
Abstract
Significance:
Generalized selenoprotein deficiency has been associated with mutations in SECISBP2, SEPSECS, and TRU-TCA1-1, 3 factors that are crucial for incorporation of the amino acid selenocysteine (Sec) into at least 25 human selenoproteins. SECISBP2 and TRU-TCA1-1 defects are characterized by a multisystem phenotype due to deficiencies of antioxidant and tissue-specific selenoproteins, together with abnormal thyroid hormone levels reflecting impaired hormone metabolism by deiodinase selenoenzymes. SEPSECS mutations are associated with a predominantly neurological phenotype with progressive cerebello-cerebral atrophy.
Recent Advances:
The recent identification of individuals with defects in genes encoding components of the selenocysteine insertion pathway has delineated complex and multisystem disorders, reflecting a lack of selenoproteins in specific tissues, oxidative damage due to lack of oxidoreductase-active selenoproteins and other pathways whose nature is unclear.
Critical Issues:
Abnormal thyroid hormone metabolism in patients can be corrected by triiodothyronine (T3) treatment. No specific therapies for other phenotypes (muscular dystrophy, male infertility, hearing loss, neurodegeneration) exist as yet, but their severity often requires supportive medical intervention.
Future Directions:
These disorders provide unique insights into the role of selenoproteins in humans. The long-term consequences of reduced cellular antioxidant capacity remain unknown, and future surveillance of patients may reveal time-dependent phenotypes (e.g., neoplasia, aging) or consequences of deficiency of selenoproteins whose function remains to be elucidated. The role of antioxidant therapies requires evaluation. Antioxid. Redox Signal. 33, 481–497.
Introduction
Selenium (Se) is an essential micronutrient that is incorporated as the amino acid selenocysteine (Sec) into human selenoproteins, encoded by 25 separate genes. Most selenoproteins function as oxidoreductases, with the Sec residue involved in catalytic activity. Selenoproteins have diverse functions ranging from maintenance of redox potential; regulation of redox-sensitive biochemical pathways; protection of genetic material, proteins, and membranes from oxidative damage; metabolism of thyroid hormones; regulation of gene expression; and control of protein folding (Table 1). However, the function of several selenoproteins is unknown (57).
Selenoproteins
SELENOP, selenoprotein P; SEPHS2, selenophosphate synthetase 2.
Biosynthesis of selenoprotein requires a UGA codon within its messenger RNA (mRNA) to be recoded as the amino acid Sec, preventing its recognition as a premature stop, possibly targeting the transcript for nonsense-mediated decay (57, 79). This process is achieved via a unique Sec-insertion machinery, comprising cis-acting SEleniumCysteine Insertion Sequence (SECIS) elements located in the 3′-UTR (untranslated region) of selenoprotein mRNAs and the UGA codon, interacting with trans-acting factors (SECIS binding protein 2 [SECISBP2], Sec tRNA [transfer RNA], specific eukaryotic elongation factor [EEFSEC], and Sec-tRNA[Ser]Sec) (Fig. 1) (4, 24, 33, 58, 85).

In contrast to other amino acids, selenocysteine does not have an aminoacyl-tRNA synthetase, but is synthesized on its own tRNA, encoded by TRU-TCA1-1 (Fig. 1) (5). This tRNA was originally named the human opal suppressor gene and a truncated pseudogene that is not expressed also exists (68). Delivery to the ribosome and subsequent cotranslational insertion of Sec is mediated by a multiprotein complex (81), which includes EEFSEC and SECISBP2 as well as other factors. The specialized elongation factor EEFSEC (rather than general elongation factors eEf1a and EF-Tu, which delivers all other aa-tRNAs) delivers Sec-tRNA[Ser]Sec to the ribosome acceptor site (85). SECISBP2 interacts with the SECIS element, a stem–loop structure present in the 3′-UTR of every selenoprotein mRNA (12, 13, 23). The SECIS elements within each selenoprotein mRNA are distinct, but share common structural features, consisting of two helices separated by an internal loop, a GA quartet of non-Watson–Crick base pairs, and an apical loop, resulting in the adoption of a “kink-turn” structure (21, 33, 41, 89, 90). All selenoprotein mRNAs contain a single SECIS element, except for selenoprotein P (SELENOP), which has two, tandemly repeated SECIS elements. This configuration coincides with human SELENOP being the only selenoprotein that contains more than one selenocysteine residue (57). The SECIS element is required for Sec incorporation, whereas other motifs in some selenoprotein mRNAs (e.g., Sec redefinition element, proximal stem loop element) have been described as contributing to the translation process (15, 22, 47, 48, 62).
The EEFSEC
The biological importance of selenoproteins is highlighted by the fact that both Trsp (mouse tRNA[Ser]Sec) and Secisbp2 null mice are embryonically lethal (19, 78). Mutations in individual human selenoproteins and their consequences have recently been reviewed in detail elsewhere (35): selenoprotein N (myopathy), glutathione peroxidase 4 (respiratory failure and skeletal defects), thioredoxin reductase 2 (associated with familial glucocorticoid deficiency and dilated cardiomyopathy), and thioredoxin reductase 1 (generalized epilepsy); two reports describe patients with a complex, hereditary spastic paraplegia and mutations in SELENOI, which catalyzes the transfer of phosphoethanolamine from CDP-ethanolamine to diacylglycerol to produce phosphatidylethanolamine (2, 46). Here, we describe mutations in genes (SEPSECS, SECISBP2, and TRU-TCA1-1) encoding three components of the selenocysteine insertion pathway, affecting general incorporation of selenocysteine into selenoproteins and their clinical consequences.
