
Abstract
Significance:
Glutathione (GSH) represents the most abundant and the main antioxidant in the body with important functions in the brain related to Alzheimer's disease (AD).
Recent Advances:
Oxidative stress is one of the central mechanisms in AD. We and others have demonstrated the alteration of GSH levels in the AD brain, its important role in the detoxification of advanced glycation end-products and of acrolein, a by-product of lipid peroxidation. Recent in vivo studies found a decrease of GSH in several areas of the brain from control, mild cognitive impairment, and AD subjects, which are correlated with cognitive decline.
Critical Issues:
Several strategies were developed to restore its intracellular level with the
Future Directions:
We address how GSH-coupled nanocarriers represent a promising approach for the functionalization of nanocarriers to overcome the blood/brain barrier (BBB) for the brain delivery of GSH while avoiding cellular toxicity. It is also important to address the presence of GSH in exosomes for its potential intercellular transfer or its shuttle across the BBB under certain conditions. Antioxid. Redox Signal. 35, 270–292.
Introduction
Currently, 50 million people suffer from dementia worldwide, and about two-thirds of them are affected by Alzheimer's disease (AD) (11). AD is clinically characterized by cognitive impairment and memory loss disrupting daily life. Generally, these clinical signs appear a few decades after the initiation of cellular damages in the brain. The sporadic form of AD accounts for over 95% of cases, while the familial form represents only 5%. Both forms share the same neuropathological hallmarks with the presence of the amyloid-beta (Aβ) plaques and the neurofibrillary tangles (NFTs), which spread through the brain as the disease progresses. Synaptic and neuronal loss, astrogliosis, neuroinflammation, and oxidative stress are also observed in several areas of the brain (208).
Multiple lines of evidence suggest that the Aβ cascade, with the overproduction and structural modifications of the Aβ peptide to form harmful oligomers, is central in AD. Indeed, the presence of metal ions (iron, zinc, and copper) and reactive oxygen species (ROS) can accelerate the oligomerization and aggregation of the Aβ peptide (35), which causes the formation of oxidative markers with deleterious effects in the vicinity of the Aβ plaques (139). The mechanisms underlying the decrease in Aβ clearance remain to be clarified, but could involve the failure of microglial cells and macrophages to adequately degrade the extracellular Aβ deposits and the apolipoprotein E (ApoE). The APOE4 isoform does not alter Aβ synthesis, but can dramatically increase Aβ deposition in AD animal models (87). Unlike Aβ, there is little overlap between ApoE-immunoreactive neurons and NFT-positive neurons (81). Collectively, ApoE4 is considered a major causative or contributing factor for AD by acting as a chaperone for Aβ, which affects the clearance and deposition of Aβ, ultimately contributing to Aβ plaque formation, promoting tau-induced neurodegeneration and brain atrophy (186, 187).
NFTs are constituted by highly phosphorylated tau proteins that are present in axons and the best-established functions of tau are to stabilize microtubules. In AD, no tau mutations have been identified but its hyperphosphorylation negatively regulates its binding to microtubules leading to paired helical filament (PHF-tau) formation and the disruption of the axonal transport. Interestingly, NFT density, but not Aβ plaques, is correlated with cognitive decline in AD (76). Tau hyperphosphorylation requires the activation of different kinases mainly glycogen synthase kinase-3β (GSK-3β), which was found to be activated by oxidative stress [see recent review by Rana and Singh (172)].
Oxidative stress is considered the main contributor to aging and represents one of the major risk factors for AD (29). It shares common mechanisms between the divergent pathogenic pathways of AD and its progression. The implication of oxidative stress in the AD physiopathology emerged from abundant cellular and animal model data and strengthened by abundant studies in patients (23, 163, 170, 171, 190). Cells regulate oxidative stress by the action of housekeeping nonenzymatic and enzymatic antioxidants. Among them, glutathione (GSH) represents the most abundant and the main antioxidant with multiple functions. In this review, we summarize how GSH homeostasis is tightly linked to other redox mechanisms, its important roles in the brain, its multiple functions notably in the detoxification of proteins and lipid peroxidation, and how it can be involved in the pathophysiology and therapeutics of AD in innovative tools as a ligand of nanocarriers.
GSH Synthesis and Homeostasis
GSH is a small tripeptide consisting of the amino acids
As its cytosolic pool, the mitochondrial matrix and intermembrane space (IMS) pools of GSH are mostly in a reduced state. The IMS pool of GSH, which is ∼10–14 mM, is in equilibrium with the cytosolic GSH due to the presence of porins that allow free passage of reduced GSH (GSH) and oxidized GSH (GSSG) across the outer mitochondrial membrane (31). In contrast to the mitochondrial, cytosolic, and nucleus, the ER GSH/GSSG ratio pool is lower than in the cytoplasm and ranges from 3:1 to 1:1 (181). This is quite low compared with the cell overall ratio, which is greater than 30:1 and usually 100:1. Extracellular GSH/GSSG concentrations, ranging from 0.14 to 2.80 μM, are also usually 100 to 1000 times less than the intracellular ones being between 1 and 11 mM (181, 194), while the extracellular concentration of its cysteine precursor is much higher, with values ranging from 40 to 50 μM (140).
Cellular GSH homeostasis is regulated by its biosynthesis, oxidation, reduction, protein glutathionylation, adduction with toxic compounds, and export from the cells. The GSH biosynthetic capacity of various cells and tissues throughout the body is under the control of multiple factors, including the intracellular substrate availability (L-cysteine), the activity of GCL, the rate-limiting enzyme in GSH synthesis, and the feedback inhibition of GSH on γ-glutathione synthetase (γ-GCS). GCL is composed of a catalytic (GCLC) and a modifier (GCLM) subunit, which are regulated at multiple levels. The posttranslational modifications of the GCLC subunit by phosphorylation or by the lipid peroxidation product 4-hydroxynonenal can also affect the activity of the enzyme (15). In addition, the γ-GCS activity can also be modulated by phosphorylation and nitrosation (74). Once synthesized, GSH can undergo transport across biological membranes, notably across the plasma membrane for intercellular distribution. Some studies have suggested the presence of GSH transporters (62) and the first one identified was the multidrug resistance-associated proteins, a subclass of the ATP-binding cassette transporter superfamily. However, these efflux pumps have a broad substrate specificity with a low affinity for GSH than for GSH conjugates (17).
At basal levels of oxidative and nitrosative stress, GRX activity and NADPH availability are sufficient to maintain the ratio [GSH]/[GSSG] > 100 (6, 157). However, under stress conditions or in the presence of other factors, which limit the GRX reaction (e.g., glucose-6-phosphate dehydrogenase deficiency may limit NADPH synthesis), GSSG may accumulate, leading to the cellular shift of the thiol redox status that activates oxidant response transcriptional elements (185). Also, GSSG may be preferentially secreted from the cells (5) and degraded extracellularly because GSSG is not taken up by cells.
The impairment of the ratio GSH/GSSG and consequently of the cell redox balance may affect the activity of some redox-sensitive transcription factors such as the nuclear factor erythroid 2-related factor (Nrf2), the major regulator of the antioxidant response element (ARE) pathway, defined as the Nrf2-ARE system (125) (Fig. 2). Genes coding for several key enzymes linked to GSH metabolism, including the catalytic and regulatory subunits of GCL, the GSH synthase, the Xc - system, GRX, and glutathione peroxidase (GPX) familiesm are transcriptionally regulated by Nrf2 (104, 203) (Figs. 2 and 3). Hence, Nrf2 represents a key transcriptional regulator of GSH metabolism and homeostasis.

