
Abstract
Significance:
Chronic kidney disease (CKD) can be regarded as a burden of lifestyle disease that shares common underpinning features and risk factors with the aging process; it is a complex constituted by several adverse components, including chronic inflammation, oxidative stress, early vascular aging, and cellular senescence.
Recent Advances:
A systemic approach to tackle CKD, based on mitigating the associated inflammatory, cell stress, and damage processes, has the potential to attenuate the effects of CKD, but it also preempts the development and progression of associated morbidities. In effect, this will enhance health span and compress the period of morbidity. Pharmacological, nutritional, and potentially lifestyle-based interventions are promising therapeutic avenues to achieve such a goal.
Critical Issues:
In the present review, currents concepts of inflammation and oxidative damage as key patho-mechanisms in CKD are addressed. In particular, potential beneficial but also adverse effects of different systemic interventions in patients with CKD are discussed.
Future Directions:
Senotherapeutics, the nuclear factor erythroid 2-related factor 2–kelch-like ECH-associated protein 1 (NRF2-KEAP1) signaling pathway, the endocrine klotho axis, inhibitors of the sodium–glucose cotransporter 2 (SGLT2), and live bio-therapeutics have the potential to reduce the burden of CKD and improve quality of life, as well as morbidity and mortality, in this fragile high-risk patient group. Antioxid. Redox Signal. 35, 1426–1448.
CKD as a Public Health Priority
Chronic kidney disease (CKD) has been defined as “abnormalities of kidney structure or function, present for >3 months, with implications for health” according to KDIGO guidelines (94). Patients with CKD are classified using different biomarkers of kidney function [e.g., estimated glomerular filtration rate [eGFR] and albuminuria as assessed by albumin-to-creatinine ratio (94)], because of their well-established association with CKD progression to end-stage kidney disease (ESKD) and mortality (55, 119). Importantly, mortality is extremely high in patients with ESKD requiring renal replacement therapy (RRT) (87, 195). According to a recent European Renal Association–European Dialysis and Transplant Association (ERA-EDTA) registry annual report, patients aged 20–44 years on RRT live only one-third of the expected remaining lifetime of the age-matched general population (107).
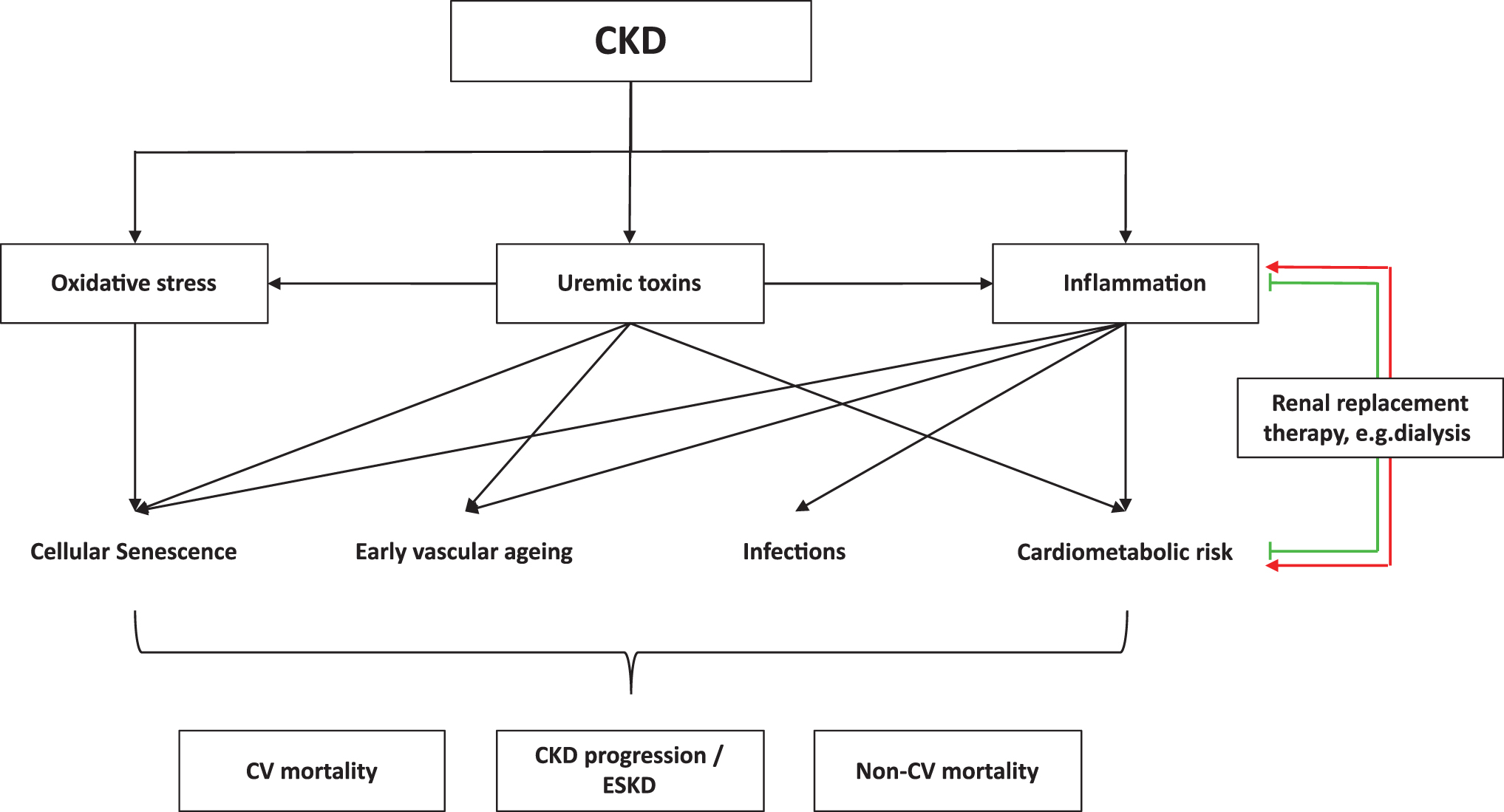
Compared with the general population, patients with CKD have a highly accelerated and premature aging process, a complex constituted by several adverse components, including vascular disease, chronic inflammation, osteoporosis, periodontal disease, depression, sarcopenia, and other maladies (34). Early vascular aging (EVA) in particular, resulting in increased arterial stiffness and endothelial dysfunction, is believed to be a crucial patho-mechanism linking CKD with mortality (88a). Further, oxidative stress is also associated with endothelial dysfunction through different mechanisms (150, 244, 249). Collectively, there is a vicious cycle comprising oxidative stress, inflammation, CKD, and premature cardiovascular disease (CVD) (169). As a consequence, therapeutic aims for reducing EVA and CVD mortality in CKD should include control of inflammation, reduction of oxidative stress, and improvement of endothelial dysfunction (Fig. 1) (31). It has also been reported that patients with asymptomatic proteinuria exhibit low-grade inflammation linked to endothelial dysfunction (158). As EVA in CKD can be both a cause and a consequence of the underlying renal disease, factors contributing to this pro-senescence milieu are important potential treatment options in the uremic milieu.
