
Abstract
Dr. Sue Goo Rhee is recognized as a Redox Pioneer because he has published five articles in the field of antioxidants and redox signaling that have been cited >1000 times and 69 of his articles in this field have been cited between 100 and 1000 times. Dr. Rhee is known for his discovery of the first three prototypical members of the phospholipase C family, and for the discovery of the ubiquitously expressed peroxiredoxins. Peroxiredoxin catalyzes the thiol-mediated reduction of H2O2. These enzymes protect cellular molecules from oxidative damage. Importantly, they also regulate cell signaling by modulating the intracellular levels of H2O2 that are induced by signaling agonists. He elucidated the mechanism by which the peroxiredoxins participate in signaling by H2O2: Dr. Rhee demonstrated that growth agonists such as epidermal growth factor induce a transient elevation of intracellular H2O2 that oxidize the catalytically essential cysteine residue of protein tyrosine phosphatases. The oxidation inactivates the phosphatases, allowing enhanced protein tyrosine phosphorylation to mediate cell signaling. In addition, he established that peroxiredoxins are exquisitely regulated through phosphorylation, glutathionylation, and hyperoxidation of their active site cysteine to cysteine sulfinic acid. Dr. Rhee showed that cysteine oxidation to its sulfinic acid derivative is not irreversible as previously thought. The reduction of hyperoxidized peroxiredoxin is catalyzed by sulfiredoxin. His further investigations implicated cyclic hyperoxidation and reduction of peroxiredoxin in the regulation of certain circadian rhythms.

I would like to express my gratitude to all of my colleagues, postdoctoral fellows, and students who made invaluable contributions to my research programs at the National Institutes of Health in Bethesda, Maryland, USA and at Ewha Womans University and Yonsei University School of Medicine in Seoul, Korea. I also want to thank Drs. P. Boon Chock and Rodney L. Levine, my long time colleagues at NIH, for nominating me as a Redox Pioneer.
—Professor Sue Goo Rhee
Background, Training, and Development
Dr. Rhee received his BS degree in 1965 from Seoul National University, Korea. After 2 years military service, he came to the United States for graduate study. He received his PhD in organic chemistry under the direction of John J. Eisch at the Catholic University of America in Washington, DC. In 1973, he accepted a postdoctoral fellowship in the laboratory of P. Boon Chock, a newly appointed senior investigator in the laboratory of biochemistry of the National Heart, Lung, and Blood Institute of the National Institutes of Health. Earl Stadtman headed the Laboratory of Biochemistry where the main focus of study was the regulation of glutamine synthetase in Escherichia coli. By the time Rhee joined Chock's laboratory, Stadtman's group had discovered that glutamine synthetase activity in E. coli is regulated by a bicyclic cascade mediated through reversible adenylylation (covalent attachment of an adenosine monophosphate moiety to a specific tyrosine residue). They showed that both adenylylation and deadenylylation were catalyzed by adenylyltransferase. The direction of the reaction was controlled by uridylylation (attachment of a uridine monophosphate moiety to a specific tyrosine residue) of PII (40). Rhee measured kinetic and equilibrium constants to quantitatively describe the adenylylation–deadenylylation cycle (37). He was promoted to tenured senior investigator in 1979. For several years, he continued studying the E. coli glutamine synthetase system, including purification and cloning of uridylyltransferase, and showed that uridylyltransferase catalyzes both uridylylation and deuridylylation of PII (11).
In the mid 1980s, Rhee began to move away from studying E. coli glutamine synthetase in favor of yeast glutamine synthetase. The yeast enzyme is extensively regulated in response to nitrogen status as in E. coli, but this is not mediated by nucleotidylation nor phosphorylation. During purification of yeast glutamine synthetase, a striking observation led Rhee's group to discover a family of unconventional antioxidant enzymes. He later named these enzymes “peroxiredoxins,” as described hereunder (34).
About the same time, Rhee was also investigating signal transduction initiated by cell surface receptors in mammalian cells. To this end, he discovered, purified, and cloned the first three prototypical members of the phospholipase C (PLC) family, enzymes that catalyze the hydrolysis of phosphoinositol bisphosphate, in response to transmembrane signals. Hydrolysis generates two second messengers, inositol 1,4,5-trisphosphate and diacylglycerol. These, respectively, induce Ca2+ release from endoplasmic reticulum and activate protein kinase C (32, 41). Rhee's group then established the mechanisms underlying the activation of PLC in cells stimulated by activation of various cell surface receptors. Through his seminal study, PLC-β was identified as the first effector enzyme activated by Gαq and PLC-γ as the first substrate of receptor protein tyrosine kinases (19, 32, 44). Together with the pathway leading to the activation of adenylyl cyclase to produce cAMP, the PLC activation mechanisms form a text book case of a receptor signaling pathway (32). Rhee also purified and characterized inositol 1,4,5-trisphosphate 3-kinase, which catalyzes the first step of the synthesis of inositol polyphosphate (9). In 1995, he was appointed as the founding chief of the laboratory of cell signaling in the National Heart, Lung, and Blood Institute.