SECISBP2
SECISBP2 is an obligated limiting factor for selenoprotein synthesis, as first shown by the absence of selenoprotein synthesis in SECISBP2-depleted cell lysates, with restoration of production by repletion with SECISBP2 (23, 24). Human SECISBP2 is a large (854 amino acids, 120-kDa) protein, with the first 400, amino (N-) terminal, residues being dispensable for its function in vitro (25, 26) (Fig. 2). In contrast, the carboxy-terminal (C-terminal) region (amino acids 399–784) is both necessary and sufficient for SECIS-binding and Sec incorporation in vitro and contains several functional domains. The Sec incorporation domain (SID), located centrally in SECISBP2, is not essential for SECIS binding but required for Sec incorporation. The RNA-binding domain (RBD) contains an L7Ae-type RNA interaction motif identified in a large family of ribosomal proteins (e.g., RPL30, SUP1, eRF-1, and 15.5-kD/Snu13p) (4, 5, 16, 25, 26), which interacts with the “kink-turn” structure adopted by the SECIS-element. The RBD mediates interaction with the SECIS element (34, 89) and 28S ribosomal RNA (25, 49, 56, 59). A domain amino-terminal (N-terminal) to the L7Ae module, referred to as either the bipartite, SID, or K-rich region, is involved in specific recognition of SECIS elements and other regulatory RNA motifs, thereby also controlling selenoprotein expression levels (17, 30, 83). SECISBP2 also contains several other functional motifs, as shown in Figure 2 (69).
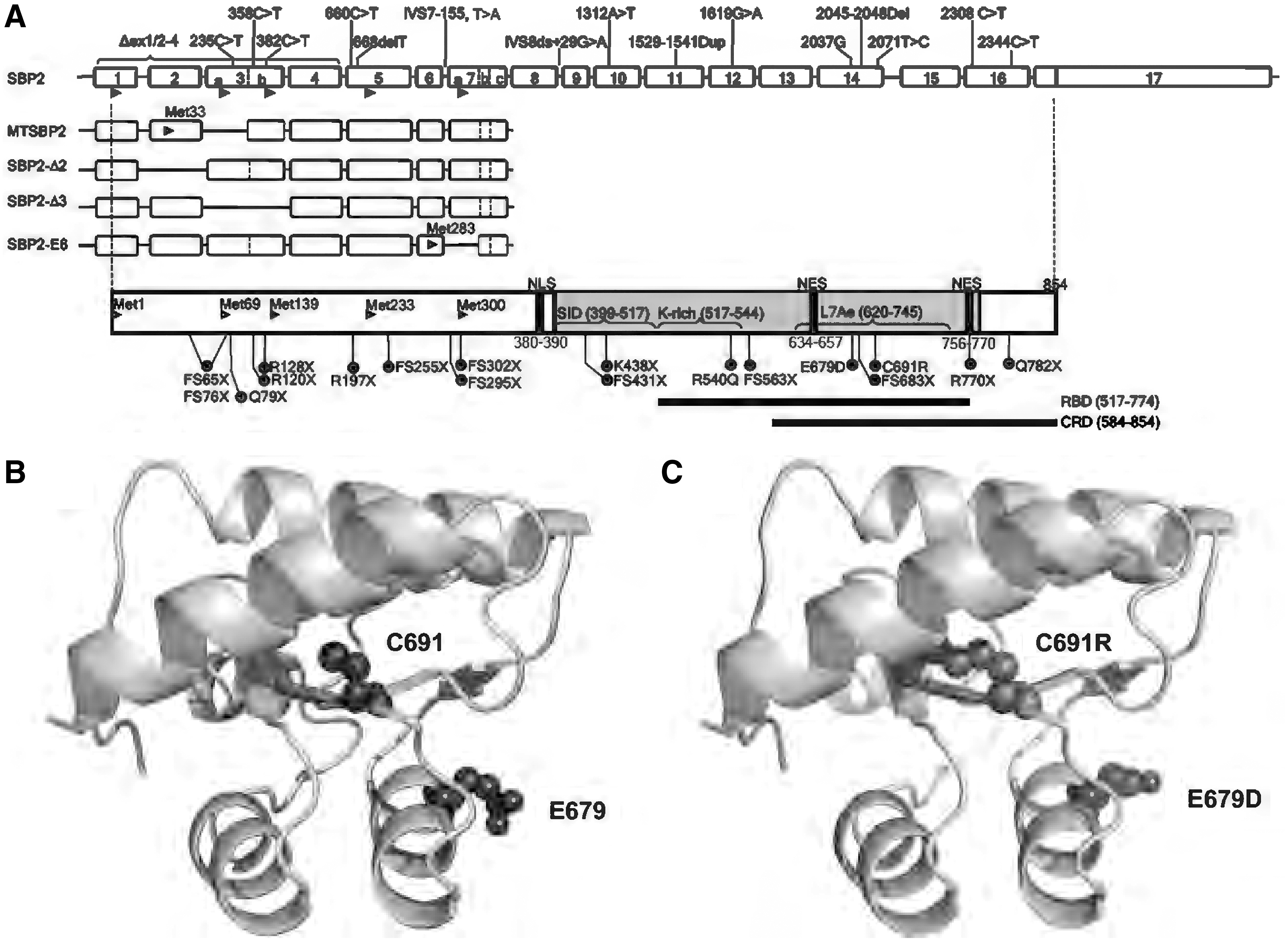
Alternative splicing events in the 5′-region of human SECISBP2, with use of alternative initiation of translation from downstream ATG start codons in exons 2, 3a, 3b, 5, and 7, generates five different protein isoforms, each containing a varying N-terminal protein sequence (Fig. 2) (24, 70). These alternate splicing events alter content of the dispensable N-terminal region, but not the essential C-terminal domain, within protein isoforms. Nevertheless, it is possible that the alternately spliced isoforms do play a role in regulation of SECISBP2-dependent Sec incorporation and selenoprotein expression in vivo. During protein synthesis, dynamic interaction of SECISBP2 with the ribosome and SECIS element is essential for recruitment of the EEFsec/Sec-tRNA[Ser]sec complex to the UGA codon, enabling the incorporation of Sec into the polypeptide.