In this review, we summarize current concepts of inflammation and oxidative stress in CKD as crucial pathophysiological mechanisms (Fig. 2) of the uremic phenotype and provide a perspective on possible future treatment options for treating all three components, that is, CKD, inflammation, and oxidative stress.

The Many Facets of CKD
The main role of the kidney is to maintain the homeostatic balance of a variety of solutes in the blood and to excrete undesirable components. This activity comes at a significant metabolic cost: The kidney and the heart have the highest specific resting metabolic rate of all major organs, roughly twice higher than that of the brain or liver (232). To carry out their function, the kidneys are highly vascularized and receive roughly a quarter of the cardiac output (139). In addition, the health of the vasculature and the integrity of the endothelium are also crucial for the proper function of the kidney and its ability to filter the blood (245). It is, therefore, unsurprising that the kidney is an especially vulnerable target for the homeostatic imbalances that accompany CKD: chronic inflammation, oxidative stress, EVA, and accumulation of uremic toxins (31, 33). Oxidative stress stands out as a major contributor to many adverse facets of CKD (Figs. 1–3) and will be discussed in detail.

Chronic inflammation
Inflammation is an essential response mechanism that allows the body to cope with a variety of external (e.g., pathogens) and internal (e.g., damaged or cancerous cells) threats (8). In acute inflammation, a triggering stimulus leads to an inflammatory response involving (i) the release of cytokines and chemokines, most notably interleukins (IL)-1 and IL-6, interferon gamma, and tumor necrosis factor that drive both a localized and a systemic response; (ii) the recruitment and proliferation of immune cells, particularly macrophages and neutrophils; and (iii) the eventual clearing of the threat, followed by a return to baseline and tissue repair (8). However, if clearing fails, or the inflammatory response cannot be switched off, the resulting chronic inflammation leads to tissue dysfunction and damage, including fibrosis, stem cell depletion, and increased cellular senescence (8, 188).
Indeed, chronic inflammation is bi-directionally linked to CKD (26) (Fig. 1). On the one hand, the uremic milieu drives uremic inflammation, which shares common features with the chronic low-grade inflammation associated with aging, known as “inflammageing”; on the other hand, chronic systemic inflammation leads to dysfunction in the kidneys and can further precipitate fibrosis and the progression of CKD (92, 139, 208). At the same time, uremic inflammation has been mechanistically associated with premature aging, contributing to processes such as telomere attrition, mitochondrial dysfunction, and dysregulated nutrient sensing (102). In a large proportion of patients with advanced CKD, a systemic inflammatory response is detectable, and the prevalence increases with the progression of CKD stage (26, 102). At the same time, certain components of the immune system, particularly the adaptive immune system, are impaired in a process resembling immunosenescence (42, 127). Patients with ESKD have a lower relative abundance of lymphoid cells, and their T and B cells are more prone to activation-induced apoptosis (127).
A plethora of mechanisms are involved in CKD-related immune disruption. Increased blood concentration of several cytokines and inflammatory markers, such as IL-1, IL-6, and C-reactive protein, mostly released by endothelial cells and circulating monocytes, is associated with CKD (26, 139). This is due to both limited clearance and increased production of these solutes, which can result from factors such as endothelial dysfunction, oxidative stress, increased cellular senescence, greater permeability of the lining of the gastrointestinal tract (periodontitis and gut dysbiosis are comorbidities in CKD), calcium (Ca++) phosphate (Pi) overload, sodium accumulation in tissues, and buildup of uremic toxins in the blood (26, 42). The large endothelial surface of the highly vascularized kidneys makes them particularly sensitive to local pro-inflammatory effects (139). Endothelial activation can impair local vasodilatory ability, increase reactive oxygen species (ROS) production, and exacerbate the already physiologically hypoxic state of the renal medulla (139).
The nuclear factor-κB (NF-κB) deserves special mention in the context of inflammation as a key activator of the upregulated uremic inflammatory response, and it is known to be sensitive to activation by increased ROS levels (102) and mitochondrial dysfunction (24). NF-κB is also responsive to inflammatory cytokines, generating a potential positive feedback loop that can sustain inflammation over time (102). The NLRP3 inflammasome also plays an important role as a signaling nexus for the activation of NF-κB, responding to such stimuli as increased ROS, release of mitochondrial DNA (mtDNA), extracellular ATP, and more (139). Further direct effects of inflammation on oxidative stress are discussed later in the “Oxidative Stress” section. An increase in advanced glycation end-products (AGEs) can also upregulate NLRP3 and NF-κB by binding to the AGE receptor (RAGE), thus contributing to CKD progression (243). Interestingly, the central nervous system has recently been shown to play an inhibitory role in the inflammatory response via a reflex mediated by the vagus nerve (10).