Summary of Dr. Rhee's Major Contributions to the Field of Redox Signaling
Dr. Rhee discovered a family of peroxiredoxins that are highly expressed in mammalian cells. Like catalase or glutathione peroxidase, peroxiredoxins scavenge H2O2 through a thiol–disulfide catalytic cycle involving an intermediate sulfenic acid. In addition, peroxiredoxins undergo reversible inactivation to allow local accumulation of H2O2. This H2O2 then mediates cell signaling through oxidation of specific targets. Furthermore, Rhee showed that peroxiredoxin III, found exclusively in mitochondria, undergoes oscillatory hyperoxidation and reduction, with the reduction catalyzed by sulfiredoxin. Rhee's further investigation led to his proposal that the oscillatory process may regulate circadian rhythms.
Description of Key Finding 1
Discovery of the peroxiredoxin family
While studying the regulation of glutamine synthetase in Saccharomyces cerevisiae in 1983, Kangwha Kim, a postdoctoral fellow in Rhee's laboratory, noticed that the yeast enzyme, which had been purified to apparent homogeneity, lost activity and underwent partial degradation when stored in a buffer containing dithiothreitol. Evidence suggested that the glutamine synthetase degradation was not due to protease contamination. Around this time, Earl Stadtman and Rodney Levine in the Laboratory of Biochemistry were investigating the effect of oxidative modification on protein degradation (26). Influenced by their studies, Rhee's group investigated whether reactive oxygen species were the culprit. Studies revealed that the damage to yeast glutamine synthetase was actually caused by hydroxyl radicals (HO•). They were being generated from O2 through the Fenton reaction, catalyzed by Fe2+ produced through a dithiothreitol-mediated reduction of trace amounts of Fe3+ in the buffer (22).
The fact that glutamine synthetase in crude extracts of yeast remained active in the presence of dithiothreitol and contaminating Fe3+, while the purified enzyme was rapidly inactivated, suggested the existence of a yeast component that protected glutamine synthetase from inactivation. This protective component was purified and found to be a 25-kDa protein (21). Rhee's group subsequently purified another protector protein from bovine brain on the basis of its ability to protect glutamine synthetase from the dithiothreitol–Fe3+ oxidation system. They cloned genes that encode the yeast and rat proteins and found similar amino acid sequences for the two protector proteins (6).
The 25-kDa protector proteins did not resemble any antioxidant known at the time: All known antioxidant enzymes were dependent on a cofactor such as a heme, flavin, manganese, or selenium, none of which was found in the 25-kDa yeast and rat proteins. Critical information needed for the characterization of the 25-kDa proteins came from studies of a seemingly unrelated enzyme, bacterial alkyl hydroperoxide reductase (AhpC), which had been shown in the laboratory of Bruce Ames to catalyze the conversion of alkyl hydroperoxides to their corresponding alcohols at the expense of nicotinamide adenine dinucleotide phosphate (NADPH) (13). Initial comparison of the amino acid sequences of 25-kDa proteins and AhpC exhibited similarity only within a small NH2-terminal portion of the proteins. However, when Rhee's group resequenced the AhpC gene, similarity became apparent over their entire sequences, suggesting that the 25-kDa proteins also might be peroxidases (6).
Soon thereafter, Rhee's group found that the 25-kDa yeast protein reduces H2O2 and contains a cysteine residue at its active site. The electrons for the reduction of H2O2 were found to come from NADPH via thioredoxin reductase and thioredoxin. Serendipitously, dithiothreitol included in the homogenization buffer had provided the electrons in the initial studies. Rhee initially referred to this 25-kDa protein as thiol-specific antioxidant and subsequently as thioredoxin-dependent peroxidase, before settling on the name of peroxiredoxin, chosen because not all members of this family relied on thioredoxin as the electron donor (5).
A database search by Rhee's group subsequently found >25 protein sequences from a variety of organisms that showed similarity to the yeast/rat 25-kDa protein (6). None of these proteins had known antioxidant or other biochemical functions. Alignment of the sequences of the peroxiredoxin family members present in the database in 1994 revealed two highly conserved cysteine residues corresponding to Cys47 and Cys170 of yeast peroxiredoxin (6). The NH2-terminal cysteine [later designated the peroxidatic cysteine (CP)] was conserved in all family members and the COOH-terminal cysteine [designated the resolving Cys (CR)] was present in most but not all members. On the basis of the location or absence of the CR, peroxiredoxin enzymes were classified into 2-Cys, atypical 2-Cys, and 1-Cys peroxiredoxin subfamilies (34). Peroxiredoxin enzymes are present in multiple isoforms in most organisms (e.g., three in E. coli, five in S. cerevisiae, and nine in Arabidopsis thaliana).
In the early stage of peroxiredoxin studies, it was difficult to explain how the CP–SH could be oxidized specifically by H2O2, given that many other proteins also contain cysteine residues. The unusual reactivity of peroxiredoxin toward H2O2 was later explained by the crystal structure of a peroxiredoxin, in which two basic amino acid residues interact with the sulfur atom of CP–SH, thus promoting ionization of the thiol to the thiolate anion (8). It was known that only the thiolate anion was oxidized by peroxides. Structural studies carried out later in several laboratories revealed an extensive hydrogen bond network that lowers the activation energy for breaking the O–O bond of peroxides during the oxidation of CP-S− to sulfenic acid (CP-SOH) (12).