Homozygous or compound heterozygous mutations in SECISBP2 have been described in 13 individuals from 11 families (Table 2). Disruption of SECISBP2 function prevents appropriate Sec incorporation into selenoproteins during their biosynthesis, resulting in a multisystem disorder due to deficiency of diverse selenoproteins (Table 3). The biochemical signature that identifies SECISBP2-deficient patients consists of low circulating selenium (reflecting low plasma SELENOP and GPX3) and abnormal thyroid hormone levels due to diminished activity of deiodinases (Table 3) (31, 75). Most cases present in childhood due to growth retardation with raised circulating free thyroxine (FT4), normal to low free triiodothyronine (FT3), and raised reverse triiodothyronine (T3) levels, reflecting deficiency of all three deiodinase enzymes.
Human SECISBP 2 Mutations: Genetics and Effect
N/A, not available; SECIS, SEleniumCysteine Insertion Sequence; SNP, single nucleotide polymorphism.
Human SECISBP 2 Mutations: A Multisystem Disorder with a Thyroid Signature
DBA, delayed bone age; FT4, free thyroxine; H, high; IQ, intelligence quotient; L, low; N, normal; N/H, high-normal; N/L, low normal; OGTT, oral glucose tolerance test; rT3, reverse triiodothyronine; Se, selenium; T3, triiodothyronine; T4, thyroxine; TSH, thyroid stimulating hormone; TT3, total triiodothyronine; TT4, total thyroxine; TSH, thyroid stimulating; hormone.
Muscle weakness, due to progressive rigid spine muscular dystrophy, affecting axial and proximal limb muscles with raised creatine kinase (CK) levels and fatty infiltration on imaging, is similar to that seen in patients with mutations in selenoprotein N. In one patient (proband E), male azoospermic infertility was described, reflecting loss of testis selenoproteins (mitochondrial GPX4, thioredoxin reductase, and selenoprotein V) required for spermatogenesis (75).
Significantly decreased expression levels of antioxidant selenoenzymes are associated with increased levels of cellular reactive oxygen species (ROS). Clinical consequences of raised cellular ROS include skin photosensitivity, progressive sensorineural hearing loss, and possibly increased total body adipose tissue mass paradoxically associated with enhanced systemic insulin sensitivity (75).
Reduced red blood cell and total lymphocyte counts, with impaired mononuclear cell cytokine secretion and T cell proliferation (similar to findings in T cell-specific Trsp null mice) (80), were recorded in one case, proband E (75). Although other hematological and immune cell phenotypes have not been formally evaluated, immunodeficiency is neither a reported feature in other SECISBP2 mutation cases nor seen in mouse models of selenoprotein deficiency. Additional age-dependent phenotypes such as neurodegeneration, premature aging, or neoplasia may emerge but have not been described hitherto.
Oral selenium supplementation in some SECISBP2 patients raised total serum Se levels, but without clinical effect (9, 20, 28) or altering synthesis (circulating GPX's, SELENOP) or action (thyroid hormone metabolism) of selenoproteins (77). Treatment of probands C and F with T3 alone (thyroxine [T4] was not effective in one case) or in combination with growth hormone (proband G) resulted in an improvement in growth, development, and bone maturation. Treatment of proband G with a combination of alpha tocopherol (vitamin E) and T3 resulted in the most promising response, with decreased serum levels of lipid peroxidation products, altered FT4 and FT3 concentrations, and increased circulating white blood cells and neutrophils, all of which were reversed after treatment withdrawal (74). These observations suggest that treatment with antioxidants, to counteract the effects of elevated cellular ROS, is the best available therapeutic option for this disorder.
Most SECISBP2 mutations identified to date cause premature stop codons, resulting in the absence of full-length SECISBP2 protein. However, elegant minigene experiments have shown that, for premature stops located in the N-terminal part of the protein, initiation of translation from alternative, downstream ATG codons in exons 5 (Met233) and 7 (Met300) permits low-level synthesis of shorter SECISBP2 isoforms (28, 75). Some premature stop mutations are situated downstream of Met300 and may completely eliminate synthesis of functional protein. However, stop mutations (e.g., R770X, Q782X), distal to the RBD, might generate C-terminally truncated proteins whose RNA binding and nuclear localization functions remain, partially, intact (Fig. 2). In a patient with defective mRNA splicing due to an intronic mutation IVS8ds +29G>A (31), it has been shown that levels of normally spliced transcript are only reduced by 50% and if a similar mechanism operates with other splice site mutations, this suggests some preservation of normally spliced SECISBP2 mRNA in such cases.