It is worth noting that dialysis treatment fails to adequately remove solutes of the size of most cytokines and is, therefore, unable to fully correct the pro-inflammatory uremic milieu (26). In addition, dialysis itself has a pro-inflammatory effect (123) (Fig. 1). This is due to a variety of factors: technical, such as vascular access and limited biocompatibility of the membranes and surfaces (44), as well as microbial, such as contamination of dialysis solutions or catheters with either live microorganisms or with microbial components (including the highly pro-inflammatory endotoxins) (102, 123). Better clinical practice and the use of improved materials can, to a degree, reduce this pro-inflammatory effect (42, 59). Novel approaches that improve antioxidant defenses can also be helpful. For instance, it has been recently shown that vitamin E-loaded membrane dialysers may contribute to reducing the signs of inflammaging associated with dialysis (180).
Oxidative stress
Oxidative stress is another prominent feature of CKD (Figs. 2 and 3); the high metabolic rate of the kidney makes this organ particularly sensitive to oxidative stress and can exacerbate oxidative damage, along with an impaired vascular phenotype, inflammation, fibrosis, and proteinuria (74). In general, an antioxidant defense prevents the accumulation of ROS and reactive nitrogen species (RNS). However, when there is an imbalance between generation of oxidant compounds and antioxidant defense mechanisms, oxidative damage occurs (168). At a systemic level, oxidative stress can contribute to renal dysfunction, vascular diseases, alteration in immune system, metabolic complications, and anemia (Fig. 2) (123). Patients on dialysis have a markedly increased oxidative stress compared with patients with CKD who are not on RRT due to increased ROS formation and reduced antioxidant defenses (61, 167).
As the primary site of redox biochemistry and ROS production, mitochondria are a natural target for ROS damage, with consequences that include mtDNA damage, reduction of mitochondrial mass, disruption of the mitochondrial membrane integrity, leakage of ROS and pro-apoptotic factors, as well as loss of membrane potential, possibly impairing the cell's energetic metabolism or leading to cell death (Fig. 3) (187, 210). Dysfunctional mitochondria are major drivers of the intermediate inflammatory phenotype that drives premature aging in CKD and other burden of lifestyle diseases (196). Thus, Galvan et al. (64) have demonstrated significantly increased mitochondrial ROS in the kidneys of mice with diabetic CKD, compared with glucose tolerant control mice, using an innovative in vivo approach. As mitochondria regulate major cellular processes, such as cell proliferation and differentiation, as well as cell death (209), ROS production by damaged mitochondria can induce cell death and inflammation (209). Indeed, mitochondrial dysfunction and reduced levels of PPARγ co-activator 1α (PGC-1α), the master regulator of mitochondrial biogenesis, are strongly linked to CKD (19, 54). In addition, a retrograde signaling pathway allows dysfunctional mitochondria to induce specific gene expression changes in the nucleus (131).
Apart from mitochondrial dysfunction, peroxisomes, that is, intracellular organelles that contribute to redox homeostasis (225), also become dysfunctional in several models of acute kidney injury and CKD (Fig. 3) (209, 225); and Vasko et al. (225) have recently proposed a dysregulated mitochondria–peroxisome axis as an important mediator of cellular redox homeostasis. Collectively, diminished antioxidant defense capacity (e.g., in peroxisomes) in combination with increased mitochondria ROS lead to a disturbed redox homeostasis in CKD (Fig. 3).
In addition, mitochondria are closely connected to the endoplasmic reticulum (ER) (175). As both ER stress and mitochondrial dysfunction are linked to an increased activation of pro-inflammatory NF-κB signaling (102, 209), the ER-mitochondrial axis (175) might also be involved in an impaired redox homeostasis in CKD (Fig. 3).
The cellular metabolic and redox homeostasis crucially depends on the pyridine nucleotides NAD+/NADH and NADP+/NADPH (Fig. 3) (71, 166). Evidence of a defective cellular redox status in CKD has been reported by Canestrari et al. (14) in red blood cells (RBC). In more detail, NADPH in RBC was significantly decreased, whereas RBC oxidized glutathione, that is, glutathione disulfide, was increased in patients with ESKD compared with controls (14). In contrast, RBC glutathione (GSH) levels were unchanged in patients with CKD and controls (14). Importantly, hemodialysis (HD) further increased RBC oxidized glutathione levels, indicating that not only reduced renal function per se but also dialysis impairs cellular redox homeostasis in RBC (14). Mechanistically, protein expression and activity of GSH-S-transferase in RBC is increased in patients with ESKD compared with control subjects (14, 63), most likely through uremic retention solutes/uremic toxins (63). Translationally, intensified daily vs. conventional HD lowered distinct uremic retention solutes, thereby improving redox status in ESKD (62). It should be noted that most of the studies investigating cellular redox status focus on RBC. However, accumulating evidence indicates that redox homeostasis in CKD is also impaired in other organs/cell types, including muscle (15), endothelial cells, and aortic smooth muscle cells (173).