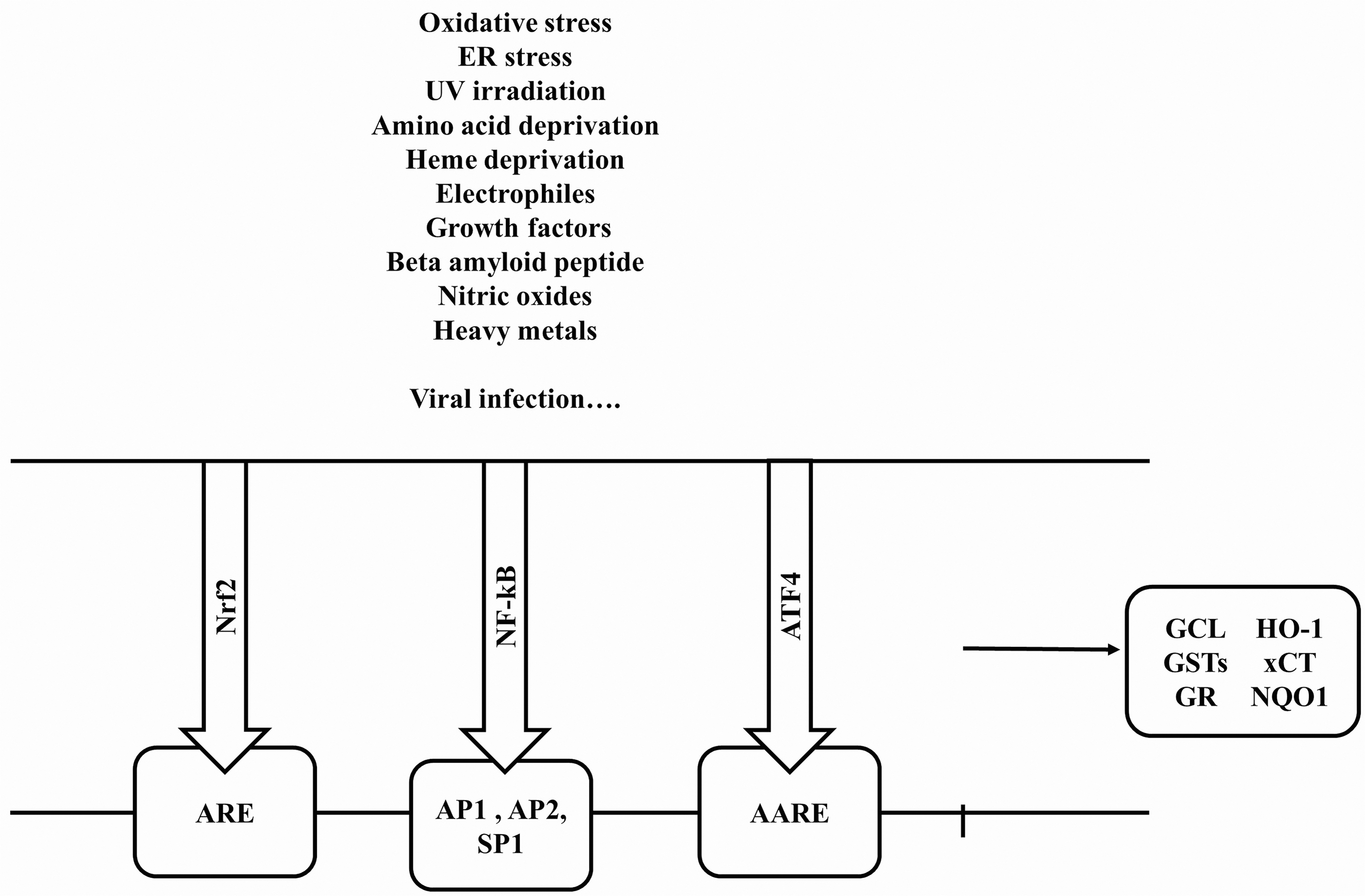
The nuclear factor-κB (NF-κB) is another redox-sensitive transcription factor involved in GSH synthesis (141). Compared with Nrf2, NF-κB is a rapid response factor. The 5′-flanking region of the human γ-GCSH gene contains putative NF-κB, activator protein-1 (AP-1), AP-2, antioxidant, electrophile response elements (ARE/EpRE), and Sp-1 binding sites within 1500 bp of the transcription start site (149) (Fig. 3).
The activating transcription factor 4 (ATF4) also plays a role in GSH metabolism (82). ATF4 is a member of the ATF/cAMP response element-binding protein (CREB) group of the bZIP transcription factor family. Its binds to a variety of genes involved in amino acid import, GSH biosynthesis, and resistance to oxidative stress (82). ATF4 also plays a key role in regulating the basal GSH levels in neuronal and non-neuronal cells, induces the expression of the specific subunit xCT of the glutamate/cystine antiporter system Xc - in response to cystine starvation (179). Basal xCT expression is determined by ATF4, whereas Nrf2 represents an inducer of xCT (178, 188) (Fig. 3). Thus, the intracellular GSH/GSSG concentrations are much higher than the extracellular ones being mainly under the reduced GSH form, and the cellular GSH synthesis and homeostasis depend on multiple factors and the stress conditions.
Cellular Functions of GSH
As one of the most important small-molecule antioxidants, GSH plays a central role in maintaining cellular redox homeostasis (181). Indeed, GSH and GSH-associated metabolism represent the major line of defense for the protection of cells from oxidative and other forms of toxic stress. GSH can scavenge free radicals, reduce peroxides, form adduct with electrophilic compounds (for instance, acrolein), thereby eliminating both ROS and their toxic by-products.
Many cellular redox couples work together to maintain the redox environment. However, the intracellular GSSG/2GSH couple is the most abundant and represents an important indicator of the redox environment, while the extracellular redox state is mainly maintained by cysteine/cystine (Cys/CySS) (97). Changes in the redox potential of various thiols, disulfides, GSSG/2GSH could be considered a series of nanoswitches, a cellular switchboard correlated with the biological status of the cells. Thus, by changing the reduction potential of redox couples, a series of nanoswitches are activated that could move the cell from proliferation to various stages of differentiation into apoptosis or necrosis. Necrosis represents the complete loss of the ability to activate and/or respond to changes in these nanoswitches.
GSSG/2GSG together with NAD+/NADH and NADP+/NADPH (219) play a key role in the regulation of the intracellular redox environment with a more reducing environment associated with cell proliferation, and a more oxidizing environment associated with differentiation. GSH plays also a role in signal transduction, in apoptosis, and in the expression of some genes such as those encoding for heat shock proteins, two components of AP-1 (Fos and FosB), positive regulators of NF-κB (TRAF6, FADD, EDG2, and tumor necrosis factor-a [TNF-α]), and of death receptor pathways (14, 42, 67, 96, 213). Thus, as the most abundant redox buffer in cells, GSH plays an important role in controlling the oxidoreductive nanoswitches, the status of the protein thiol/disulfide equilibrium, and thereby drives the cell biological status.
Glutathion, Thioredoxin, and Glutaredoxin Pathway Cross Talk
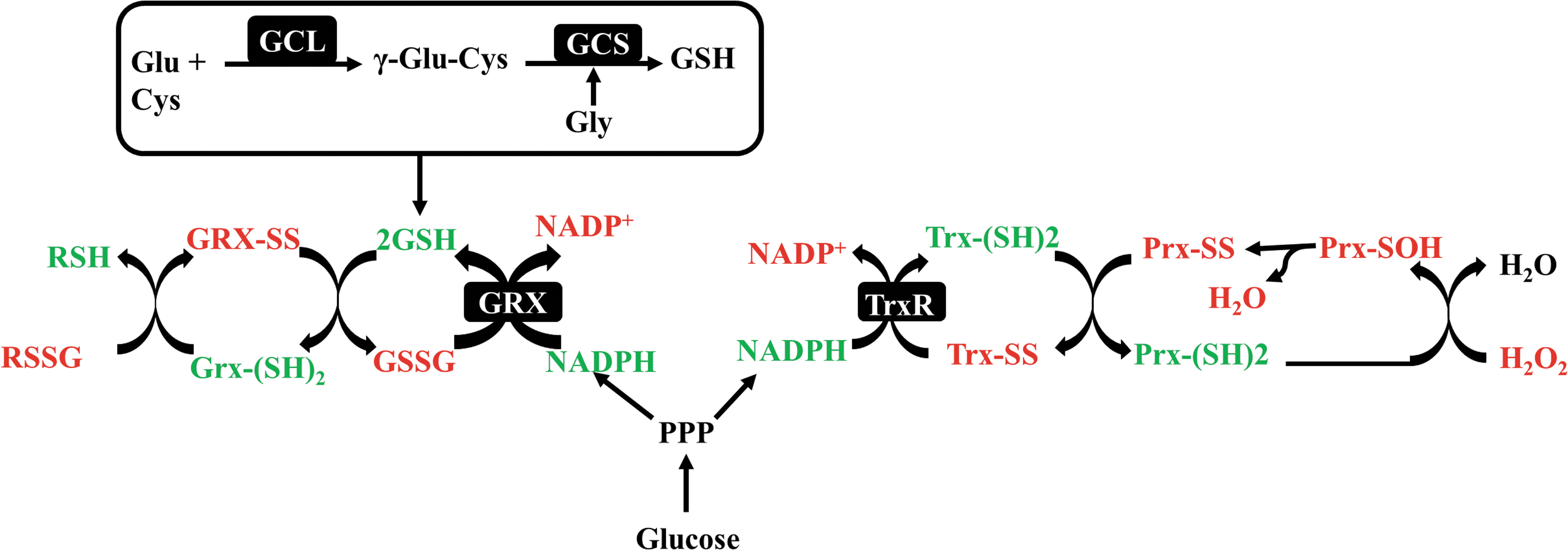
The three redox systems NADP+/NADPH, GSSG/2GSH, and TrxSS/Trx(SH)2, the major cellular thiol-dependent antioxidant, are not functionally isolated systems (Fig. 1). The Trx and GSH systems with NADPH are thermodynamically connected although the Trx levels are 100- to 1000-fold less than GSH (181). The thioredoxin (Trx) pathway composed of Trx, Trx reductase (TrxR), and the GSH/GRX system is two cellular disulfide reductase systems dependent on the NADPH/NADP+ ratio (Fig. 1). Trx is a ubiquitous small (12 kDa) selenoenzyme with a -CGPC- motif within its active site, which usually develops intramolecular disulfides in contrast to GSH that forms intermolecular disulfides. There are three forms of mammalian TrxRs: cytosolic TrxR1, mitochondrial TrxR2, and testis-specific TrxR3. The reduction of the disulfide back to the dithiol form is catalyzed by TrxR and by NADPH (86). In addition to the GSH/GSSG and Trx systems, mammal cells also express the Trx glutathione reductase, also known as TXNRD3/TrxR3 a fusion protein of TrxR and Grx domains, able to reduce Trx, GSSG, and GSH-mixed disulfide (121). Trx and GSH systems have some overlapping functions allowing maintenance of redox regulation, but in many cases, cross talk occurs between both systems where components of one system serve as a backup for the other (59). For instance, in stressed neurons, GSH can serve as a backup molecule for TrxR to keep Trx in a reduced state (28). In contrast, Trx2 has also been demonstrated to reduce GSSG, which further stresses the importance of cross talk between both systems (148).
