
Abstract
Significance:
In humans, imbalances in the reduction–oxidation (redox) status of cells are associated with many pathological states. In addition, many therapeutics and prophylactics used as interventions for diverse pathologies either directly modulate oxidant levels or otherwise influence endogenous cellular redox systems.
Recent Advances:
The cellular machineries that maintain redox homeostasis or that function within antioxidant defense systems rely heavily on the regulated reactivities of sulfur atoms either within or derived from the amino acids cysteine and methionine. Recent advances have substantially advanced our understanding of the complex and essential chemistry of biological sulfur-containing molecules.
Critical Issues:
The redox machineries that maintain cellular homeostasis under diverse stresses can consume large amounts of energy to generate reducing power and/or large amounts of sulfur-containing nutrients to replenish or sustain intracellular stores. By understanding the metabolic pathways underlying these responses, one can better predict how to protect cells from specific stresses.
Future Directions:
Here, we summarize the current state of knowledge about the impacts of different stresses on cellular metabolism of sulfur-containing molecules. This analysis suggests that there remains more to be learned about how cells use sulfur chemistry to respond to stresses, which could in turn lead to advances in therapeutic interventions for some exposures or conditions.
Introduction
The element sulfur (S) is found in two proteinogenic amino acids, L-cysteine (Cys) and L-methionine (Met), as well as in a diverse group of other biologically important organic and inorganic small molecules. The ability of S to donate and accept electrons, and to access hypervalent states through bonding in its empty d-orbital, allows it to form bonds with other S atoms as well as with carbon, nitrogen, some metals/metalloids, and up to four oxygen atoms. This property, for example, allows S residues to participate in structural, yet nonetheless reversible, disulfide bonds within or between some proteins. More importantly for this discussion, however, it underlies Biology's expansive utilization of S in molecules and situations in which transient bonding is used as a conduit for electron transfers; that is, reduction and oxidation (redox) reactions. Indeed, S is probably the atom most often used to directly alter the oxidation state of oxygen atoms within our cells!
Within the active forms of Cys and Met used for protein synthesis and found participating in most cellular processes, the S is in a fully reduced state, with either the thiol-form in Cys or thioether-form in Met. To synthesize Met or Cys, cells need access to a source of S in its fully reduced state. Whereas many plants and microbes have sulfate reducing enzymes that allow them to use abundant sources of oxidized inorganic S to synthesize these S-amino acids, animals cannot reduce these oxidized forms (82). In animals, S-amino acids come entirely from dietary protein. Moreover, Met is essential in all metazoans, meaning that animals are incapable of synthesizing Met from other dietarily available S-containing nutrients, so a healthy human diet must provide an adequate source of Met (Fig. 1, box labeled [1]) (82, 118). Cys, on the contrary, can be synthesized from Met via the trans-sulfuration pathway (Fig. 1, box labeled [2]) and is only essential in humans in specific situations (discussed below). Whereas both Met and Cys are needed for protein synthesis, as compared with Met, Cys is more abundant in proteins, is a more broadly used precursor for the biosynthesis of other products in cells, and unlike for Met, many of these products are excreted from cells (Fig. 1). Thus, in addition to its incorporation into protein, Cys is the critical upstream S-containing precursor for the biosynthesis of glutathione (GSH), coenzyme A (CoA), hydrogen sulfide (H2S), hypotaurine, taurine, iron–sulfur (Fe–S) clusters, much of the sulfate in adenosine-5′-phosphosulfate and phosphoadenosine-5′-phosphosulfate (APS and PAPS, respectively), and several other S-containing molecules in cells (Fig. 1).
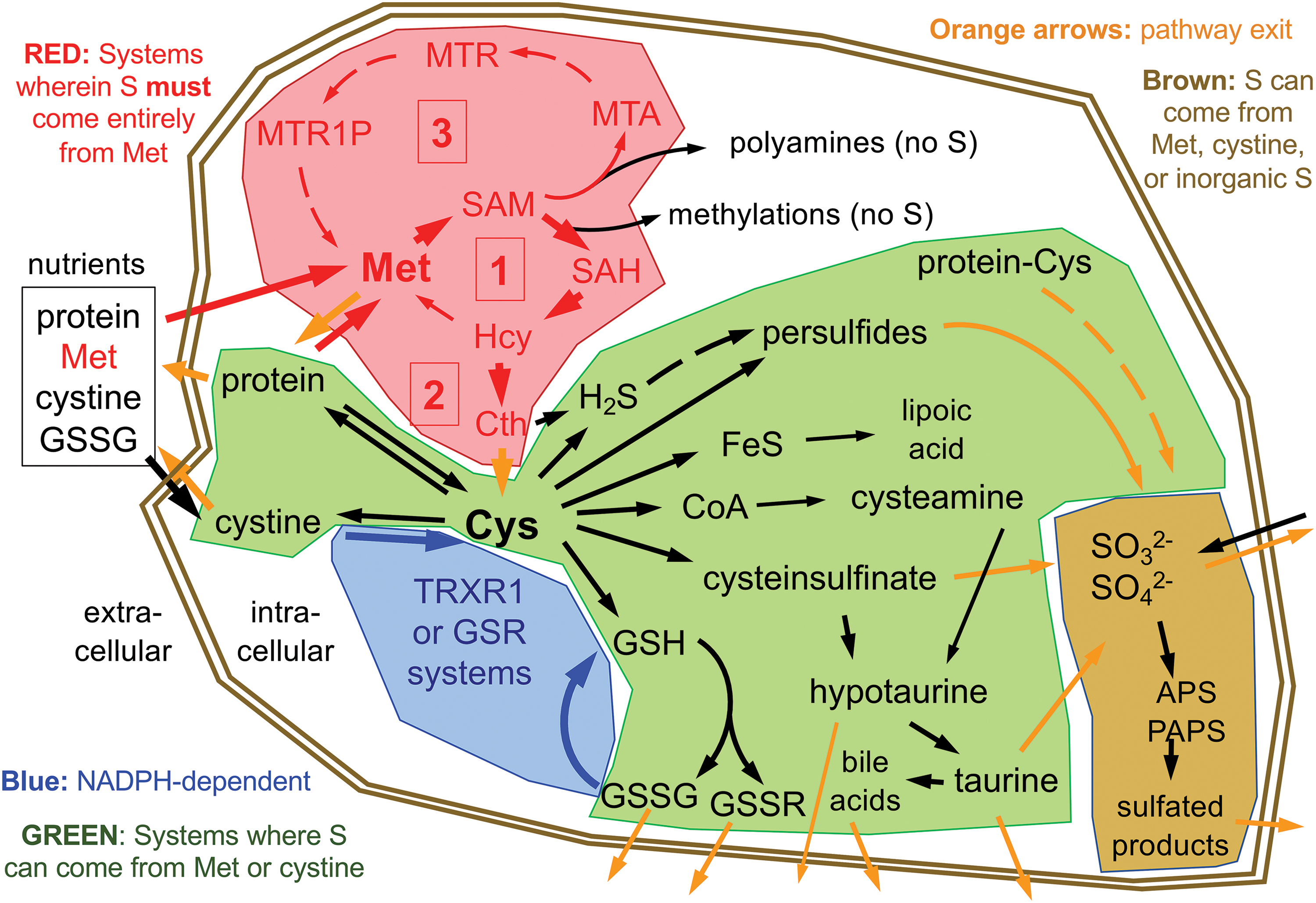
Diverse stresses on living cells or systems will interact profoundly with cellular S-containing molecules and therefore with S-metabolism. Most evident among these, perhaps, is oxidative stress, but it also includes other electrophilic stresses, reductive stress, toxin/drug/metal exposures, other energetic metabolic imbalances, carcinogenic stress, and likely other cellular or systemic insults. Interestingly, cancers can often experience overlapping combinations of many or all of these stresses, in particular during therapeutic interventions. Although each of these stresses can impact structural S-based bonds, this article will focus on the transient S-based bonds underlying the redox reactions affected by representative stresses.