Besides GSH metabolism, NAD+ levels are reduced in CKD due to an impaired biosynthesis and augmented consumption (Fig. 3) (71, 166). Interestingly, the transcriptional coactivator PGC-1α promotes NAD+ biosynthesis via the de novo pathway (Fig. 3) (217). Importantly, NAD+ is also a substrate for the sirtuin family of enzymes that mediate processes, including metabolic flux and epigenetic regulation (71). Further, NADPH oxidase (NOX) enzymes are one of the major sources of ROS besides mitochondria, and their activity in the kidney and vascular endothelium can impair blood flow, reduce NO synthesis, and cause hyperhomocysteinemia and kidney damage (109, 229).
Although there is convincing evidence that oxidative stress induces inflammation, the evidence of the adverse effects of a pro-inflammatory milieu on oxidative stress is less strong. In a uremic environment, peripheral blood mononuclear cells are activated and show a pro-inflammatory expression signature (251). Infiltrating leukocytes in the kidney can not only release large amounts of pro-inflammatory cytokines, but they also contribute to a respiratory burst by promoting myeloperoxidase-enhanced production of superoxide (74, 168, 178).
Hypoxia is another factor that is bi-directionally linked to both increased inflammation and oxidative stress in CKD. Thus, hypoxia induces inflammation by several mechanisms but, conversely, inflamed tissue can also become hypoxic (45). Similarly, oxidative stress and hypoxia contribute to each other, most likely through uremic toxins (159) and mitochondrial dysfunction (76). It is interesting to note in this context that sodium–glucose cotransporter 2 (SGLT2) inhibitors (179) and the hypoxia-inducible factor-stabilizing Roxadustat (also known as FG-4592) (122) exert beneficial renal effects, at least in part, via attenuating renal hypoxia.
Although a certain level of ROS and RNS is unavoidable and has a physiological protective role, the elevated production of ROS and RNS, paired with the lower antioxidant defense observed in CKD, leads to a redox imbalance and a toxic amount of oxidative stress (33, 36, 74). Due to their high reactivity, ROS can damage a variety of macromolecules, including DNA, proteins and lipids, as well as cell organelles (Fig. 3) (109).
DNA damage is of particular concern due to the potential long-term consequences of telomere attrition, DNA mutations, epigenetic dysregulation, and DNA damage signaling (131, 160). Among the four bases found in DNA, guanine (G) is the one most sensitive to oxidation (165); oxidation products of G such as 8-hydroxydeoxyguanosine (8-OH-dG) and 8-oxodeoxyguanosine (8-oxo-dG) are the most common oxidative DNA lesions, as well as established biomarkers of oxidative DNA damage (Fig. 3) (123). 8-OH-dG has been associated with mortality in CKD and with carcinogenesis (35, 123), and both 8-OH-dG and 8-oxo-dG are increased in dialysis patients (177, 240). Importantly, 8-oxo-dG pairs preferentially with adenine, rather than cytosine, providing this DNA lesion an additional potential mutagenic mechanism beyond the general DNA damage response (DDR) (177). Interestingly, the stacked G repeats found in telomeric regions are even more sensitive to oxidation and may, thus, act as a redox-sensitive alarm system through which oxidative stress can cause accelerated telomere attrition and cellular senescence (Fig. 3) (131). Given that mitochondria have reduced protection and DNA repair mechanisms compared with the nucleus, mtDNA is particularly exposed to oxidative damage (19, 177). Indeed, 8-oxo-dG lesions have been shown to be twice as numerous in mtDNA compared with nuclear DNA (91). In accordance with these data, Fazzini et al. have recently demonstrated that mtDNA copy number as a marker of mitochondrial dysfunction and oxidative stress is an independent predictor for all-cause mortality in 4812 patients from the German Chronic Kidney Disease study (50).
Oxidative stress can damage proteins and lipids through a variety of mechanisms, including ROS, RNS, reactive carbonyl species, and through the action of reducing sugars, generating a diverse class of compounds that can be categorized as advanced oxidation protein products (AOPPs), AGEs, and advanced lipoxidation end-products (ALEs) (Fig. 3) (59, 226). The AGEs in the extracellular matrix (ECM) can impair tissue function, in the case of the vasculature contributing to vascular stiffening and the progression of CVD (102, 218). The AGEs can be recognized by the advanced glycation end products receptor 1 (AGER1), RAGE, and soluble RAGE (sRAGE) (201). AGER1 mediates AGE detoxification and exerts an antioxidant and anti-inflammatory role, sRAGE acts as a decoy for RAGE, and full-length RAGE activates a number of downstream pro-oxidative and pro-inflammatory effects (201). Diabetes, CKD, and inflammatory diseases have been shown to downregulate AGER1 and upregulate RAGE, potentially exacerbating inflammation and oxidative stress (201, 206, 227).
Tyr, Cys, and Met residues are particularly prone to oxidation, but other residues such as Lys, Arg, Thr, and His are also susceptible (59, 226). Several oxidation products, including oxidated Tyr and the lipid peroxidation products malondialdehyde (MDA) and F2-isoprostanes, have been found to be increased in the plasma of dialysis patients (33, 59, 123). In more detail, lipid peroxidation contributes to cellular damage and tissue degeneration in CKD. Metabolites, including 4-hydroxy-2-nonenal (HNE), MDA, and F2-isoprostanes, are end-products from the oxidation of unsaturated fatty acids or arachidonic acid and can react with the earlier-mentioned amino acid residues of proteins, thereby creating ALEs (Fig. 3) (53). Several of these lipid peroxidation metabolites have been classified as uremic toxins (38) and show direct adverse effects in CKD. As an example, arachidonic acid-based F2-isoprostane is not only associated with markers of renal function and being increased in HD (67) but also dose-dependently reduces GFR and renal plasma flow in rats (207). Further, F2-isoprostane is associated with markers of inflammation (67) and directly contributes to an adverse vascular phenotype that is similar to the clinical challenges observed in patients with CKD. Indeed, this metabolite induces vascular smooth muscle cell (VSMC) vasoconstriction (58), as well as endothelial dysfunction (248) in vitro. Importantly, the causal effects of lipid peroxidation radicals on renal function have recently been demonstrated by Kruger et al. (110), who have shown that lipid radicals directly impair podocyte motility and cytoskeletal structure through redox-sensitive RhoA signaling (Fig. 3).
