
Abstract
Significance:
Hydropersulfides (RSSH) and related polysulfide species (RSnR, n > 2, R = alkyl, H) are highly biologically prevalent with likely important physiological functions. Due to their prevalence, many labs have begun to investigate their possible roles, especially with regards to their protective, redox, and signaling properties.
Recent Advances:
A significant amount of work has been performed while delineating the chemical reactivity/chemical properties of hydropersulfides, and it is clear that their overall chemistry is distinct from all other biologically relevant sulfur species (e.g., thiols, disulfides, sulfenic acids, etc.).
Critical Issues:
One way to predict and ultimately understand the biological functions of hydropersulfides is to focus on their unique chemistry, which should provide the rationale for why this unique functionality is present. Interestingly, some of the chemical properties of RSSH are strikingly similar to those of selenols (RSeH). Therefore, it may be important to consider the known functions of selenoproteins when speculating about the possible functions of RSSH species.
Future Directions:
Currently, many of the inherent chemical differences between hydropersulfides and other biological sulfur species have been established. It remains to be determined, however, whether and how these differences are utilized to accomplish specific biochemical/physiological goals. A significant aspect of elucidating the biological utility of hydropersulfides will be to determine the mechanisms of regulation of their formation and/or biosynthesis, that is, based on whether it can be determined under what cellular conditions hydropersulfides are made, more meaningful speculation regarding their functions/roles can be developed.
Introduction
Recent studies indicate that hydropersulfides (RSSH) and related polysulfides are highly prevalent and implicate them as important physiological effectors, mediators, and/or signaling species (although the exact physiological roles and functions are currently a matter of speculation and remain unestablished). (Note: RSSH will be used to denote both the protonated RSSH and anionic RSS− species and distinctions will be made only when necessary). For example, Ida et al. (32) discovered that levels of small-molecule hydropersulfides such as glutathione (GSH) hydropersulfide (GSSH) and cysteine (Cys-SH) hydropersulfide (CysSSH) are particularly high in mammalian cells, tissue, and plasma, with some tissues containing >100 μM levels of GSSH. Moreover, numerous proteins were also found to contain hydropersulfides on cysteine residues. Before this, Mustafa et al. (52) reported that as much as 10%–25% of many liver proteins were “sulfhydrated” [an unfortunate term (72) that inaccurately describes the formation and/or presence of an RSSH moiety] under physiological conditions. Further, Longen et al. (47) reported that cells treated with H2S donors contained numerous protein persulfides and that there is a preference for hydropersulfide formation on specific cysteines in proteins with multiple cysteine residues. Doka et al. also reported that tissues and whole cells contain high steady-state levels of protein hydropersulfides (18).
One of the most recent and exciting discoveries regarding both small-molecule hydropersulfides as well as protein hydropersulfides is the report that Cys-SSH can be made from cysteine (Cys-SH) by the cysteine t-RNA synthetase (CARS) and can be either released, resulting in free intracellular Cys-SSH, or incorporated into proteins, translationally resulting in proteins containing Cys-SSH (2). Indeed, in some cells, this process appears to be the major pathway for Cys-SSH generation. Although further work is required to determine the mechanisms of regulation of the CARS system for biosynthesizing and incorporating Cys-SSH into proteins, this article may represent the discovery of the 22nd amino acid (in eukaryotes). Interestingly, the established 21st amino acid is selenocysteine (Sec) and is synthesized from serine on a t-RNA before translational incorporation (48, 73). Importantly, there are striking chemical similarities between the selenol (RSeH) function of Sec and the hydropersulfide function of Cys-SSH, which will be discussed in depth later.
Hopefully, the examples just cited provide sufficient evidence of the prevalence of hydropersulfides in biological systems. To be sure, studies performed much earlier alluded to the existence of hydropersulfides (specifically Cys-SSH) in mammalian proteins. For example, in the 1970s, Massey's lab reported that both xanthine oxidase (49) and aldehyde oxidase (10) contained a Cys-SSH residue that was important to their activities (although this has been debated and likely not the case, other related proteins have been found to contain the Cys-SSH moiety, vide infra). The examples just cited should also serve to indicate that Cys-SSH generation and incorporation into proteins and peptides is intentional and purposeful and not a random or artifactual event. That is, Cys-SSH, and the species derived and/or downstream of its biosynthesis, undoubtedly serve a specific and important function that cannot be accomplished by other biological functionalities (e.g., thiols, sulfenic acids, Sec, etc.).
Thus, it is the tenet of this review that the evolutionary process has selected the hydropersulfide function as a means to accomplish specific and important biochemistry not present in other biochemical entities and that selection in this regard is based entirely on the unique and novel chemistry of the hydropersulfide functional group. Therefore, it is important to first discuss the chemical reactivity and chemical properties of the RSSH functional group before speculating about its biological function.
The Biologically Relevant Chemistry of RSSH
Since RSSH (e.g., Cys-SSH) is likely generated from the corresponding thiol RSH (e.g., Cys-SH), it will be important to compare and contrast the chemical differences between RSH and RSSH to begin to determine the reasons this conversion is made (and under what conditions this is likely to occur). It will also be important to compare and contrast the chemical differences between RSSH and other established RSH-derived species such as disulfides (RSSR) to determine whether RSSH chemistry is distinct from these species as well.
RSH and RSSH oxidation states
One of the most important aspects of the conversion of RSH to RSSH is that this represents an oxidation. That is, the formation of RSSH from RSH requires an oxidant or oxidizing step. This is easily understood by simply assigning oxidation states to the sulfur atoms. The oxidation state of the sulfur atom of RSH is 2− (RS2−H), whereas the oxidation state of the same sulfur atom when RSH is converted to RSSH is 1− (RS1−SH). Importantly, the oxidation states of both sulfur atoms of RSSH are the same as the sulfur atoms of a dialkyl disulfide RSSR (all S1−). It is well established that the oxidative conversion of RSH to RSSR is an important biological process (often accomplished by oxidants such as H2O2) with potential roles in protein folding, cell signaling, etc. (33, 78) and from a redox perspective the conversion of RSH to RSSH is analogous. Thus, it is important to remember that RSSH is generated from RSH under oxidizing conditions (e.g., biological oxidative stress). This fact will become important later when discussing the possible biological utility of RSSH species.
Dual reactivity of RSSH
In biological systems, RSH (or more specifically, RS−) can act as a nucleophile and/or reducing agent. When RSH is oxidized to the corresponding disulfide RSSR, nucleophilicity is lost and this species becomes electrophilic and oxidizing—electrophilic because it can react with other nucleophilic RSH species and oxidizing since this reaction results in the oxidation of RSH (Reaction 1, thiol sulfur shown in bold).
