation for studying the impact of senescence on aging is the lack of a reliable marker to detect senescent cells. One of the recently tested strategies consists of a newly synthesized compound—GL13 or SenTraGor—that reacts with lipofuscin (present at high levels in senescent cells) (70) and, through the presence of biotin, allows enhancement of immunohistochemical enzymatic detection. This reaction offers a reliable sensitive means for tracing and measuring cellular senescence in tissue specimens (44, 84). However, whether GL13 is a reliable marker applicable to all senescence contexts is poorly understood. A multimarker workflow such as the detection of lipofuscin combined with the increased levels of p16INK4a and DDR markers might lead to the recognition of senescent cells with higher accuracy (55).
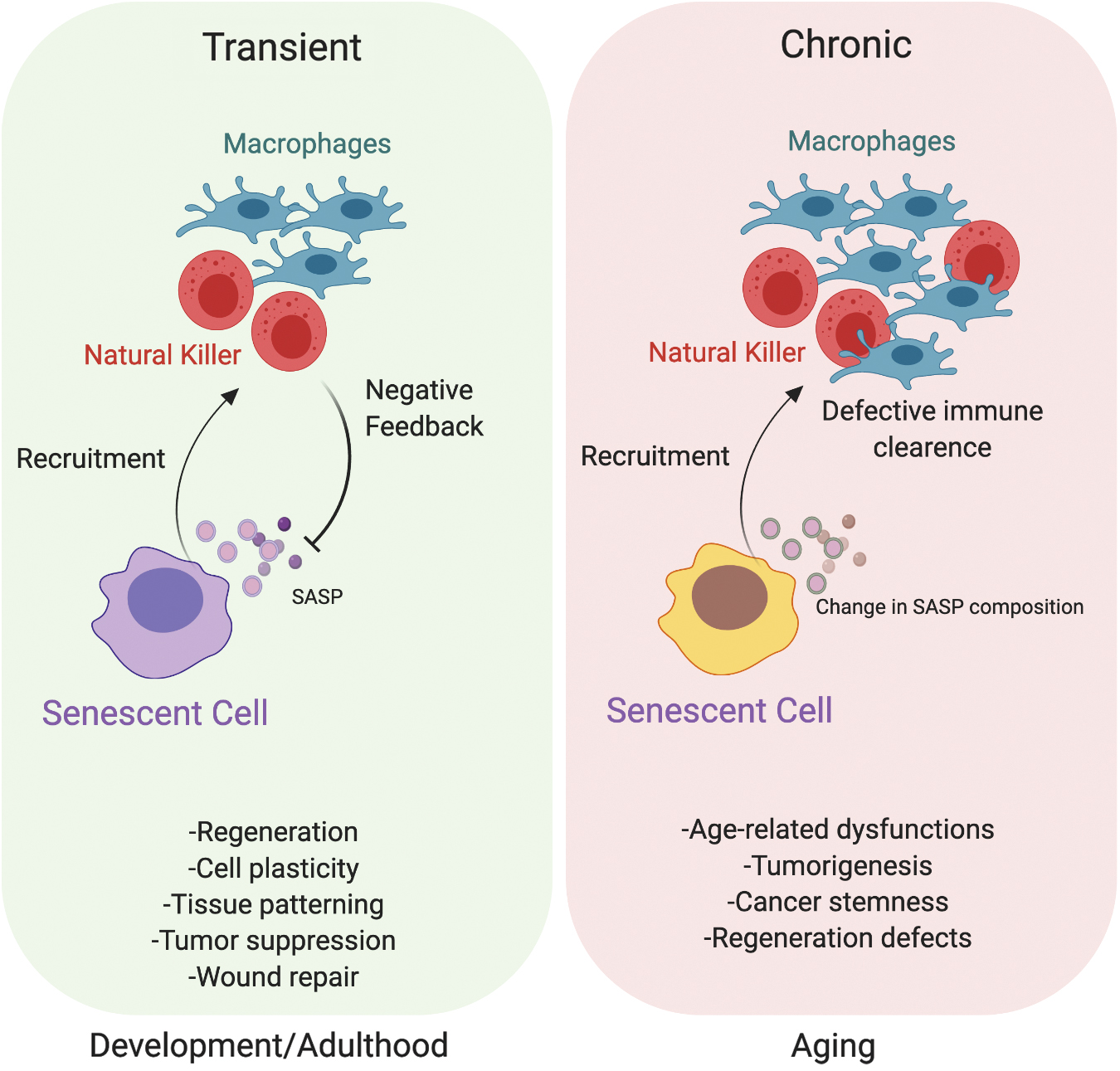
It is possible to distinguish different types of senescence depending on its physiological context: in the development stage, in adulthood, and in the elderly (Fig. 5). In the former, cells experience acute senescence and secrete signaling molecules through the SASP, promoting morphogenesis. Eventually, immune clearance and apoptosis drive changes in cellularity to favor developing tissues. In the adult, senescence is beneficial as an anticancer barrier and to support the regenerative response. Later on, at the onset of aging, the senescent state becomes chronic, due to dysfunction of the immune response that is progressively impaired in the ability to clear senescent cells, and is fueled by the progressive accretion of DNA damage. At this stage, senescent cells contribute to tumorigenesis and tissue dysfunction (27) (Fig. 5).