As in the case of inflammation, dialysis not only fails to fully correct the oxidative stress associated with CKD, but, in fact, it also contributes to it through biocompatibility issues and fluid contamination risk (59). Renal transplantation, on the other hand, appears to correct both the pro-inflammatory and the pro-oxidant status (59, 192). Of particular interest in the context of CKD is albumin, a key plasma protein that is often depleted in the uremic milieu and regarded as a key part of the plasma's antioxidant defense, with roles that include plasma thiol and homocysteine (Hcy) homeostasis, Cys and GSH metabolism, and binding of plasma solutes (59, 164). Oxidative damage can modify the Cys-34 residue of albumin and impair the functionality of this protein (164).
The body possesses a range of protective mechanisms that can counteract oxidative stress through enzymatic and nonenzymatic antioxidants, but these mechanisms are often impaired in CKD (33). The list of antioxidant enzymes includes superoxide dismutase, catalase, and others regulated by an antioxidant response element (ARE) (33). The nuclear factor erythroid 2-related factor 2–kelch-like ECH-associated protein 1 (NRF2-KEAP1) axis is a key player in managing the antioxidant response that is downregulated in CKD (197). In the presence of oxidative stress, the transcription factor NRF2 is released from KEAP1, translocates to the nucleus, and acts as transcription factor regulating the expression of >300 ARE-containing genes involved in cellular stress and damage repair (197, 200). Glyoxalase-1, the main enzyme responsible for detoxification of methylglyoxal and other AGEs, contains an ARE, but the extent to which it may be regulated by NRF2 is still a matter of debate (136, 218). Some important nonenzymatic antioxidants include GSH, as well as a range of dietary compounds or elements including vitamin C, E, polyphenols, zinc, and selenium (33). Being fat soluble, vitamin E is particularly important in the prevention of lipid peroxidation, and supplementation has shown positive results in patients with CKD (167). Importantly, a deficiency in dietary antioxidants has been reported in patients with CKD (33).
Early vascular aging
EVA is a prominent feature of CKD that is associated with increased cardiovascular risk (42b, 199). Its causes have not been fully elucidated, but the existing evidence points to a key role for calcification driven by Ca++ and Pi homeostasis in the blood, as well as inflammation and allostatic load (186). Physiological levels or Ca++ and Pi are sufficiently high to induce spontaneous precipitation of calcium phosphate; however, this process is usually prevented by plasma proteins, primarily fetuin A, that bind crystallization nuclei into calciprotein particles (CPP) and prevent their growth (112). In CKD, however, the plasma concentration of Pi is increased due to dysregulation of the klotho-fibroblast growth factor 23 pathway that is responsible for Pi excretion and homeostasis (113). At the same time, fetuin A levels drop due to persistent inflammation, leading to a loss of the major protective mechanism (12, 102, 142). As a result, CPPs accumulate and are taken up by VSMC, causing stress, cellular senescence, and a shift toward an osteogenic phenotype. This, in turn, leads to increased deposition of crystals in the ECM and eventually media calcification and vascular stiffness (102, 230). Vascular endothelial cells also play a role in EVA. It has been shown that uremic serum induces ER stress and upregulation of the pro-inflammatory NF-κB signaling in endothelial cells (102).
Cellular senescence
One major contributing factor to the accumulation of damage within the vascular system is cellular senescence, a state of permanent growth arrest of the cell that can be caused by a number of triggers, including oxidative stress, telomere erosion, persistent DNA damage signaling, and oncogene activation (22). The transition into a senescent state is mediated by the p53/p21 and/or the p16/pRb pathways (22). Senescent cells are metabolically active, but physiologically noncontributory (193). They contain damage foci and are essentially precancerous; however, they are resistant to apoptosis and survive by upregulating a number of senescent cell anti-apoptotic pathways (193). This feature enables senescent cells to survive some insults that would usually kill “healthy” cells (97). At the same time, this creates a “vulnerability” that allows specific killing of senescent cells through the use of senolytic compounds, as discussed later (193). Senescent cells exhibit a senescence-associated secretory phenotype (SASP) and release a range of factors, including pro-inflammatory cytokines and matrix metalloproteinases. Collectively, they increase inflammation, damage the ECM, and induce senescence in other cells, either through the NF-κB pathway or via a paracrine mechanism (172, 193) that generates a noncell-autonomous pro-senescence effect. Senescent cells are usually cleared by the immune system, but this process is not perfect and, as organisms age, they tend to accumulate in the body, though they remain a minority of the cells even at a very old age (9). However, due to the SASP, senescent cells play an important role in tissue dysfunction and accelerated aging associated with CKD (34).
In patients with advanced CKD, immune-senescence markers are increased and a low relative telomere length is associated with mortality (27), especially in dialysis patients (16, 27). Indeed, a number of experiments in which senescent cells were cleared, or added in animal models, points to a causal link between cellular senescence and a number of features of aging (6, 7, 97). Recent preliminary evidence suggests that senescent cell clearance may also be beneficial in humans (73).
