
Abstract
Significance:
Air pollution is a considerable global threat to human health that dramatically increases the risk for cardiovascular pathologies, such as atherosclerosis, myocardial infarction, and stroke. An estimated 4.2 million cases of premature deaths worldwide are attributable to outdoor air pollution. Among multiple other components, airborne particulate matter (PM) has been identified as the major bioactive constituent in polluted air. While PM-related illness was historically thought to be confined to diseases of the respiratory system, overwhelming clinical and experimental data have now established that acute and chronic exposure to PM causes a systemic inflammatory and oxidative stress response that promotes cardiovascular disease.
Recent Advances:
A large body of evidence has identified an impairment of redox metabolism and the generation of oxidatively modified lipids and proteins in the lung as initial tissue response to PM. In addition, the pathogenicity of PM is mediated by an inflammatory response that involves PM uptake by tissue-resident immune cells, the activation of proinflammatory pathways in various cell types and organs, and the release of proinflammatory cytokines as locally produced tissue response signals that have the ability to affect organ function in a remote manner.
Critical Issues:
In the present review, we summarize and discuss the functional participation of PM in cardiovascular pathologies and its risk factors with an emphasis on how oxidative stress, inflammation, and immunity interact and synergize as a response to PM.
Future Directions:
The impact of PM constituents, doses, and novel anti-inflammatory therapies against PM-related illness is also discussed.
Introduction
The World Health Organization (WHO) estimates that 4.2 million premature deaths per year are caused by ambient air pollution worldwide, rendering it as one of the most important environmental health risks (157a). On a global scale, 9 of 10 people breathe air that is contaminated by different pollutants, such as gases (carbon monoxide, sulfur dioxide, nitrogen oxides, and ozone) and airborne particulate constituents (157a). The latter, referred to as particulate matter (PM), is a heterogeneous mixture of solid and liquid particles suspended in the air, varying in concentration, size, chemical composition, and surface area, originating from natural and anthropogenic sources (13, 16, 102). Among the different compounds in polluted air that are harmful, numerous epidemiological studies point out a central role of PM in mediating cardiovascular disease (59).
Physicochemical Characteristics, Elemental Composition, and Experimental Models of PM
Beside its complex chemistry and diverse composition, PM is mainly categorized by the mean particle size (Fig. 1): PM with an aerodynamic diameter below 10 μm (PM10) enters the upper respiratory airways where it predominantly exerts local effects. PM with a diameter of 2.5 μm or less (PM2.5) has the ability to penetrate deeper into the alveolar space and therefore a greater biological activity. In addition, PM2.5 is more likely to cause systemic complications (119). Accordingly, epidemiological studies have demonstrated that PM2.5 correlates more strongly with death from cardiopulmonary diseases compared with PM10 (38). PM2.5 therefore is considered major risk factor for cardiac disease, such as myocardial infarction (137), heart failure (135), and stroke (9, 62).
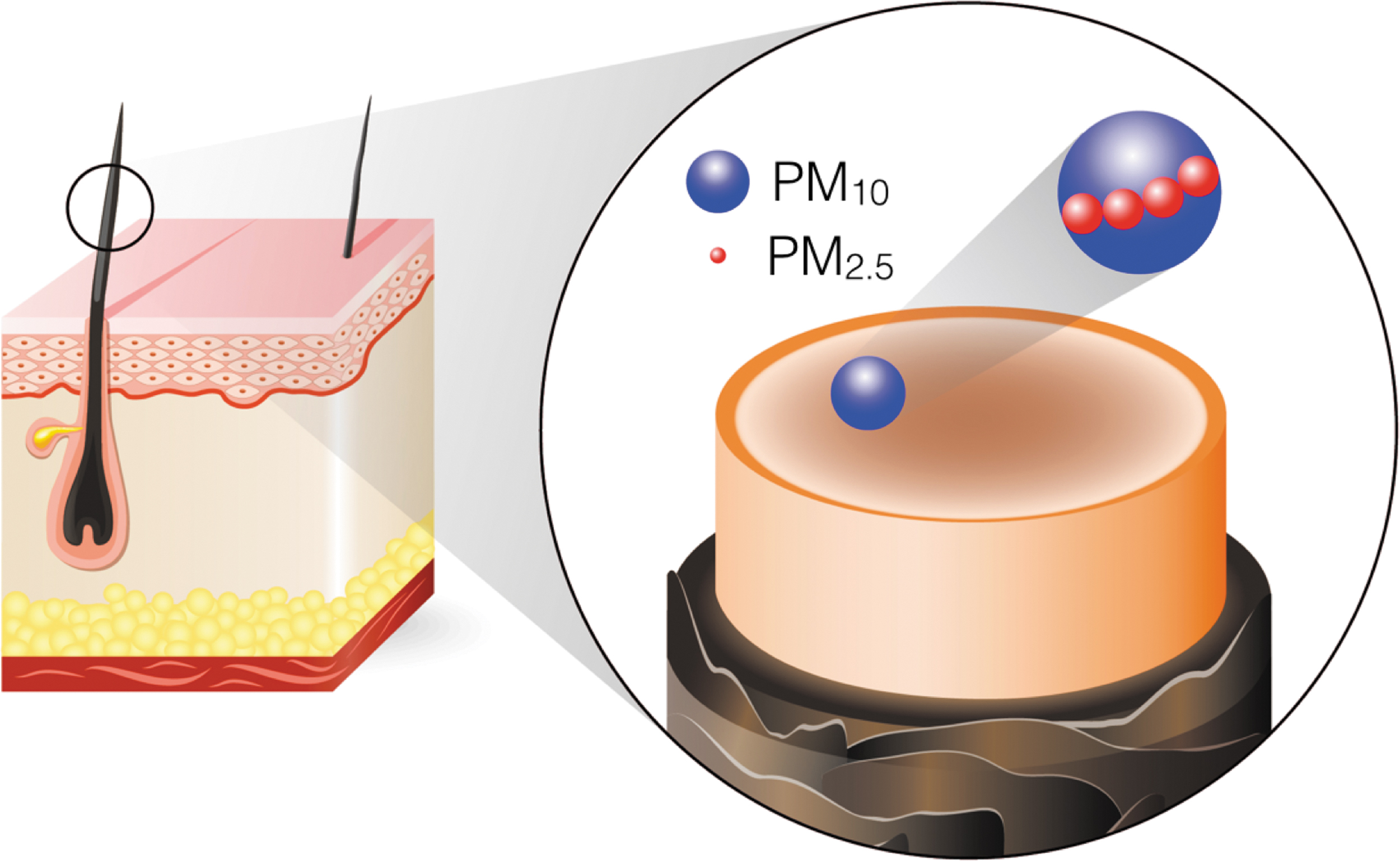
Coarse particles in PM10 mostly originate from natural sources, such as dust and agricultural practices, while fine particles, which predominate in PM2.5, largely arise from anthropogenic sources, such as diesel and gasoline exhaust, fossil fuel combustion during power generation, and industrial processes (128). The larger surface-to-mass ratio of fine PM2.5 carries more toxic compounds on the surface than PM10. The delivery of surface-adsorbed compounds to lower respiratory airways provides one potential explanation for the increased toxicity of PM2.5 in the clinical setting (119). In addition, it has been proposed that nanoscale PM can surpass the respiratory epithelia and translocate into the bloodstream and peripheral organs, where it may exert local effects (111).