Developmental senescence
Recent studies demonstrate that programmed senescence also occurs during embryonic development to orchestrate tissue growth and patterning (82, 107). By performing whole-mount staining for SA-β-Gal activity on mouse embryos between embryonic days (E) 9.5 and 17.5, senescent cells have been shown to accumulate from E 9.5 in different regions of the body, such as developing limbs, the tip of the tail, and the closing neuronal tube (107), as well as mesonephros and the endolymphatic sac of the inner ear (82), to later disappear at E 17.5. This developmental senescence patterning is altered in p21Waf1-deficient mice, demonstrating that this process is p21 dependent. Intriguingly, developmentally programmed senescence has been shown to be p53, p16INK4a, and DNA damage independent. Macrophage infiltration follows the appearance of senescence in the embryo to clear senescent cells that enter apoptosis, therefore enabling tissue remodeling (82, 107). Hence, senescence in the embryo likely plays a relevant role in controlling cellular growth, the balance among different cell types, and tissue patterning. Senescence and apoptosis share this competence and both can be considered mutually compensatory during the developmental stages (27, 33). The challenging hypothesis that programmed senescence and its role in driving tissue development appear before its tumor suppression function is supported by the fact that development of senescence has been detected in mice, chicks, frogs, axolotls, and zebrafish (33). Moreover, the lack of DNA damage markers and independence from p16INK4a in developmental senescence poses this process at a more ancestral evolutionary state. This suggests the new perspective that cellular senescence evolves during embryonic development as a tissue-remodeling mechanism and later regulates tissue regeneration and damage response in adult organisms.
Plasticity and reprogramming-associated senescence
Recent observations provide evidence that senescence also plays a role in cellular plasticity and stemness (90). Overexpression of Ras in primary mouse keratinocytes, beside the typical features associated with cellular senescence, induces a stem signature at the global transcriptional level. By taking advantage of coculture experiments, the authors show that transient exposure of primary mouse keratinocytes to the SASP triggers tissue regeneration through activation of stem cell function. This is confirmed in vivo, where induction of senescence in the liver leads to stem cell marker increase in neighboring cells within the tissue. Conversely, a prolonged incubation period with paracrine factors secreted by senescent cells evokes keratinocyte cell cycle arrest (90).
In vivo overexpression of the four factors OCT4, SOX2, KLF4, and c-MYC (OSKM) induces, although with low efficiency, pluripotency in vivo (1). This process not only induces cellular reprogramming but also drives DNA damage accumulation and senescence. Intriguingly, reprogrammed cells are detected in close proximity of the senescent one. In this context, IL-6 is secreted by senescent cells through the SASP and becomes the molecular determinant that fuels OSKM-mediated reprogramming (81). However, senescence-associated stemness might acquire a detrimental function. This is the case of chemotherapy-induced senescence in lymphoma cells, which confers a tissue stem cell signature and higher clonogenic growth potential to B cell lymphoma only when cells experience the senescence program (80). These data suggest that senescence might impart a cell-autonomous feature, which consists of high aggressiveness once the cell escapes the senescent state, eventually explaining the outcomes associated with relapse tumors.