Uremic toxins and their effects on the kidneys and vascular function
In advanced CKD, several organic compounds that accumulate in the body (219) produce an adverse response to the biological system (222). In recent years, a large number of such compounds, that is, uremic toxins, has been identified and subsequently categorized into small water-soluble molecules, protein-bound molecules, and middle molecules (38, 138, 223). For a variety of these uremic toxins, adverse cardiometabolic associations have been described with the most convincing clinical implications for the protein-bound uremic toxins indoxyl sulfate (IS) and p-cresyl sulfate (PCS). Both these toxins increase CpG hypermethylation and decrease mRNA and protein expression of the aging-associated klotho gene in proximal tubule epithelial cells (204). Further, they induce oxidative stress by a variety of mechanisms, including increased NOX activity and ROS production (234), and diminish antioxidative activity, that is, reduced superoxide scavenging activity in the kidneys (157) or levels of antioxidants (43). Moreover, IS and PCS have been shown to reduce mitochondrial mass by mitophagy (205). In addition, several other uremic toxins also attenuate mitochondrial function in human conditionally immortalized renal proximal tubule epithelial cells in vitro (143). IS further mediates pro-senescent effects through the induction of ROS (190), whereas the anti-oxidant N-acetylcysteine inhibits IS-induced activation of p53 in vitro (190).
Besides these progeric effects on the kidney, uremic toxins also affect vascular function in CKD (154). Thus, IS induces protein expression of p53, p21, and senescence-associated β-galactosidase also in human aortic VSMC in vitro and N-acetylcysteine suppresses IS-induced effects (141). Further, AST-120, which adsorbs uremic toxins and their precursors within the gastrointestinal tract (4), reduces the expression of these senescence and oxidative stress markers in the area of aortic calcification in uremic rats (141). Uremic toxins further impair vascular function through endothelium-dependent effects (153). Thus, IS significantly increases ROS production in human umbilical vein endothelial cells in vitro (85, 161), where it enhances IL-1β-induced oxidative stress. Both N-acetylcysteine and apocynin pretreatment inhibit the additive adverse effects of IS and IL-1β (183). Further, oxidative stress and uremic toxins upregulate microRNA-92a in endothelial cells, resulting in endothelial dysfunction and atherosclerosis (182). These data are further supported by a translational study using AST-120 in patients with CKD, which demonstrated improved endothelial function as assessed by flow-mediated endothelium-dependent vasodilatation (247).
Besides these studies predominantly investigating protein-bound molecules, larger middle molecules, as well as small water-soluble compounds, could also significantly contribute to the development of CKD complications by mediating a pro-inflammatory and oxidative stress phenotype. Using an innovative scoring system, middle molecules and small water-soluble compounds scored lower for their toxicity and overall experimental and clinical evidence compared with protein-bound molecules (224). Among the most relevant members of both groups were asymmetric dimethylarginine (ADMA), trimethylamine (TMA)-N-oxide (TMAO), uric acid, as well as β2-microglobulin, ghrelin, and parathyroid hormone (224). As an example, the uremic toxin ADMA is related to an inflammatory (228) and oxidative stress (236) phenotype and associates with an adverse outcome in patients with CKD (128). Middle molecules, such as pro-inflammatory cytokines, adipokines, and other hormones (224), also contribute to increased inflammation and oxidative stress, as well as other cardiometabolic risk factors (39). Small water-soluble compounds can be removed by dialysis (224), whereas dialytic removal of middle molecules is more difficult in conventional HD (164). Importantly, different dialysis strategies, for instance using protein-leaking dialyzers that can remove larger solutes, can decrease markers of inflammation and oxidative stress compared with standard dialyzers (60), supporting the hypothesis that the full spectrum of uremic toxins can contribute to oxidative stress and the pro-senescent phenotype in both kidneys and the vascular system, with most pronounced effects for protein-bound toxins. Thus, the inhibition of uremic toxins is a potential treatment option in CKD, potentially attenuating cardiovascular morbidity and mortality.
CKD and Allostatic Load
Individual trajectories of age-related health and disease progression are affected by interindividual differences in exposomes (i.e., physical and social environment, psychological, lifestyle, and nutritional risk factors acting independently, cumulatively, or synergistically with an individual's genome and epigenome over their life course). The “burden of wear and tear” is manifest as both physiological and molecular allostatic (over)load (Fig. 4). The maintenance of physiological homeostasis in the face of allostasis, thus, becomes increasingly difficult as uremic toxins accumulate in the course of CKD (186).