Basal Mammalian S Metabolism
Dietary sources and systemic distribution
The predominant sources of all S in humans and other animals come from dietary protein. Uniquely among nutritional sources of S, Met is both essential and can generally serve as the sole source of nutritional S, if needed. In most diets, cystine (the oxidized disulfide form of Cys usually found in foods) is also a source of S. As long as sufficient Met is available in the diet, dietary Cys is not required in adult humans; however, dietary Cys is important during postnatal growth and development, making Cys a “semiessential” amino acid (96). Inorganic sulfate or sulfite from dietary sources can be utilized by human cells in a limited set of reactions, and some S-containing trace nutrients such as lipoic acid can be obtained directly from nutritional sources. Like all nutrients, sources of nutritional S enter the circulation via the portal vein and transit directly to the liver. In liver hepatocytes, considerable metabolism of S-based nutrients occurs, which are detailed below. Unsurprisingly, liver plays the predominant role in intermediary metabolism of S-amino acids, in particular Cys, by excretion of S-containing molecules into the peripheral circulation.
Unlike the reducing conditions found within the cytosol of most if not all living cells, extracellular fluids in the body, including blood plasma and extravascular interstitial fluids, are moderately oxidizing and will promote spontaneous oxidation of most free thiols, such as that within Cys. Moreover, abundant sulfhydryl oxidases in plasma, such as Qsox, catalytically oxidize free Cys into its homodimeric disulfide form, cystine (126). As a result, cells do not have access to substantial extracellular sources of free Cys. Rather, Cys is typically obtained from assimilation of cystine followed by intracellular reduction by the cytosolic NADPH-dependent disulfide reductase systems to yield 2Cys (discussed below), or from other abundant thiol- or disulfide-containing molecules in plasma. These include reduced glutathione (glutathione-thiol or GSH), its oxidized homodimeric form (glutathione disulfide [GSSG]), or oxidized glutathione conjugates (GSSX). Of particular importance is glutathionylated serum albumin (19, 129). Most of the glutathione in plasma is thought to come from hepatocytes as GSSG or GSSX; however, a substantial portion likely comes from erythrocytes as GSH (40). Importantly, however, there are no known transporters that can allow GSH, GSSG, or other glutathionylated products to enter the cytosol of cells. Rather, γ-glutamyl transpeptidase (GGT) on the cell surface hydrolyzes the γ-glutamyl bond of the glutathione moieties releasing the glutamate and, in combination with ubiquitous dipeptidases, also liberating
The S demands for synthesis of low abundance low turnover S-molecules, such as lipoic acid, CoA, FeS clusters, and polyamine synthesis (also called “methionine salvage”) pathway intermediates, represent a minor portion of total S metabolism and are not likely to impact S availability for the high-demand stress responses considered here. The different nutritional S sources, predominantly Met, cystine, and sulfite/sulfate, each enter cells with their sulfurs in different redox states, which determines how each can be utilized.
Sulfate/sulfite metabolism
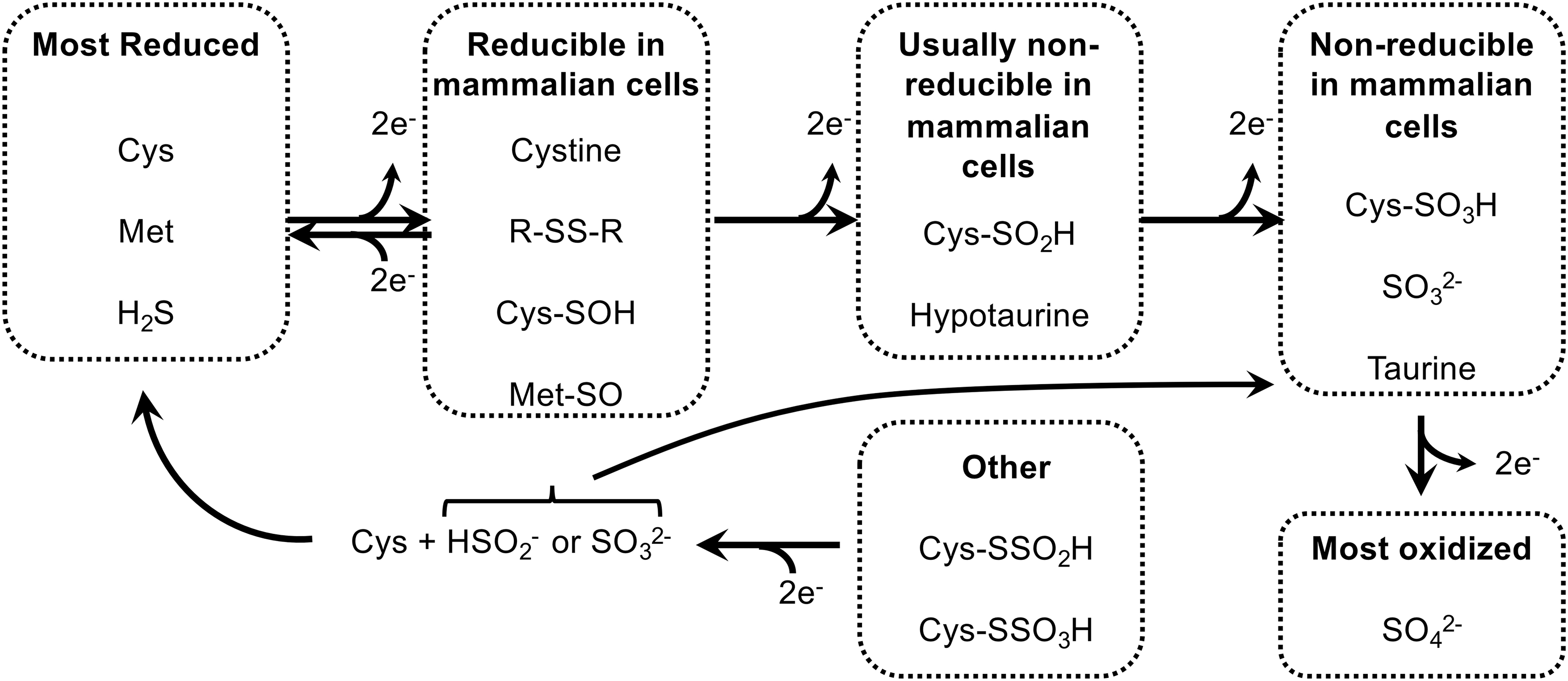
Inorganic sulfate is in the most oxidized state of the nutritional S-sources that mammalian cells can use; sulfite is one 2-electron state more reduced than sulfate (Fig. 2). Either sulfate or sulfite can be used to synthesize APS for production of PAPS (133). Subsequently, the oxidized S group from PAPS can be used in diverse reactions catalyzed by the sulfotransferase (SULT) family of enzymes. These enzymes function in the production of extracellular matrix molecules (e.g., heparin sulfate and chondroitin sulfate), sulfated steroids (e.g., estrone sulfate), bile acid sulfates, and sulfation-based phase-2 drug metabolism/detoxification reactions (22, 59). However, mammalian cells are unable to reduce inorganic sulfate or sulfite, nor are they able to reduce the oxidized S residues within any organosulfates. Therefore, dietary sulfite/sulfate cannot be used to generate S-amino acids. The inverse, however, is not true. Specific mechanisms, and likely also some spontaneous oxidation reactions, within our cells will allow S residues from more reduced species, for example within S-amino acids, to be oxidized to generate disulfides, sulfoxides, sulfenic acids, sulfinic acids, sulfonic acids, and inorganic sulfite or sulfate (101, 120). H2S may also act as a primary S source for generation of highly oxidized S species, although the specific mechanisms for formation of thiosulfate (S2O3 2−), S0, and sulfite from H2S are still debated (8, 53). As H2S is primarily derived from trans-sulfuration of Cys and Met, dietary S-amino acids can fully supply cellular oxidized sulfur needs for PAPS production and sulfation in the absence of nutritional inorganic sulfate/sulfite.
Cys and cystine metabolism