It is important to note that RSSH has the properties of both reduced RSH and oxidized RSSR, and these are largely dependent on its protonation state (59). As will be discussed in more detail later, the deprotonated RSS− species is very nucleophilic and reducing (akin to RS−) whereas the protonated species RSSH is electrophilic and oxidizing (akin to RSSR). Thus, the pK a of RSSH becomes an important factor in predicting its chemical biology. Discussions of the pK a, nucleophilicity, electrophilicity, reducing potential, as well as other aspects of RSSH chemistry are given later.
One important aspect of the dual nucleophile/electrophile reactivity of RSSH is the fact that under certain conditions (e.g., physiological conditions) both the RSSH and RSS− forms can exist simultaneously. This means that RSSH under these conditions is inherently unstable since the nucleophilic RSS− should readily react with the electrophilic RSSH species, giving numerous products depending on the site of nucleophilic attack (and the likelihood of subsequent reactions). Thus, it should always be considered that this chemistry can occur under biological conditions and the generation of other sulfur species can potentially confound experimental interpretation.
RSSH versus RSH pK a
The pK a of RSH varies significantly depending on the nature of the R group and the environment (e.g., the nature of the surrounding residues in a protein). For example, the pK a of free cysteine in aqueous solution is 8.2 whereas in a protein a cysteine residue can have a pK a < 6 (77). This lowering of the pK a is due in part to protein-mediated stabilization of the anionic RS− species. In solution, the pK a of a free hydropersulfide (not incorporated into a protein) is significantly lower than that of the corresponding free thiol, typically 1–2 pK a units lower (21), although differences as great as 4 pK a units have been proposed (16). The conversion of a protein cysteine residue to a protein hydropersulfide may be expected to also have an effect on the pK a, but as indicated earlier for protein cysteines, the magnitude (and possibly the direction) of the pK a change will, undoubtedly, be a function of the protein environment. Regardless, protein effects notwithstanding, RSSH species are inherently more acidic than the corresponding RSH. Thus, it may be expected that the ratio of RS−/RSH will be significantly less than the RSS−/RSSH ratio for the same R group. Since anionic species are more nucleophilic than the corresponding protonated species, it may also be expected that conversion of RSH to RSSH will result in a higher concentration of the more nucleophilic anionic species.
RSSH versus RSH nucleophilicity
The earlier discussion indicates that conversion of RSH to RSSH yields a stronger acid, thus increasing the relative levels of the more nucleophilic anionic species. It should also be recognized that RSS− is inherently more nucleophilic compared with the corresponding RS− species. At least some of the enhanced nucleophilicity can be attributed to an alpha effect, the effect that occurs when a nucleophilic electron pair has an adjacent atom that also possesses an electron pair. In these cases, the nucleophile with an adjacent electron pair exhibits increased rates of reaction with electrophiles. For example, hydroxylamine (NH2OH) is significantly more nucleophilic than ammonia (NH3), although it is a much weaker base (19). The origins of the alpha effect are not clearly established and have been proposed to be due to a variety of effects, including transition state stabilization, ground state destabilization of the nucleophile, solvation effects, and product stability, among others (23).
The kinetic enhancements associated with an alpha effect are highly variable and can depend greatly on the nature of the electrophile. For example, for oxygen-based nucleophiles it is reported that HOO− reacts with phosphate esters (e.g., Sarin) ∼50-times faster than HO− (41) whereas with addition reactions to nitriles, the rate enhancement can be as high as 66,000 times faster (76). To be sure, it is not established that an alpha effect associated with sulfur atoms can be as significant as that reported for oxygen atoms. Cuevasanta et al. (16) have, however, established the existence of an alpha effect with sulfur-based nucleophiles as they have reported that RSSH can be more reactive compared with RSH by ∼20-fold toward disulfides. As with oxygen-based nucleophiles, there is likely to be a wide range of rate enhancements associated with the conversion of RSH to RSSH depending on the nature of the electrophile and the polysulfur species.
Electrophilicity of RSSH and RSSR
As previously mentioned, the protonated RSSH species is electrophilic, akin to the electrophilicity of RSSR. As with RSSR, nucleophilic cleavage of the S-S bond in RSSH occurs only with “soft” nucleophiles such as thiols, phosphines, and cyanide (CN−) (and not with “hard” nucleophiles, including oxygen- or nitrogen-based nucleophiles) and can occur on either sulfur atom, although there appears to be a slight preference for nucleophilic attack at the terminal sulfur atom (59). Although it is intriguing to speculate that RSSH can be more electrophilic than RSSR since RSSH is less sterically crowded (for nucleophilic attack at the terminal sulfur, RSSH) and HS− is a superior leaving group compared with RS− (for nucleophilic attack at the internal sulfur, RSSH) (59), this has yet to be addressed experimentally.
RSSH versus RSH as a reductant
Along with the increased nucleophilicity of RSSH versus RSH, it has also been established that RSSH is a significantly better one-electron reductant compared with RSH. For example, the reduction of ferric cytochrome c (Fe3+Cyt c) to ferrous cytochrome c (Fe2+Cyt c) (Reaction 2) is facile with RSSH but not with RSH or H2S (24).
Moreover, Everett and Wardman (21) have shown that RSSH can be a good antioxidant by quenching a reactive one-electron oxidant via either one-electron donation or hydrogen atom transfer (Reactions 3 and 4).
Importantly, the oxidized RSSH species, RSS·, is not reactive as an oxidant and typically only dimerizes to RSSSSR (8) even in the presence of the paramagnetic species O2 or NO (8, 14). The lack of reactivity and relative stability of RSS· is due to the delocalization of the unpaired electron onto the adjacent sulfur atom, a stabilizing effect not available to the unpaired electron of a thiyl (RS·) radical species. Thus, it is the stability of RSS·that explains, to a significant degree, the ability for RSS−/RSSH to act as a good hydrogen atom/electron donor. By comparison, RSH is a much poorer hydrogen atom donor compared with RSSH as indicated by the differences in the S-H bond dissociation energies (BDEs). The BDE for the S-H bond in RS-H is 92 kcal/mol whereas the BDE for RSS-H is 70 kcal/mol (6), consistent with RSS·being much more stable compared with RS·and supporting the fact that RSSH is a superior H-atom donor compared with RSH. Also, the reduction potential for the RS·/RS− couple is 0.92 V versus a normal hydrogen electrode (NHE) (pH 7) (11) whereas the calculated reduction potential for the RSS·/RSS− couple is significantly lower, 0.68 V versus NHE (pH 7) (39). These values also indicate that RSS− is a superior reducing agent compared with RS−.