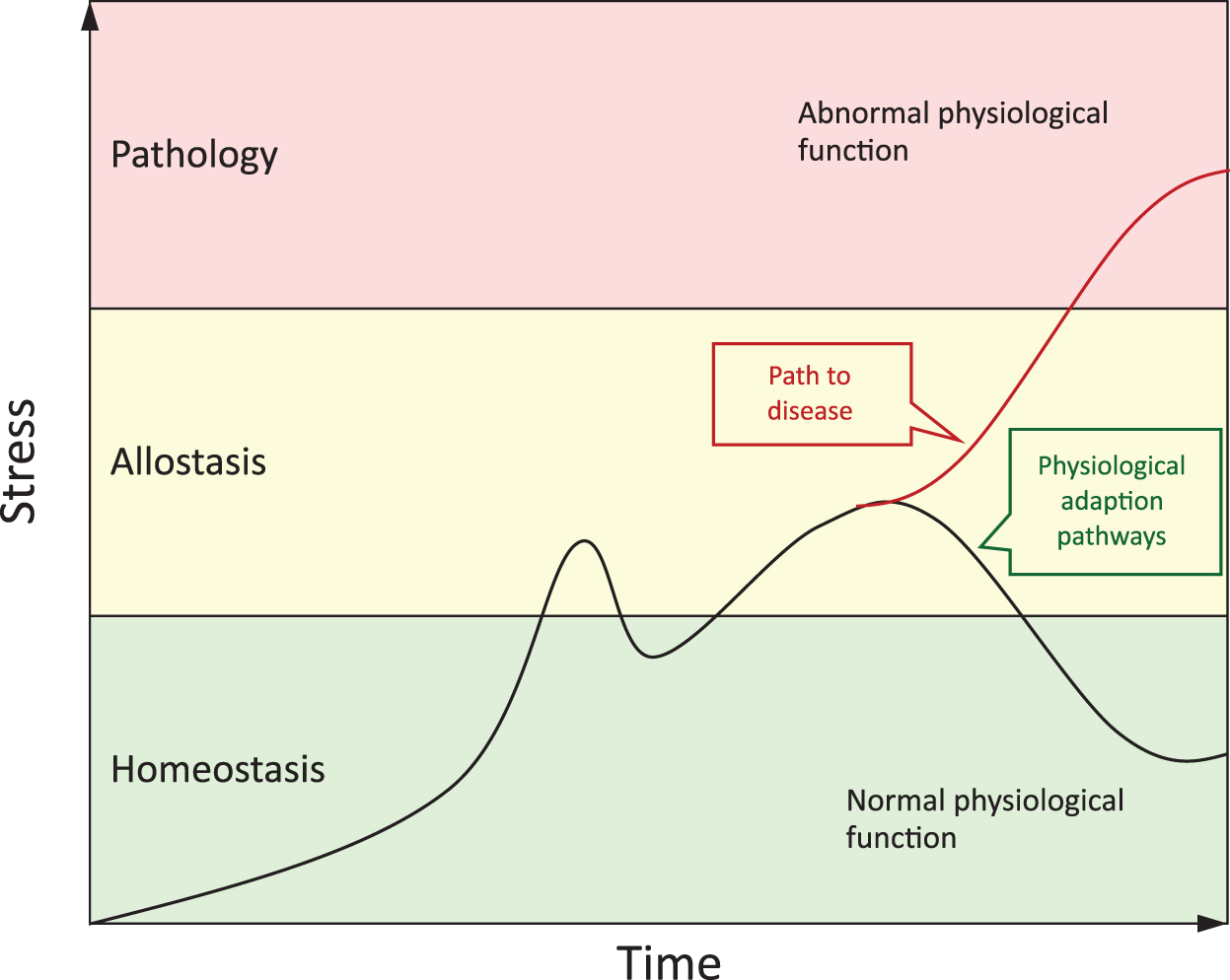
Consequently, CKD can be viewed as a burden of lifestyle disease that is part of a “diseasome of ageing” (156) that shares common underpinning features and risk factors with the aging process. In addition to chronic inflammation, oxidative stress, and EVA, both the aging process and CKD are also associated with sarcopenia, frailty, cognitive dysfunction, diminished NRF2 expression, and an altered gut microbiome (103, 186, 189, 197). The interplay between the exposome and the genome is thus mediated via the epigenetic landscape of aging and reflected in changes in gene expression (186). In particular, changes in the methylome (i.e., DNA methylation) are of particular interest, as they provide a direct mechanistic link between the foodome (i.e., individual diet, its composition, and the chemical structure of the ingredients), the gut microbiome, and the epigenome (130, 186).
The role of the gut microbiome
Maintenance of the methylome depends on a limited natural synthetic capacity for generating methyl donor metabolites, supplemented by the acquisition of methyl donor metabolites and cofactors of the one-carbon metabolic pathway, such as choline and betaine, through diet and the microbial metabolic activity in the gut (132). The gut microbiota has gained significant attention in recent years and is increasingly seen as a “supplementary organ” with complex interactions with the rest of the body, relevant for health and disease (139). A range of gut microbes can metabolize compounds to generate (i) betaine that feeds into methyl donor group production and (ii) TMA, which, in turn, gets oxidized by the liver into TMAO (186). TMAO has been shown to have pro-atherogenic and pro-inflammatory properties and may play a central role in inflammaging, age-related epigenetic changes, as well as CKD and CVD (41, 100, 140). Another microbial gut metabolite of note is butyrate, which has been shown to inhibit histone deacetylases that are involved in the regulation of chromatin (51). Importantly, epigenetic dysregulation is a hallmark of both aging and CKD (188).
A relatively novel finding is the association of CKD with intestinal dysbiosis and increased gut permeability, which can increase the leakage of bacterial DNA, endotoxins, and potentially whole microorganisms into the bloodstream (102, 137, 184). The gut microbiome has been shown to play an important role also in directly interacting with the immune system (118). In particular, the loss of anti-inflammatory taxa may contribute to the increase in inflammation associated with CKD (25). Alkyl catechols, produced by certain bacteria from plant phenolic compounds, can upregulate NRF2 and, thus, may also play a role in improving antioxidant defenses and provide increased resilience in patients with CKD (197).
Novel Treatment Approaches
As the landscape for potential therapies directed against the aforementioned mechanisms is broad, we have selected five groups of therapies for discussing potential treatment options (Fig. 5). Thus, senotherapeutics are discussed as senescence is closely related to oxidative stress and inflammation (22). NRF2 is an attractive treatment target controlling oxidative stress and inflammation in many burden of lifestyle diseases, including CKD (28). We further present kidney-secreted klotho as a potential renal target for the treatment of inflammation (124, 252), oxidative stress (96), and premature aging (115) in CKD. Moreover, we describe SGLT2 inhibitors as one of the most interesting and already approved pharmaceutical compounds for CKD and associated complications. Finally, live biotherapeutics are discussed, with special emphasis on the foodome and gut microbiome (130).