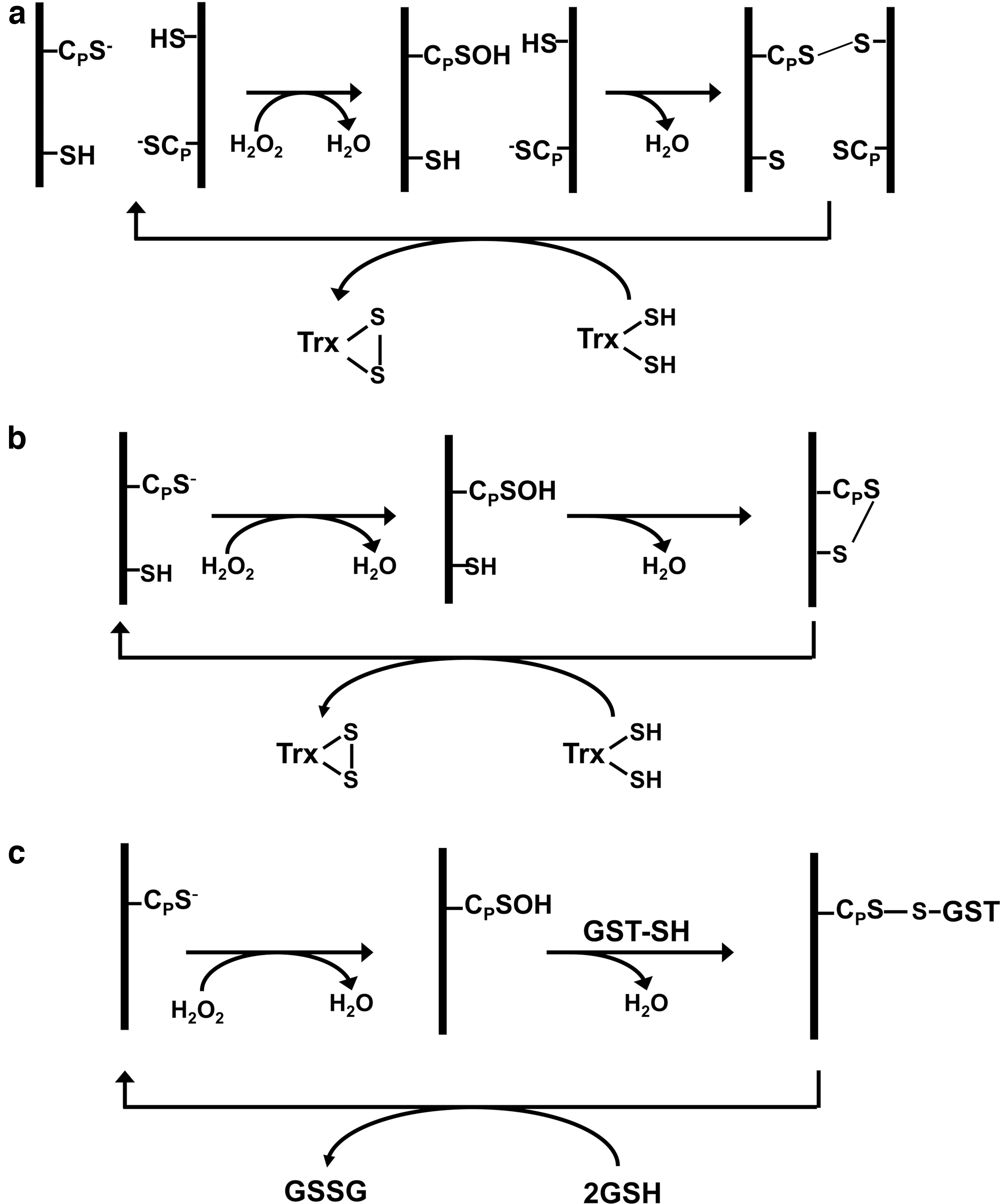
Rhee's group showed that most mammalian cells abundantly express six peroxiredoxin isoforms, four 2-Cys peroxiredoxin isoforms (peroxiredoxin I to peroxiredoxin IV), one atypical 2-Cys isoform (peroxiredoxin V), and one 1-Cys peroxiredoxin isoform (peroxiredoxin VI). Rhee's laboratory elucidated the catalytic mechanisms of peroxiredoxins I–V (34), and the reaction mechanism of peroxiredoxin VI was established by Fisher (10). All three types of peroxiredoxin enzymes share a common first step, in which the CP-S− is oxidized to CP–SOH (Fig. 1). In peroxiredoxins I–IV, the CP–SOH in the NH2-terminal region of one subunit of the homodimer is attacked by CR-SH located in the COOH-terminal region of the second subunit, resulting in the formation of an intersubunit disulfide bond that is subsequently reduced by thioredoxin (Trx) (Fig. 1a). In peroxiredoxin V, both CP and CR are present in the same polypeptide and, therefore, form an intrachain disulfide bond that is also reduced by thioredoxin (Fig. 1b). Peroxiredoxin VI lacks a CR residue, and the CP–SOH is resolved by a CR–SH provided by a glutathione S-transferase (Fig. 1c).