The pathogenicity of PM2.5 has also been linked to its chemical composition. Harmful compounds, such as polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), as well as the transition metals iron, nickel, vanadium, and other components (110), are absorbed on the surface of PM. These redox-active compounds can participate in Fenton and Fenton-like chemical reactions and induce oxidative damage to macromolecules (24). The latter observation explains why oxidative stress pathways are considered a major cause of PM pathogenicity (77, 128).
To experimentally evaluate the biological effects of PM2.5, several PM-surrogates have been developed and tested in humans and different animal models. Frequently used model particles include concentrated air particles (CAP), which are obtained from ambient air particulate sampler devices and represent a mixture of airborne breathable particles from regional or local environments, such as urban areas (69, 89). To interrogate the effect of specific chemical components of PM, particles enriched in certain pollutants have been established as well. Diesel exhaust particles (DEPs) are generated from the incomplete combustion of fossil fuel in diesel engines and are composed of a carbonaceous core with adsorbed carcinogenic and redox-active volatile hydrocarbons, such as PAHs and PCBs (47, 152). In addition, residual oil fly ash (ROFA) particles represent the inorganic residue that remains after the incomplete oxidation of carbonaceous materials that are used for power generation and industrial processes. ROFA particles are rich in soluble transition metals and are therefore particularly useful to study PM-induced effects on redox metabolism and inflammation (24, 46). In addition, synthetic nanoparticles have been developed to test the specific contribution of individual metals (96). Moreover, standardized PM surrogates, such as the ones provided by the U.S. National Institute of Standards and Technology, are commercially available and have been used as reference PM (136).
Epidemiology of PM-Related Illness
Numerous epidemiological studies have established a clinical link between air pollution and cardiometabolic diseases. It is now recognized that a short-term exposure (days to weeks) to air pollution increased morbidity and mortality rates of pre-existing cardiovascular disease, while long-term exposure (months to years) may also provoke de novo cardiorespiratory disease [for a comprehensive review of these epidemiological studies, see Brook et al. (14)]. Analyses from ∼61 million U.S. citizens in the Medicare population show that a short-term increase in environmental PM2.5 by 10 μg/m3 is associated with a 5.0% increase in all-cause daily mortality (36). On longer term, a 10 μg/m3 increase in PM2.5 increases all-cause mortality up to 7.3% in the subsequent year (37). Similar findings have been recently reported in the framework of the multicenter European Study of Cohorts for Air Pollution Effects (8).
It is estimated that 3.3 million deaths yearly are caused by air pollution PM2.5 (82), mostly as a consequence of aggravated cardiovascular disease (14). Accordingly, the American Heart Association and the European Society of Cardiology classify ambient PM2.5 as a major cardiovascular risk factor (16, 115). Specifically, an acute exposure to PM2.5 of hours to a few weeks has been associated with an increased incidence of myocardial infarction, stroke, cardiac arrhythmias, and worsening of pre-existing heart failure (14). A long-term exposure may additionally increase the risk for the development of several cardiometabolic conditions, including obesity, diabetes mellitus, and the metabolic syndrome (107). Patients with pre-existing cardiovascular pathologies are under a particular high risk, as rates for recurrent myocardial infarction increase by 64% with every 10 μg/m3 increment in PM2.5 levels after a first event (23). A higher age and a lower socioeconomic status have been identified as risk enhancers in PM-exposed individuals (75). Actual recommended limits of PM2.5 range between 10 and 35 μg/m3 (Table 1). Still, PM2.5 concentrations may reach up to 200 μg/m3 depending on the region (145).
Current Air Quality Regulations for PM2.5 in Different Regions of the World
EC, European Commission; EPA, Environmental Protection Agency (United States); MEP, Ministry of Ecology and Environment (China); NAAQS, National Ambient Air Quality Standards; WHO, World Health Organization.
Inhaled PM Impairs Redox Metabolism and Fuels an Inflammatory Response in the Lung
After its penetration into the lungs, inhaled PM—especially the ultrafine, nanoscale particles—reach bronchioles and alveoli where it may accumulate or break the air/blood barrier. PM particles have been detected in the blood and peripheral tissues, such as in the kidney and liver, in hamster (10, 114) and humans (111), by using radioactively marked particles and two-photon microscopy (83). Ambient black carbon particles have also been detected in the fetal side of human placenta by femtosecond pulsed illumination (12), which might explain some of the reported developmental and epigenetic complications of PM. In vivo effects observed after intravenous injections of PM argue for a systemic action of circulating PM (112), but it remains unclear which, when, and under which circumstances PM translocates into the blood (79).