The reduced thiol group of Cys spontaneously oxidizes in most extracellular environments. In blood plasma, a common stable product of this oxidation is the cysteine-mixed disulfide of albumin; however, the most common nutritional form of Cys is the disulfide bond-linked homodimer of Cys, cystine (129, 132). After uptake through amino acid transporters (e.g., SLC7A11, also called xCT), cellular utilization of nutritional cystine requires cytosolic reduction of the disulfide bond to generate two molecules of Cys. The only systems capable of catalyzing this reaction are the cytosolic disulfide reductase systems; that is, the GSH system or the cytosolic thioredoxin-1 (TRX1) system (Fig. 1) (6, 82). Reducing power for the reaction comes from one of the two cytosolic NADPH-dependent disulfide reductase enzymes, glutathione reductase (GSR) or thioredoxin reductase-1 (TRXR1). These enzymes are able to obtain 2 electrons of reducing power from NADPH through a flavin cofactor and use this to reduce their respective substrates, generating the reduced form of their substrates (two GSH or TRX1-dithiol) and oxidized NADP+ as products (7). The relative contributions of each system, and of the different redoxin effectors of each system, including TRX1, other thioredoxin-related proteins (TRPs), or various members of the glutaredoxin (GRX) family, remain largely undefined. Biochemical and genetic studies suggest, however, that there is considerable redundancy and promiscuity in the systems that catalyze this reaction (83, 93).
Cys constitutes 2%–3% of the amino acids within proteins of mammalian cells, and this results in a concentration of cytosol-accessible protein-thiols in the low-millimolar range (41, 47). The redox activity of the thiol-S makes Cys residues particularly common in enzyme active sites, regulatory sites, and in positions where they can participate in disulfide bond formation. In addition to its roles in proteins, however, Cys is a “metabolic hub” for biosynthesis of other S-containing small molecules in cells (Fig. 1) (117, 119). Most abundant among these is the tripeptide GSH (γ-L-glutamyl-L-cysteinylglycine), which is also present at low-millimolar concentrations in cells (80). Importantly, whereas the predominant redox roles of GSH involve considerable recycling between the reduced GSH and oxidized GSSG forms, other often major roles for GSH, including hepatic export of GSSG as a systemic source of amino acids for intermediary metabolism and many GSH-S-transferase (GST)-catalyzed conjugation activities involved in detoxification reactions, result in loss of GSH from the cell. Unlike Cys, a substantial fraction of the plasma glutathione is in the reduced GSH form, with GSSG and protein-glutathione–mixed disulfides accounting for the remainder (129). Despite both protein and GSH contributing fairly equivalently to the total pools of solvent-accessible thiols within cells, the rapid turnover (43, 81) and frequent excretion of GSH from cells (71), as compared with proteins, suggest that a considerably greater amount of Cys might be committed to GSH biosynthesis than to protein synthesis. Other metabolites either synthesized from Cys or obtaining their S from Cys include CoA, Fe–S clusters, H2S, hypotaurine, taurine, and sulfate. Whereas due to their cellular retention and/or low cellular concentrations, CoA and Fe–S clusters are not likely to consume substantial amounts of Cys, taurine and likely also H2S and sulfate can be abundantly excreted, and therefore might drive substantial consumption of cellular Cys (38, 130).
Met metabolism
The S in Met is in the same fully reduced state as that in Cys; however as a thioether, it is refractory to spontaneous oxidation as compared with the thiol-S found in Cys (83). For that reason, cells are able to obtain Met in its fully reduced form directly from nutritional sources. Met is the initiator amino acid for translation of nearly all proteins in mammalian cells and, even though the initiator Met is often post-translationally removed, due largely to the use of Met internally in proteins, it constitutes 1%–2% of the total steady-state amino acids in animal protein (41). Like Cys, Met also acts as a metabolic hub, although to a lesser extent (120). Specifically, Met enters the Met cycle, which provides the methyl source for most cellular methylation reactions. The Met cycle, which is the transit of Met to S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH),
As its name implies, trans-sulfuration effectively transfers the fully reduced Met-derived S from Hcy to Cys. In this two-step process, all other Met-derived atoms are discarded to other metabolic pathways as α-ketobutyrate and ammonia, and a new amino acid skeleton is obtained from L-serine (Ser). Thus, in addition to providing an “outlet” to prevent toxic overaccumulation of Met-cycle intermediates, this pathway provides cells with a route of de novo Cys biosynthesis. In hepatocytes and some other cell types, this pathway is exceptionally robust. In hepatocytes, ∼1/2 of the S in Cys comes from Met via this route (87). Importantly, liver is largely responsible for intermediary metabolism of Cys, and it provides Cys to the body by excretion of GSSG into the blood. In addition, red blood cells provide a source of circulating free GSH (40). Likely dependent on the respective nutritional availability of cystine versus Met, the S in the GSSG excreted from liver can likely be fully derived from either Met or cystine.