Coordination chemistry of RSSH and RSH
RS− species are well-known ligands for metals, forming especially strong complexes with “soft” metals/Lewis acids (since sulfur species are “soft” Lewis bases) (56). Considering the enhanced nucleophilicity associated with RSS− compared with RS−, it may also be expected that RSS− could be a superior metal ligand as well. Although metal complexes with RSS− are much less known compared with RS−, some complexes have been reported. For example, there are reports of RSS− complexes with copper, iridium, platinum, titanium, tungsten, iron, ruthenium, and zinc (20, 26, 31, 35, 62
–65, 81). Importantly, Galardon et al. (26) examined Zn2+ coordination and demonstrated that the RS− and RSS− ligands are capable of exchanging and in the case of t-butyl-S− versus t-butyl-SS−, the thermodynamic preference was determined to be coordination of the RSS− species. To be sure, this thermodynamic preference for RSS− may not be entirely due to RSS− being an inherently “better” ligand for Zn2+ since steric effects, undoubtedly, play a significant role (26). This group also pointed out that a metal-persulfide complex presents more reaction pathways compared with metal-thiolate complexes (considering only the reactivity of the metal center and the ligating sulfur species). That is, an M-SSR complex (M = metal) possesses three possible electrophilic sites for reactions with nucleophiles/Lewis bases, the metal and the two sulfur atoms, whereas an M-SR complex will react typically with nucleophiles/Lewis bases only at the metal center (26). Thus, the reaction of nucleophiles with M-SSR complexes can, in theory, give a variety of possible products from attack at: (i) the metal center (Reaction 5), (ii) the coordinating sulfur atom (Reaction 6), and (iii) the noncoordinating sulfur atom (Reaction 7). However, with M-SR complexes only nucleophilic attack at the metal center (displacing the RS− ligand) is expected (Reaction 8).
Similarity between RSSH and RSeH chemical biology
Hopefully, it can be gleaned from the earlier discussion that RSSH chemistry is distinct from that of RSH. Importantly, many of the chemical properties associated with biological thiols (e.g., nucleophilicity and reducing potential) are enhanced with hydropersulfides and there is some evidence that RSSH species are also adept at coordinating metals similarly, if not better, compared with RSH. It is especially noteworthy that many of the chemical properties of the RSSH functionality are similar to that of RSeH. The similarities between RSSH and RSeH become more striking when they are both compared with the chemistry of RSH. Using Cys-SH, Cys-SSH, and Sec as examples, both Sec and Cys-SSH are more acidic compared with Cys-SH (pK a of 5.2 for Sec, approximately <7 for Cys-SSH, and 8.2 for Cys-SH), indicating a higher percentage of the nucleophilic anionic species for Sec and Cys-SSH. The redox couples for Sec (RSe·, H+/RSeH, E°′ = 0.43 V), Cys-SSH (RSS·, H+/RSSH, E°′ = 0.68 V), and Cys-SH (RS·, H+/RSH; E°′ = 0.92 V) indicate that both Sec and Cys-SSH are superior reductants compared with Cys-SH (39, 55). Both Sec and Cys-SSH are also superior nucleophiles compared with Cys-SH. For example, Sec (the anionic RSe− species) reacts with RSSR ∼15-times faster than Cys-S− and Cys-SS− reacts with RSSR ∼20-times faster than Cys-S− (16, 54). Finally, both Sec and Cys-SSH are better hydrogen atom donors compared with Cys-SH, as indicated by their relative BDEs [BDEs for X-H, (X = S or Se) in RS-H, RSS-H, and RSe-H are 92, 70, and 74 kcal/mol, respectively (6, 53)]. Thus, it is reasonable to say that aspects of the biologically relevant chemistry of RSSH and RSeH are enhanced compared with RSH (e.g., acidity, nucleophilicity, and reducing potential) (Table 1). It is, however, important to reiterate that RSSH does possess several chemical properties that are not present in RSeH species; RSSH can be electrophilic whereas RSeH cannot (vide infra) and RSSH is capable of reacting with other RSH species, transferring the hydropersulfide functionality (vide infra, Reaction 9 below).
Chemical Properties of RSH, RSeH and RSSH
For the radical RS·, RSS·, and RSe· species (pH 7).
X = S for RSH; X = SS for RSSH; and X = Se for RSeH.
pK a values for RSSH have been reported to be one to four orders of magnitude lower than the corresponding thiol (16, 21).
Based on reaction with electrophilic disulfide (16).
This is a calculated value (39).
This value is based on HSS-H (6).
Based on reaction with electrophilic disulfide [at pH 10 so this represents a direct comparison of RSe− and RS− (54)].
BDE, bond dissociation energy; RS, thiyl radical; RSeH, selenol; RSH, thiol; RSS, perthiyl radical; RSSH, hydropersulfide; RSSR, disulfide.
Another biologically important aspect of selenium chemistry is that Sec resists permanent, irreversible biological oxidative modification (58, 68). Oxidation of RSH to, for example, sulfinic (RS(O)OH) acid represents a predominantly irreversible modification [i.e., these species are not readily reduced back to the corresponding thiol under biological conditions, except for select peroxiredoxins (79)]. However, the corresponding seleninic acid (RSe(O)OH) is readily reduced by biological thiols back to the selenol, RSeH. Interestingly, highly oxidized RSSH species, for example a perthiosulfonic acid (RSS(O)OH), can be reduced back to the corresponding RSH under biological conditions (note, however, that the reduction is not back to the original RSSH species) and therefore RSSH is also resistant to irreversible oxidative modification (50). Thus, irreversible biological oxidative modification of RSeH and RSSH is more difficult compared with RSH and represents another strong similarity between RSeH and RSSH. Figure 1 schematically depicts some of the similarities between RSSH and RSeH when compared with RSH.