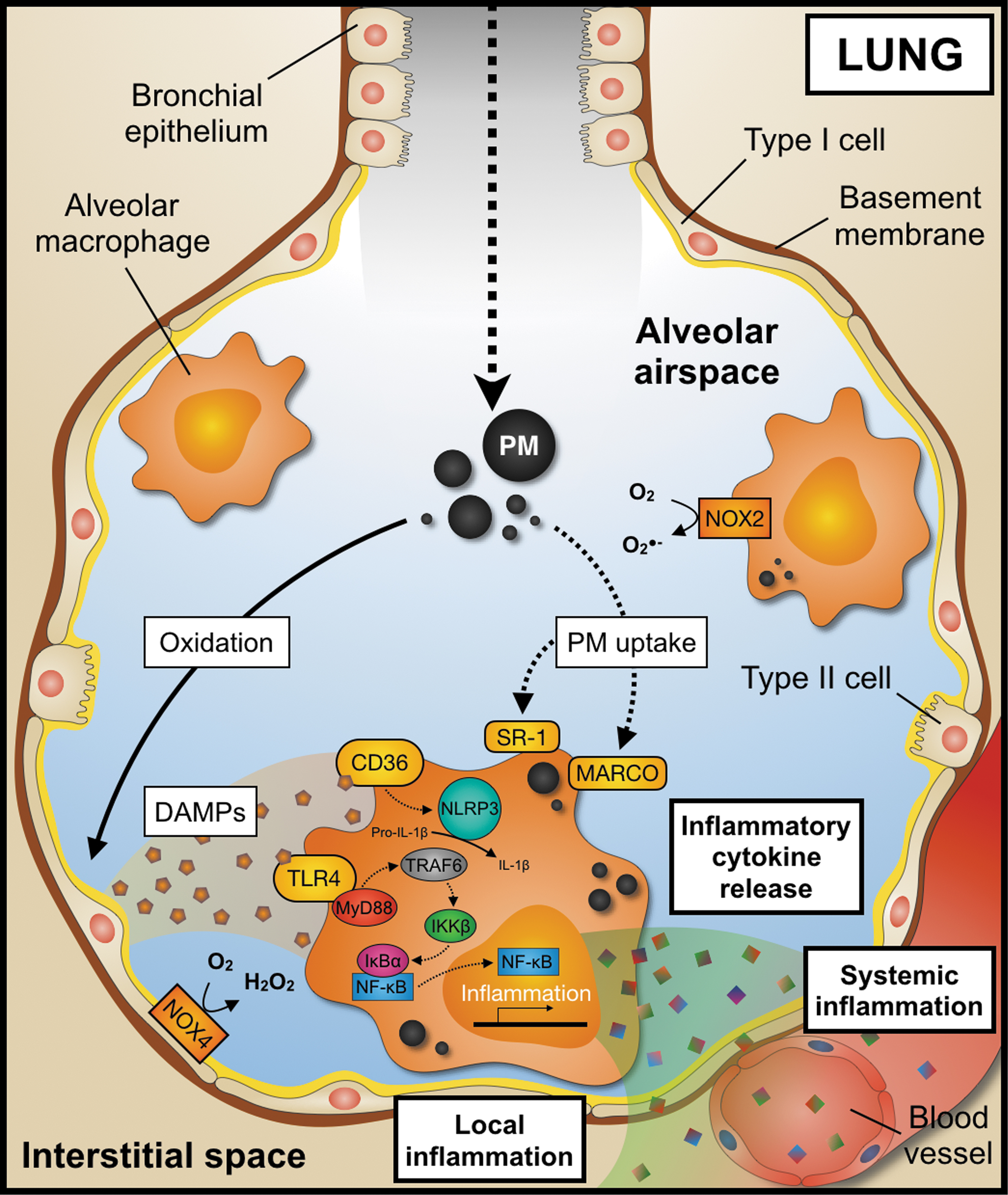
While the deposition of PM in the upper airway represents a likely cause of lung disease, such as asthma, infection, and cancer (73), a fraction of PM is taken up by tissue-resident phagocytes, in particular by alveolar macrophages (2) that usually maintain lung homeostasis through the removal of exogenous materials and microorganisms by phagocytosis. The uptake of PM is complex and may proceed via different routes. Phagocytosis of PM may be mediated by scavenger receptors, such as class A scavenger receptor (49), the macrophage receptor with collagenous structure (66, 105), toll-like receptors (TLRs), and scavenger receptor CD36 (129, 136), which initiates proinflammatory signaling events (102). In addition, PM induces oxidative stress pathways in the lung (97) by direct reactive oxygen species (ROS) generation from PM redox-active compounds and by an increased activity of oxidases triggered by an activation of phagocytes. The latter has been shown to proceed by NADPH oxidases (NOXs) homologue 2 and 4 in inflammatory (74, 98) and epithelial cells (3, 81). These oxidative processes generate damage-associated molecular patterns (DAMPs), such as oxidized phospholipid palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC), that are preferentially taken up by TLR4 (74). In addition, scavenger receptor CD36 senses other PM2.5 oxidation by-products, such as 7-ketocholesterol (129). Collectively, these pathways trigger subsequent proinflammatory signaling cascades. Typical examples include, but are not limited to, signaling events mediated through TLR4 and myeloid differentiation primary response 88 protein (136), tumor-necrosis-factor (TNF) receptor-associated factor-6 (70), inhibitor of nuclear factor kappa-B kinase subunit beta(IKKβ), nuclear factor of kappa-light-chain-enhancer of B cell inhibitor alpha, and the transcription factor nuclear factor of kappa-light-chain-enhancer of B cells (NF-κB). Activation of these pathways induces a plethora of proinflammatory events, such as the secretion of proinflammatory cytokines including interleukin (IL)-1β, TNF-α, IL-6, and monocyte chemoattractant protein (MCP)-1 (105).
The T cell cytokines interferon-γ, IL-10, IL-17, and IL-21 have been also shown to be secreted by CD4+ and CD8+ T cells incubated with PM. Interestingly, lymphocyte activation seems to occur only in the presence of macrophages (95), suggesting a cross talk between both cell types. Secretion of the proinflammatory and atherosclerosis-promoting cytokine IL-1β (39) may occur as a consequence of macrophage activation by oxidative processes, and the intracellular accumulation of inorganic compounds that favor activation and assembly of the nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 inflammasome (128). Whether impaired redox metabolism and generation of DAMPs represent the cause or the consequence of local inflammation due to PM inhalation is still a matter of debate.
NOX2 has been proposed to act as a functional link between lung inflammation and the redox system (74, 98). The superoxide anion produced by this enzyme during the inflammatory response may represent a key event of PM lung injury, which is supported by the observation that increased extracellular superoxide dismutase (ecSOD) activity protects from pulmonary inflammation in mice exposed to ROFA (48). Moreover, overexpression of ecSOD in the lung prevents systemic PM effects, such as vascular inflammation and insulin resistance (56). The contribution of alternative superoxide anion sources, such as the mitochondrial respiratory chain complexes (98) and other oxidases (19) in PM-induced lung injury, still needs to be clarified. The release of the proinflammatory cytokine IL-8 from human alveolar epithelial cells incubated with PM has also been reported (71, 120), suggesting additional direct effects of PM on stromal cells. Increased secretion of proinflammatory cytokines in the lung has been observed in acute (101, 105) and chronic (74) models of PM exposure.
Collectively, it is now accepted that PM accumulation and its uptake by tissue-resident macrophages initiate a proinflammatory response in the lung. Secreted cytokines perpetuate local inflammatory pathways but are also secreted in the blood circulation to remotely act on peripheral tissues or the bone marrow (74). We and others recently showed that the therapeutic depletion of lung macrophages abolishes some of the detrimental effects of PM (102, 108, 124), suggesting a central role of alveolar macrophages in the oxidative stress and inflammatory cellular response after PM inhalation (Fig. 2).
Systemic Oxidative and Inflammatory Effects of PM Exposure
Several studies have demonstrated an increase of proinflammatory cytokines in the blood, such as of IL-1β, TNF-α, IL-6, and MCP-1 (20, 58, 124, 132, 144), as well as of C-reactive protein (CRP) (32, 85), high-sensitive CRP (63, 67), endothelial cell adhesion markers, for example, soluble intercellular adhesion molecule (ICAM)-1 and soluble vascular cellular adhesion molecule(VCAM) -1 (124), and markers of thrombogenicity (158). In addition, increased plasma markers of oxidatively modified macromolecules, such as of F2-isoprostanes (131) or 8-hydroxy-2′-deoxyguanosine (28), as well as of an impaired antioxidant defense system (33), have been reported in humans exposed to PM. In addition, it has been shown that microvascular ROS production increases after PM-exposure by subsequent NOX2 (161) and myeloperoxidase activation (117).