The GSH-glutaredoxin (Glrx) system is another disulfide reduction system in cells. Glrxs, a part of the Trx protein family, can catalyze the reduction of substrates via a monothiol mechanism. In mammalian cells, there are two major forms of Glrx: Glrx1, present in the cytosol, and Glrx2, located in the mitochondria and nuclei (123). Glrx1 accepts electrons from GSH, while Glrx2 can obtain electrons from both GSH and TrxR2 (95).
Although peroxiredoxins, GSH-PX, and catalase are more efficient to degrade H2 O2 than GSH (122), their activities and concentrations in the brain are lower than GSH (43, 50, 197). In brain cells under oxidative conditions, GSH, Trx, and Glrxs are important redox regulatory pathways and the dysregulation of their crossroad could be important events in AD pathogenesis (8, 13). Thus, these disulfide reductase systems represent critical antioxidant systems actively involved in the maintenance of the redox balance, an activity essential for the critical biological events in various areas of the brain.
Regulation and Functions of GSH in the Brain
Important roles of the antiporter glutamate/cysteine Xc - on the GSH synthesis and homeostasis
The brain, one of the most metabolically active organs, is highly vulnerable to oxidative stress (37). Thus, the optimal redox homeostasis is crucial to limit oxidative-induced damages leading to some neurodegenerative disorders. In the brain, GSH is the most abundant thiol-containing molecule and one of the most important antioxidants with concentrations being around 2–3 mM (200). However, its distribution was found to be variable with the highest level in the cortex, followed by the cerebellum, hippocampus, and striatum, and is lowest in the substantia nigra (98). Its low levels in the susbtantia nigra dopaminergic neurons probably contribute to the oxidation of dopamine and early nigral degeneration in Parkinson's disease (56).
In the brain, GSH is mainly synthesized locally because, due to its rapid metabolization in the blood (109), its plasma levels are much lower (2–20 μM) than those in the brain. Also, due to its hydrophilic property, its permeability through the blood/brain barrier (BBB) by passive diffusion is very limited (41) and it is as yet unclear whether a direct GSH transport system exists at the BBB.
Astrocytes, as secretory cells of the brain (212), have a high capacity of production, storage, and release of GSH into the extracellular medium in response to oxidative stress, via the multidrug resistance transporter protein (57, 85). In the extracellular space, GSH could be degraded by the membrane-associated enzyme γ-glutamyl transpeptidase into γ-glutamyl moiety and the dipeptide cysteine glycine (CysGly) could be hydrolyzed to glycine and cysteine. This latter is oxidized to form cystine, its dimeric form. CysGly and cystine are, respectively, taken up by the peptide transporter PepT2 and the Xc
- system in astrocytes for the GSH resynthesis (57, 58, 188, 210) (Fig. 4). For this, cystine is reduced to cysteine by GSH or TRX-reductase 1 (TRX-R1) (130). The Xc
- system is an amino acid antiporter that mediates the import of
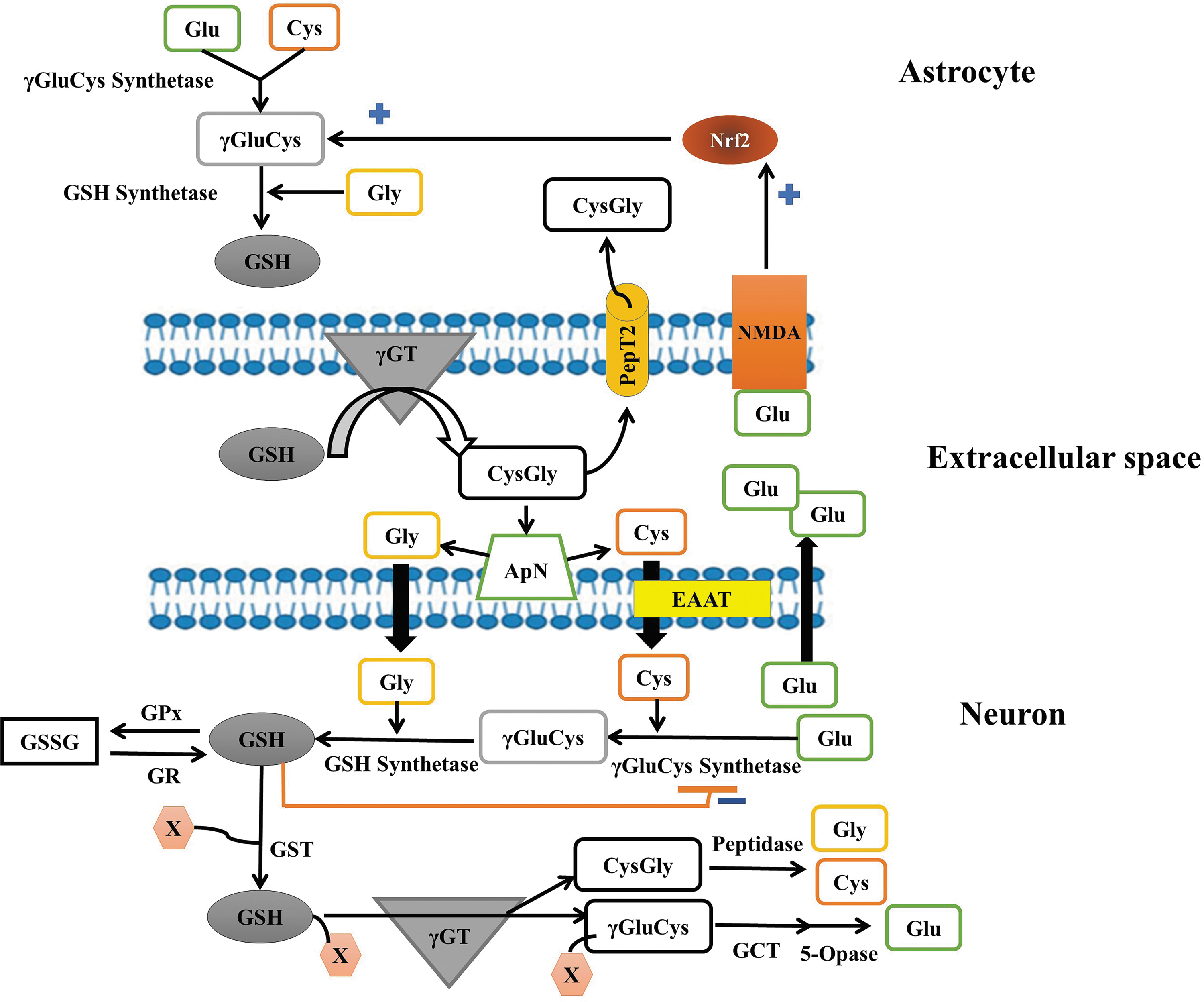
Thus,
Neurons do not express cystine transport and depend on cysteine transport for the synthesis and maintenance of the intracellular GSH levels. For these cells, cysteine is taken up by the excitatory amino acid transporter 3 (EAAT3/EAAC1) (Fig. 4) and their survival, activities, and the maintenance of their intracellular GSH levels are greatly dependent on this transporter. Thus, neuronal cells in culture can only survive when they are cocultured with the Xc - system expressing cells to provide cysteine to neuronal recipient cells (84, 92). However, many cell types, particularly neurons, express xCT, the light chain subunit of the Xc - system, encoded by the SLC7A11 gene under the regulation of the redox-sensitive transcription factor Nrf2 (178). Nrf2 and the ATF4 are activated under amino acid depletion, ER stress, or oxidative stress and induce cystine transport (178, 217). The Xc - system was found to be implicated in memory and behavior. For example, mice deficient for the xCT subunit have lower extracellular hippocampal glutamate, impaired spatial memory, which was more pronounced in younger mice, and susceptibility to limbic seizures (49). Recently, Xc - was shown to contribute to the neuroprotective and the improvement of the cognitive impairments of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine in AD (155).