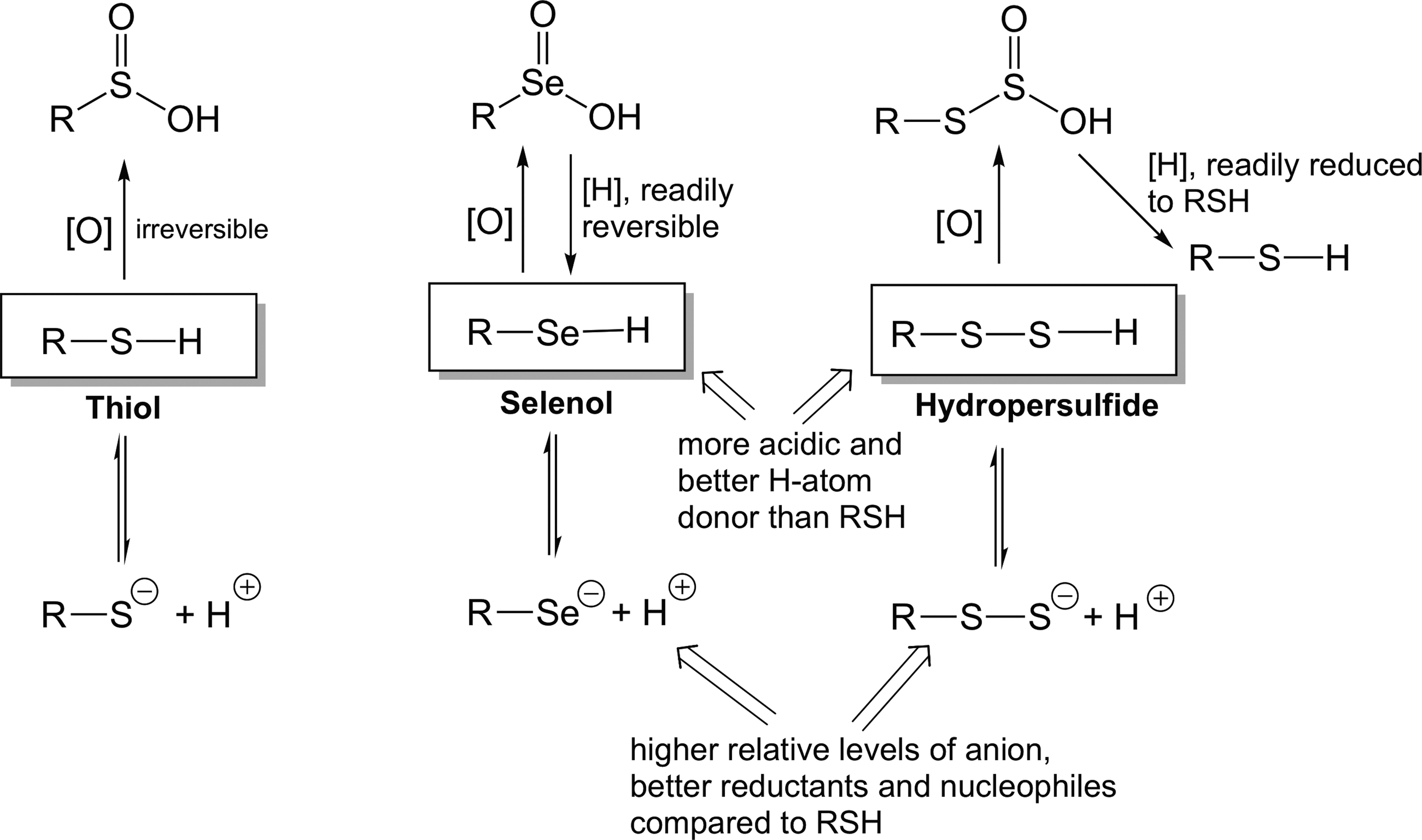
The similarity in chemistry between RSeH and RSSH (especially with regards to a comparison with RSH) may indicate that some of the biological functions of RSSH may be analogous to the functions of RSeH (vide infra). One important difference, however, is the fact that RSSH are potentially impermanent species whereas RSeH species are not. That is, RSSH can be readily reduced back to the corresponding RSH species via, for example, reaction with a thiol (Reaction 9 or Reactions 10 and 11) whereas there is no equivalent reaction for RSeH (since it is already a fully reduced species).
Thus, RSSH can be viewed as having RSeH-like properties in a functional group that can be regulated via synthesis-degradation. However, the Sec is a more permanent species that cannot be regulated equivalently. Therefore, arguments regarding the biological necessity and utility of Sec [e.g., Nauser et al. (54) and Reich and Hondal (58)] can be reasonably applied to RSSH.
Possible Biological Functions of RSSH
The biological function and utility of RSSH species will, undoubtedly, be due to their unique chemistry that is distinct from other existing sulfur (or chalcogen) species. As generally discussed earlier, much of the chemistry of RSSH is similar to that found with RSH, except that the reactivity in many regards is significantly enhanced. Also, RSSH (the protonated species) is electrophilic, akin to RSSR. Figure 2 provides a general schematic depicting some of the previously mentioned changes in the chemical properties when RSH is converted to RSSH, some of which are proposed to be important to the biological functions of RSSH (vide infra).

Possible protective functions of RSSH
The enhanced and/or distinct chemical reactivity of RSSH versus RSH is, undoubtedly, the basis for its biosynthesis and presence. That is, replacement of RSH with RSSH (by either post-translational modification or translational incorporation, vide supra) allows access to reactivity that is not adequate or present in the usual RSH function. The increased nucleophilicity and reducing capacity of RSSH predicts that it would be more reactive with electrophiles and oxidants. Thus, it is reasonable to predict that RSSH could be a cellular response to the presence of electrophilic and/or oxidative stress. Indeed, the idea that RSSH can protect against oxidative and electrophilic stresses is supported by several reports. One of the earliest examples of this was by Greiner et al. (28), who found that the phosphatase PTEN was protected from irreversible oxidative inhibition by H2O2 when in the hydropersulfide form. Ida et al. (32) found that cystathionine gamma-lyase (CSE) overexpression leads to increased levels of intracellular RSSH species (such as Cys-SSH and GSSH), which correlated with an increased resistance to cell death caused by oxidative stress (H2O2). Using an H2O2-triggered hydropersulfide donor, Powell et al. (57) report RSSH-mediated cellular protection from H2O2 toxicity and expound on the potential therapeutic value of these types of compounds. Ezerina et al. (22) have also reported that the antioxidant and protective effects of N-acetyl cysteine (NAC) may be due to the generation of RSSH from NAC metabolism. Moreover, it is reported that cells treated with sulfane sufur donor polysulfides, which results in increased levels of intracellular per- and poly-sulfides, inhibit pro-inflammatory pathways in macrophages and in mice they can protect against endotoxin shock (83). Finally, Bianco et al. (7) reported that HEK293T cells treated with cysteine trisulfide (thiocystine, Cys-SSS-Cys) led to an increase in intracellular Cys-SSH and GSSH, which correlated with a significant protection against electrophilic stress caused by N-ethylmaleimide, an effect that has been also seen with heavy metals and other electrophiles [reviewed in Shinkai and Kumagai (66)].
The electrophile trapping by nucleophilic RSSH and the antioxidant (i.e., reductive) properties of RSSH provide convenient and logical mechanisms for the biological effects mentioned earlier. However, as also discussed earlier, RSSH is electrophilic and, as such, can modify thiols and potentially cause oxidative stress. Although the pK a of RSSH is likely to be lower than 7, indicating a predominance of the nucleophilic and reducing anion, RSS−, at high overall levels of RSSH a significant amount of the protonated and electrophilic RSSH will be present. Thus, it may be expected that intracellular levels of RSSH need to be controlled to avoid RSSH-mediated oxidative stress. In support of this idea, cells treated with Cys-SSS-Cys, which results in high levels of intracellular RSSH species, were found to export Cys-SSH (45). To be sure, it is also speculated that the export of Cys-SSH may protect cells from extracellular electrophilic or oxidative stressors before they have the opportunity to impact intracellular targets.