Three missense SECISBP2 mutations (R540Q, C691R, and E679D) have been described: The R540Q mutation localizes to the K-rich region within the RBD (Fig. 2), with the R540Q mutant exhibiting reduced binding to SECIS elements in GPX1 and DIO2 mRNAs, correlating with diminished GPX1 and DIO2 enzyme activity in patient-derived primary cells and the mouse model. Detailed analyses suggest that R540Q mutant SECISBP2 fails to bind only a subset of SECIS-elements, consistent with the K-rich region mediating the recognition of specific (type I and type II) SECIS elements (14); a mouse model revealed a possible tissue-specific pattern of SECISBP2 protein stability, correlating with varying loss or preservation of expression of different selenoproteins (14, 31, 96).
The C691R SECISBP2 mutation, also located in its RBD, is expected to affect RNA binding. Homology modeling, based on the crystal structure of the spliceosomal 15.5 kDa protein (87), suggests that the mutation of cysteine to a bulky and charged arginine residue (75) may destabilize its hydrophobic core, disrupting local protein structure (Fig. 2B, C). In vitro assays, showing enhanced proteasomal degradation of the C691R mutant SECISBP2 protein, confirmed this (75). A mouse model suggests that the C691R mutant SECISBP2 is unable to bind RNA and is non-functional (96). The E679D SECISBP2 mutation is predicted to be deleterious (PolyPhen-2 algorithm score of 0.998) and also located in the RBD and may, therefore, affect its RNA binding function, but this has not been investigated in detail (37) (Fig. 2).
Knowledge that knockout (KO) of Secisbp2 in mice is embryonic lethal (78), with no evidence for an alternative Sec-incorporation mechanism in humans, suggests that there is some residual SECISBP2 activity in all patients. All patients described to date are expected to harbor at least one allele that directs the synthesis of SECISBP2 at either reduced levels or that is only partially functional, in combination with either a mutant or a shorter form of the protein synthesized from Met300 (Table 2). A limited number of patients, mostly compound heterozygous for different SECISBP2 mutation combinations, with limited knowledge of phenotypes in heterozygous relatives, have been described, making it difficult to assess the effect of a specific SECISBP2 mutation or its correlation with the severity of phenotype. However, since SECISBP2 is rate limiting for Sec incorporation, a significant reduction in functional SECISBP2 protein levels will result in diminished but not complete loss of selenoprotein synthesis. A further variable is that differences in the architecture of SECIS elements within different selenoprotein mRNAs may dictate the extent to which reduced SECISBP2 protein limits their biosynthesis and expression levels in vivo, as suggested by elegant experiments testing SECISBP2 with luciferase reporter genes containing different SECIS elements (60, 84). Future studies, undertaking ribosomal RNA (96) or selenoprotein expression profiling in secisbp2 mutant mouse models and in vitro reconstitution experiments with different mutant SECISBP2 proteins and SECIS element containing luciferase reporter genes (84), may help us better understand the effect of specific SECISBP2 mutations. In turn, greater understanding of how different mutations affect selenoprotein expression may enable better prediction of clinical outcome or targeting of therapy in patients.
TRU-TCA1-1
Selenocysteine is synthesized on its own tRNA (Fig. 1), tRNA[Ser]Sec (encoded by TRU-TCA1-1) that has several unique features, being longer (90 nucleotides vs. usual 78), with an atypical long acceptor- and D-stem with a few modified bases (57) that distinguishes it from other tRNAs (Fig. 3A). Its promoter region, containing both tRNA and U small nuclear RNA (snRNA) gene regulatory elements (18, 39, 44) forming a new class of RNA polymerase III transcribed genes, also differs from other tRNA genes (Fig. 3B) and consists of four regulatory elements: an activator element, containing an SPH motif, but is octamer independent; the proximal sequence element; an extended TATA-motif, essential for efficient transcription; and an internal B box (64, 65).