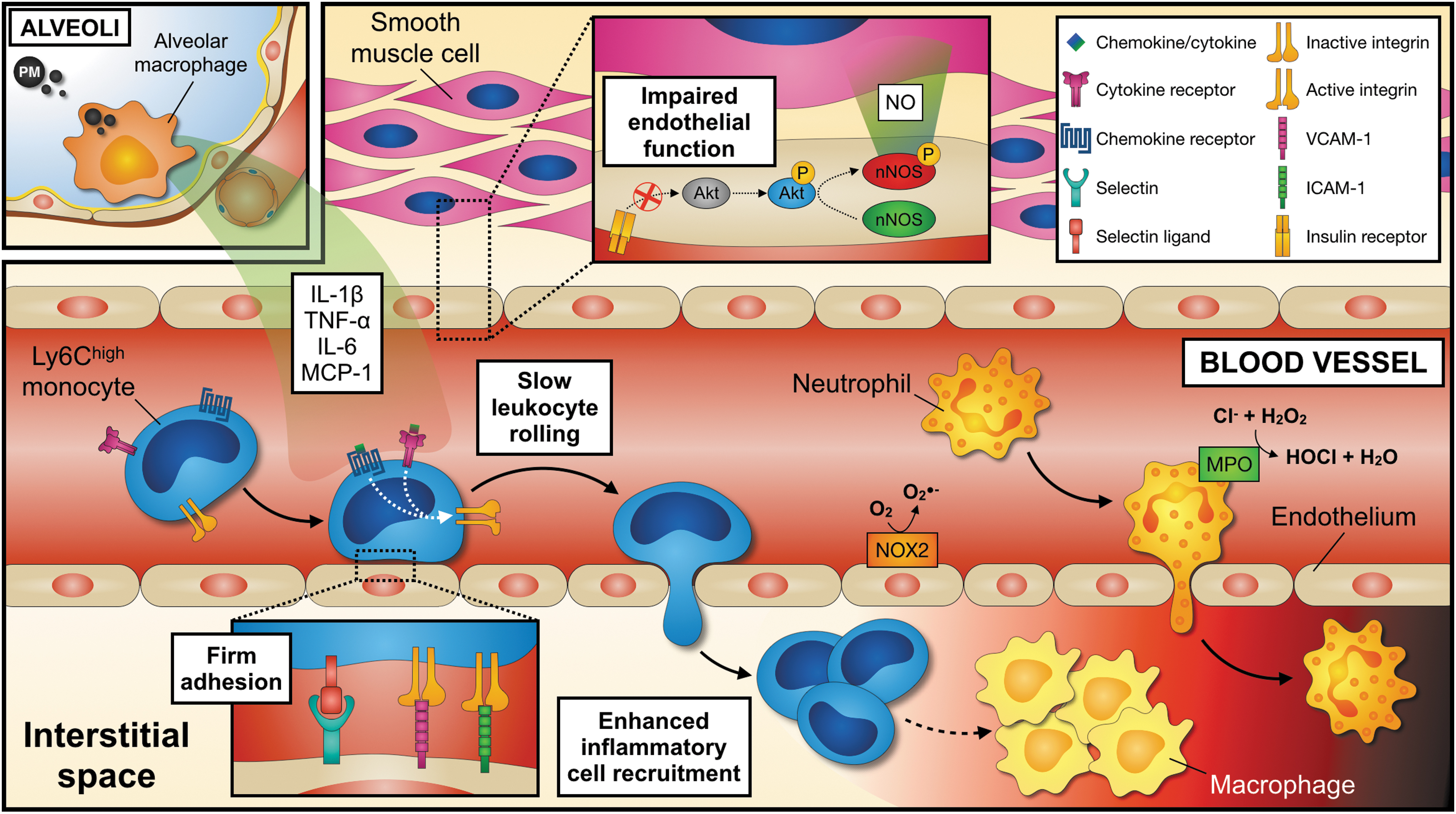
If circulating oxidatively modified molecules are arguing for a systemic oxidative stress response or for a translocation of PM into the blood circulation is not entirely clear. It also remains a matter of discussion whether PM effects are confined to tissue-resident cells in the lung (102, 108, 124) or whether PM translocates into the bloodstream and directly acts on peripheral cells and tissues. In this line, it has been established that proinflammatory leukocytes accumulate in the microcirculation (74), adipose tissue (139), atherosclerotic plaques (103), and infarcted myocardium (102), as a direct result of experimental PM exposure (Fig. 3) and increased expression of endothelial and leukocyte adhesion molecules (102) that promote trafficking of leukocytes to peripheral tissues (117, 139, 159).
Impact of Air Pollution PM on Cardiovascular Disease
Atherosclerosis, a chronic inflammatory disease of middle- to large-sized arteries, represents the main underlying vascular pathology that leads to the buildup of collagen-, lipid-, and cell-rich plaques in the intima of affected vessels (156, 157). Its consequences, myocardial infarction and stroke, are mostly caused by the acute erosion or rupture of atherosclerotic plaques and an immediate arterial thrombosis that leads to vessel occlusion and ischemic tissue injury. In the heart, an acute occlusion of coronary arteries is followed by myocardial ischemia, apoptosis of cardiomyocytes, adverse cardiac remodeling, myocardial fibrosis, and a loss of ventricular function (35).
In addition to individual and genetic risk factors, several environmental factors have been proposed to trigger an unstable plaque phenotype with decreased collagen content and increased lipid and cellular infiltrates that predisposes to rupture. Several clinical observational studies have suggested air pollution as a trigger of myocardial infarction (122, 123, 125). In the largest associative study performed so far, Pope et al. (125) established a positive association between increasing concentrations of airborne PM and the risk of acute coronary syndromes throughout an 11-year follow-up (93). Of more than 12,000 cardiac events, risk of myocardial infarction was increased by 4.5% with every increment of 10 μg/m3 in PM2.5. In addition, several preclinical studies have suggested that the extent of myocardial tissue damage after short- or long-term PM exposure worsens the outcome after myocardial infarction in animal models (102, 149). Yet, the precise mechanisms remain a matter of debate.
Four major pathways at the interface of air pollution, myocardial infarction, and subsequent heart failure were identified so far. First, effects of PM on the progression of underlying atherosclerotic disease and the induction of unstable plaque phenotypes; second, the induction of prothrombotic states that promotes arterial thrombosis; third, an aggravation of the detrimental inflammatory and immune response that accompanies myocardial infarction; and fourth, direct effects on the myocardium, including impaired bioenergetic capacity that triggers the death of cardiomyocytes and favors adverse tissue remodeling, as well as altered cardiac redox metabolism (52).