Senescence and muscle regeneration
Recent studies have demonstrated that senescent cells also transiently accumulate upon tissue injury and improve tissue remodeling during the repair process (39, 69, 72, 76). The role of senescence during tissue repair is thought to contribute to the secretion of microenvironmental signals during the regenerative process as well as to eliminate undesired cells once tissue homeostasis has been reached. By taking advantage of the p16-3MR mouse model, the Campisi group has shown that during skin wound healing, there is a transient accumulation of p16INK4a-positive senescent cells and that their elimination by ganciclovir treatment delayed tissue regeneration, demonstrating a relevant role of senescent cells during tissue repair (39). The authors further showed that the majority of senescent cells during skin wound healing are endothelial cells and fibroblasts and that they act through secretion of PDGF-A to promote efficient tissue regeneration. The characterization of the identity of senescent cells in tissues will provide significant insights into how senescent cells contribute to tissue repair.
In skeletal muscle, a role of cellular senescence in tissue repair and aging has only been shown recently. Skeletal muscle is a low turnover tissue with high regenerative potential in which the rate of cell loss and replacement in healthy conditions is very low compared with high turnover tissues such as epithelia, skin, and blood; however, throughout the life span of an organism, skeletal muscle is able to repair itself and maintain its function (43). The regenerative potential of skeletal muscle is exerted by MuSCs, tissue-resident stem cells that in homeostatic conditions exist in a quiescent state in their own niche underneath the basal lamina (45, 94). In response to stress or injury, MuSCs emerge from quiescence, proliferate, self-renew, and give rise to both more copies of stem cells and to committed progenitors that ultimately differentiate to repair the tissue (96). However, during aging as well as in chronic conditions, such as degenerative diseases (muscular dystrophies) and cancer, skeletal muscle mass and fitness progressively decline, with detrimental effects on patient quality of life. This is due to changes in metabolism, proteostasis, and impairment in tissue repair. These conditions are also associated with significant changes in tissue composition, which include increased inflammation and fibrosis, reduction in capillary density and perfusion, and impaired tissue innervation. During aging, these changes result in age-associated atrophy, also defined as sarcopenia that significantly contributes to frailty of the elderly (Fig. 6). Skeletal muscle is also exposed to systemic signals emanating from the circulation, that is, inflammatory cells and cytokines or factors secreted by distant organs and reaching skeletal muscle by traveling through the circulation. One clear example is cachexia, a condition associated with cancer and other human chronic conditions, in which there is a severe loss of skeletal muscle mass due to systemic signals emanating from a distant tumor, metastatic cells, inflammatory cells, or distant organs. This example underscores the need for improving our understanding of systemic signals and interorgan communications that affect skeletal muscle as it will shed light on the relevant molecular pathways and identify novel potential targets for interventions.