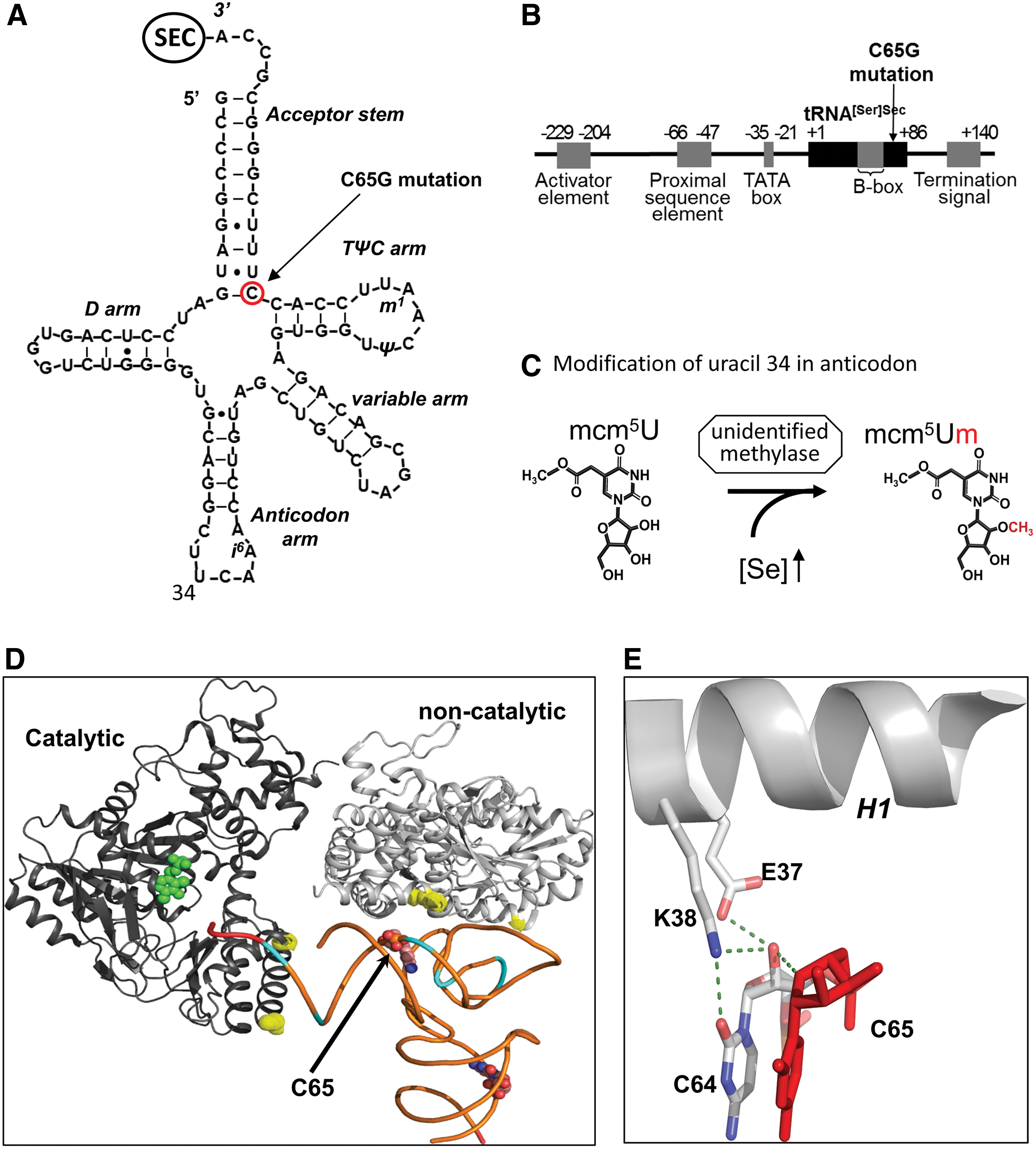
Two major isoforms of Sec-tRNA[Ser]Sec have been identified, containing either 5-methoxycarbonyl-methyluridine (mcm5U) or its methylated form 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um) at position 34 (Fig. 3C). Uridine 34 is located in the anticodon loop and its methylation may contribute to stabilization of the codon
A single patient, homozygous for a single nucleotide change (C65G) in TRU-TCA1-1 has been identified (Fig. 3A) (76). The proband exhibits a similar phenotype to that seen in SECISBP2 mutation patients. However, a comparison of cellular selenoprotein expression profiles in the two disorders has revealed differences, with the expression of relative housekeeping selenoproteins (e.g., TXNRDs, GPX4) being more preserved than in SECISBP2 cases. In contrast, the expression of stress-related selenoproteins (e.g., GPX1, GPX3) was similarly reduced in both disorders. In primary cells from the TRU-TCA1-1 mutation patient, lower total tRNA[Ser]Sec expression with a disproportionately greater diminution in Sec-tRNA[Ser]Sec mcm5Um levels was observed, with decreased i6A modification at position 37 suggesting that its post-transcriptional maturation is impaired. The mutation had no effect on tRNA[Ser]Sec aminoacylation with serine or Sec synthesis or its interaction with O-phosphoserine tRNA:Sec tRNA synthase (SEPSECS). Low levels of tRNA[Ser]Sec in the proband were insufficient to direct normal synthesis of stress-related selenoproteins, but they were not rate limiting for adequate synthesis of some housekeeping selenoproteins and similar, differential preservation of selenoprotein synthesis has been observed in murine tRNAsec mutant models (19, 57).
The human SEPSECS
In summary, these observations indicate that reduction in Sec-tRNA[Ser]Sec levels, with particular deficiency of the Sec-tRNA[Ser]Sec mcm5Um subtype, contributes to the selective pattern of selenoprotein deficiency seen in the proband. The precise mechanism mediating the reduction in mutant Sec-tRNA[Ser]Sec presence remains unclear, with defective post-transcriptional modification of mutant Sec-tRNA[Ser]Sec or instability of the mutant Sec-tRNA[Ser]Sec
SEPSECS
Human SEPSECS was initially identified as an autoantigen (soluble liver antigen/liver pancreas) in autoimmune hepatitis (52). Subsequent studies in which mammalian cell extracts were treated with autoimmune hepatitis patients' serum showed that SEPSECS co-precipitated with Sec-tRNA[Ser]Sec, as part of a ribonucleoprotein complex (40). This led to the identification of SEPSECS as the enzyme that catalyzes the conversion of O-phosphoserine-tRNA[Ser]Sec to Sec-tRNA[Ser]Sec, using selenophosphate as a donor substrate (81, 93) (Fig. 1).