Possible functions of the one-electron redox properties of RSSH
Clearly, RSSH is a superior one-electron reductant compared with RSH. As previously discussed, the increased ability for RSSH to donate an electron is due to the increased inherent stability of RSS· versus RS·. Although RS·has biological utility as an oxidant, it is typically not considered a readily accessible and common redox intermediate. For example, a cysteinyl radical (Cys-S·) species in ribonucleotide reductase has been shown to act as a potent oxidant that is capable of abstracting a hydrogen atom from ribose, a critical step in the generation of deoxyribose for the biosynthesis of DNA [e.g., Stubbe and van der Donk (71)]. The high reduction potential of the Cys-S·, H+/Cys-SH couple (0.92 V, vide infra) and strong S-H BDE of Cys-SH (92 kcal/mol, vide infra) are consistent with the ability of Cys-S· to perform this chemistry. On the other hand, RSS· is not a strong oxidant and RSSH/RSS− is a decent reductant (Table 1). Thus, redox chemistry that is not possible with RSH may become possible when RSH is converted to RSSH. Based on the differences in the redox chemistry of RSH versus RSSH, it has been postulated that an RSH to RSSH conversion can constitute a “redox gate” (4). That is, a catalytic electron transfer process cannot occur when a protein contains a Cys-SH function (since Cys-S· is too oxidizing and Cys-S− is a poor reductant, causing the system to get stuck in the Cys-S− form). However, oxidation of RSH to RSSH allows an electron transfer process to occur (since RSS·is not a strong oxidant and RSS− is a reasonable reductant). In this way, electron transfer processes are “gated” via oxidative processes that lead to RSSH generation from RSH (Fig. 3). To be sure, this is currently pure speculation and there is no evidence that this process occurs. However, the chemical differences between RSH and RSSH predict this phenomenon as a redox regulatory possibility.

Interestingly, some of the first reports of possible protein hydropersulfides were from Massey's lab and their presence in the redox active molybdenum (Mo) proteins xanthine oxidase and aldehyde oxidase was reported (10, 49). Although the presence of an RSSH/RSS− species in either enzyme is debatable (many favoring the more likely presence of a molybdenum sulfide, MoS, species instead) [e.g., Wahl and Rajagopalan (75)], it remains possible that with other Mo enzymes the MoS species can be in equilibrium with an Mo-SSCys complex, vide infra (30). As already discussed, the reducing capabilities of RSSH species also predict that they will readily react with oxidizing species and therefore can serve as efficient antioxidants (e.g., Reactions 3 and 4) (21).
Cys-SSH biosynthesis and clues to RSSH function
Interestingly, there are numerous pathways for the biosynthesis of RSSH species (70). This fact alone points to the possibility of diverse functions for RSSH that can be differentially regulated and/or compartmentalized. Two of the most studied and established pathways involve the enzymes CSE and cystathionine beta-synthase (CBS), both of which are known primarily as H2S-producing enzymes but generate Cys-SSH as an intermediate to reductive H2S formation (82). Previous work indicates that in some cells CSE is the major Cys-SSH producing enzyme [e.g., Ida et al. (32)]. Therefore, this discussion will focus on the CSE expression and regulation. To be sure, this focus is not meant to discount the importance of the other enzymes in RSSH biosynthesis [e.g., CBS, CARS, mercaptopyruvate sulfur transferase (70), etc.], but it is done to be illustrative and concise and since a comprehensive treatment of all of these pathways is beyond the scope of this review.
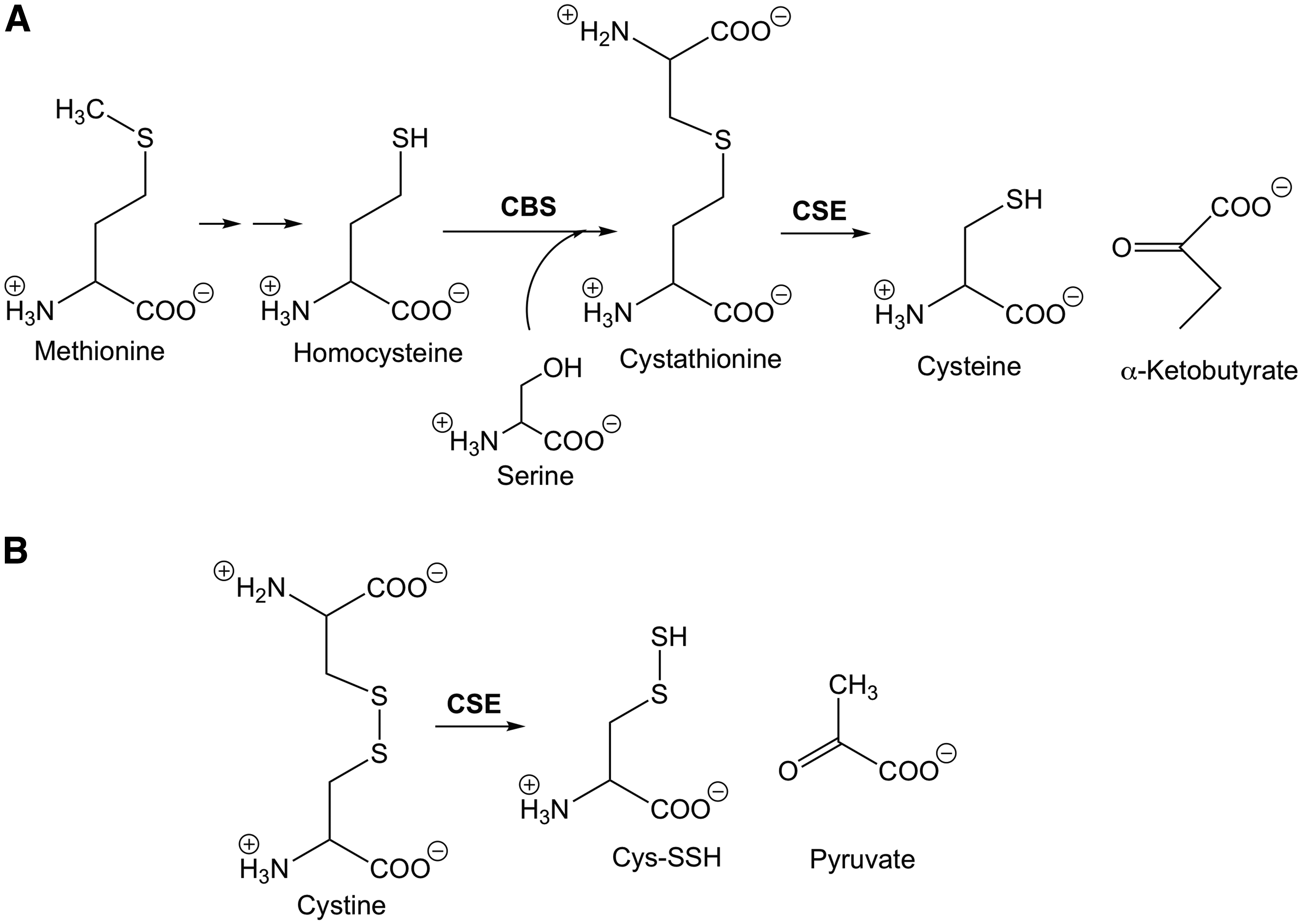
CSE is a somewhat promiscuous enzyme in that it is capable of catalyzing reactions of multiple substrates. It is probably best known (and understood) as a critical player in the transsulfuration pathway, which converts methionine into cysteine (70). In this pathway, CSE converts cystathionine (an adduct between homocysteine and serine) to cysteine and α-ketobutyrate (Fig. 4A). As an H2S- and RSSH-producing enzyme, CSE is also capable of converting cystine (Cys-SS-Cys) to Cys-SSH and pyruvate (82) (Fig. 4B). The reason that CSE is considered an important H2S biosynthetic enzyme is that the generation of Cys-SSH can lead to the formation of H2S via a reductive process such as further reaction with a thiol (Reaction 10). Regardless, it is important to note that Cys-SSH generation from CSE precedes H2S formation.