Description of Key Finding 2
Reversible hyperoxidation of 2-Cys peroxiredoxins and its significance
While studying the kinetics of a yeast peroxiredoxin and human peroxiredoxin I, Rhee and his colleagues observed that their peroxidase activity gradually decreased with time. They subsequently found that this inactivation was due to selective oxidation of CP–SH to the sulfinic acid (CP–SO2H) (53). The Cys–SOH generated as an intermediate during catalysis thus occasionally underwent further oxidation to Cys–SO2H, a reaction that cannot be reversed by thioredoxin and thus leads to peroxiredoxin inactivation. The presence of H2O2 alone was not sufficient to induce the oxidation of CP–SH of peroxiredoxin I to CP–SO2H. All of the catalytic components (H2O2, thioredoxin, thioredoxin reductase, and NADPH) were required, indicating that hyperoxidation occurred only when peroxiredoxin I was engaged in the catalytic cycle. Kinetic analysis indicated that the enzyme was hyperoxidized at a rate of 0.07% per turnover, about 1 out of 1000.
In 2000, Hyun Ae Woo, a graduate student in Rhee's laboratory, showed that the sulfinic peroxiredoxin I produced during exposure of cells to H2O2 was gradually reduced back to the thiol form after removal of H2O2 (47). This was surprising because oxidation to the sulfinic state was thought to be an irreversible process in cells. Subsequently, Michel Toledano's laboratory identified sulfiredoxin as the enzyme responsible for the reduction of sulfinic peroxiredoxin in yeast and showed that this reaction utilized ATP to activate the reduction process (3) (Fig. 2).

Critical cysteine residues of many other cysteine-containing proteins are also oxidized to sulfinic acid. Atypical 2-Cys peroxiredoxin (peroxiredoxin V) and 1-Cys peroxiredoxin (peroxiredoxin VI) also undergo hyperoxidation, although more slowly than 2-Cys peroxiredoxin. Rhee's group found that reduction of Cys–SO2H catalyzed by sulfiredoxin is specific to 2-Cys peroxiredoxin isoforms (48). Furthermore, prokaryotic 2-Cys peroxiredoxin enzymes were found to be insensitive to hyperoxidation, and prokaryotes did not contain sulfiredoxin (51). The inactivation of 2-Cys peroxiredoxin via cysteine hyperoxidation was suggested to be a result of structural features acquired during evolution to support regulation of cellular function by H2O2 (51).
To this end, hyperoxidation of a yeast 2-Cys peroxiredoxin was shown to convert the dimeric protein to a decamer. The decamer undergoes further aggregation and gains a protein chaperone function (14). This gain of chaperone activity concomitant with the loss of peroxidase activity after 2-Cys peroxiredoxin hyperoxidation was also observed in other organisms, including mammalian cells (38).
The detection of hyperoxidized peroxiredoxin I had initially relied on complex proteomics analysis involving two-dimensional polyacrylamide gel electrophoresis followed by mass spectrometry. Rhee's group subsequently devised a simple immunoblot assay for the detection of hyperoxidized 2-Cys peroxiredoxins by employing antibodies that bind to the conserved sequence surrounding CP–SO2H of these enzymes (49). This simple assay made it possible to link the seemingly wasteful inactivation–reactivation cycle of 2-Cys peroxiredoxins to circadian rhythms. Daily timekeeping by living organisms is largely driven by transcriptional–translational feedback loops that give rise to rhythmic expression of clock genes. The remarkable story of the circadian oscillation of peroxiredoxin–SO2H started with the discovery by O'Neill's laboratory of the hyperoxidation of peroxiredoxin as a transcription-independent circadian biomarker in a green algae and in red blood cells (28, 29). This was followed by the discovery in Rhee's group of the feedback control of steroidogenesis via peroxiredoxin III hyperoxidation in the mitochondria of adrenal gland (17).