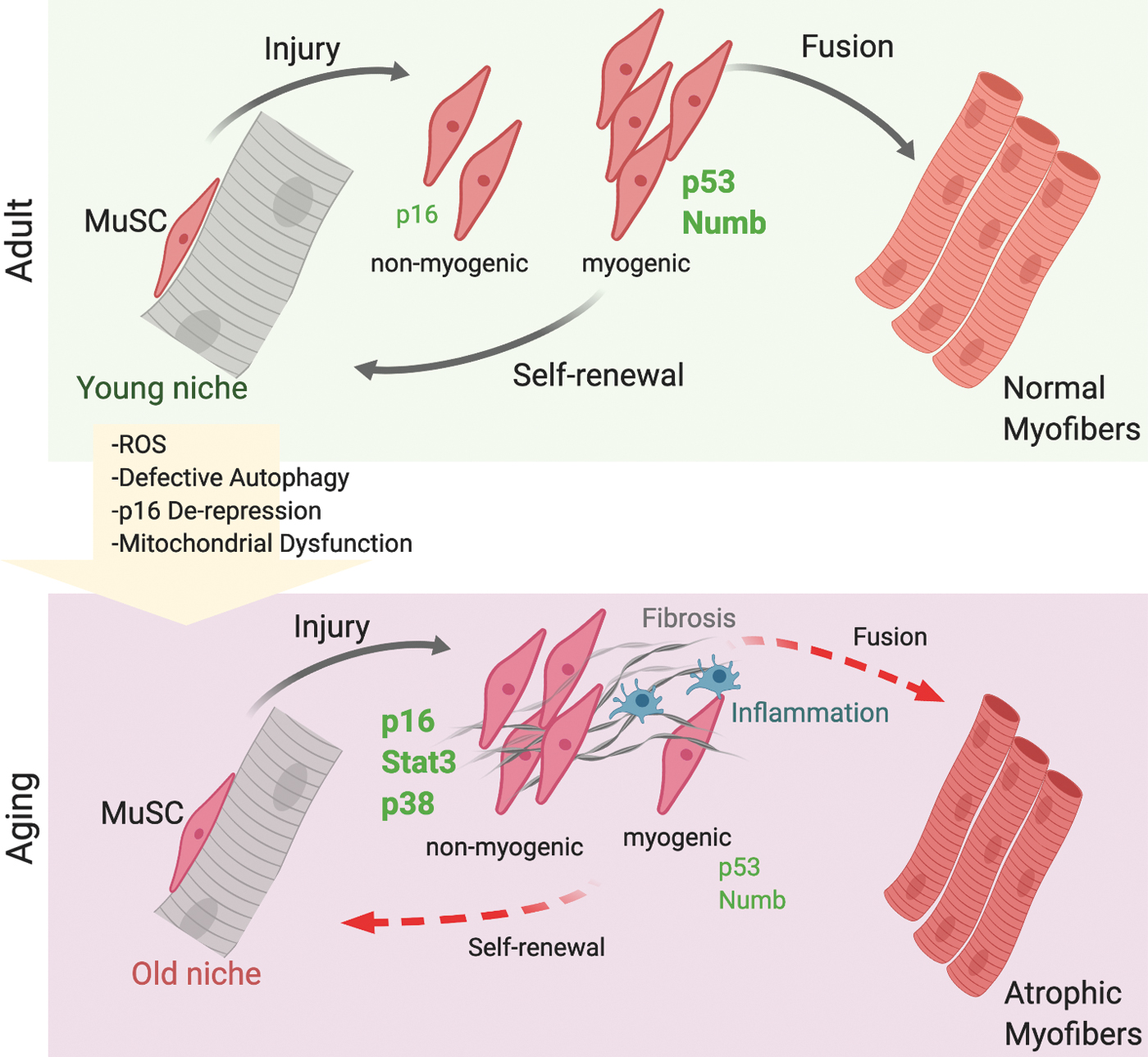
A role of cellular senescence in skeletal muscle during regeneration has only been recently reported. Work by the Tajbakhsh group provided evidence of accumulation of senescent cells during skeletal muscle repair (76). The findings described two types of senescence during tissue regeneration: a transient senescence of nonmyogenic cells during physiological tissue repair, mainly endothelial cells and macrophages partially dependent on p16INK4a, but p53 independent, and a persistence senescence in myogenic cells lacking Numb, which is p53 dependent and can be rescued by antioxidant treatments (Fig. 6). These findings highlight the unappreciated complexity of senescence in the context of skeletal muscle repair: cells of different lineages in the tissue microenvironment can undergo senescence, and might exhibit distinct features and effects on their neighboring cells, as well as senescence programs in different cell types have different genetic requirements. The application of single-cell technologies as well as in vivo clonal lineage tracing in future studies will enable dissecting this complex heterogeneity to comprehensively map which cell types undergo senescence, what are the dynamics of the process, and what is the impact of this process on skeletal muscle repair and maintenance.
During skeletal muscle aging, seminal studies by the Munoz-Canoves group have provided evidence that MuSCs transition from a quiescent state to senescence (106). This process is characterized by accumulation of reactive oxygen species, impaired mitochondrial function, derepression of p16INK4a , and defective autophagy (54). Indeed, the authors have shown that the autophagic process is impaired in aged MuSCs and that promoting autophagy can rescue the defect. Similarly, they have shown that impairing autophagy in young MuSCs through genetic deletion is sufficient to trigger the entry of MuSCs into a senescent state. Understanding how autophagy is impaired in aged skeletal muscle and what are the upstream signals affecting the autophagic process will provide novel tools for therapeutic interventions for extending the MuSC tissue regenerative function and health span in aged contexts.
We and others have previously shown that during aging, the signal transducer and activator of transcription 3 (STAT3) signaling pathway activity is elevated, consistent with increased expression of its upstream regulator and SASP factor IL-6, and that its transient inhibition by pharmacological interventions promotes MuSC expansion and tissue repair (88, 110). More recently, we identified the secreted factor, Fam3a, as a direct downstream effector of STAT3 signaling in MuSCs and provided evidence that Fam3a mediates STAT3-dependent MuSC myogenic commitment through regulation of mitochondrial respiration (98). While there is clear evidence that the beneficial effects of STAT3 inhibitors on skeletal muscle repair are mediated by expansion of MuSCs and enhanced tissue repair, understanding whether these interventions also act by impacting cellular senescence in aged muscle will provide key insights into their mechanisms of action.