CSE expression can be regulated by the transcription factor ATF4 (17), which controls genes involved in numerous stress responses (29). Overall, activation of the ATF4 signaling pathway is considered a “master” transcriptional stress response, important in endoplasmic reticulum stress, nutrient (amino acid) limitation, hypoxia, and oxidative stress (80). Interestingly, ATF4 also controls the expression of xCT, an important protein of the Cys-SS-Cys uptake transporter Xc − (43). Thus, activation of the ATF4 transcriptional signaling pathway can lead to an increased expression of Cys-SS-Cys uptake and CSE-mediated conversion of Cys-SS-Cys to Cys-SSH. It can be hypothesized, therefore, that Cys-SSH generation is a cellular response to stress. Before the recent realization that Cys-SSH was prevalent and possessed unique and intriguing chemical properties (compared with RSH), the interpretation of these events (i.e., the stress-induced increased expression of xCT and CSE) was done differently. An increased expression of xCT as a stress response would be construed as a cell's desire to increase intracellular Cys-SH levels for the purposes of increasing GSH biosynthesis [e.g., Lewerenz et al. (44)]. Since the major form of extracellular cysteine is in the oxidized Cys-SS-Cys form, increased intracellular Cys-SH requires the uptake of the oxidized Cys-SS-Cys first, followed by reduction to Cys-SH and then incorporation into GSH. The increased expression of CSE can be interpreted similarly; increased Cys-SH generation from methionine (via homocysteine intermediacy) could also increase GSH biosynthesis [e.g., Dickhout et al. (17)].
One potential problem with the idea that Cys-SS-Cys uptake is for the specific purpose of increasing Cys-SH levels to be used for the synthesis of the antioxidant and reducing GSH is the fact that a reducing equivalent is required to make a reducing agent. That is, Cys-SS-Cys must be reduced to Cys-SH to make the reducing GSH species. From an electron counting perspective, this process is a “wash” (no net gain in reducing equivalents), something that will not benefit an oxidatively stressed cell. GSH certainly has specific systems built around it (such as GSH transferase, GSH peroxidase, etc.) that may justify its biosynthesis in spite of the fact that GSH formation from Cys-SS-Cys does not increase the net reductive capacity of the cell. However, it is intriguing to speculate that increased Cys-SS-Cys uptake, via increased expression of xCT, and increased CSE expression instead increases the levels of intracellular Cys-SSH, which protects cells from oxidative/electrophilic stress. As previously discussed, RSSH species are oxidized (at the same oxidation state as RSSR, Cys-SS-Cys) and therefore do not require a reducing equivalent in their biosynthesis. Moreover, RSSH species are more reducing than the corresponding RSH, meaning that a potent reductant is made without “wasting” a reducing equivalent, as would be the case for GSH biosynthesis from Cys-SS-Cys. Again, this idea is purely speculative, but consistent with the chemistry previously discussed and consistent with the idea that RSSH species are potentially protective to cells against oxidative/electrophilic stress. These ideas are schematically depicted in Figure 5. Clearly, other RSSH biosynthesis pathways need to be examined in this regard. Especially important will be an in-depth examination of the regulation of CARS-mediated Cys-SSH generation and translational incorporation into proteins.

Changes in enzyme activity of protein persulfides
As discussed earlier, the conversion of RSH to RSSH dramatically reduces the pK a (and therefore increases the percentage of the anionic species) and increases the inherent nucleophilicity. Thus, it is possible that RSSH formation from RSH will increase the activity of cysteine proteins that utilize a critical cysteine function as a nucleophile. One of the first presumed examples of this was reported by Mustafa et al. (52), who indicated that treating glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with H2S resulted in hydropersulfide formation at the catalytic cysteine residue and an increase in catalytic activity. Since the catalytic activity of GAPDH requires a nucleophilic cysteine, it is not unreasonable to speculate that an increase in activity is due to the increased nucleophilicity of a catalytic hydropersulfide. However, it should be realized that hydropersulfide formation in GAPDH increased the Vmax and had no effect on the Km, which is more consistent with the idea that H2S simply increased the percentage of active enzyme (i.e., reduced inactive oxidized catalytic cysteine residues to the reduced and active form) and did not necessarily increase the catalytic efficiency. Indeed, subsequent work indicates that hydropersulfide formation on GAPDH results in an inhibition of activity (34). More recently, it was reported that hydropersulfide generation in the E3 ubiquitin ligase Parkin results in an increase in activity, implicating hydropersulfide formation as a protective strategy against Parkinson's disease (74). To be sure, numerous examples of inhibition of enzyme activity resulting from hydropersulfide formation on catalytic cysteine residues have been reported [e.g., Francoleon et al. (24) and Jarosz et al. (34)]. This may not be surprising since the catalytically important residues of most enzymes may be optimized for a single sulfur atom in the active site (i.e., the sulfur on a catalytic cysteine) and the addition of an additional sulfur atom may result in a nonoptimal geometry/structure. Of course, there is always the possibility that conversion of a catalytically important Cys-SH for one reaction to a Cys-SSH species actually changes the reaction and/or substrate preference. Regardless, there appears to be no general trend with regards to the effects of hydropersulfide formation on cysteine enzyme activity.