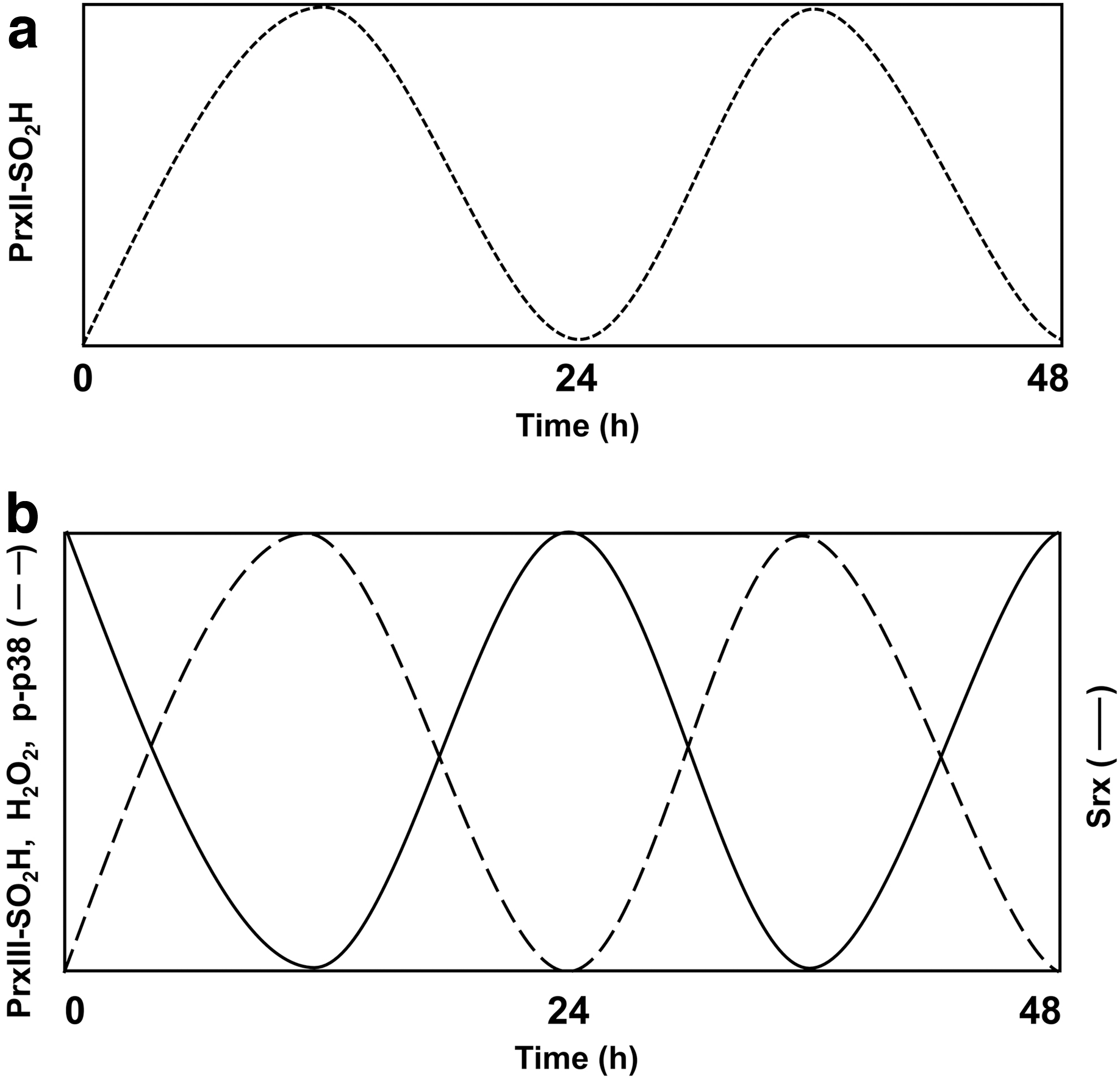
In 2005, Rhee returned to Korea to lead a newly established research institute at Ewha Womans University and was named a National Honor Scientist of Korea. In Korea, Rhee's group showed that peroxiredoxin II undergoes an oscillatory hyperoxidation in red blood cells and that the rising phase of the oscillation is driven by H2O2 produced from the autoxidation of hemoglobin during O2 transport. The decreasing phase is attributable to degradation of peroxiredoxin II–SO2H by the 20S proteasome in the red blood cells (7) (Fig. 3a). Rhee's group also found that peroxiredoxin III, which is exclusively localized in the mitochondria, undergoes an oscillatory hyperoxidation in the adrenal gland, brown adipose tissue, and the heart (17, 18, 36) (Fig. 3b). The rising phase of the peroxiredoxin III–SO2H oscillation is driven by H2O2 produced by cytochrome P450 during the conversion of cholesterol to corticosterone in mitochondria, whereas the decreasing phase is due to reduction of peroxiredoxin III–SO2H catalyzed by sulfiredoxin. Rhee's group showed that the oscillation results in the release of mitochondrial H2O2 into the cytosol followed by an increase in the level of phosphorylated p38 mitogen-activated protein kinase (MAPK), both of which oscillate approximately in parallel with peroxiredoxin III–SO2H (Fig. 3b). The level of sulfiredoxin in mitochondria also undergoes oscillation with a phase opposite to that of peroxiredoxin III–SO2H (Fig. 3b). Rhee proposed that peroxiredoxin III–SO2H formation and H2O2 release represent a circadian process that couples cholesterol metabolism (adrenal gland) or energy metabolism (brown adipose tissue and heart) to local clocks (35, 36).
Description of Key Finding 3
Intracellular messenger function of H2O2 and the localized regulation of peroxiredoxin
Through 1996, Rhee's research efforts were largely focused on the receptor-mediated activation of PLC, with only moderate attention being devoted to the identification and biochemical characterization of mammalian peroxiredoxin members. This pattern was slowly reversed as Rhee became interested in the potential signaling role of H2O2. Rhee reasoned that the role of multiple peroxiredoxin isoforms was not simply to scavenge peroxides, because cells are already equipped with efficient antioxidant enzymes such as catalase and glutathione peroxidase. He postulated that multiple peroxiredoxins would regulate the concentration of H2O2. This is analogous to the regulation of the local concentration of cyclic nucleotides via multiple isoforms of cyclic nucleotide phosphodiesterases. In support of this proposal, Yun Soo Bae, a postdoc in Rhee's laboratory, showed that epidermal growth factor (EGF) induced a transient accumulation of H2O2 in A431 cells. Blocking this accumulation resulted in the inhibition of EGF-dependent tyrosine phosphorylation of several proteins, including PLC-γ1 (2). Further, production of H2O2 had been detected in a variety of cell types stimulated with transforming growth factor-β1, interleukin-1, tumor necrosis factor-α, platelet derived growth factor (PDGF), or angiotensin II (33). Additionally, in 1999 David Lambeth's group identified a new type of NADPH oxidase isoform that could be coupled to the activation of growth factor receptors in nonphagocytic cells (42).