It is interesting to note that the Nrf2-target genes in neurons are quite different from those in astrocytes. For instance, in neuronal cells, the Nrf2 activator tert-butylhydroquinone (tBHQ) induced the upregulation of genes related to mitochondrial activation, calcium binding and transport, cell adhesion, integrin signaling synaptic activity (106), the catalytic and regulatory subunits of GCL, Srxn1 (sulfiredoxin), and xCT, which are dynamically regulated by the synaptic activity (159). In astrocytes, most of Nrf2-dependent upregulated genes are detoxification enzymes, including glutathione-S-transferases (GSTs), NAD(P)H:quinone acceptor oxidoreductases (NQO1), ferritin, GSH synthetase, and those involved in glucose metabolism-related genes. In addition, neuronal Nrf2 is less responsive to tBHQ than astrocytes probably because neuronal cells express 30- to 1000-fold less Nrf2 protein (24, 93, 188) and contain greater Cul3-dependent Nrf2 degradation capacity than astrocytes (4, 93). Thus, the Nrf2 induction is considered the major mechanism by which astrocytic cells protect nearby neuronal cells.
Thus, to survive, neurons receive strong antioxidant support from surrounding glial cells, particularly astrocytes (24, 53, 57). Although the GSH synthesis in neuronal cells is highly dependent on the GSH precursors provided by astrocytes, neuronal cells are also able to control GSH homeostasis by reducing GSH through different pathways such as GRX, Trx, and peroxiredoxins (Figs. 1 and 4).
Cross talk between neurons and astrocytes contributes to maintain GSH homeostasis in the brain
The interaction between astrocytes and neurons is important for neuronal GSH homeostasis.
For instance, the chronic activation of astrocytic NMDA receptors, a Ca2+ subtype of ionotropic glutamate receptor, can activate Nrf2 (93). Indeed, the application of NMDA directly to astrocytes triggers Cdk5-mediated Nrf2 phosphorylation and induction of Nrf2-dependent gene expression with the capacity to confer neuroprotection on nearby neurons (93). In contrast, NMDA receptor blockade in vivo causes a reduction in brain GCL expression, activity, and thus GSH levels (21). These results strongly suggest that the neuronal glutamate release could control astrocytic Nrf2 (93). Accordingly, stimuli that promote the synaptic activity or neuronal depolarization can increase the astrocytic nuclear accumulation of Nrf2 (78). Increased synaptic activity causes an immediate elevation of GSH utilization (83) and the capacity of neurons to synthesize and recycle GSH (21). In contrast, the blockade of the neuronal activity is associated with neurodegeneration, which can be ameliorated by supplying the brain with a cell-permeable form of γ-GC, the product of GCL catalysis (21).
In microglial cells, GSH plays also an important role. In both human microglia and astrocytes, the depletion of GSH by the treatment with
Neuronal and astrocytes have clearly distinct homeostatic mechanisms related to GSH. However, it is not clear to what extent neuronal activity influences the transcriptome of the surrounding glial cells. A better understanding of reciprocal interactions between neuronal/glial cells is required to gain a better picture of brain redox homeostasis. The cooperative nature of the interaction is depicted in Figure 4.
Role of GSH in the S-Glutathionylation of Proteins
Under oxidative or nitrosative stress conditions, GSH can protect cells against toxic proteins by forming a mixed disulfide between reactive thiols and GSH, a process called S-glutathionylation. Similar to S-nitrosylation, protein S-glutathionylation is a well-controlled reversible reaction that alters protein functions in physiological conditions in the brain. S-glutathionylated proteins could be deglutationylated by the Glrx enzymes. Although several proteins, including Trxs (73), protein disulfide isomerase (161), and sulfiredoxin (64), have deglutathionylase activity under different conditions, Glrxs are considered to be the major deglutathionylase enzymes due to their high affinity and selectivity for glutathionylated proteins (95).
Protein S-glutathionylation induced an important signal in neuronal cell death via the effects of abnormal protein polymerization and protein degradation. For example, actin was reported to be constitutively glutathionylated in the brain (196) and the Glrx1-catalyzed deglutathionylation of actin is crucial for its polymerization, a key event required to maintain the cellular dynamics, and to protect neurons from the accumulation of disarranged actin filaments (160).
In AD, there is a significant increase in the level of S-glutathionylated in the inferior parietal lobule tissues compared with the level observed in age-matched controls (153). For instance, S-glutathionylated tau protein rapidly undergoes polymerization to form filaments (54). Redox proteomics in the AD brain has identified specific S-glutathionylated proteins, including those involved in glucose and energy metabolism (153), deoxyhemoglobin, α-crystallin B, glyceraldehyde phosphate dehydrogenase, and α-enolase (45, 46). However, S-glutathionylated can also protect the integrity of the sulfhydryl groups of some proteins such as p53 from an irreversible modification that occurs in the progression of AD, leading to its permanent inactivation (52, 211). Selective glutathionylation of p53 in the AD brain could prevent the formation of aggregates involved in oxidative stress conditions and neurodegeneration (52). For instance, in the transgenic mice model of AD (APP/PS1), the S-glutathionyl levels in the blood and brain increased with aging compared with wild-type mice with proteins at 8.4–20.3 and 13.2–37.2 kDa being most susceptible to S-glutathionylation (222). Thus, the accumulation of abnormal protein S-glutathionylation could potentially be considered early biomarkers to predict AD.
The GSH Depletion Is a Key Player for Oxytosis and Ferroptosis
GSH depletion can induce different forms of cell death in the brain (Fig. 5). GSH depletion is a common feature of apoptotic cell death triggered by a wide variety of stimuli, including activation of death receptors or stress inducers (66), and excessive GSH depletion can switch apoptotic to necrotic cell death (206). The combination of the depletion of GSH or its inhibition, the accumulation of ROS production, lipid peroxidation, lipooxygenase activation, and calcium influx can induce neuronal cell death through the pathway named oxytosis (20). On the contrary, the accumulation of lipid peroxidation by-products triggered by the inhibition of GSH biosynthesis and the GSH-dependent antioxidant enzyme GSH peroxidase 4 (GPX4), the exacerbation of iron causes cell death by ferroptosis. Oxytosis and ferroptosis are distinct from apoptosis, classic necrosis, autophagy, and other forms of cell death. Both oxytosis and ferroptosis represent very similar (or even the same) forms of regulated cell death (112) characterized by ROS generation and GPX4 inactivation (162). Thus, the central endogenous suppressor of ferroptosis is the selenoenzyme GPX4, which requires GSH as substrate (1). Both oxytosis and ferroptosis are also prevented by radical scavengers such as ferrostatin-1, liproxstatin-1, and endogenous vitamin E (34, 39, 55, 91, 111, 126, 128, 198) (Fig. 6).