Crystal structures of the archaeal and murine Sepsecs apo-enzymes as well as human wild type and mutant SEPSECS complexed with Sec-tRNA[Ser]Sec have been solved, suggesting that SEPSECS is a distinct member of the fold type I family of the pyridoxal phosphate-dependent enzyme family (7, 38, 68a, 73). The human structure shows a complex containing an SEPSECS tetramer binding two Sec-tRNA[Ser]Sec molecules through their long acceptor-TΨC arms, with the non-catalytic SEPSECS dimer mediating RNA
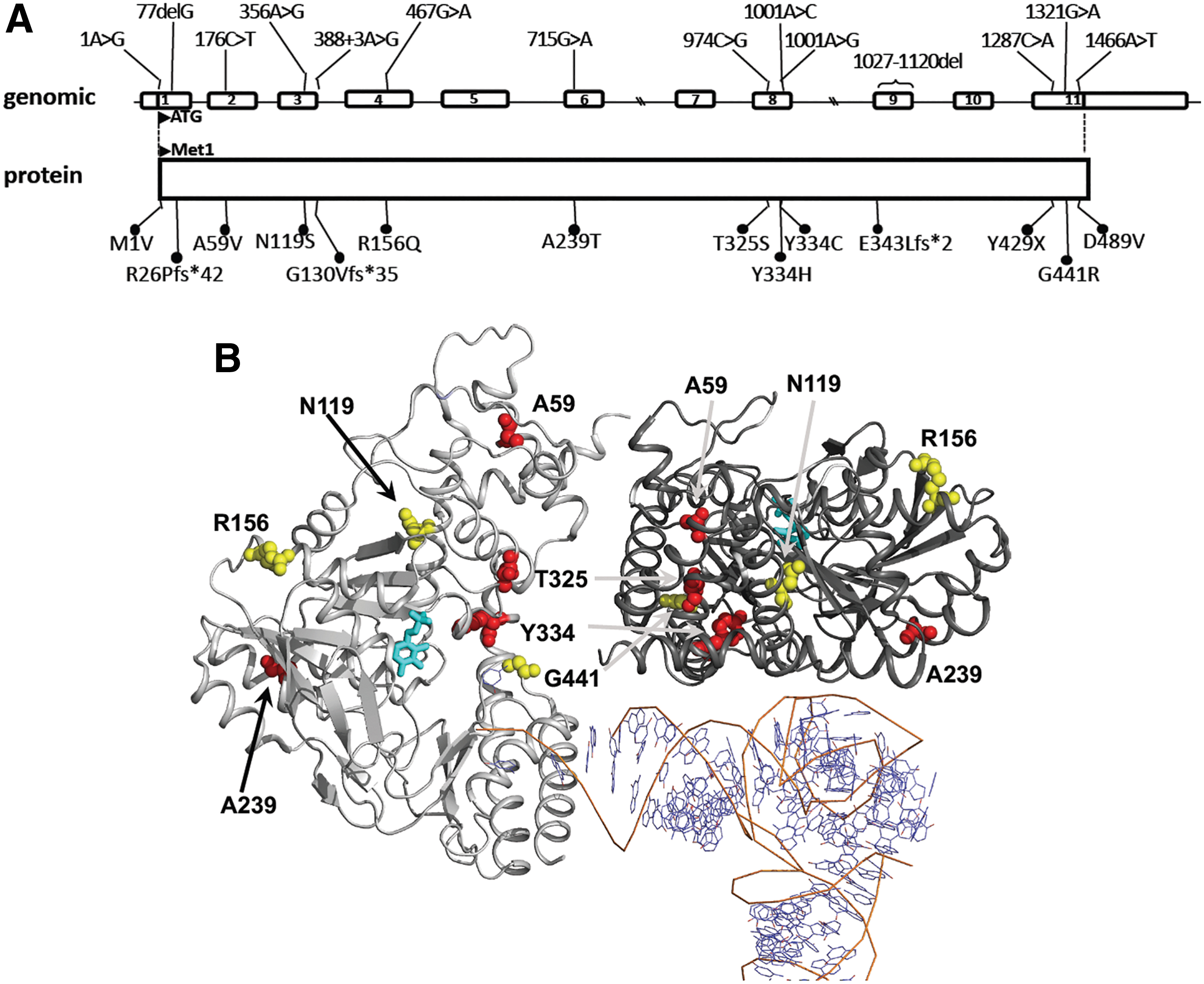
Homozygous and compound heterozygous mutations in SEPSECS (Table 4, Figs. 4, and 5) are associated with profound intellectual disability, global developmental delay, spasticity, epilepsy, and hypotonia with progressive microcephaly due to cortical and cerebellar atrophy on magnetic resonance imaging (1). Additional phenotypes described in other patients include axonal neuropathy, optic atrophy, and early onset epileptic encephalopathy with burst suppression (6, 67, 72). The timing of presentation in patients with SEPSECS mutations is variable, ranging from severe prenatal onset to delayed postnatal presentation and a mild, late onset phenotype in three patients (50, 86). This disorder is classified as autosomal recessive pontocerebellar hypoplasia type 2D (PCH2D, OMIM No. 613811), also known as progressive cerebellocerebral atrophy (PCCA) (1, 11). Mice homozygous for a SEPSECS mutation (Y334C) present with Sedaghatian-type spondylometaphyseal dysplasia and die shortly after birth, in contrast to humans with the same mutation. This divergence in phenotype may be due to species differences, dietary environment, or the genetic background of the mouse line used (36).