These findings led several groups including Rhee's to propose that H2O2 serves as an intracellular messenger (30, 31, 33). However, little was known about the molecules through which H2O2 acted to propagate the signal. Nor was it known how the H2O2 was removed in a timely manner to terminate the signal. Having learned through studies on peroxiredoxin enzymes that protein sulfhydryl groups with a low pK a can be selectively oxidized by H2O2, Rhee postulated that proteins with low pK a cysteines were the target of H2O2. He focused on protein tyrosine phosphatases, because the reversible inactivation of protein tyrosine phosphatase family members could explain why the production of H2O2 was required for growth factor-induced tyrosine phosphorylation, as shown by Toren Finkel's and Rhee's groups (2, 43). All protein tyrosine phosphatases share a Cys-X5-Arg signature motif (where X is any amino acid), in which the invariant, low-pK a cysteine residue functions as a nucleophile in catalysis.
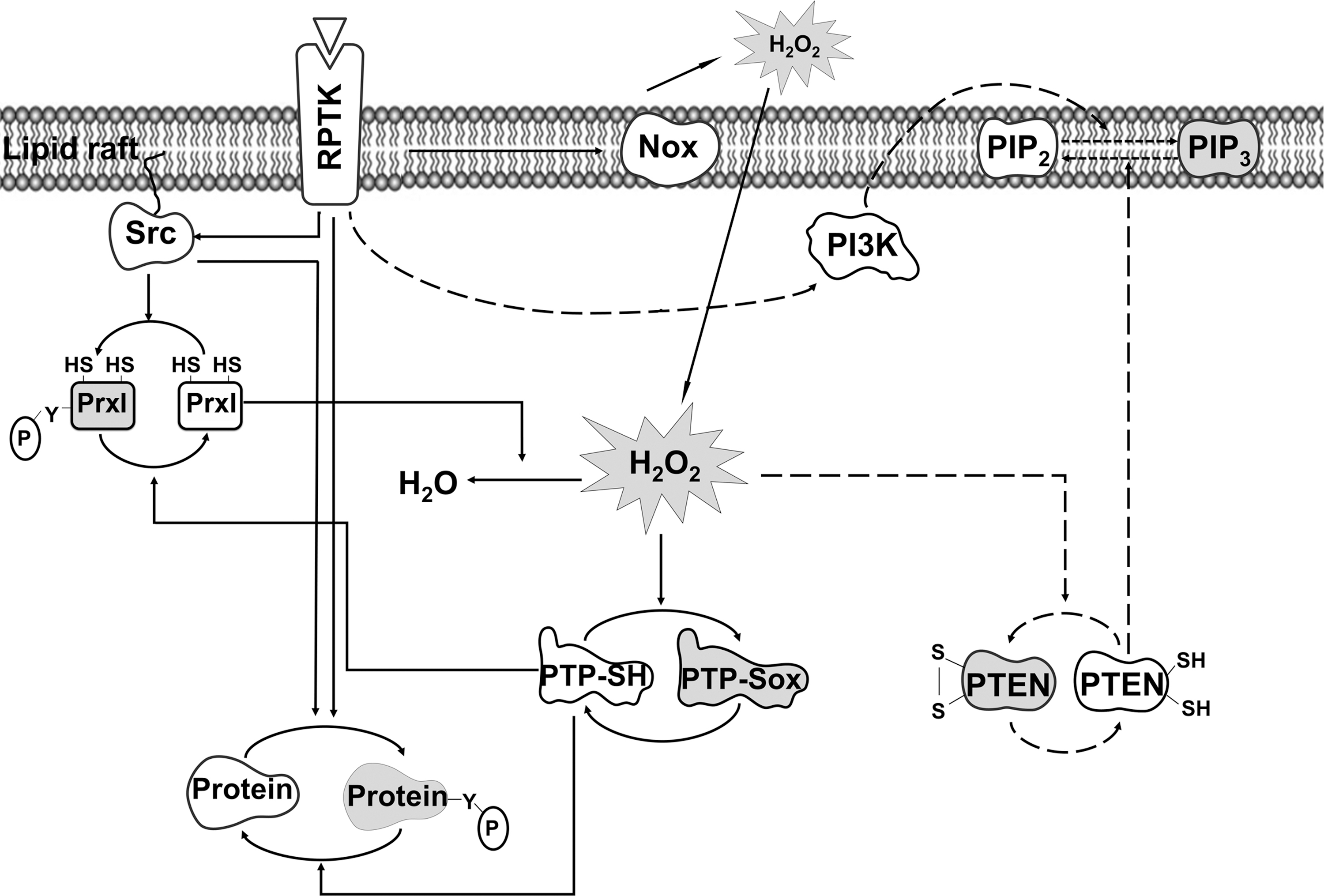
By taking advantage of the fact that reduced but not oxidized thiol can be alkylated with 14C-labeled iodoacetic acid, Seung Rock Lee in Rhee's laboratory showed that protein tyrosine phosphatase1B is reversibly oxidized by H2O2 produced in response to EGF stimulation in A431 cells (25). Rhee's group also showed that the tumor suppressor protein phosphatase and tensin homolog (PTEN), another member of the protein tyrosine phosphatase family, is an effector of H2O2 in PDGF-stimulated cells (23). On the basis of these findings, Rhee proposed that the activation of protein tyrosine kinases and phosphoinositide 3-kinase in growth factor-stimulated cells was not sufficient to increase the steady state level of protein tyrosine phosphorylation and phosphatidylinositol 3, 4, 5-trisphosphate (PIP3). Rather, concurrent inhibition of protein tyrosine phosphatases and PTEN by H2O2 was also required for these effects (30) (Fig. 4). Subsequently, not only protein tyrosine phosphatases but also many other signaling proteins including protein kinases, ion channels, and transcription factors have been shown to be regulated through the reversible oxidation of their cysteine residues by H2O2 (39).