Besides trafficking S from the Met cycle to Cys, the trans-sulfuration pathway is the predominant source of cellular H2S production in mammals (61, 114). Since metazoans cannot synthesize Cys from H2S, this has been generally considered a one-way outlet of S from the pathway; however, more recent advances in understanding the relationships between H2S, protein persulfidation, and the enzymatic activities involved in catalyzing sulfide-based chemistry in mammalian cells hint at potentially far more complex and physiologically important fates for cellular H2S (see the S-Persulfidation in Mammalian Cells section and other papers in this issue).
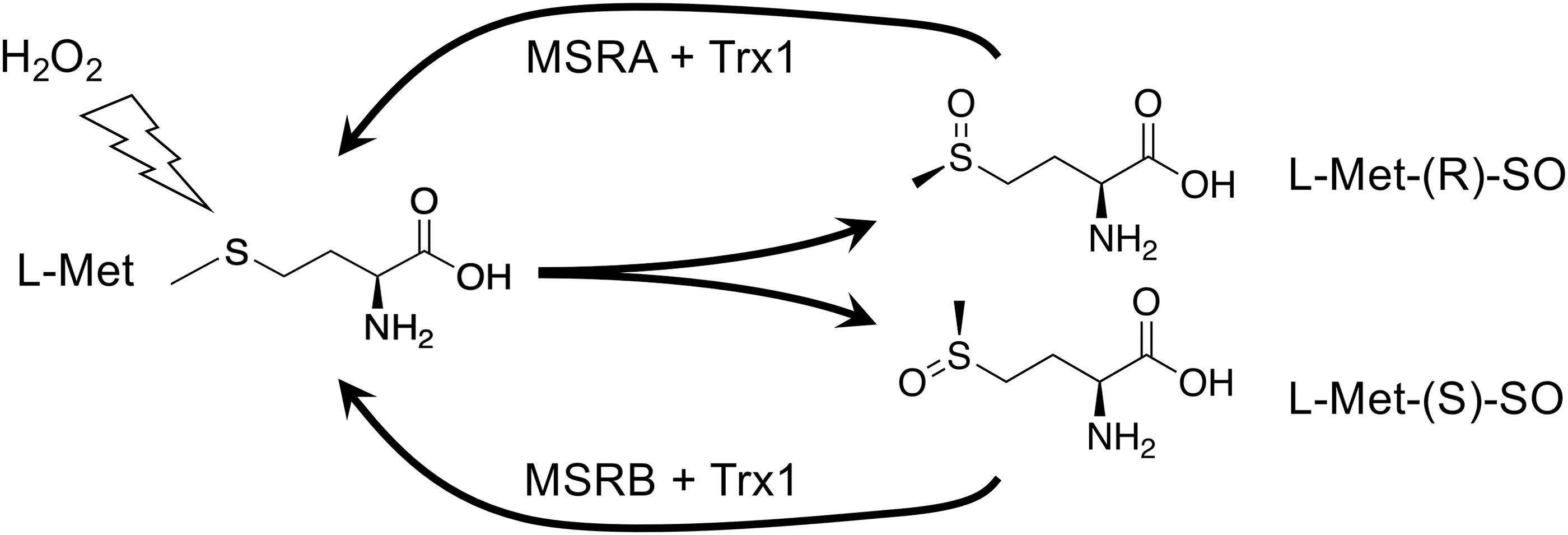
Although the thioether of Met is resistant to oxidation, exposure to oxidants can result in a 2-electron oxidation to Met-sulfoxide (Met-SO), which can adopt either of two diastereoisomeric forms: the R- or the S-form (Fig. 3). Although the R- and S-forms will occur randomly upon oxidation of free Met, the context of a Met within a protein or complex might influence which form will be adopted. Met-SO can be reduced back to Met by the TRX1-dependent Met-SO reductase (MSR) enzymes of which, interestingly, MSRA will only reduce the R-isomer and MSRB will only reduce the S-isomer (70, 123). MSRA and MSRB can be differentially expressed and differently subcellularly localized, and it has been proposed that different accumulation or repair of Met-SO diastereomers could serve regulatory or signaling functions in cells (92).
Impacts of Stress on S Metabolism
Oxidative stress and endogenous antioxidant systems
The predominant source of reactive oxygen species (ROS) in most cells under most conditions is the mitochondrial electron transport chain (ETC) (17). In addition, ROS can arise as collateral by-products of energetic metabolism (e.g., β-oxidation of fatty acids), anabolic metabolism, detoxification or anabolic activities of cytochrome p450 (CYP) enzymes, as primary products of NADPH-oxidases (NOX), or as a result of toxin-driven redox cycling by diverse NADPH-dependent enzymes (76, 79, 95, 113). Another major source of oxidative stress arises extracellularly, largely from either defensive or pathological production of ROS by activated inflammatory cells (111).
For the classical case of mitochondrially generated ROS, a small amount of incompletely reduced oxygen as superoxide radical (O2 −•) is collaterally generated by the ETC even under normal conditions. This O2 −• is rapidly dismutated to hydrogen peroxide (H2O2) by abundant superoxide dismutases (SODs). Conditions like ETC disruption by various toxins or ischemia/reperfusion injury can dramatically increase the production of O2 −• by the ETC. The efficacy of SODs renders H2O2 the predominant species in both the mitochondria and the cytosol (109, 110, 112). Each compartment has its own similar systems for detoxifying H2O2; for simplicity, we will focus here on only the cytosolic systems.
Cells efficiently eliminate cytosolic H2O2 via highly active and abundant peroxidases: the peroxiredoxins (PRXs) and glutathione peroxidases (GPXs), each of which obtain reducing power from the disulfide reductase systems (35, 111, 136). These peroxidases utilize cysteine thiols either intermolecularly (e.g., in “1-Cys PRXs”), intramolecularly (in “2-Cys PRXs”), or within a Cys-L-selenocysteine (Cys-Sec) pair in their active sites (e.g., in most GPXs, in which the GPX-Sec residue collaborates intramolecularly with a Cys from GSH or a GRX) (91). After each catalytic cycle, the oxidized enzymes require 2 electrons of reducing power to return the peroxidase to its active state. This reducing power is most often acquired from other thiol-based reductants, such as TRX1 or GRX/GSH. As discussed above, the upstream source of this reducing power is NADPH, which fuels the disulfide reducing systems via GSR or TRXR1 (82).