Effects of PM on Experimental Atherosclerosis
Atherosclerosis is a complex pathology that involves both classical cardiometabolic risk factors, such as smoking, hypertension, diabetes, dyslipidemia, and a chronic, low-grade inflammatory and immune cell response. Mechanistically, atherosclerosis is caused by the accumulation of naive and oxidized lipids, mainly low-density lipoprotein (LDL)-cholesterol in the intima of arteries, and the buildup of atherosclerotic plaques that obstruct the inner lumen and may cause critical tissue ischemia (87). Exposure to ambient PM2.5 and PM10 correlates with the extent of atherosclerotic lesions in human associative studies that have used intima-media-thickness and coronary calcium scores in imaging as clinical surrogate parameters for atherosclerosis (29, 68, 76). How PM exposure modulates atherosclerosis has mainly been tested in the atherosclerosis-prone Ldlr- and Apoe-deficiency mouse models, in hyperlipidemic rats and rabbits (157). These studies have mostly revealed an overall proatherosclerotic effect after exposure to PM2.5 (4, 26, 51, 138, 140, 146, 164), PM10 (26, 141, 146), DEP (103), and ultrafine particles (UFP) (76) with dosages ranging from 29 to 300 μg/m3.
Main histomorphologic findings have suggested increased accumulation of macrophages in atherosclerotic lesions, enhanced lipid depositions, as well as increased atherosclerotic lesion size and signs of a less stable plaque phenotype (76, 138, 140, 141, 164). Enhanced lipid accumulation and formation of lipid-laden foam cells in a model of PM exposure seem to be dependent on the uptake of oxidized lipids by scavenger receptor CD36 in developing foam cells, likely as a result of enhanced oxidative processes (129). PM increases circulating oxidized LDL (oxLDL) levels (17, 26, 142) and promotes the uptake of LDL by the endothelial lectin-like oxLDL receptor (LOX-1) (94). In addition, PM enhances proinflammatory gene expression in the lung, plaques, or systemically (26, 51, 103, 146, 164). Some of these effects may be a direct result of amplified oxidative stress pathways, which is suggested by the increase of markers of systemic lipid peroxidation and of antioxidant genes, such as nuclear factor erythroid-2-related factor 2, hemeoxygenase-1, catalase, and SOD (4, 103), as well as of protein tyrosine nitration in aortic plaques (5, 138). Some of these effects seem to be mediated by PM redox-cycling organic components, mainly PAHs (4, 76). Enhanced systemic lipid peroxidation seems to impair anti-inflammatory and antioxidant mechanisms and may contribute to atherosclerosis development in mice exposed to DEP and UFP, as well as in humans (4). In contrast, two studies have failed to prove a modulation of atherosclerotic disease with PM2.5 in a short-term model of 6 weeks at a PM concentration of 27 μg/m3 (51) and after carbon black exposure (27).
Modulation of Immune Cells by PM
The observation that PM exposure regulates the numbers of circulating neutrophils (141), monocytes, platelets (164), and of anti-inflammatory T-regulatory cells (147) suggests that leukocytes may be one the effectors of PM-induced lung injury. Whether this proinflammatory leukocyte response is mediated by a de novo generation of leukocytes in the bone marrow as recently suggested (74), or by enhanced infiltration of leukocytes as result of increased adhesion receptors on leukocytes and their subsequent emigration to inflamed tissue (102), has not been clarified in detail yet. Moreover, studies from the lung tissue registry at the Centre for Heart Lung Innovation have suggested that a lipid-lowering therapy with statins reduces both PM depositions in tissues and in blood vessels. Because statins have anti-inflammatory properties beyond their lipid-lowering effects (88), it is to speculate whether inflammation itself could directly affect the clearance of PM depositions by macrophages (65). In addition, it has been suggested that PM affects microbial diversity in the gut of atherosclerotic animals, which may alter immune cell functioning and contribute to its proatherogenic effects (42).
Effects of PM on Hemostasis and Thrombosis
The formation of vessel-occluding arterial thrombosis on eroded or ruptured atherosclerotic plaques occurs within seconds. Arterial thrombosis is mostly caused by the aggregation of thrombocytes under high arterial shear but is also promoted by enhanced plasmatic coagulation, which is pronounced in smokers (116). PM seems to affect the hemostatic system by several direct and indirect effects. First, PM promotes endothelial dysfunction, impairs vasomotor function, induces platelet activation and aggregation pathways, and increases the expression of prothrombotic mediators on the endothelium, such as of tissue factors (6, 15, 140). Second, PM reduces endogenous fibrinolysis in humans—an effect that occurs within 1 h of an acute exposure to DEP and persists up to 6 h (6, 15, 104). Third, it was shown that PM induces venous thrombosis and plasmatic hypercoagulation after myocardial infarction by enhanced expression of coagulation factor VII and VIII, and fibrinogen (40, 113, 149), as well as of platelet coagulation factor III (140) and diminished thrombolytic tissue plasminogen activator release (104). Fourth, it has been demonstrated that PM directly enhances arterial thrombus formation, likely by increasing the reactivity of platelets (41, 112, 143).
In conclusion, PM promotes thrombosis by the modulation of prothrombotic, antifibrinolytic, and cellular pathways. These effects seem to be partially induced by oxidative stress (116), driven by transition metals in PM that are able to participate in Fenton and Fenton-like chemical reactions (133). Because intranasal and intravenous DEP delivery modulates coagulation factors directly, the PM thrombogenic effect has been suggested to occur partially independently from the local inflammatory response in the lung (143). Other reports, however, have demonstrated that the elimination of proinflammatory mediators, lung macrophages, or cytokines in models of cell depletion and knockout mice abolished PM prothrombotic effects in mice (108). Collectively, these findings make it likely that inflammation and oxidative stress pathways synergize to induce thrombosis and may partially overlap depending on the animal model and the source and composition of the PM used. More detailed studies will be required in the future to dissect the exclusive and mutual roles of inflammation and thrombogenicity.
Effects of PM on Myocardial Infarction and Cardiac Remodeling
We and others have recently shown that PM impairs myocardial remodeling and increases infarct size in rodents after surgical induction of myocardial infarction (21, 31, 102). These effects may be explained by locally secreted inflammatory cytokines in the lung or direct effects of potentially circulating PM on cardiomyocytes. Because a depletion of lung macrophages abolished the inflammatory response after PM and prevented excessive myocardial tissue injury, a relevant proportion of PM-related effects seem to be mediated by alveolar macrophages (102). These and other findings suggest a remote effect of PM-related inflammation on postmyocardial infarction tissue healing with elevated proinflammatory cytokines, such as TNF-α, IL-6, and MCP-1, in the lung and the circulation. As a result, endothelial cells are activated and increase surface expression of the adhesion- and rolling-receptors, VCAM-1, ICAM-1, and selectins, respectively. Consequently, neutrophils and inflammatory Ly6Chigh monocytes are recruited to the injured myocardium, increase local inflammation, and drive adverse cardiac remodeling. This stepwise inflammatory cascade (Fig. 4) is in line with the finding that increased leukocyte recruitment into the myocardium is associated with a worse outcome following myocardial infarction (109). In addition to circulating cytokines, PM directly aggravates endothelial dysfunction by PI3K/Akt/endothelial nitric oxide synthase (eNOS)-dependent pathways, providing an additional link between insulin signaling and vascular inflammation (16, 56).