Human SEPSECS Mutations: Genetics and Effect
EOEE-BS, early onset epileptic encephalopathy with burst suppression; PCH2D, pontocerebellar hypoplasia type 2D.
The effect of SEPSECS mutations on selenoprotein expression has only been studied in four patients (Family E, G, F; 6). In brain tissue levels of TXNRD1, TXNRD2, GPX1, and GPX4 proteins are reduced, correlating with increased cellular oxidative stress. However, selenoprotein deficiency is not generalized with normal TXNRD levels in a patient's fibroblast and muscle cells, suggesting that residual SEPSECS activity preserves selenoprotein synthesis in some tissues or existence of alternative pathway(s). Consistent with findings in human tissues Sepsecs mutant mice exhibit decreased Gpx4 expression in neurons but not hepatocytes (36).
Although the selenium content of brain is not high (95), it is kept very stable (10, 66), exemplified by the fact that in systemic selenium deficiency circulating SELENOP delivers this trace element preferentially to this organ at the expense of other tissues (66). This may explain why a reduction in SEPSECS activity could have a greater effect on brain development and function compared with other tissues.
It is remarkable that an overt central nervous system (CNS) phenotype is not reported in patients with SECISBP2 and TRU-TCA1-1 defects, but with most individuals with these mutations being children the possibility of a late onset neurological phenotype cannot be discounted. However, a neurodegenerative phenotype has been described in brain-specific Gpx4, Trsp, and Secisbp2 KO mouse models, suggesting that Se deficiency and/or reduced redox capacity has a larger impact on the mouse brain compared with the human brain (78, 91, 92).
Biochemical hallmarks of selenoprotein deficiency in SECISBP2 and TRU-TCA1-1 mutation cases include low circulating selenium and abnormal thyroid hormone levels, reflecting deficiency of circulating selenoproteins (SEPP, GPx3) or all three deiodinase enzymes, respectively (31, 75). In SEPSECS mutation patients, low serum selenium is not reported to be part of the phenotype and thyroid status has only been partially investigated in four patients, documenting either normal thyroid hormone (Family H; 72) or normal T4 but elevated thyroid stimulating hormone (TSH) levels (Family E, G, F; 6). However, more detailed investigation, with measurement of T3 or reverse T3 levels, which would be abnormal with decreased deiodinase enzyme activity, has not been undertaken in SEPSECS mutation cases (6). Myopathic features with raised CK levels, abnormal mitochondria, cytoplasmic bodies, and increased lipid accumulation in muscle have been documented in one SEPSECS mutation case (Family H; 72), with broad-based gait and postural instability suggesting muscle weakness in another patient (Family O, 86). Similar findings have been noted in adult SECISBP2 mutation patients, reflecting deficiency of selenoprotein N and altered redox capacity in skeletal muscle (75). Overall, these observations suggest that some SEPSECS mutation patients can exhibit phenotypes associated with more global deficiency of selenoproteins. It is also possible that severity of neurological problems in patients has precluded detailed investigation and ascertainment of non-neurological phenotypes.
The availability of the crystal structure of human SEPSECS
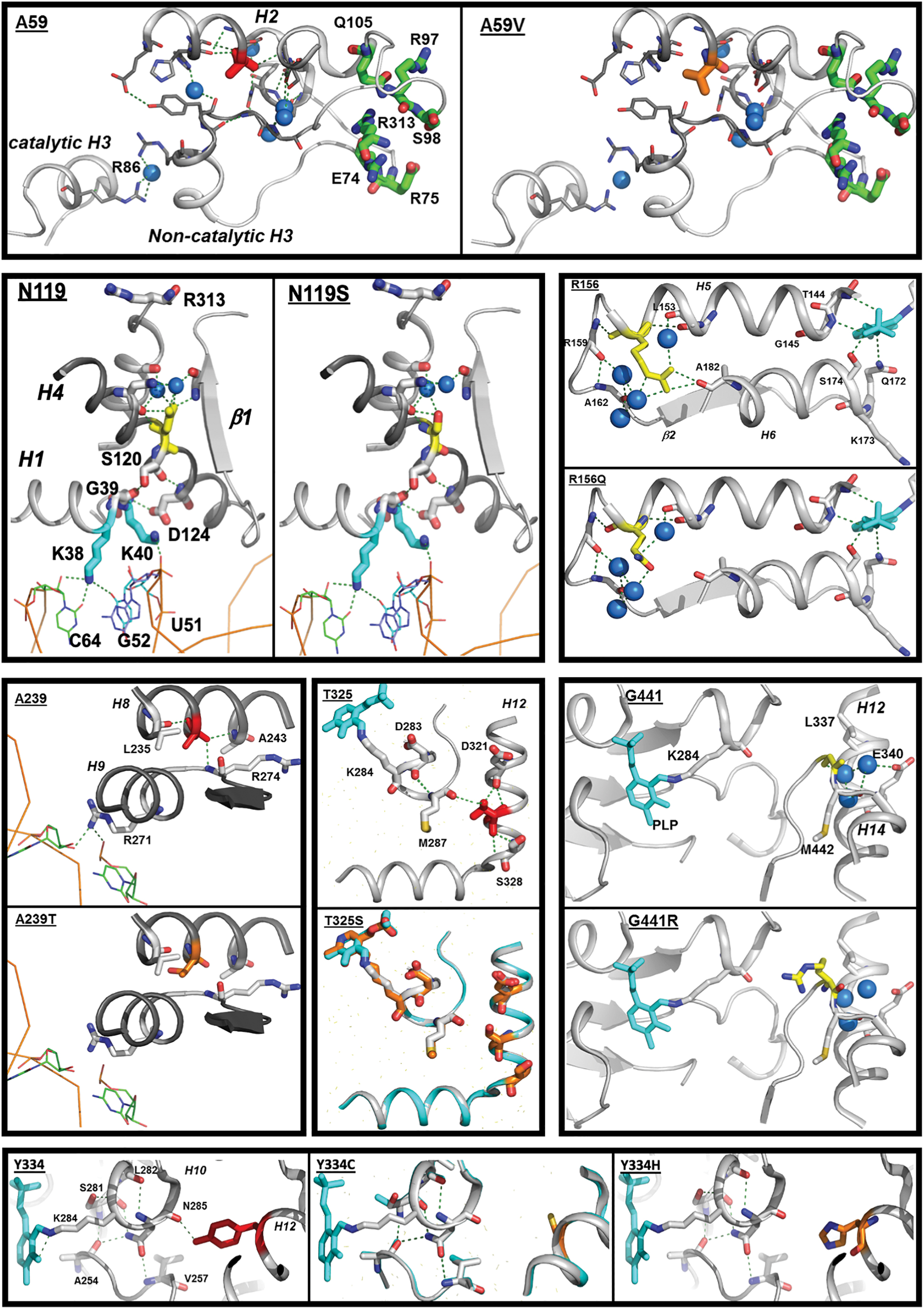