Other stresses
Electrophilic stress
Whereas small inorganic nonmetallic oxidizing molecules are typically called “oxidants,” metallic or somewhat larger organic oxidizing molecules are more commonly referred to as “electrophiles.” Both groups will readily accept electrons from other molecules, and therefore qualify as oxidants and electrophiles. Moreover, both are primarily detoxified by S-based reductase systems in cells. However, based on their other molecular properties, each of them is generally targeted by different detoxification enzymes, and this activity can have very different impacts on the cells and on S metabolism pathways. For example, H2O2 is considered an oxidant, and is detoxified by reduction to water and molecular oxygen. In the case of detoxification of H2O2 by GPXs, the reducing power for this comes initially from cellular GSH pools (16, 35, 127). In contrast, organic electrophiles such as reactive quinones are more often detoxified by conjugation to GSH by GSTs, followed by excretion of the glutathionylated product out of the cell (49). Lipid-hydroperoxides (ROOH) are relatively large organic electrophiles, yet they are often detoxified by direct enzymatic reduction by GPX4, for which the reducing power comes from GSH (35). Heavy metals (e.g., Au) and metalloids (e.g., As), on the contrary, are often monoatomic oxidants, yet they are more likely to be primarily detoxified by glutathionylation (104). Although both GPX- and GST-driven detoxification reactions use GSH, each has distinct impacts on the GSH system and on supporting metabolic pathways. GSH-driven reduction by GPXs generates intracellular GSSG, which, as long as the intracellular disulfide reductase systems are intact, consumes NADPH and fully regenerates the 2GSH without loss of the component atoms of the GSH. This reaction, therefore, consumes cellular energy sources to maintain NADPH:NADP+ pool ratios, but does not result in loss of cellular S (35). GSH conjugation and export, by contrast, irreversibly removes S from the cell (14, 27), which must be replaced by acquisition of new S-amino acids from extracellular sources (83).
Reductive stress
The need for cells to maintain a balanced redox environment, not only avoiding excessive accumulation of oxidants but also avoiding an overly reducing environment, is often overlooked. Oxidative activities of cells play critical roles in all aspects of physiology, and if the intracellular milieu becomes excessively reducing, it might interfere with these activities (15, 143). Among these activities is redox signaling, wherein oxidants need to react with regulatory protein-thiols as a part of the signaling mechanisms that coordinate cellular activities. Indeed, it is easy to artificially overexpress antioxidant systems in cells, showing that it would be a small matter for cells to have increased antioxidant activities. However, evolution did not give us a more reducing cytosol. Why? Likely because any benefits from increased resistance to oxidative stress would be negated by the detriments of the resultant reductive stress.
Excessive accumulation of GSH can cause reductive stress (143). However, a diverse battery of metabolic and regulatory systems in cells generally prevent this. For example, the heterodimeric rate-limiting enzyme for GSH biosynthesis, γ-glutamyl-Cys ligase (GCL), is strictly regulated to keep GSH levels balanced. GCL, itself, is catalytically inhibited by GSH; this feedback prevents excessive GSH accumulation (36, 103). Moreover, both the catalytic (GCLC) and modulatory (GCLM) subunits of GCL are regulated by the stress-responsive nuclear factor erythroid 2-related factor 2 (NRF2) pathway (85), such that under conditions of oxidative or electrophilic stress, GSH biosynthesis capacity is increased to restore the redox balance (see Master-Regulation of S Metabolism in Stress section). Recent evidence shows that cells also use export of GSSG from subcellular compartments (90, 97) or from the cytosol (28, 86, 89) to eliminate excessive GSSG, thereby modulating both total levels and the redox status of glutathione pools.
Drug/toxin/xenobiotic stress
Many drugs, toxins, and other xenobiotics interact with the cellular disulfide reductase systems. However, in terms of understanding how this impacts the cells, much can be inferred by assessing whether the end-result of this activity consumes cellular NADPH, cellular S, or both (Fig. 4). Compounds that uncouple mitochondrial electron transport, such as rotenone, can induce production of O2 −• → H2O2 (Fig. 4A) (84). Typically, this will be remediated by GPX and PRX responses, which consume recyclable disulfide reducing power (from GSH or TRX1, respectively), supported by GSR and TRXR1 using electrons obtained from NAPDH (35). Upstream of this, the NADPH-regenerating systems typically consume resources that must be diverted from energetic or anabolic processes (glucose, malate, or isocitrate) (21, 82). By contrast, drugs or toxins that are conjugated to S-containing molecules (e.g., glutathionylated by GSTs or sulfated by SULTs) and excreted, for example aflatoxin-B1, will irreversibly consume Cys/GSH and tax cellular S-amino acid stores, but this will not substantially consume NADPH (Fig. 4B) (24, 45). Some electrophilic drugs or toxins, for example, organoarsenites (As3+) or organomercurial compounds, are glutathionylated yet also covalently bind with high affinity to Cys or Sec residues of GSR, TRXR1, and other critical enzymes, thereby irreversibly disrupting activity of these enzymes (104). Importantly, most of these metals are cytotoxic at low doses, suggesting that consumption of cellular S-stores is not a major determinant of toxicity. Rather it is likely the dominant disruption of enzymatic activities in the cells is critical (Fig. 4C). Redox-cycling drugs or toxins, such as doxorubicin or paraquat, will interact with NADPH-dependent reductases in the presence of molecular oxygen to catalytically generate large amounts of O2 −• (37). This redox cycling will consume cellular NADPH stores and, after the O2 −• is dismutated to H2O2, additional NADPH will be consumed by GSR or TRXR1 to support peroxidase-driven reduction of the H2O2 (Fig. 4D).