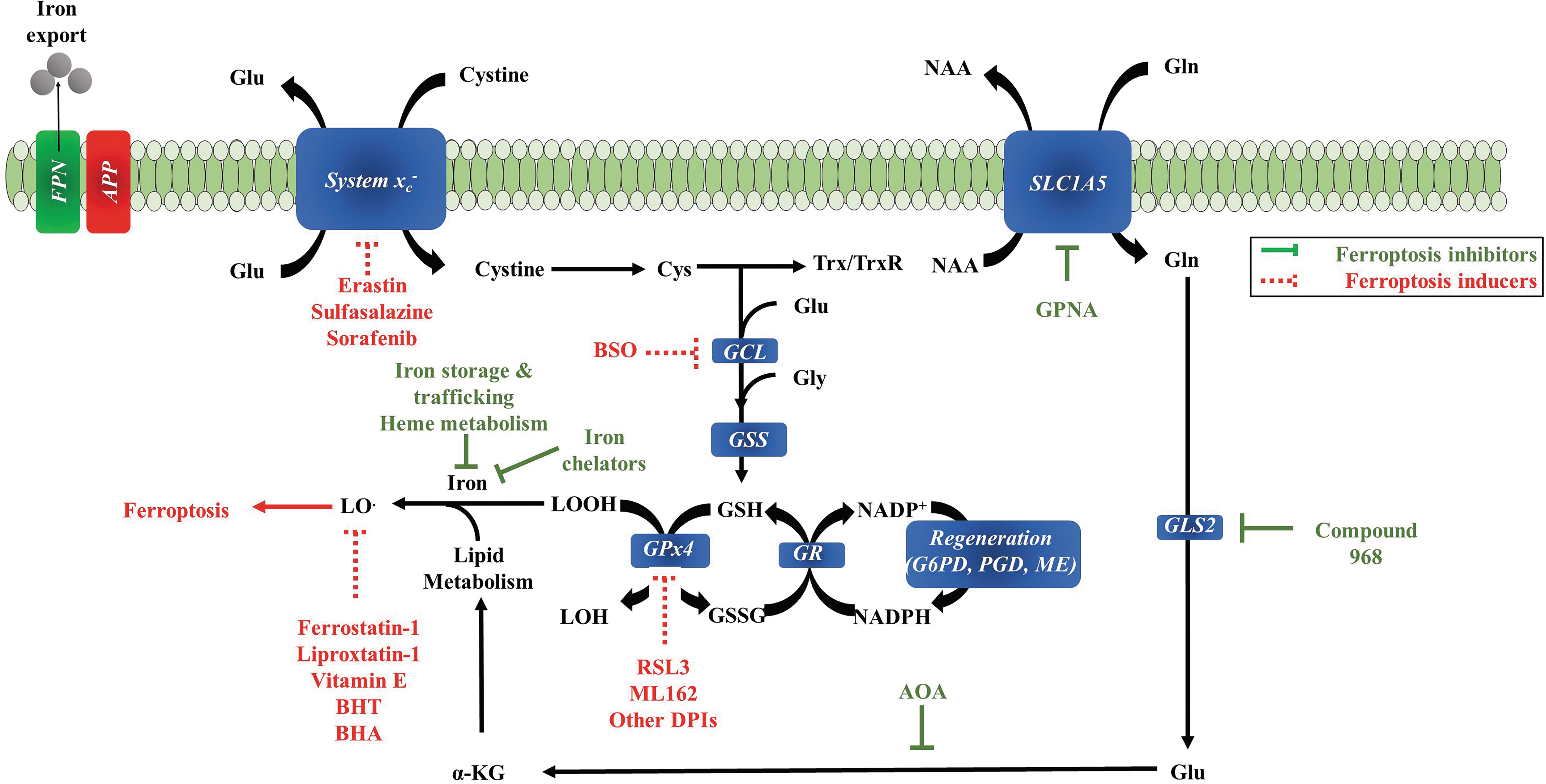
Another key initiating step for ferroptosis is the inhibition of the cellular uptake of cystine. As the Xc - system is among the cystine transport systems into cells, its inhibition can induce cell death by ferroptosis (150). Thus, ferroptosis appears similar in several aspects to glutamate toxicity, a phenotype observed in certain neuronal cell lines treated with high concentrations of glutamate to inactivate the system Xc - and deprive cells of cystine/cysteine.
The concentration of iron is another factor that control ferroptosis. In the brain, higher concentrations of iron are preferentially found in the nucleus accumbens, substantia nigra, deep cerebellar nuclei, and parts of the hippocampus (77, 192). Lipid peroxidation, a hallmark feature of ferroptosis, is considered an early event in the pathology of AD (173, 190). The conditional ablation of GPX4 in the forebrain (cerebral cortex and hippocampus) of mice (Gpx4BIKO) resulted in AD-like cognitive impairment (spatial learning and memory) accompanied by hippocampal neurodegeneration, elevated lipid peroxidation (enhanced 4-hydroxynonenal adducts observed in the cerebral cortex), and neuroinflammation (80). These phenotypes were further exacerbated in mice fed with a diet deficient in the lipid-soluble antioxidant tocopherol (80). These observations are in line with the depletion of GSH in the frontal cortex and hippocampus, which correlates with the decline in cognitive functions (129, 131). Taken together, these data indicate that ferroptosis is one of the consequences of the GSH decline in AD (80, 126, 129, 136, 198).
Roles of GSH in AD
As described above, brain GSH levels decrease with age and a loss of GSH can impact on the cognitive function. A decrease in GSH is also associated with microglial activation and endothelial dysfunction, both of which can contribute to the impairments of brain function. Here, we describe that several changes in brain function in AD are directly or indirectly linked to GSH metabolism.
There are very limited data available in the determination of GSH in post mortem tissues from the AD brain. Altogether, these studies indicate that GSH levels in frontal, occipital lobes, or hippocampus were broadly not altered in AD (16, 101, 164, 199). One of the limitations of these studies was the absence of the determination of the ApoE genotype because we and others have demonstrated that the levels of GSH and other oxidative markers were lower in the hippocampus only in ɛ4 allele of ApoE carriers (145, 171). This reduction appears to be specific to the hippocampus because it is not reduced in the frontal cortex (170). Using in vivo proton resonance spectroscopy to measure GSH levels in the hippocampi and frontal cortices from control, mild cognitive impairment (MCI), and AD subjects, an AD-dependent decrease in GSH was found in both regions that correlated with the decline in cognitive functions (129, 131). Moreover, levels of hippocampal GSH could discriminate between healthy controls and MCI subjects, while cortical GSH levels could discriminate between MCI and AD patients. Very recently, the quantification of the in vivo brain GSH levels in the cingulate cortex (CC) showed a significant GSH depletion in AD and MCI control patients (189). Moreover, receiver operator characteristic analysis of GSH level in the CC differentiated between MCI and normal control groups with an accuracy of 82.8% and 73.5% between the AD and normal control groups (189). However, we cannot exclude the possibility that the decrease of GSH could promote adaptative changes of neurons to better tolerate subsequent stress to maintain cognitive health, as a “neurohormseis” concept (137, 176), because cellular stress response activates prosurvival pathways, which are under the control of protective genes called vitagenes (GSH, heat shock proteins, sirtuins, Trxs…) (32, 48),
In cells, tissues, and plasma, the reactivity of GSH leads to several forms such as glutathionylation, the formation of adducts with electrophilic compounds such as by-products of lipid peroxidation (i.e., acrolein). Thus, the determination of its levels in tissues does not consider its different forms and redox states. Therefore, the analysis of the global thiol–disulfide redox status in tissues and cells is a challenging task because of the following: (i) thiols easily undergo auto-oxidation during the preanalytical phase of sample preparation; (ii) disulfides can be reduced back to thiols enzymatically during cell manipulation; and (iii) basal levels of disulfides (GSSG and oxidized peroxiredoxin [PSSG]) are very low compared with GSH. Also, a large part of ROS/reactive nitrogen species-mediated signal transduction relies on modifications of cysteine thiol (-SH) within proteins. Irreversible thiol modification (sulfonic acid, -SO3H) has been considered a hallmark for various pathological conditions and usually leads to permanent functional loss and degradation of the protein. Reversible thiol modifications, such as S-nitrosylation (-SNO), S-sulfenylation (sulfenic acid, -SOH), S-glutathionylation (-SSG), disulfide formation (-S-S-), S-sulfhydration (-S-SH), and sulfinic acid (-SO2H), are involved in redox signaling (Fig. 7).
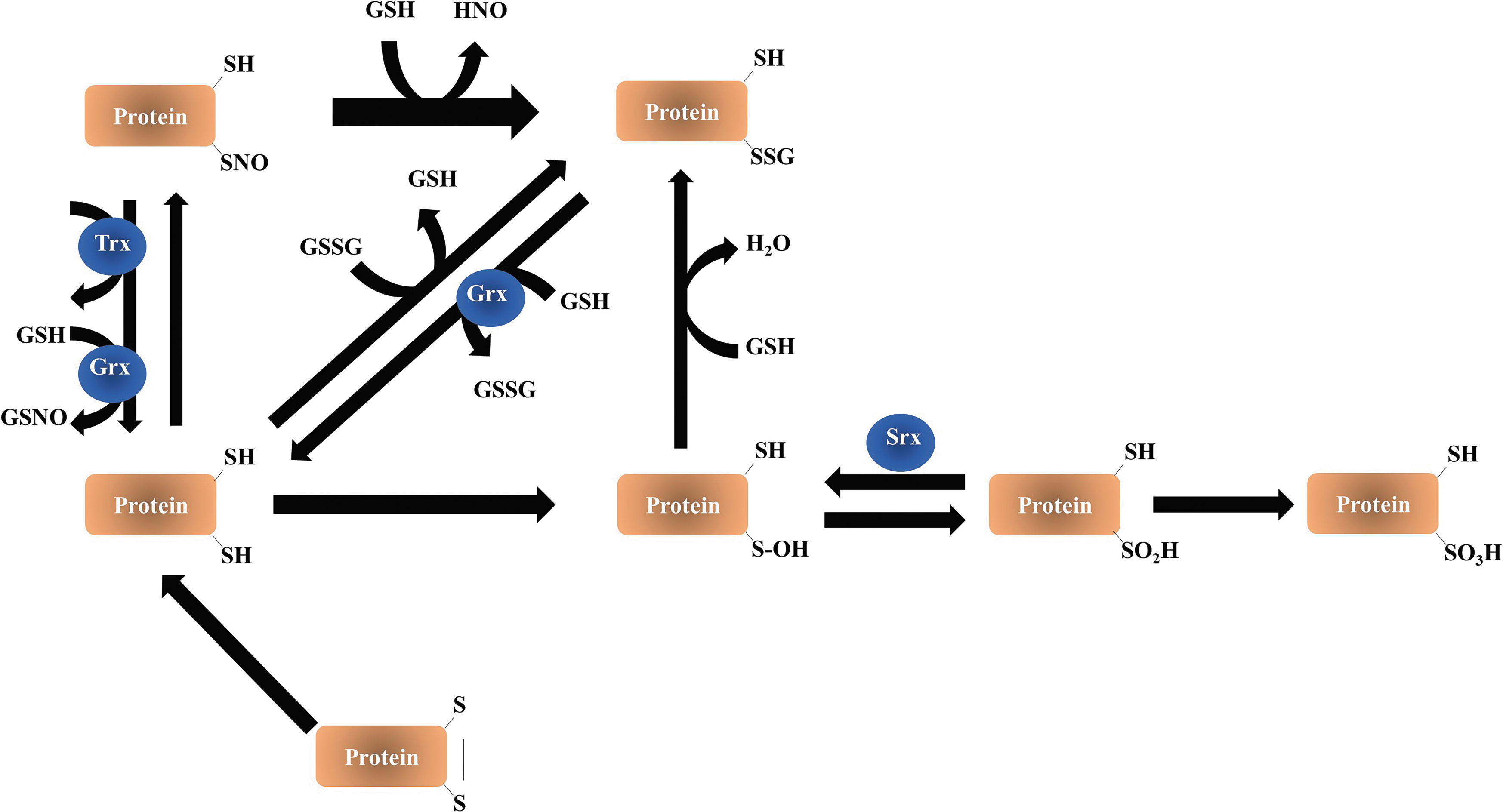
GSH disulfide, formed upon oxidation, is referred to as “oxidized GSH” (GSSG), but further oxidation products are also formed from GSH, for example, sulfonates. Other forms of disulfide are of the mixed type, a major class of biologically interesting ones being GSH-cysteinyl disulfides on proteins. Thus, proteins can be glutathionylated or, as sometimes referred to, thiolated. These reactions demonstrate that P-SH contributes to the cellular antioxidant network, thereby influencing its redox environment.