Even though the target thiols of H2O2 effector proteins often have a low pK a, they react with H2O2 several orders of magnitude slower than CP–SH of peroxiredoxin (45). Furthermore, peroxiredoxins are abundant proteins with a high-affinity binding site for H2O2. Rhee thus felt that it was necessary to explain how the effector proteins can overcome such a competitive disadvantage and be oxidized by H2O2 that is produced transiently. Rhee launched an investigation into how the kinetic disadvantage of H2O2 effector proteins relative to peroxiredoxin can be overcome. He hypothesized that peroxiredoxin molecules near these effector proteins must be transiently inactivated to allow the effectors to react with H2O2. His studies uncovered two examples of such a scenario.
One is the reversible inactivation of mammalian peroxiredoxin I through phosphorylation on tyrosine-194. This is mediated by Src-family protein tyrosine kinases in cells stimulated by growth factors or by activation of T cell and B cell receptors (50). Only ∼0.3% of total peroxiredoxin I was found to be phosphorylated in PDGF-stimulated NIH 3T3 cells, because phosphorylation is confined to peroxiredoxin I molecules associated with lipid rafts in the plasma membrane. The spatially confined inactivation of peroxiredoxin I thus provides a means for generating a favorable H2O2 gradient around lipid rafts, where signaling proteins are concentrated, while preventing the global accumulation of H2O2 (Fig. 4).
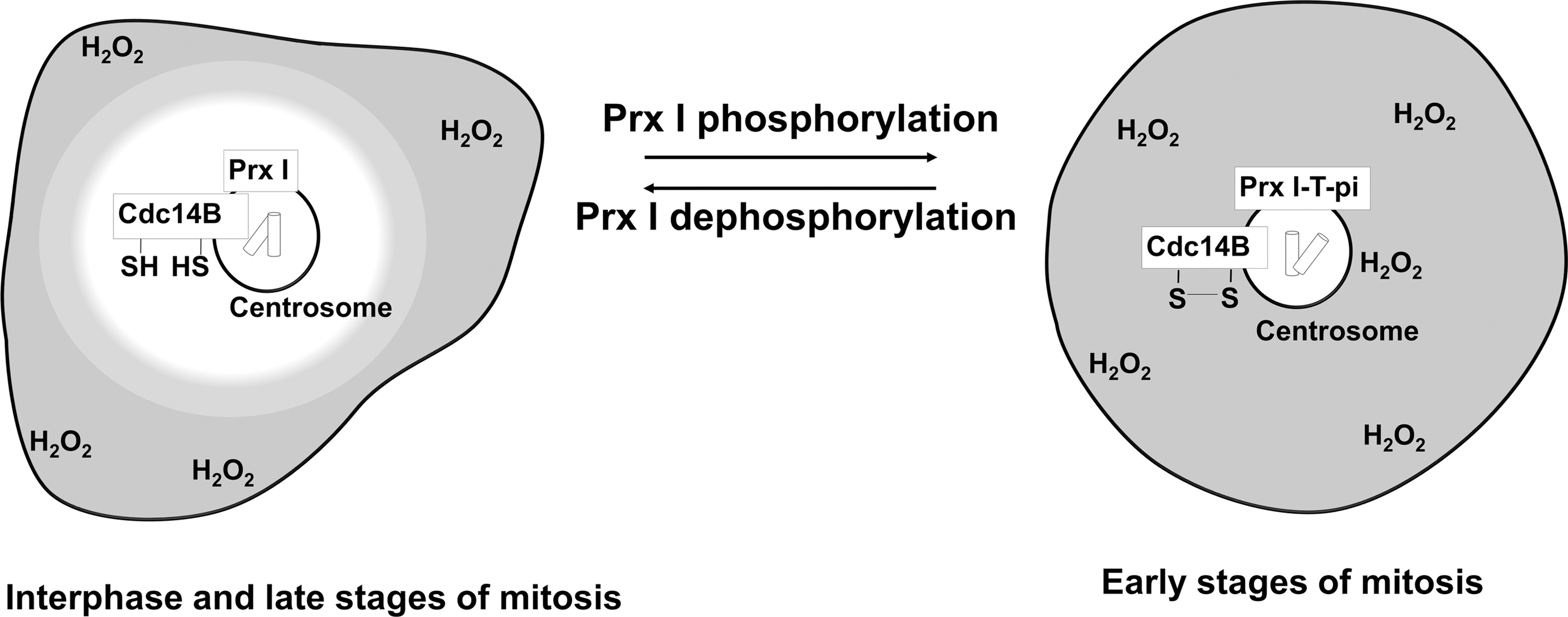
The second example is the reversible inactivation of peroxiredoxin I through phosphorylation on threonine-90 by cyclin-dependent kinase1 (Cdk1) during cell cycle progression (Fig. 5). The threonine-phosphorylated peroxiredoxin I was found at the centrosome during early stages of mitosis but not during interphase or late mitotic stages (27). The accumulation of H2O2 around the centrosome that results from the inactivation of centrosome-associated peroxiredoxin I facilitates oxidative inactivation of centrosome-bound dual specificity phosphatase cell division cycle 14B (Cdc14B), a protein tyrosine phosphatase family member. Inactivation prevents premature degradation of mitotic activators (27).