As a culminative example, some drugs, such as acetaminophen (paracetamol), can have even broader impacts on these metabolic systems (Fig. 4E) (99). Upon entering liver hepatocytes, much of the acetaminophen is conjugated to sulfate by SULTs or to glycogen-derived sugars by UDP-glucuronyltransferase and exported by multidrug resistance (MDR) or ATP-binding cassette (ABC) organic ion transporters, thereby removing oxidized sulfur and depleting energetic resources (glycogen) (56). At higher doses, these systems become exhausted and excess acetaminophen is chemically reduced by CYP enzymes, thereby consuming NADPH. The product of this reaction, N-acetylbenzoquinoneimine (NAPQI), is a highly electrophilic quinone that is aggressively glutathionylated by GSTs. Subsequently, the conjugated product is excreted by ABC/MDR exporters, thereby irreversibly consuming cellular GSH reserves and depleting S-amino acids. At still higher doses, GSH pools can become exhausted, allowing free NAPQI to accumulate (34, 58). In these conditions, NAPQI also inhibits the active site residue in TRXR1 (56) and likely with nucleophilic active sites in other redox enzymes. This interferes with the cells' ability to regenerate disulfide reducing power and it might induce redox cycling, which would consume more NADPH and catalytically generate O2 −• → H2O2. Subsequently, reduction of the H2O2 by peroxidases will require disulfide reducing power and therefore more NADPH, although these activities might already be compromised by inactivation of TRXR1 and depletion of GSH. In severe overdose situations, hepatic glycogen, GSH, S-amino acids, and NADPH all become severely depleted; TRXR1 becomes unable to reduce TRX1-disulfide, peroxidases cannot be supported, and O2 −• production increases. This “perfect storm” can result in necrotic hepatocyte death and acute liver failure. Indeed, this is the leading cause of acute liver failure in the United States and Europe (12). However, even under such severe overdose conditions, hepatocytes that have not yet died can often be rescued by administration of N-acetyl-L-cysteine (NAC), and in the clinic NAC treatment saves the lives of many acetaminophen-overdose patients (99). Whereas NAC is often referred to as an “antioxidant” or a “ROS scavenger,” it likely does not serve as a direct antioxidant because the reaction of the NAC thiol with H2O2 is too slow to be biologically relevant (68 M −1s−1), in particular under severe oxidative challenge (138). Although much remains to be resolved about the complex roles of NAC in cells (30), in the case of acetaminophen overdose, one likely role of NAC is to provide the hepatocytes with an alternate source of Cys that can be assembled into GSH to support continued glutathionylation and export of the NAPQI, and possibly also to allow continued trafficking reducing power from GSR to GPXs (2, 42, 56). In addition, recent studies have shown that NAC can act as a potent Cys-persulfidating agent, which might protect protein thiols from overoxidation, not only in the context of GSH depletion but more universally under diverse oxidative stress conditions as well (discussed below) (30).
Metabolic stresses
The interplay between the disulfide reductase systems and metabolism is finely tuned by a complex network of feedback mechanisms that are still being resolved. As suggested above, the relative availability of Cys versus Met sources in the diet will influence flux through S-amino acid metabolic pathways (54, 78). However, most mammalian cell types have only modest trans-sulfuration activity, and likely rely on having a ready supply of extracellular cystine and intracellular disulfide reducing power to support their intracellular Cys demands. Intermediary metabolism by liver hepatocytes provides a constant supply of extracellular cystine to the whole body. Thus, the hepatocytes maintain circulating levels of plasma GSSG/GSSX, and erythrocytes provide circulating GSH (see the Dietary sources and systemic distribution section). Within the hepatocytes, GSH is synthesized using Cys, which can be acquired from either dietary cystine and reduce to Cys by the NADPH-dependent disulfide reductase systems, or from dietary Met via their robust NADPH-independent trans-sulfuration pathway (83, 87, 101, 118).
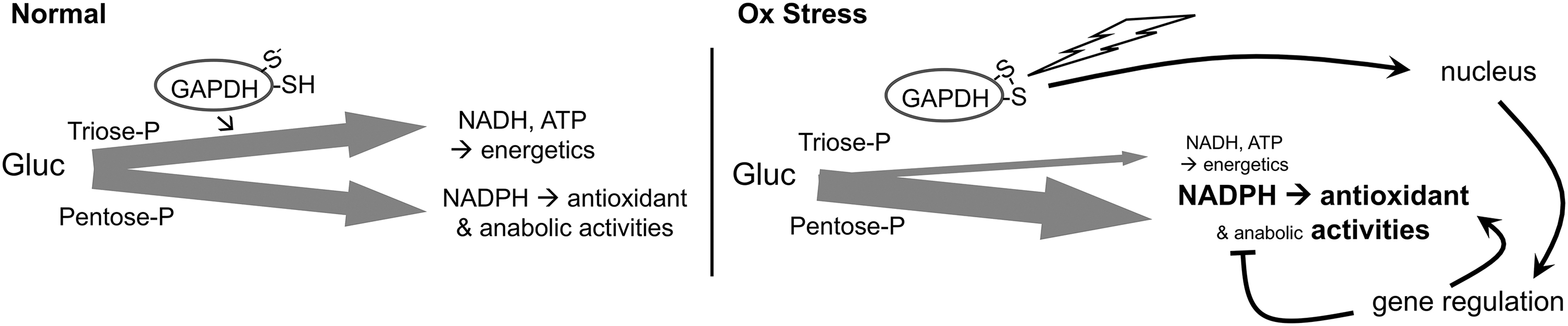
β-oxidation of fatty acids, which becomes elevated with high-fat diets, and glycolysis, which is elevated in many cancers, are each associated with increased production of ROS, increased oxidative damage, and increased demands on the GSH and TRX1 systems (4, 48, 55, 57, 76, 115). During oxidative stress, GSR and TRXR1 likely compete with other NADPH-dependent enzymatic systems to obtain the reducing power needed to maintain redox homeostasis. Favoring GSR and TRXR1 under these conditions, the active site Cys of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is highly redox reactive and functions as an oxidant-sensitive switch (94). Relatively subtle increases in H2O2 will preferentially oxidize this Cys, which inactivates the glycolytic activities of GAPDH and converts the protein into a nuclear-localized transcription factor (Fig. 5). With the disruption of glycolysis, more glucose is shunted to the pentose phosphate pathway, resulting in increased production of NADPH. Moreover, as a transcription factor, oxidized GAPDH realigns expression of diverse genes, resulting in a global metabolic rewiring by which NADPH-consuming anabolic activities are attenuated and redox homeostasis activities are elevated (52).
Carcinogenic stress
Transformation to a cancer phenotype and the characteristics of the tumor microenvironment are associated with diverse stresses on the cancer cells that can influence, or be influenced by, S metabolism activities. Most of these are redundant with stresses already mentioned above, but in cancer these can often occur in combination, leading to greater deviations from normal cell physiology and larger impacts on S metabolism. Metabolic realignments in transformed cells, such as the preferential switch to glycolysis, are often associated with increased levels of ROS in these cells, which, in turn, is expected to increase their reliance on the disulfide reductase-based antioxidant systems (63, 128). Conditions of hypoxia, nutrient restriction, or inflammation within the tumor microenvironment can further increase metabolic and oxidative stresses. The replicative activity of cancer cells is associated with proliferative stress, and therapeutic measures taken to combat the cancer can be associated with electrophilic stress, oxidative stress, metabolic stress, and drug/xenobiotic stress. Whereas different cancers might exhibit diverse combinations of these stresses, in some cases potentially targetable liabilities in S amino acid metabolism might be found. For example, many cancers upregulate the glutamate/cystine antiporter, xCT, to acquire cystine, which might be a vulnerability that can be targeted therapeutically (see the S-Overoxidation in Mammalian Cells section). By contrast, however, some other cancers are critically dependent on glutamine anaplerosis during glucose starvation and overexpression of xCT or cystine supplementation can drive glutamate exchange and potentially starve the tumor (66, 67, 88, 128).