Another challenge is the compartmentation of GSH and GSSG, which may be at a disequilibrium steady state to each other. Measurement of the total content of GSH and GSSG in cells would represent an overall redox of the cytosol, not the redox environment of the various compartments such as the ER, nucleus, or mitochondria. However, the mtGSH pool plays an important role in AD because its depletion specifically can account for the increased susceptibility to the Aβ peptide. First, mtGSH levels determine the sensitivity of brain mitochondria to Aβ-mediated oxidative stress and the release of apoptotic proteins (63). Second, the depletion of the mtGSH pool induced neuroinflammation and neuronal damages (63). Collectively, these results suggest that a possible therapeutic approach to slow disease progression could be to replenish mtGSH (133).
The determination of oxidative stress markers and antioxidants in blood from subjects with AD or those with MCI highlights potential interactions between peripheral redox changes and the brain pathology. The extent of oxidation in peripheral blood is greater than the changes in the brain for most markers [see review by Schrag et al. (184)]. For instance, an increase of oxidative stress in serum, erythrocytes, and circulating lymphocytes with lower GSH/GSSG ratio in lymphocytes (7) and low-molecular-weight thiols was observed in MCI and AD [(25, 44) and review by Schrag et al. (184)]. A decrease in GSH/GSSG ratio may be due to the reduction of GSH synthesis, GSH depletion, or a lower regeneration of GSH from GSSG, or a combination of all. The challenges when dealing with fluids such as blood to assess the redox environment is relatively complex because the levels of GSH are different from one cell type to another. The decrease in GSH content in red blood cells from male AD patients was associated with reduced activities of GCL and GSH synthase with no change in the amount of oxidized GSH or the activity of GRX.
Oxidative markers are higher in blood than in the brain and raise the possibility that systemic derangements leading to a pro-oxidative environment may be upstream of brain injury in AD. The observation of increased risk of dementia in patients who have systemic proinflammatory/pro-oxidative diseases such as diabetes, obesity, and hypercholesterolemia supported this hypothesis.
In the following sections, we describe two potential mechanisms of action implicated in AD pathophysiology requiring GSH: one is related to inflammation and the formation of advanced glycation end-products (AGEs) and the second is related to the accumulation of acrolein, a by-product of lipid peroxidation, both can exacerbate each other.
Roles of GSH in the Detoxification of AGEs
AGEs arise from the nonenzymatic addition of sugars to proteins. Glycation can directly affect the protein structure, function, and aggregation. Although the enhanced formation of glycated proteins was initially associated with diabetes, it is now recognized that glycation of proteins is also increased in normal aging as well as in AD (30, 51, 214).
Glucose is often associated with AGE formation, but methylglyoxal (MG) or glyoxal (GO) could highly contribute to the generation of the intra- and extracellular AGEs because MG is more reactive than glucose (51). Thus, the most potent glycating agent and the major source of AGEs is the dicarbonyl MG, a by-product of glycolysis, glucose auto-oxidation, and lipid peroxidation. In mammalian cells, MG and GO are formed nonenzymatically from the triose phosphate glycolytic pathway intermediates, dihydroxyacetone phosphate and glyceraldehyde 3-phosphate (165).
The main pathway for the removal of MG is the glyoxalase system, which is composed of two enzymes, glyoxalase-1 (GLO-1) and glyoxalase-2 (GLO-2), and GSH (205) (Fig. 8). GLO-1 is the rate-limiting enzyme for the system and is dependent on GSH. GLO-1 and GLO-2 successively convert MG into S-D-lactoylGSH and
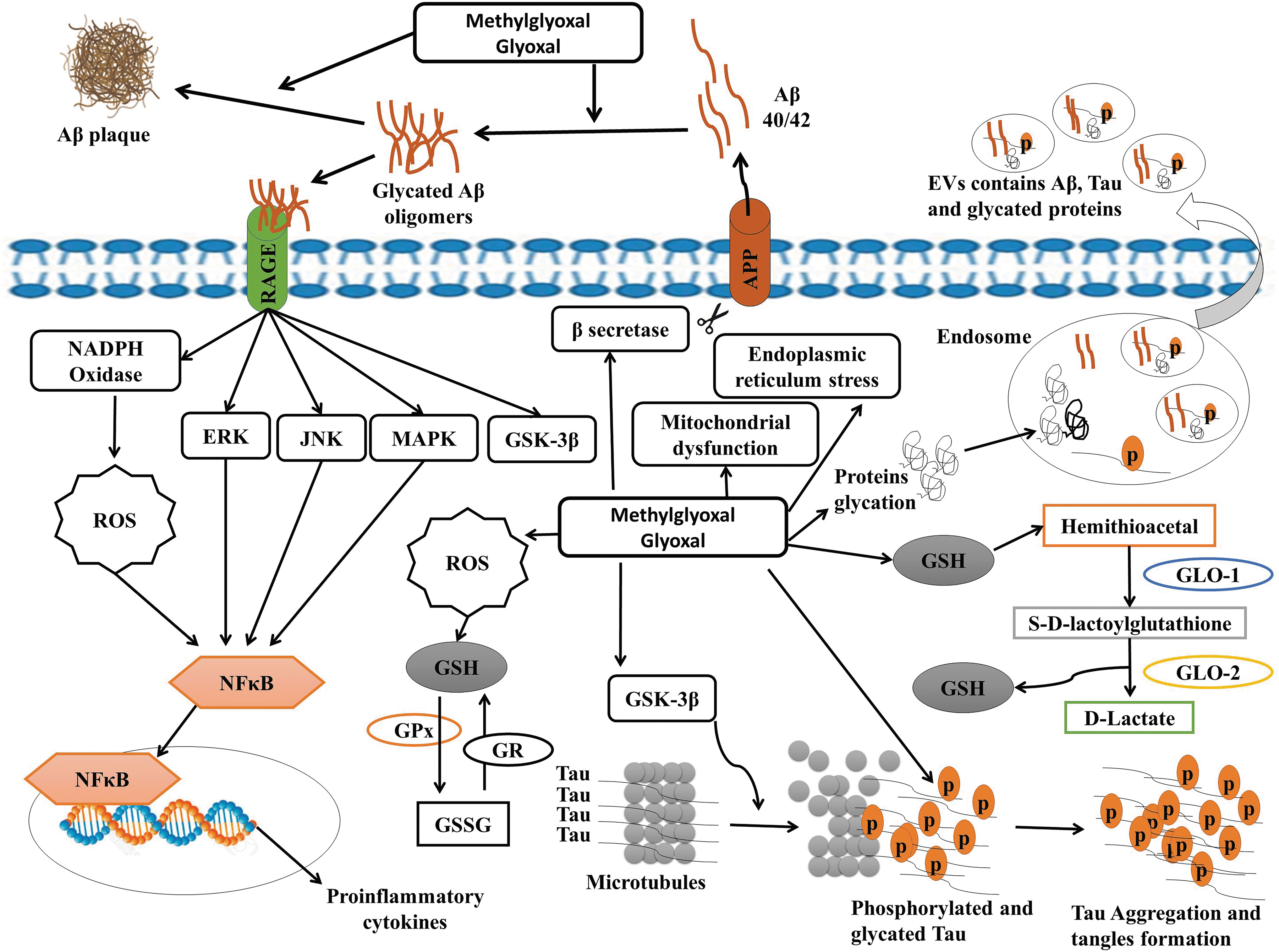
Interestingly, higher serum levels of MG-modified proteins (22) or pentosidine (216) were associated with a faster rate of cognitive decline in nondemented elderly subjects (>80 years). Recently, we found that the serum levels of a by-product of MG and GO, N-(1-carboxymethyl)-L-lysine (CML), were negatively correlated with the clinical cognitive scores, they were higher in the early stage of AD while the levels of pentosidine, another AGE, remained unchanged (79). We have also demonstrated that the levels of CML in the circulating extracellular vesicles (EVs) were lower in the moderate stage of AD (79). This accumulation of CML is probably due to the reduction of the glyoxalase system during the development of AD. This reduction was not attributable to low levels of the limiting enzyme GLO-1 because it is upregulated in the early and middle stages of AD (108) but probably caused by the decrease of its essential cofactor GSH in AD. Also, GSSG was found to be able to inactivate GLO-1 through covalent modification (27). Interestingly, in APP/PS1 mouse, the administration of ψ-GSH, a synthetic cofactor of glyoxalase, completely reversed the development of the spatial memory and long-term cognitive/cued-recall impairment, the Aβ deposition, and oxidative stress indicators (171). Therefore, the restoration of the glyoxalase system through GSH is a plausible mechanism for the attenuation of oxidative stress in AD.