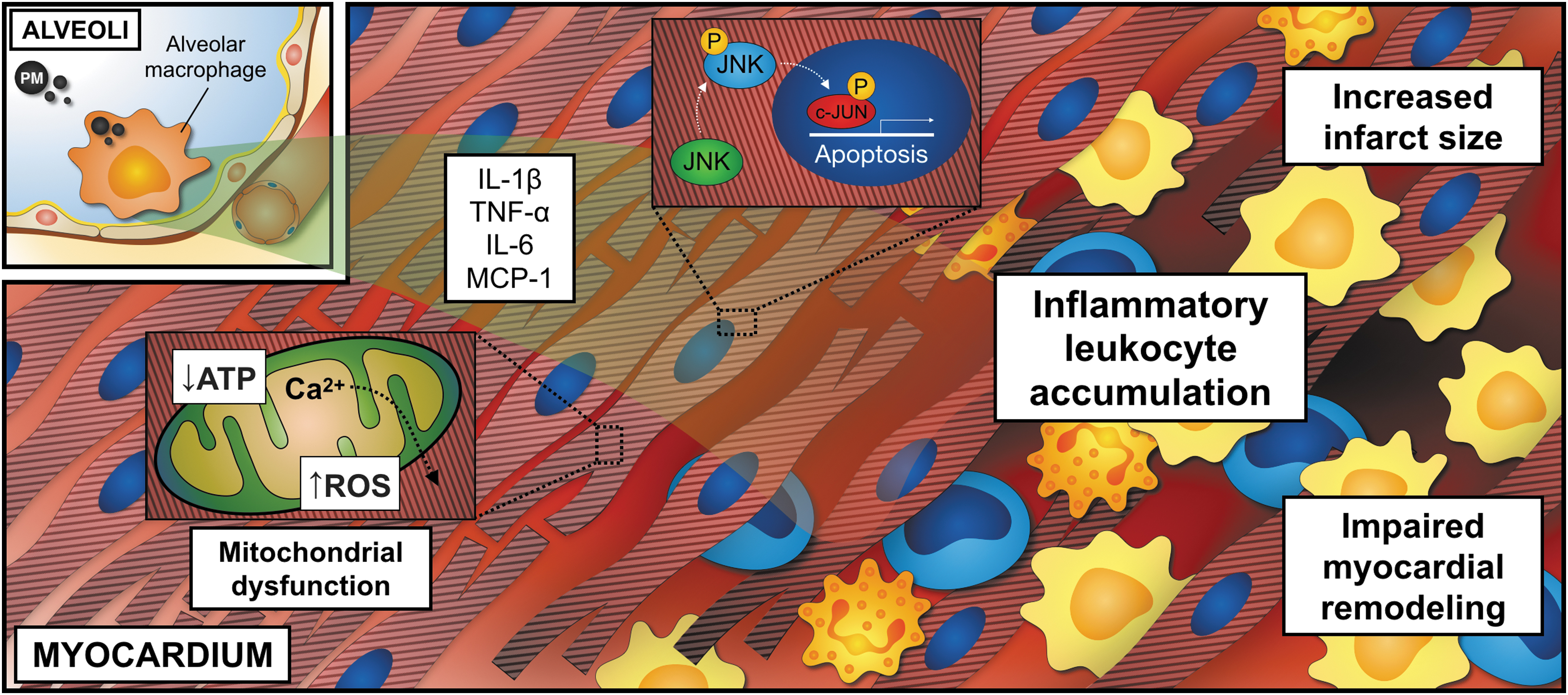
Locally deposited PM may also exhibit direct effects on cardiomyocytes by promoting inflammatory and apoptotic signaling through c-Jun N-terminal kinase, p53, and caspases in rats (149), or in the setting of heart failure and left ventricular hypertrophy induced by transverse aortic constriction (163). Notably, ventricular effects of PM seem to be modulated by adenosine monophosphate-activated protein kinase (AMPK). In AMPK−/− mice, exposure to ROFA induces ventricular dysfunction even in the absence of any surgical intervention (148). Likewise, impaired mitochondrial respiration and cardiac oxygen consumption, as well as decreased inner mitochondrial membrane potential and ATP synthesis, were observed after PM inhalation (100).
In mice exposed to ROFA, increased circulating TNF-α concentration in the blood and impaired myocardial calcium homeostasis seem to weaken myocardial contractile function and relaxation (99, 101). Decreased expression of cardiac sarco/endoplasmic reticulum calcium-ATPase 2a, a major regulator of cytosolic calcium concentration that uses ATP for calcium transport into the sarcoplasmic reticulum during relaxation, may contribute to the observed weakened cardiomyocyte shortening following CAP incubation (154). In addition, a pathogenic role has been proposed for the proinflammatory chemokine MCP-1 that is produced by PM-stimulated lung epithelial cells and contributes to contractile myocardial dysfunction in vitro (50).
Interestingly, PM-induced oxidative stress pathways seem to have the potential to directly alter contractile function and calcium homeostasis in cardiomyocytes: In one study, exposure to DEP reduced sarcomere shortening, which was prevented by the mitochondrial targeted antioxidants Tiron and MitoTEMPOL. Moreover, DEP incubation reduced cardiomyocyte calcium handling ability after β-adrenergic stimulation with isoproterenol (50). PM exposure also promotes myocardial apoptosis in the setting of otherwise only mild hypoxia (86) and may also trigger ventricular arrhythmias (14, 134). Both effects seem to depend on PM-induced ROS release and calcium/calmodulin-dependent protein kinase II activation (78). In this scenario, inappropriate mitochondrial calcium handling could be the either the cause or consequence of myocardial bioenergetic dysfunction and ROS release (11). Taken together, these observations provide a plausible explanation for increased cardiac mortality and aggravated heart failure after myocardial infarction in individuals exposed to PM.
Effects of PM on Angiogenesis
Numbers of circulating endothelial progenitor cells (EPCs) are indicative of vascular health and are frequently decreased in patients at high cardiovascular risk (64) and with the metabolic syndrome (72). Following PM exposure, blood EPCs decrease in humans (118), without impaired EPC viability or defective recruitment to sites of injury, stem cell niches, or the lymphatics (54). The decrease of EPCs after PM exposure has been explained by an impaired mobilization from the bone marrow due to blunted vascular endothelial growth factor (VEGF) signaling (54). Moreover, bone marrow-derived EPCs from mice exposed to CAP fail to improve tissue recovery when injected into unexposed mice subjected to hind limb ischemia/reperfusion injury (53). Thus, impaired EPC function may contribute to cardiac injury after myocardial infarction. Mechanistically, impaired angiogenic and vascular EPC repair capacity after PM exposure is associated with the downregulation of several genes involved in vascular development, such as Mmp9 (matrix metalloproteinase-9, MMP-9), Nos3 (eNOS), and Vegfb (VEGF-B) (53).