S-Overoxidation in Mammalian Cells
In addition to oxidant-driven disulfide formation, which is reversible by the NADPH-driven disulfide reductase systems, biological “overoxidation” reactions can occur (Fig. 2). In these, Cys-thiols within proteins or small molecules can react with successive oxidants to first yield a sulfenic acid (Cys-SOH), then a sulfinic acid (Cys-SO2H), and finally a sulfonic acid (Cys-SO3H) (83). Cys-sulfenic acids are highly unstable species that usually rapidly react with another thiol to yield H2O and a disulfide. However, when concentrations of an oxidant are high, a second oxidation of the sulfenic acid to the sulfinic acid form can occur (46). Historically, it had been thought that neither the sulfinic- nor the sulfonic form could be reduced back to a Cys-thiol in mammalian cells. However, the discovery of the enzyme sulfiredoxin (Srx) revealed that some Cys-sulfinic acids can be reduced back to the thiol. Srx uses both ATP and reduced dithiols from TRX1 in a slow 4-electron reaction to repair an overoxidized Cys-sulfininc acid within the active site of 2-Cys PRXs back to the Cys-thiol form (13, 18, 139). This discovery led to the proposed “floodgate model” in redox signaling, wherein a burst of H2O2 could generate a Cys-sulfinic acid on PRX in a rapid two-step process, which would transiently inactivate the PRX peroxidase activity and allow H2O2 to accumulate locally and oxidize Cys residues on other proteins as a signaling molecule (140). Srx will then slowly repair the PRX-sulfinic acid, thereby restoring peroxidase activity and shutting off the signaling event (102). Recently, an unexpected diversity of other potential protein-sulfinic acid targets that can be repaired by Srx, at least in vitro, were reported, including oxidatively damaged PTPN12, DJ-1, and others (5). A mechanism to reduce Cys-sulfonic acid in mammalian cells has not been demonstrated, supporting the current understanding that this is, indeed, an irreversible modification. The fate of Cys-sulfonic acid in damaged proteins is also poorly understood; but it is presumed that proteins with Cys-sulfonic acids, being irreversibly damaged, are degraded by the proteasome. This will release Cys-sulfonic acid, which can be converted to taurine or further oxidized to liberate sulfate (116, 120). There is also evidence of some chaperones stably carrying Cys-sulfonic acid modifications, which might be critical to their specialized activities (72).
Importantly, severe oxidative stress is not the only means of inducing Cys-overoxidation in cells. The enzyme Cys-dioxygenase (CDO) catalyzes the irreversible conversion of free Cys + O2 → Cys-sulfinic acid. Deamination converts this to hypotaurine. Hypotaurine can further oxidize to the sulfonic acid form taurine, and then further oxidize to release inorganic sulfate (117). These regulated overoxidations play several roles in physiology. First, taurine is an important osmolyte in blood plasma that is secreted predominantly by the liver and, to a lesser extent, the kidney (130, 131). Taurine is also conjugated onto bile acids within hepatocytes in many animal species, which likely plays important roles in digestive processes (131). Overoxidation of Cys in liver provides an amino acid-derived source of sulfate that can be assembled into PAPS for important anabolic and defensive SULT reactions (see Dietary Sources and Systemic Distribution section). Finally, it has more recently been recognized that Cys overoxidation might provide an important “outlet” that prevents excessive accumulation of Cys in cells, thereby rectifying conditions of potential reductive stress (see Other Stresses section). Indeed, some cancer cells are critically addicted to CDO activity for modulating intracellular Cys levels, which represents a potentially targetable liability to these cancers (64, 65).
S-Persulfidation in Mammalian Cells
Another important Cys modification in cells is Cys-persulfidation (Cys-SSH) (11, 51). Protein-Cys-persulfides are readily reduced by cellular redoxins (25, 26, 29, 135), yielding a Cys-thiol and H2S. This modification is therefore a likely regulator of enzyme activity and signaling pathways, although the extent of this use of protein-persulfidation remains largely undefined (60). Liberation of H2S as a result of regulation of specific individual proteins is unlikely to be substantial; however, some studies suggest that protein persulfides might be more abundant than expected, in which case their redoxin-catalyzed conversion to thiols could generate large amounts of H2S in cells or subcellular compartments. For example, the mitochondrial Cys-tRNA synthetase, CARS2, has Cys-persulfidation activity, and this could lead to extensive cotranslational insertion of Cys-persulfide into diverse proteins (3). It has also been proposed that cells might generate a reserve of certain proteins with persulfide modifications on critical Cys residues that, although perhaps inactive in their normal roles, could be activated to thiols by redoxins after oxidative stress-mediated overoxidation to support stress recovery (discussed further below) (25). The reader is directed to other papers in this issue for updates on mechanisms and activities of protein persulfidation.
The contributions of H2S in generating Cys-persulfides on proteins or small molecules remain unclear. The S in H2S is in the same reduced state as thiols and cannot react with a thiol to generate a persulfide unless an oxidant is added. However, if H2S is present coincident with a 2-electron thiol-oxidation, for example by H2O2, the unstable Cys-sulfenic acid intermediate could be resolved with the H2S to form a Cys-persulfide rather than with another thiol to form a disulfide (Fig. 6, steps a–c). Due to the combined nucleophilicity of the two S residues in a Cys-persulfide, the terminal S more readily ionizes as compared with the Cys-thiol (pKa ∼4.5 as compared with pKa ∼8.2, respectively), so at physiological pH the Cys-persulfide will be predominantly in the highly reactive perthiolate-anion state (Cys-SS−) (25). Whereas the reaction of most Cys-thiols and H2O2 at physiological pH is slow (137, 138), a Cys-perthiolate anion is likely to be dramatically more reactive with H2O2, forming Cys-persulfenylate (Cys-SSO−) and possibly Cys-persulfinylate (-SSO2 −) and -persulfonylate (-SSO3 −) anions upon subsequent reactions with additional equivalents of H2O2 (Fig. 6, step d) (11, 51). Cys-SSO3 − can also be generated by the reduction of a disulfide with SO3 2− (9, 75). Interestingly, unlike Cys overoxidation, which yields typically irreversible sulfinic and sulfonic acids, the persulfide forms have a disulfide bond linking the overoxidized terminal S to the Cys. Recent work shows that this disulfide bond remains a reducible substrate for TRX1, thioredoxin-related protein of 14 kDa (TRP14), and GRXs (Fig. 6, step e) (25). The redoxin-catalyzed reduction of an overoxidized Cys-persulfide, whether generated on proteins cotranslationally by CARS2 or on any molecule by thiol oxidation in the presence of H2S, yields, in each case, the native Cys-thiol-containing molecule and liberates the inorganic overoxidized S species (25, 31). Although the overoxidized inorganic S have been irretrievably lost from the pool of metabolites that could contribute S to Met or Cys in the future, in doing so it could have removed up to four molecules of H2O2 from the cell. Such nonrecycling use of S amino acids to reduce H2O2 might seem unfavorable under normal conditions, when the disulfide reductase-fueled GPXs and PRXs are able to remove H2O2 without loss of S. However, under conditions wherein these systems are disrupted or overwhelmed (28, 82, 98, 104), “sacrificial” production of H2S to support a persulfide-based overoxidation mechanism to eliminate H2O2, as depicted in Figure 6, could provide a stopgap measure for limiting oxidative damage.