The binding of AGEs to their receptors for advanced end-products (RAGE) induced the activation of multiple downstream mechanisms, which further affect neuronal functions, such as lipid metabolism impairment, activation of protein kinase-C (PKC), oxidative stress, inflammation, and apoptosis through the activation of the transcription factor NF-κB (75), inhibition of AKT-mediated activation of GSK-3, Erk1/2, and p38 kinases, as well as the upregulation of RAGE (114) (Fig. 8). In the brain, RAGE activation has been shown to induce inflammation, ROS formation, and impaired microglia (105), astrocytes (26), cerebrovascular (113), and neuronal cell viability (218). Thus, the depletion of GSH through AGEs and their precursors MG or GO can also directly or indirectly increase inflammatory responses and reduce neuronal viability (65). Besides, the formation of AGEs due to the depletion of GSH could induce the glycation of phospho-tau and Aβ oligomers, which exacerbate the in vitro and in vivo phospho-tau and Aβ toxicity (60, 94, 115). Thus, the depletion of GSH can indirectly contribute to neurodegeneration through the glyoxalase pathway, which represents an interesting pharmacological target to prevent or attenuate the toxic effects induced by AGEs or AGE derivatives in AD.
Roles of GSH in the Detoxification of Acrolein and Implication in AD
Acrolein is an α, β-unsaturated aldehyde with a molecular weight of 56 Da (CH2 = CH-CHO) and two reactive and functional groups: the aldehyde group and the carbon/carbon double bond. It is the strongest electrophile among the unsaturated aldehydes and therefore displays strong reactivity with nucleophile compounds such as GSH (61). Indeed, acrolein can form adducts with amino acid residues such as lysine, histidine, and cysteine (61, 135). As cysteine is more nucleophilic than lysine and histidine, it is thus the immediate target for acrolein.
We have previously demonstrated that acrolein induces rapid depletion of the neuronal and glial levels of GSH, reaching 50% only after 30 minutes but rebounded to control levels after 24 h (47, 152, 191). These data demonstrate the important role of the intracellular thiols in the removal of toxic aldehyde products resulting from lipid peroxidation or polyamine metabolism.
The depletion of GSH induced by acrolein due to alkylation reactions could be responsible for the modification of the ratio GSH/GSSG and thus the cellular redox state toward a more oxidizing condition. The thiol-acrolein adducts are very stable with a dissociation constant being 10 to 10,000 times lower than those with other aldehydes (61, 70, 103). Thus, thiol-acrolein can accumulate and induce cell death (47, 152, 191). The reaction between acrolein and GSH occurs spontaneously at neutral pH (2), at the third carbon, generating a glutathionylpropionaldehyde (GS-propionaldehyde). This compound can in turn elicit superoxide anion or hydroxyl radical formation in the presence of xanthine oxidase and be responsible for the induction of lipid peroxidation (2). GS-propionaldehyde can also be metabolized by aldehyde dehydrogenase (142). Glutathiolation can facilitate the aldehyde reduction by the aldose reductase, an enzyme implicated in the cellular response against oxidative stress (169).
It is now well recognized that by-products of lipid peroxidation such as 4-hydroxynonenal, F4-isoprostane, and acrolein are involved in AD pathogenesis (36, 47, 88, 134, 143, 144, 146, 147, 151, 152, 170, 171, 182, 184, 190, 195). In MCI and AD brain, levels of acrolein were found to be significantly higher in vulnerable brain regions such as the hippocampus and amygdala (120, 215). In AD, acrolein is associated with proteins detected in NFTs and dystrophic neuritis surrounding senile plaques (33) and levels of acrolein-conjugated protein in plasma and CSF were significantly higher than those from control subjects (207). By reacting with GSH, acrolein alters the ratio GSH/GSSG supporting the formation of S-glutathionylation or S-glutathiolation. Accordingly, an increase in S-glutathionylation proteins was found in the inferior parietal lobule of the AD brain (153). Thus, in AD, the depletion of GSH synthesis could render cells particularly susceptible to acrolein toxicity (12, 118).
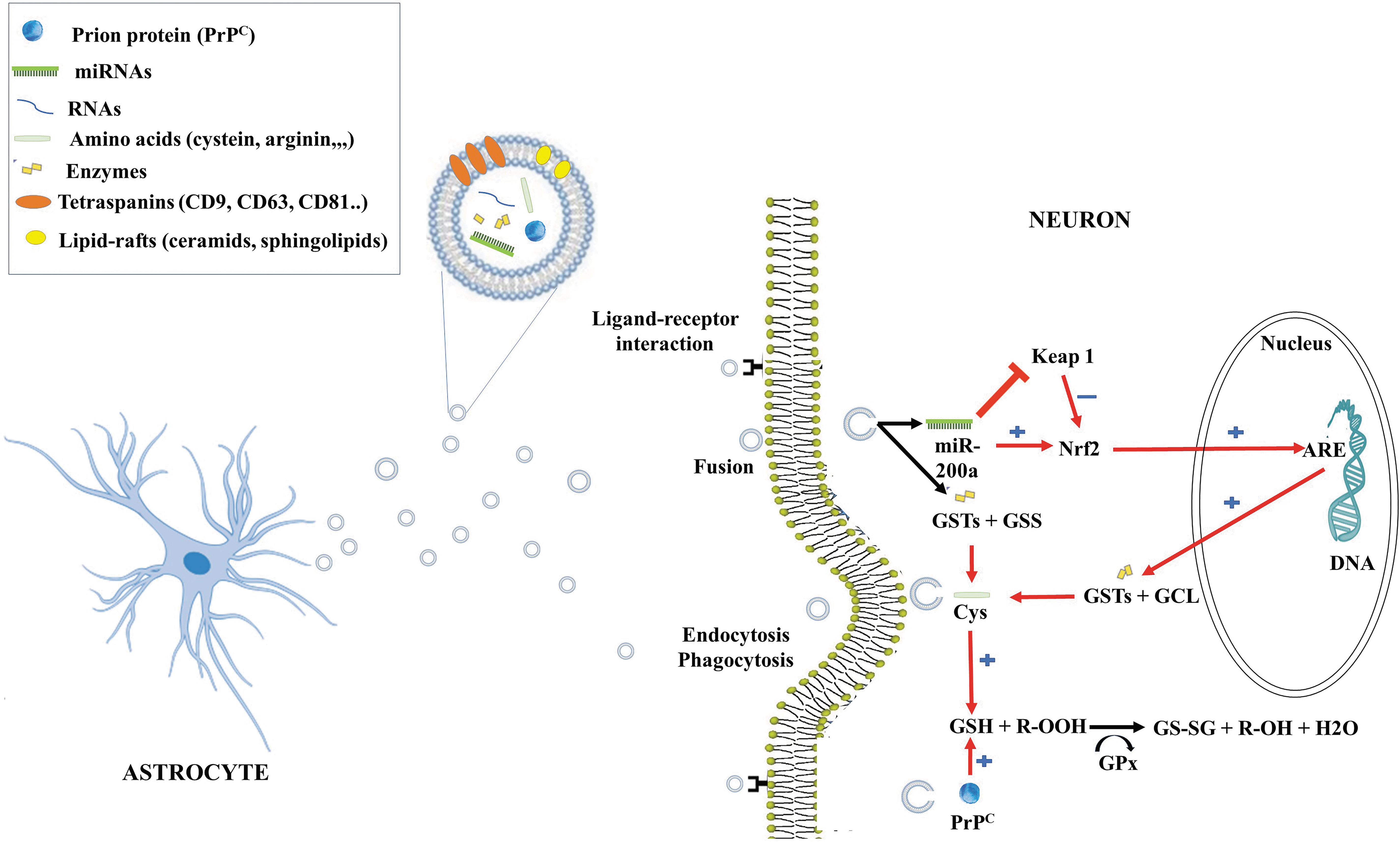
Role of EVs/Exosomes in the Secretion of GSH
Astrocytes are known to actively release divergent secretory organelles, including synaptic-like microvesicles and exosomes. Exosomes are released from astrocytes notably in response to oxidative and heat stress (201) and also in pathological conditions. Proteomic analysis revealed that exosomes released from different types of cultured cells contain various proteins. It is interesting to note that five target genes of the Nrf2-ARE are among the top 50 most commonly identified proteins in exosome preparation. Peripheral and platelet-derived EVs can, respectively, contain GSH synthase and GRX (102, 116, 166) but their content in neuronal or astroglial-derived EVs remains to be determined (Fig. 9). Also, the demonstration of the presence of GSH in exosomes will be interesting and could represent a new mechanism for the intercellular transfer of GSH.