Interestingly, PM-induced impairment of EPC homeostasis can be prevented by lung-selective ecSOD overexpression, suggesting that oxidative stress may be one of the causes leading to a derangement of EPC function (53). Since the administration of insulin sensitizers to CAP-exposed mice prevents vascular inflammation, oxidative stress, and defective EPC mobilization (55), metabolism may represent a powerful link between distinct PM-induced pathologies. Down this line, it was shown that decreased vascular insulin sensitivity after PM exposure precedes systemic insulin resistance, an effect driven by the activation of the NF-κB pathway, but not the MAPK/ERK pathway, and vascular PI3K/Akt/eNOS insulin signaling (56).
Regulation of Adipose Tissue Inflammation and Metabolism by PM Exposure
The metabolic syndrome, a cluster of cardiovascular factors comprising dyslipidemia, hypertension, insulin resistance, obesity, and hepatic steatosis, is associated with a variety of cardiovascular pathologies, including atherosclerosis, myocardial infarction, and stroke (106). Because the metabolic syndrome is accompanied by a chronic, low-grade inflammatory response in adipose tissue, the liver, and other key metabolic organs (153), it was proposed that PM may affect cardiovascular disease already at the stage of risk factors. Indeed, PM exposure shows a strong association with type-2 diabetes mellitus with a relative risk increase up to 63% with every 10 μg/m3 increment in air PM2.5 (127). Furthermore, several experimental studies have established an overall impactful role of PM in the development of obesity, insulin resistance, or other components of the metabolic syndrome, such as dyslipidemia.
Exposure to PM in rodent models of diet-induced obesity or genetically induced obesity in diabetic KK-Aγ mice increases visceral obesity and aggravates insulin resistance (90, 91, 139). Impaired Akt and eNOS phosphorylations are common findings in aortic sections from CAP-exposed mice and might partially explain this observation (56, 139). In contrast, some studies demonstrated that PM may only provoke an obese phenotype in rodents under a standard low-fat diet (150, 159). This suggests that the diet-induced obese phenotype may overwhelm the obesity-promoting effect of PM.
PM increases the expression of various inflammatory mediators in obese adipose tissue and the circulation, including TNF-α, IL-6, E-Selectin, ICAM-1, PAI-1 (139, 150, 159), C–X–C motif chemokine 5 (CXCL5), and MCP-1 (150) (Fig. 5). Likewise, PM promotes the accumulation of adipose tissue macrophages and their polarization toward a proinflammatory phenotype in adipose tissue (90, 139). This effect appears to coincide with an egress of inflammatory Ly6Chigh monocytes from the bone marrow and increased numbers in the periphery, their recruitment into adipose tissue, and their subsequent differentiation into CCR2+ inflammatory macrophages (22, 90, 91). This process has been described to be partially dependent on TLR4 and NOX2 mechanisms triggered by oxidized PAPC and the IL-1 receptor-associated kinase signaling pathway (74, 159). Although it is currently unclear whether a modulation of leukocyte recruitment into adipose tissue is a requirement of adipose tissue inflammation (155), these findings demonstrate that PM has the potential to initiate and to maintain tissue inflammation in the context of obesity and oxidative processes.
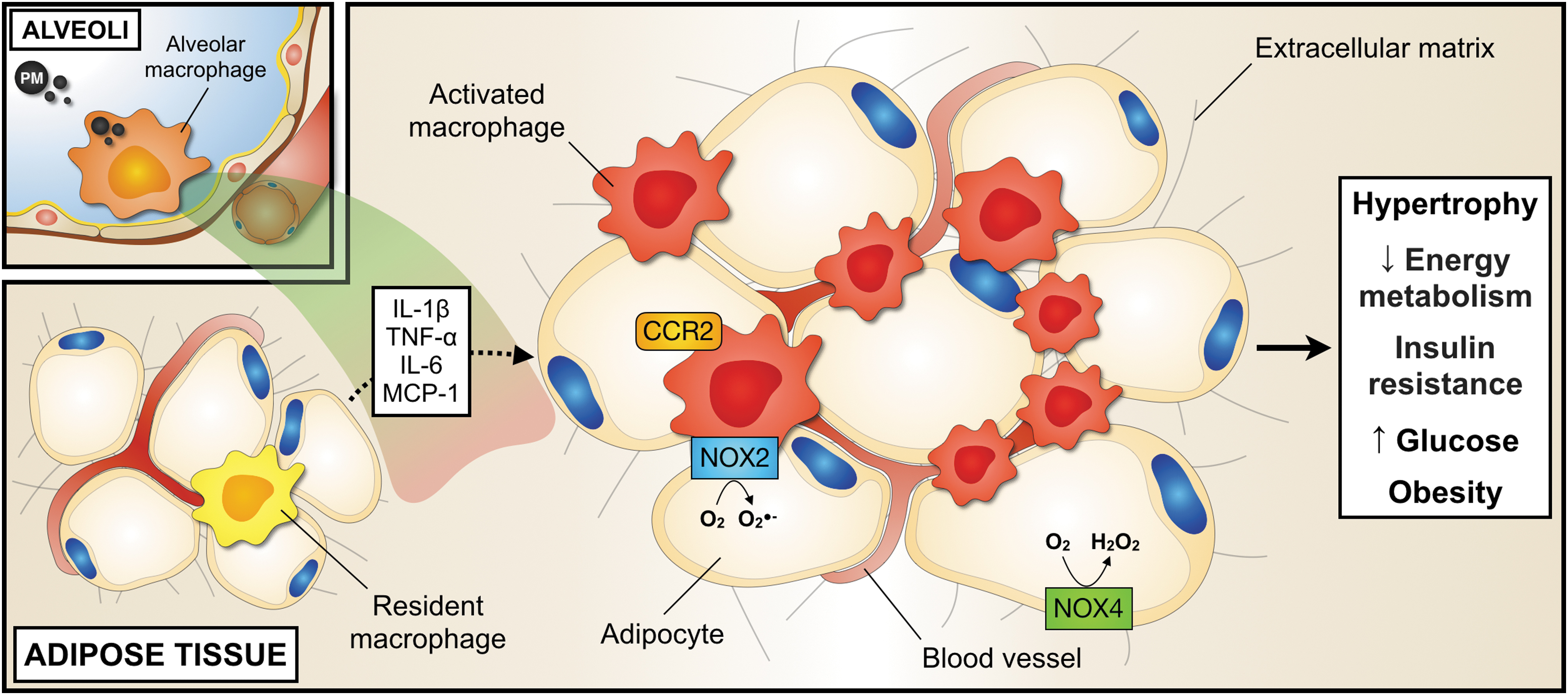
The observation that gene expression involved in de novo lipid synthesis, such as acetyl-CoA carboxylase-2, fatty acid synthase, and diacylglycerol acyltransferase-2, increases after PM inhalation in rodents (91) has further sparked the idea that an impairment of thermogenesis and impaired energy metabolism (90) may contribute to the obesity-promoting effect of PM. It was also suggested that PM may broadly affect neuroendocrine regulation by glucagon-like peptide-1 (150), NF-κB- and IKKβ-dependent inflammatory pathways in the central nervous system (90, 162), in addition to enhanced inflammation in the gut and liver (84, 92). These effects appear to be partially mediated by epigenetic changes (25, 160), for instance, in the gene coding for the satiety-signal leptin. Leptin signaling and leptin receptor methylation have accordingly been suggested to be responsible for the transfer of an obese phenotype to offspring after a maternal PM exposure (25, 160). Adipose tissue hypertrophy and inflammation following PM exposure seem to be gender-specific (25). Moreover, it is still not clear whether epigenetic imprinting may be a specific consequence of PM or of the adipose phenotype (7). It also remains unknown whether the inflammatory response in metabolic key organs is a cause or consequence of PM-induced impaired metabolism.