RSSH and metals/metalloproteins
As discussed earlier, RSSH has the potential to interact with metals and metalloproteins as either a ligand or a reductant. One of the most established functions of RSSH in this regard is in the protection from heavy metal toxicity. It is predictable that RSSH will coordinate “soft,” heavy metals readily. Indeed, the Kumagai lab has postulated that reactive polysulfides such as the inorganic polysulfide S4 2− react with and protect cells from Cd2+ (3). Also, knockdown of CSE in bovine aortic endothelial cells results in a dramatic decrease in intracellular per- and poly-sulfides, which correlated with an increase in Cd2+-mediated signaling (67). This result is interpreted as a persulfide-mediated inactivation of Cd2+ in the cells and a protection against Cd2+ toxicity. Similarly, persulfides have been implicated in the detoxification of methylmercury (CH3Hg) via the formation of the relatively nontoxic bis-methyl mercury sulfide (CH3Hg-S-HgCH3) (1).
The possible role(s) of hydropersulfides in the function and/or regulation of metalloproteins is a relatively unexplored area. There are, however, some examples that illustrate the potential importance of RSSH in this regard. As discussed earlier, Massey et al. proposed a possible function of persulfides in the activity of the molybdenum-containing enzymes xanthine oxidase and aldehyde oxidase (although this has been debated, vide supra). The bacterial SoxAX cytochromes involved in sulfur oxidation have been shown to possess a heme-persulfide complex that is presumably important to catalysis (although the exact role of the Cys-SS− ligand is not firmly established) (5, 9, 37). Another bacterial protein, carbon monoxide dehydrogenase, also contains a crucial Cys-SS− associated with an Fe-Ni-sulfur cluster and is proposed to be important for cluster assembly, stability, or redox chemistry (38).
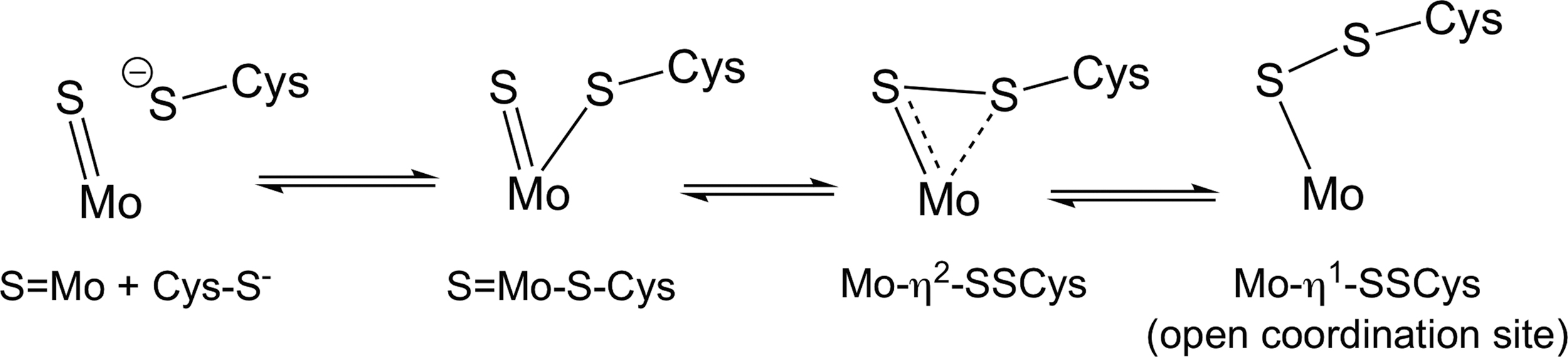
As alluded to previously, several Mo-containing enzymes (many of which are prokaryotic) have been reported to possess a Cys-SS− that coordinates to the Mo metal center. For example, the crystal structure of the bacterial nitrate reductase NapAB appears to possess a Cys-SS-Mo complex (15). Computational studies predict that the formation of the Cys-SS-Mo complex can occur via a “sulfur-shift” mechanism whereby a vicinal or coordinated Cys-S− residue can interact with MoS to give an η2-Cys-SS− and/or an η1-Cys-SS− ligand (12, 13, 30) (Fig. 6). The catalytic utility of the presumed sulfur shift may be for the purposes of opening up a coordination site for substrate binding, although this is not established. Based on the discussion herein, it will also be interesting to consider how a thiolate-to-persulfide ligand conversion may alter the redox chemistry associated with the entire molybdenum complex, allowing an important redox step to occur. Interestingly, a bacterial NADP+-dependent formate dehydrogenase, which is also an Mo enzyme, contains a Sec and has been predicted to perform similar sulfur(selenium)-shift type chemistry (13). To be sure, other examples of hydropersulfide-containing proteins (especially Mo-enzymes) have been reported [e.g., Lehrke et al. (42)]; however, an exhaustive treatment of this area is beyond the scope of this review.
As briefly discussed earlier, the redox chemistry of RSSH with heme proteins has been examined and it is clear that RSSH is a far superior reductant compared with RSH for proteins such as cytochrome c or metmyoglobin (8, 24). In a recent and more thorough study of the reaction of RSSH with hemeproteins, the Galardon group (25) reported that RSS− is capable of forming a detectable complex with the ferric heme of N-acetylated microperoxidase 11 whereas the reaction of RSS− with metmyoglobin (FeIIIMb) resulted in reduction to deoxymyoglobin (FeIIMb), which under aerobic conditions binds O2 to give oxymyoglobin (O2FeIIMb). The physiological utility of this chemistry remains to be determined.
It is likely that other important metalloproteins will be discovered to possess important persulfides that function as redox active moieties and/or metal ligands. Particularly intriguing are the Zn2+-containing proteins since cysteine is so important in the binding, structure, and regulation of Zn-proteins and Cys-SS− coordination to Zn2+ is expected. Indeed, a recent report indicates that persulfides are readily made in a Zn-finger protein and persulfide generation dramatically affects the RNA binding of this protein (40).
Electrophilic RSSH species
As previously discussed, RSSH is both electrophilic and nucleophilic depending on the protonation state. When protonated, RSSH possesses chemistry akin to RSSR and, therefore, can react with “soft” nucleophiles (such as thiols, phosphines, etc.) as an electrophile. Indeed, it is the electrophilicity of RSSH that allows it to S-sulfurate other thiols (Reaction 9) or serve as a source of H2S (Reaction 10). It is important to remember that the sulfur atoms of RSSH and RSSR are at the same oxidation state and thus represent oxidized thiol species. As the most abundant thiol, there has been significant focus on the redox biochemistry of GSH. It is generally believed that the ratio of GSH to its oxidized disulfide (GSSG), GSH/GSSG, can be an important indicator/regulator of cell general cell health, status, and/or condition (36). For example, the ratio of GSH/GSSG has been correlated to cell proliferation, differentiation, apoptosis, and necrosis. Indeed, export of GSH and/or GSSG from cells can occur to modulate the GSH/GSSG ratio. Some of the redox modulation performed by GSH/GSSG can occur via GSSG-mediated formation of mixed disulfides (i.e., glutathionylation), which can either protect proteins from thiol over-oxidation or regulate function (Reaction 12) (27).