In chronic degenerative conditions such as Duchenne muscular dystrophy (DMD), a progressive degenerative disease caused by mutations in the dystrophin gene, we and others have shown evidence of senescent cells, including accumulation of DNA damage, telomere shortening, and upregulation of p16INK4a (37, 79, 95, 109). This is consistent with our recent study in which we have shown that in a mouse model of replicative senescence, decline in MuSC-mediated regeneration coincides with activation of DDR and impaired ability to differentiate into myotubes (75). We further showed that inhibition of DDR restored MuSC differentiation ability. Similar findings were observed in replicative senescent human fibroblasts, in which the DDR precluded myoblast determination protein 1 (MYOD)-mediated activation of the myogenic program. These data provided the first evidence of DDR-mediated functional antagonism between senescence and MYOD-activated gene expression. Furthermore, the mdx mouse model of DMD exhibits an increased incidence of rhabdomyosarcoma formation (24, 48, 65), and we have recently shown that in this pathological context, MuSCs can give rise to rhabdomyosarcomas. These findings indicate that in chronic conditions, decreased tissue fitness can lead to very different outcomes, tissue degeneration or cancer (17), consistent with observations in other tissues (4). However, DMD is a degenerative disease that exhibits features of both continuous tissue damage and repair as well as accelerated tissue aging, and whether senescent cells in this pathological condition reflect the transient senescence of wound healing (due to constant tissue damage and repair) or the permanent senescence reported in aged tissues and whether senescence plays a role in the development of rhabdomyosarcomas in this pathological context are still poorly understood. Addressing these questions would allow evaluating whether interventions with senolytic drugs to eliminate senescent cells in DMD muscles could be beneficial or detrimental to their pathological progression.
Future Perspectives
One standing question is whether the chronic accumulation of senescent cells in tissues during aging, compared with the transient accumulation during physiological processes such as development or wound healing, is the result of an increase in the number of cells entering the senescent state or a defective clearance, or both. The challenge to answer this question arises also from the lack of stringent tools for faithful identification of senescent cells in vivo (55, 101). Indeed, the available tools currently preclude a precise mapping of the progression of cells through senescent trajectories. A clear definition of this cellular process and the potential identification of distinct, intermediate cellular states would be extremely useful to gain insights into the depth of senescent states and also evaluate the reversibility of these events. Indeed, while there is evidence that senescence can be reversed by oncogenic events (36, 105), it is still debated whether reversibility takes place at the single-cell level or whether a selection of cells not yet in a state of deep senescence occurs.
Accumulating evidence indicates that senescent cells in vivo are highly heterogeneous due to both their microenvironmental context and their original identity. Do senescent cells during embryonic development, adult tissue repair, or in aged organisms share the same transcriptional and epigenetic landscapes? Do they share the same secretome (SASP) and effects on their microenvironment? Do senescent cells from different lineages exhibit overlapping phenotypic and functional properties? The recent development of single-cell technologies to profile individual cells at both the mRNA and protein levels (i.e., CyTOF) offers powerful means for addressing these questions and dissecting the heterogeneity of senescent populations. However, the limitations of tissue digestion and cell isolation approaches are the removal of cells from their native environment and the loss of anatomical spatial information regarding where senescent cells are located within tissues, proximity with different cell types, and exposure to specific microenvironmental stimuli. Application of novel technologies such as NanoString Digital Spatial Profiling or STARmap (114) for 3D in situ RNA sequencing or multiplex protein detection would allow single-cell analyses in intact issues, thus overcoming this limitation. Integration of single-cell profiling technologies will allow to dissect the heterogeneity of senescent cell populations as well as to perform direct comparisons of such cells from different physiological and pathological contexts or different tissues. Finally, it will inform on the impact and therapeutic benefits of senolytic drugs for translational approaches toward promoting tissue fitness and the health span.
Footnotes
Acknowledgments
Funding Information
This work was supported by grants from the National Institutes of Health (NIH) R01AR064873 to A.S. and L.L.; French Muscular Dystrophy Association (AFM-Téléthon) Research, number 20568; and the Italian Ministry of Health, number PE-2016-02363049, to L.L.