Oxidative Metabolism in PM-Induced Adipose Tissue Inflammation
ROS production by NOX enzymes has been suggested to contribute to the development of the metabolic syndrome and adipose tissue inflammation (43, 60). NOX4 is the major isoform in adipocytes that is constitutively active and upregulated in obesity (61). Since NOX4-derived ROS production promotes the differentiation of preadipocytes and proinflammatory MCP-1 expression, NOX4 seems to play a predominant role in the early stages of adipose tissue inflammation (34). In addition, NOX2 is expressed in adipose tissue macrophages, which provides an interesting link between pro-oxidative and proinflammatory mechanisms on an immune cell level (74, 121, 159). Selective inhibition of NOX2 by the gp91 ds-tat peptide ameliorated lung redox metabolism in PM-exposed mice (98). Accordingly, antioxidant treatment with 4-Hydroxy-TEMPO (TEMPOL) or lung-specific ecSOD overexpression prevented the development of vascular inflammation and insulin resistance in CAP-exposed mice (56). These findings support a mechanistic link between PM, impaired lung redox metabolism, and inflammation.
Environmental PM Levels—the Higher, the Worse?
Levels of ambient PM2.5 are monitored by a combination of multisatellite observations and ground-based PM2.5 measurements worldwide (145). These data along with independent measurements from regulatory agencies and monitoring stations provide reliable measurements, while personal and portable air monitors offer personal exposure data and may be helpful to support awareness. Most epidemiological studies conclude that the relative risk of all-cause and cardiovascular mortality already increases at widely observed environmental levels of air pollution (14, 18, 37). The exposure/response relationship suggests that at relatively low levels of PM2.5 up to 30 μg/m3, which are frequently observed in urban environments, the relative mortality risk increases in an almost linear manner, while concentrations higher than 80 μg/m3 do not further enhance this risk in a relevant manner. This observation might also explain reports showing that active smokers are not at an excessive risk for cardiovascular mortality induced by PM2.5 exposure (126). In addition, no safety threshold for PM2.5 levels has been established so far, meaning that any reduction in ambient PM2.5 levels would lead to a decrease in morbidity and mortality rates, even below current PM2.5 regulations.
Models of PM Exposure and Experimental Limitations
In addition to epidemiological studies, different human models of exposure have been developed to unravel the mechanisms of PM-related complications. Controlled exposure protocols comprise volunteers inside chambers breathing ambient air or aerosolized particles, such as CAPs obtained from versatile aerosol concentration enrichment systems and virtual impactors (44, 69, 80). CAP inhalation studies are a frequently used approach to mimic environmental PM exposure at a certain controlled air PM concentration and without the need for the generation of artificial PM surrogates. However, only a fraction of all potential PM particles is typically concentrated in CAPs, and PM chemical composition may change during sample collection and storage. Moreover, synergistic effects between PM and gaseous pollutants are not considered (151). Another problem in mimicking effects of airborne PM is the significant daily and seasonal fluctuation. Studies to investigate occupational PM exposure (45) as well as interventional epidemiological studies (30, 130) have contributed to the understanding of PM toxicity in humans and the importance of PM composition and dosage.
In animal models, intranasal and intratracheal instillation of PM suspensions to anesthetized rodents is the most frequent approach (102, 113). Low technical complexity, dose handling, and reproducibility are the main strength of these models. However, some PM characteristics might be significantly altered during instillation, and excessive upper respiratory tract irritation and nonrealistic exposure dosing and kinetics are a major concern. Although intratracheal instillation leads to a measurable PM deposition in the lung, it bypasses the upper respiratory airways and PM clearance mechanisms. Moreover, human exposure is usually correlated to cumulative dosages, while in animal models almost exclusively acute PM effects are studied.
The most appropriate model of chronic exposure is whole-body inhalation (56, 91, 139). In this setup, street air is directed into animal housing chambers. A cyclone removes particles larger than 2.5 or 10 μm and a negative pressure inside the chamber is established by an air pump that sustains a constant air flow rate (usually at 5 L/min). As a consequence, PM levels can be enriched 5- to 10-fold, which allows to reach PM2.5 levels inside the chambers ranging from 40 to 120 μg/m3. Control animals are handled in parallel and receive an identical exposure protocol with high-efficiency filters that remove pollutants from the air stream. The suitability for longer term exposure protocols at real-life biologically relevant PM2.5 levels, and the combination with knockout/transgenic mice and with animal models of disease, significantly contributes to the understanding of in vivo effects.
Concluding Remarks
It is now clear that atherosclerosis is a central pathology for a variety of cardiovascular diseases that involves predisposing risk factors, metabolic derangements, and a chronic inflammatory and immune cell response. PM has been shown to modulate this response at different levels: via a modulation of basic energy metabolism, endothelial integrity, systemic inflammation, thrombus reactivity, and myocardial tissue remodeling. It remains a controversy whether PM-induced pathogenic effects are primarily located in the lung, where an activation or damage of tissue-resident cells, including alveolar macrophages, initiates a local inflammatory response that may transform into systemic inflammation, or whether PM may translocate into the bloodstream and local tissues and directly act on tissue-resident cells.
The inflammatory response has been identified as a strong driver of PM-induced tissue injury. From an integrative perspective, oxidative damage to the tissue may represents the earliest point of impact, while inflammation can be regarded as the resulting complication that mediates remote tissue injury and thereby represents the modulator of cardiovascular risk. Yet, it remains to be tested whether anti-inflammatory treatment strategies may be suitable to abolish the excess risk of cardiovascular disease in exposed individuals. Current clinical studies are strongly limited by their observational nature. Experimental studies in rodents, on the contrary, have partially failed to proof reproducibility, which may be reflected by the tremendous heterogeneity of PM model particles that differ greatly in size, composition, biochemical properties, origins, tested dosages, and routes of exposure [a helpful overview is found in Hadei and Naddafi (57)]. Future studies will have to systematically and carefully consider these parameters.
Footnotes
Funding Information
This work was supported by research grants from the Agencia Nacional de Promoción Científica y Tecnológica (PICT 2016-3062) and by a fellowship from the German Academic Exchange Service (Deutscher Akademischer Austauschdienst) to T.M. D.W. was supported by the Berta-Ottenstein-Program for Advanced Clinician Scientists at the Faculty of Medicine, University of Freiburg.