Other Achievements
Development of blot procedures to detect H2O2-sensitive proteins
After learning that the low pK a cysteines of protein tyrosine phosphatase1B and PTEN are targets of H2O2 generated in response to stimulation with PDGF or EGF, Rhee's group developed a procedure to detect proteins with reactive cysteine residues susceptible to oxidation by intracellularly generated H2O2. This approach was based on the labeling of cysteine residues with fluorescein-conjugated iodoacetamide (52) or biotin-conjugated iodoacetamide (20) followed by blotting of the labeled proteins with antifluorescein antibodies or with streptavidin.
Identification and characterization of thioredoxin-related protein 14 proteins
Using the blot analysis method, Rhee's group identified a 14-kDa rat protein as a target of H2O2 and named it thioredoxin-related protein 14 (TRP14) (16). TRP14 constitutes a family of proteins present from bacteria to mammals. All members possess a WCPDC motif, whereas all thioredoxin subfamily proteins possess a WCGPC motif. Rhee's group demonstrated that analogous to thioredoxin proteins, TRP14 functions as a disulfide reductase, although it acts on different substrates (15).
Identification and characterization of mitochondria-specific thioredoxin reductase
Peroxiredoxin III and thioredoxin 2 were known to be mitochondria-specific proteins. However, only one isoform of thioredoxin reductase had been identified in mammals. Rhee's group purified and cloned from rat liver a second type of thioredoxin reductase, named thioredoxin reductase 2 (24). They demonstrated that thioredoxin reductase 2 is specifically expressed in mitochondria and that together, peroxiredoxin III, thioredoxin 2, and thioredoxin reductase 2 provide a primary line of defense against H2O2 produced by the mitochondrial respiratory chain.
Identification of sestrin as a positive regulator of the Nrf2 pathway
A year after the discovery of the reversible hyperoxidation of 2-Cys peroxiredoxin and its catalysis by sulfiredoxin, it was reported that sestrin also catalyzed the reduction of 2-Cys peroxiredoxin–SO2H (4). Sestrin and sulfiredoxin show no sequence similarity, although sestrin expression is induced in response to oxidative stress. Rhee's group presented several lines of evidence that challenged the putative reductase function of sestrin (46). Subsequently, his group found that the sulfiredoxin gene is one of the targets of the major antioxidant transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) (1). Rhee's group also found that sestrin promotes the autophagic degradation of Keap1 (Kelch-like ECH-associated protein 1), a repressor of Nrf2 activity, and thereby upregulates Nrf2 signaling. This causes the induction of genes for antioxidant enzymes, including sulfiredoxin. Thus, Rhee established that the antioxidant function of sestrin is not due to sulfinic acid reductase activity, but rather to its role as a positive regulator of the Nrf2 pathway.
Current Position
In 2017, Dr. Rhee retired from the positions he held as Newilhan Professor of Biomedical Research and Director of Yonsei Biomedical Research Institute, Yonsei University College of Medicine, Seoul, Korea, and was appointed a visiting professor there. He returned to the United States and accepted an appointment as a special volunteer in the Biochemistry and Biophysics Center, the successor to the Laboratory of Biochemistry where he began his research career in 1973.
Footnotes
Acknowledgments
Dr. Rhee writes, “I would like to express my gratitude to all of my colleagues, postdoctoral fellows, and students who made invaluable contributions to my research programs at the National Institutes of Health in Bethesda, Maryland, USA, and at Ewha Womans University and Yonsei University School of Medicine in Seoul, Korea. I also want to thank Drs. P. Boon Chock and Rodney L. Levine, my long time colleagues at NIH for nominating me as a Redox Pioneer.”
Funding Information
This study was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.