GSH Pathways As a Therapeutic Approach
Given the implication of GSH in different pathways related to AD, several strategies to improve its plasma and brain bioavailability have been investigated to restore its intracellular level to prevent alterations observed in AD.
GSH synthesis is often limited by
It was found that γ-GC treatment increased the activity of selected matrix metalloproteinases (MMP2, MMP9) and MMP-9 has been shown to degrade Aβ deposits, which are dependent on GSH status (10). In opposite, GSSG can inhibit the insulin-degrading enzyme, the enzyme involved in the degradation of Aβ, by reacting with one or more of its cysteines (40).
Considering the crucial roles of GSH in various biological activities, a new pharmacological approach to raise or to maintain its levels should be encouraged. To date, there is no clinical trial with GSH restoration associated with cognitive improvement. Therefore, it is necessary to study the effect of GSH restoration in MCI and AD patients to evaluate its efficacy as a preventive or therapeutic potential.
Nanotechnology at the Rescue of GSH Deficit in Brain Cells and GSH-Pegylated Liposomes/Nanoparticles As Innovative Tools to Enhance Brain Delivery of Active Compounds
Several studies suggest that the restoration of the intracellular levels of GSH may have a disease-modifying effect (167). Thus, oral supplementation has been proposed to alleviate these deficits. However, oral delivery of GSH is a pharmacological challenge because GSH is a charged peptidic hydrophilic molecule with a low cell membrane permeability and low cellular barrier translocation. Moreover, GSH is very sensitive to degradation, with a high enzymatic degradation rate in the gastrointestinal tract, decreasing strongly its oral bioavailability. However, the daily consumption of GSH supplements was effective at increasing body compartment stores of GSH (174), but no benefit of oral uptake on GSH stock level or oxidation biomarkers has been reported in clinical trials (9). The design of the new delivery systems appears as a more promising avenue to overcome these challenges. Encapsulation in nanosized particles has been proposed to improve GSH bioavailability through protection from redox reaction or microbiome and gastrointestinal tract epithelial cell enzymatic degradation. Numerous oral liposomal forms of GSH have been commercialized with positive effects on the intracellular GSH stock and neuroprotection in vitro (221), GSH plasma level, and immune function in vivo (193). Other types of GSH-loaded nanoparticles (NPs) have been proposed for the oral route but none of them reached the clinical stage and few had presented the pharmacokinetic results in animal models (71).
One of the most important limitations for drug delivery to the brain is the presence of the BBB. Nanotechnology-based GSH has also been proposed to overcome the BBB to enhance the brain delivery of GSH based on nanocarriers. These carrier systems include, for example, liposomes and polymeric NPs that can potentially protect drugs from peripheral degradation and facilitate their transport into the brain. For this, poly(ethylene glycol) (PEG) is used as a surface modifier to reduce the toxicity, increase stability, and prolong circulation time. Coating nanocarriers or liposomal surface with ligands is also used to achieve active drug transport into the brain by targeting specific receptors located on the BBB. Different studies have also used GSH for stabilizing NPs (132).
The addition of GSH to a potential substrate for a transporter expressed at the BBB (99, 100), at the surface of liposomes/nanocarriers, can potentially enhance their brain delivery. For instance, PEGylated liposomes containing GSH (GSH-PEG) were found to enhance the brain delivery and the effects of doxorubicin and methylprednisolone in animal models with brain tumors and multiple sclerosis, respectively (69, 209). The pharmacokinetics of drugs changed greatly by encapsulation in GSH-PEG liposomes with more than 40-fold prolongation of the half-life, from several minutes to almost 7 h. Thus, GSH-PEG liposomes offer a promising system for enhancing and prolonging the delivery of drugs to the brain (117). Besides, the GSH coating does not interfere with the drug release (72).
Several teams reported enhanced delivery of nanocarriers' drug cargo into the brain via interaction between GSH-decorated drug-loaded NPs and vascular endothelial cells. GSH PEGylated liposome (“G-technology”®) enhanced the penetration of a fluorescent reporter and model drug into rat brain over untargeted liposome (138, 175). Similarly, the delivery of single-domain antibody fragments directed to Aβ was found to increase in mice when associated with GSH-modified liposome (177).
We have efficiently synthesized poly-lactic-co-glycolic acid 50:50 NPs (PLGA-NPs) coated with PEG and GSH (GSH-NPs) loaded with curcumin (GSH- NPs-Cur), using thiol-maleimide click reaction. For this, the GSH moiety was installed on the backbone of PEG-PLGA using the free thiol end group of GSH. We found that GSH functionalization did not affect the drug loading efficiency, the size, the polydispersity index, the zeta potential, the release profile, and the stability of the formulation. GSH-conjugation increases the neuronal uptake of these NPs. We have recently demonstrated that the presence of reduced GSH on the surface of the formulations exhibits a better neuroprotective property against acrolein. The neuronal internalization of GSH- NPs-Cur was higher than with free curcumin. We found that GSH-conjugation modifies the route of internalization enabling them to escape the uptake through macropinocytosis toward clathrin/caveolae-mediated endocytosis and therefore avoiding the lysosomal degradation (158). The GSH-coupled nanocarriers thus represent a promising approach for the functionalization of nanocarriers to efficiently cross the BBB for the delivery of drugs to the brain while avoiding cellular toxicity. Thus, the choice of ligand for ligand-modified NPs could be one of the potential strategies when developing a novel formulation against brain disorders such as AD.
Conclusions
Aging is the primary risk factor for AD. With aging, considerable oxidative stress contributes to the progression of neurodegeneration. GSH is the major antioxidant in the body with a particularly high concentration in the brain. Several studies have demonstrated that the levels of GSH are reduced with aging and in AD. In addition to its antioxidant effect, GSH is involved in various important pathways involved in detoxification and cell survival. The regulation of its homeostasis is challenging due to the complexity of the regulation of different enzymes involved.
Preclinical work supports a potential therapeutic role for γ-GC in the treatment of diseases associated with chronic oxidative stress and the cysteine precursor derivatives are FDA approved.
In the brain, astrocytes express complex exocytotic machinery that is associated with several types of secretory vesicles involved in the secretion of a wide variety of proteins, lipids, nucleic acid, neurotransmitters, and neurotransmitter precursors. To date, little research effort has been focused on exploring the role of exosomes on the transfer of GSH or its precursors to raise and replenish intracellular GSH and particularly to neuronal cells. More research is required to better understand the role of exosomes on the protection against oxidative stress and potentially their role in neuroprotection including the transfer of GSH. Nanotechnology appears as a promising tool to increase the GSH pool in brain cells; however, several challenges are yet to be overcome. GSH has also the potential as a targeting ligand for nanocarrier translocation across the BBB to treat various brain dysfunctions (3, 68, 154).
Footnotes
Authors' Contributions
Conceptualization, writing, editing, and validation: C.R. Writing—original draft: M.H. (50%), V.H. (20%), M.R.B.K. (15%), and J.-M.R. (15%).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors gratefully acknowledge the financial support from the Research Chair Louise & André Charron on Alzheimer's disease (C.R.), the Foundation Armand-Frappier (C.R., V.H., M.R.B.K.), Fonds de Recherche du Québec–Nature et Technologies (FRQNT) (C.R.), and support from NSERC (C.R., J.-M.R.).