In silico analysis predicts that the A59V SEPSECS mutation results in steric hindrance destabilizing helix 2 and helix 3, possibly affecting its catalytic function and/or dimerization (Fig. 5). The N119S SEPSECS mutation is a conservative amino acid substitution with a probable small effect, weakening an H-bond network in the non-catalytic dimer and possibly affecting RNA interaction. Likewise, the mutation of Arg156 to Glutamine in SEPSECS is a conservative change, predicted to perturb local structure minimally and possibly reducing activity of the catalytic site (Fig. 5). The G441R SEPSECS mutation, situated close to the catalytic site, changes the side chain of this amino acid from small and neutral to large and polar, but as Gly441 is situated within a loop, its possible effect on catalytic activity might be limited. Absence of two SEPSECS mutations (M1V, D489V) from crystal structures precludes in silico analyses: M1V affects the first methionine in SEPSECS and unless an alternative start codon (e.g., position 61) is used, no protein will be generated; the D489V mutation changes the size and charge of this amino acid and can, therefore, be expected to have a major impact on protein function/stability.
Three SEPSECS mutation patients (patient I [R26Pfs*42
Conclusions
SECISBP2, Sec-tRNA[Ser]Sec, and SEPSECS are essential components of the selenoprotein biosynthesis pathway. Unsurprisingly, in patients, harboring mutations in any of these genes, the expression of most members of the selenoproteome is affected, sometimes in a tissue-specific manner, resulting in a complex, multisystem phenotype. The combination of the nature of the gene defect, genetic/ethnic background of individuals, and environmental factors (e.g., selenium and/or iodine status) might also contribute to inter-individual differences in phenotypes. Further complexity is due to the fact that most patients harbor compound heterozygous mutations with monoallelic mutations in individuals having no reported phenotype. This makes it difficult to assess the effect of a particular mutation on the Sec-incorporation pathway. Although a substantial body of knowledge regarding the individual functions of SECISBP2, Sec-tRNA[Ser]Sec, and SEPSECS exists, we do not have a comprehensive understanding of the Sec-insertion pathway. Nevertheless, most observations in patients with gene defects accord with our current knowledge of this pathway. However, it is interesting that mutations in SECISBP2 and TRU-TCA1-1 present with similar phenotypes (growth retardation and myopathy together with abnormal thyroid function), whereas the dominant phenotype in SEPSECS mutation cases is PCCA.
In all patients, some phenotypes (e.g., photosensitivity, age-dependent hearing loss, and neurodegeneration) are clearly progressive, perhaps reflecting the absence of antioxidant selenoenzymes and resulting in cumulative oxidative damage to DNA, proteins, and membrane lipids and dysfunction of redox-dependent signaling pathways. Some phenotypes can clearly be linked to deficiency of specific selenoproteins (e.g., abnormal thyroid function and DIO1,2,3; low plasma Se and SELENOP, GPX3; azoospermia and SELENOV, GPX4, and TXRND3; myopathy; and SELENON).
However, the precise role of many selenoproteins in human biological processes is unknown, making the identification of causal links between altered expression of specific selenoproteins and human disease a particular challenge. Further, the role of individual selenoproteins needs to be analyzed in the context of a complex cellular biochemical environment, where antagonistic, additive, and synergistic effects can occur. Recent advances, including analyzing selenoprotein expression in mouse models, RNA ribosome profiling (96) in vitro technologies such as CRISPR-Cas9-VLP (88), dissection of SECIS and other functional RNA elements (22, 62) using luciferase-based reporter assays (84), and modeling using crystal structures (e.g., SEPSECS
Footnotes
Funding Information
E.S. and K.C. are supported by the Wellcome Trust (210755/Z/18/Z), and K.C. is supported by the National Institute of Health Research Cambridge Biomedical Centre.