Regulation of S Metabolism in Stressed Cells
Regulation of the Met cycle and trans-sulfuration
A previous overview of regulatory circuits in the Met cycle and trans-sulfuration pathway summarizes how these pathways are differently regulated to support intermediary metabolism in hepatocytes versus cell-autonomous activities in other cell types (78). Here, we will emphasize how stresses might impact these pathways.
The first step in the Met cycle generates SAM, which is the methyl donor for most methyltransferase enzymes in cells, and which must be demethylated by one of these methyltransferases to continue through the Met cycle (Fig. 1). SAM is synthesized from Met + ATP by methionine adenosyltransferase-II (MATII, encoded by the MAT2A gene) in nonliver cells or by MATI and MATIII (homodimeric and homotetrameric forms, respectively, both encoded by the MAT1A gene) in hepatocytes. MATI and MATII are feedback inhibited by SAM, thereby adjusting Met-cycle flux to match cellular needs. Conversely, MATIII is activated by SAM, which generates feed-forward activation in support of intermediary metabolism in hepatocytes (78). SAM also activates trans-sulfuration and inhibits methylene-tetrahydrofolate reductase, the latter of which provides new methyl groups to MS for conversion of Hcy → Met. These activities effectively shunt excess Hcy toward Cys rather than back to Met. Importantly, however, there are situations, for example, Cys deficiency, wherein flux from SAM → Hcy → Cys can become restricted by the need to demethylate SAM. Evidence suggests that conversion of glycine to sarcosine by the enzyme glycine N-methyltransferase, which accounts for ∼3% of liver protein by mass, is used by hepatocytes to promote conversion of SAM → Hcy by removing excess methyl groups, preventing accumulation of SAM, thereby allowing unrestricted transit through the Met cycle (50, 54). In yeast and mammalian cells, phospholipid biosynthesis and, in more extreme situations, histone methylation, have also been shown to provide a “methyl-sink” for SAM-dependent methyltransferase activity, again ensuring rapid flux through the cycle in support of Cys biosynthesis, when needed (141).
The signals that determine Cys- versus H2S production by the trans-sulfuration pathway remain incompletely resolved. The balance between Cys versus H2S production by cystathionine-β-synthase (CBS) is influenced by the Ser:Cys ratio; when Ser is limiting, H2S production increases (74). In addition, work from the Banerjee laboratory has demonstrated a CO-dependent switch in the trans-sulfuration products (62). Exposure to CO inhibits the first enzyme in the trans-sulfuration pathway, CBS, leading to an accumulation of Hcy and depletion of cystathionine (Cth). In response, the second enzyme in the pathway, cystathionine-γ-lyase (CSE), which normally converts Cth to Cys, switches substrate utilization from Cth to Cys or Hcy, resulting in the production of H2S. Preference can be returned to Cys production with the addition of Ser, the amino acid skeleton donor for Cys biosynthesis (62).
Other major metabolic regulation of S metabolism
Some critical S-metabolites are normally kept within narrow limits by either substrate- or product-feedback regulation. As discussed already, GSH biosynthesis is repressed by the pathway product, GSH (108), a mechanism that both prevents overaccumulation of GSH leading to reductive stress and allows rapid recovery of GSH levels after depletion. Cys levels, by contrast, are feedback regulated by the reactant, Cys. Thus, CDO is activated by Cys, leading to shunting of excess Cys into overoxidized forms that can be excreted (23, 121).
Master regulation of S metabolism in stress
The NRF2/kelch-like ECH-associated protein 1 (KEAP1) pathway plays the role of master regulator of many genes involved in stress responses of S-metabolism. This pathway is post-translationally induced in response to oxidative or electrophilic exposures. The reader is directed to an outstanding recent review for more information on the mechanisms of NRF2 regulation (122); here, we will consider the impacts of this on S-metabolism pathways. Upon induction, the transcription factor NRF2 regulates a suite of ∼102 genes that coordinately defend cells against oxidants and electrophiles, including genes encoding critical enzymes on several S-metabolism pathways and genes that more peripherally impact these pathways. In the latter case, activation of NRF2 induces expression of enzymes that generate NADPH (e.g., malic enzyme and pentose phosphate pathway enzymes), and represses expression of anabolic genes that might compete with GSR and TRXR1 for NADPH (e.g., fatty acid biosynthesis pathways). In the former case, NRF2 activates expression of genes for glutathione biosynthesis enzymes (e.g., GCLC and GCLM) (73, 85, 107, 125). Another interesting target of the NRF2 pathway is heme oxygenase 1 (HO-1), which is strongly activated by NRF2. As discussed above, in the presence of free heme, induction of HO-1 will generate CO, which will both impede Cys synthesis and therefore GSH biosynthesis, and favor H2S production by the trans-sulfuration pathway (62). Although disruption of Cys/GSH biosynthesis seems at first counterproductive in face of a stress response, it is possible that the special situation of heme exposure, including release of iron leading to possible Fenton chemistry-catalyzed generation of extremely damaging radicals (134), is less hazardous if Cys/GSH biosynthesis is transiently repressed. Alternatively, it is possible that H2S, itself, is better than Cys/GSH at remediating the hazards associated with heme exposure. Future studies will hopefully resolve the rationale for how CO, H2S, Cys, and GSH are coordinated by Nfr2 and HO-1 as a stress response.
Footnotes
Acknowledgments
We thank our colleagues and collaborators for fruitful discussions on these topics.
Funding Information
Our research is supported by funding to E.E.S. from the National Institute of Health (DK123738, OD026444, AG055022, and CA215784), the Montana Agricultural Experiment Station (MONB00443), and the Montana State University Department of Microbiology & Immunology.