Since Reaction 12 is akin to Reactions 9 and 10, it can be hypothesized that an RSH/RSSH ratio may also be an important factor in cell health, status, and/or condition. That is, if RSSH levels were important in regulating thiol protein function (via simple sulfur exchange chemistry, Reactions 9 and 10), then the levels of RSSH would need to be carefully regulated, just as GSSG levels are closely regulated [e.g., Morgan et al. (51)]. Interestingly, a recent report indicates that high Cys-SSH levels in cells result in the export of Cys-SSH from the cell (45). The reason for Cys-SSH export is not known but it can be speculated that high RSSH represents an “oxidative stress” [similar to high GSSG and the onset of apoptosis/necrosis (60)]. It is also possible that export of RSSH represents a protective mechanism in which toxic extracellular oxidants or electrophiles can be scavenged before affecting a cell. It is noteworthy, however, that only Cys-SSH was found to be exported although intracellular GSSH levels were also highly elevated. Thus, high GSSH appears to be tolerated by cells (unlike GSSG). It is important to remember that although RSSH is oxidized and possesses some chemistry similar to RSSR, it is also a potent reductant, unlike the RSSR species. Thus, interpreting the ratio of the levels of thiols versus oxidized thiols (e.g., GSH/GSSG) as an indicator of cellular “redox status” becomes more difficult. In the case of GSH/GSSG, it is clear that GSH is reducing and GSSG is oxidizing and a decrease in this ratio represents a more oxidized state. However, in the case of GSH/GSSH, this is not necessarily true. Since GSSH is a more potent reductant compared with GSH, a decrease in the GSH/GSSH ratio can represent a more reduced state. At the very least, it needs to be considered that oxidation of GSH to GSSH can produce a more reducing system, something that is counter-intuitive: Oxidation of a species generates a more potent reductant.
Protein hydropersulfides and selenoproteins
As previously discussed, the chemical similarities between RSSH and RSeH are striking, especially with regards to their comparison to RSH (Table 1). Thus, it can be predicted that protein hydropersulfides can function similar to selenoproteins except that protein hydropersulfides have the potential to be reduced back to protein thiols whereas the Sec in selenoproteins is permanent. That is, the hydropersulfides on proteins can potentially be viewed as a fleeting Sec-like functional group.
The reported biochemical utility of Sec is primarily due to its high nucleophilicity and reducing capabilities. The inherent nucleophilic reactivity of R-SeH toward H2O2 is typically one to two orders of magnitude greater than a corresponding R-SH, similar to what is found with proteins such as the selenoprotein glutathione peroxidase (GPx) (69). Among the most examined selenoproteins are GPx, iodothyronine deiodonases (DIO), and thioredoxin reductases (TrxR). With GPx and TrxR, the Sec is responsible for reacting with the oxidized species ROOH and RSSR, respectively [e.g., Lu and Holmgren (48) and Reich and Hondal (58)]. In these reactions, Sec is acting as a nucleophilic reductant. With DIO enzymes, Sec also acts as a potent nucleophile that is capable of reductive deiodination of aromatic rings of thyroid hormones via the intermediacy of Sec-I species, which can be reduced back to the Sec and iodide ion (61). Thus, it appears that the increased nucleophilicity of Sec versus Cys-SH is one reason for Sec incorporation into proteins and the same can be expected of Cys-SSH incorporation into proteins. It needs to be noted, however, that species can exhibit an unequal dependence on selenium. Indeed, there are some “selenoproteinless” animals that are devoid of selenoproteins and utilize Cys-SH homologs of selenoproteins to accomplish the same biochemical tasks (46). Thus, although Sec offers some reactivity advantages over Cys-SH, it is not absolutely required in all species. Considering the fact that Sec and Cys-SSH are chemically similar, it will be interesting to determine whether or not Cys-SSH incorporation into the proteins is more prevalent in animals that are less dependent on selenium (i.e., RSSH replaces RSeH function).
As shown in Table 1, RSe−/RSeH is a good one-electron reductant. As such, Sec has the potential to quench otherwise reactive radical species and/or participate in redox processes that are not accessible to Cys-SH (53). However, to our knowledge, there have been no reports of one-electron redox Sec biochemistry to date. Regardless, it would not be surprising to see selenoproteins participate in one-electron redox process, similar to what has been proposed herein for Cys-SSH proteins (Fig. 3).
Conclusion/summary
The discovery of the significant prevalence of Cys-SSH in both small molecules and proteins is relatively recent, and much work is still required to determine the biological role of the hydropersulfide functional group. The chemistry of Cys-SSH predicts that it may serve roles similar to what has been described for the presence of Sec in selenoproteins (e.g., to serve as a superior nucleophile or reductant). The RSSH functional group has, however, one chemical property that is distinct from RSeH—it can be electrophilic as well (when protonated). This property allows RSSH to transfer an -SH group to other nucleophilic species such as a thiol and because of this RSSH is more chemically versatile compared with RSeH. That is, the biosynthesis of Cys-SSH allows the generation of other RSSH species, whereas Sec is only translationally incorporated into specific proteins (although Cys-SSH can be translationally incorporated into proteins as well). Also, RSSH can be possibly a fleeting species since it can be reduced to the corresponding RSH species. Indeed, it should be re-emphasized that conversion of RSH to RSSH represents an oxidation and, therefore, is likely to occur under cellular oxidative stress conditions, and yet RSSH is a superior reductant compared with RSH, which is a reasonable cellular response to an oxidative stress. This unusual phenomenon—the oxidation of a species generates a superior reductant—may be fundamental to the physiological utility of hydropersulfides. Moreover, under normal reducing cellular conditions (when the oxidative stress ceases), RSSH can be reduced back to RSH, thus returning to normal conditions/functions. Clearly, these ideas are highly speculative at this time and, although grounded in chemistry, will require further work to determine whether these ideas are valid. Hopefully in the near future, a clearer and more established understanding of the physiological functions of RSSH will be forthcoming.
Footnotes
Acknowledgments
The authors would like to thank Péter Nagy, Yoshito Kumagai, Masahiro Akiyama, Christopher McGinity, and David Wink for their helpful input.
Funding Information
J.P.T. acknowledges funding from the National Science Foundation (NSF) (CHE-1900285).