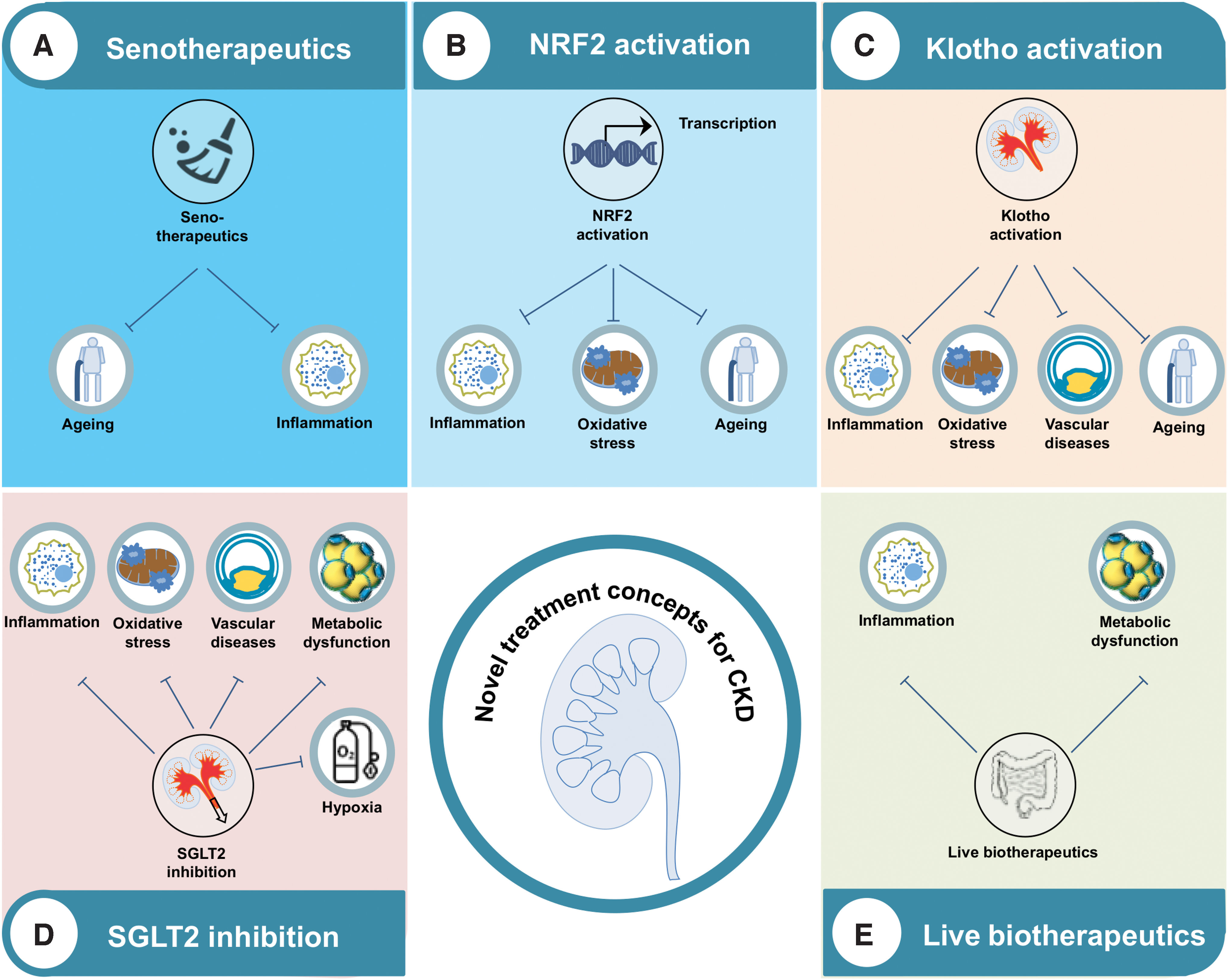
Senotherapeutics
The prominent involvement of cellular senescence in CKD suggests that drugs that can target senescent cells, termed senotherapeutics, can be a novel treatment avenue for CKD. It is worth noting here that the body can tolerate a certain amount of senescent cells without apparent harm (73, 211). It is only above a certain threshold that cellular senescence becomes a problem (73). Therefore, even if senotherapies do not get rid of all senescent cells, as long as they can provide sufficient clearance to bring the senescent load below the critical threshold, they have a chance of providing a meaningful benefit (73). Indeed, several studies have shown the benefits of senotherapy after a clearance of ∼30% of the senescent cell population (73).
Recent times have seen the development of the field of Geroscience, which champions the translational aspects of research on aging. The development of senotherapies falls under its aegis. This includes development of (i) senolytics, i.e. compounds that selectively kill senescent cells. Examples include repositioned drugs such as Dasatinib, typically used in combination with alkyl-catechols such as Quercitin, which remove the apoptotic block in senescent cells. In contrast, (ii) senostatics or senomorphics suppress the SASP without killing the senescent cells themselves (57, 73, 171).
The development of serotherapeutics, and in particular senolytics, is exciting. They have clearly demonstrated significant health benefits in preclinical—and, recently, possibly clinical—models (6, 7, 73, 90, 97). Due to the immaturity of the field, however, several caveats need noting, though it must be stressed that these do not preclude translational use of senotherapeutic agents, which would appear churlish based on preclinical data. First, the question arises as to when and where to use these agents in the life course. Antagonistic pleiotropy (237) is a concept that is pertinent in this respect, as it stipulates that what is good for you in old age is not necessarily good for you at a younger age and vice versa. Cellular senescence firmly falls under its umbrella, being onco-protective in old age, but undesirable when young as it reduces physical capability (186). The long-term effects of senotherapeutics applied under normative conditions for aging and in disease, thus, need to be established. Second, another concern is that their use may in the longer term lead to depletion of the body's regenerative capacity and or contribute to adverse changes in the epigenetic landscape, both of which are hallmarks of aging. Third, different senotherapeutic agents have different levels of efficacy in different cell types, making it unclear which treatment—or combination of treatments—would be best suited as a therapy in the context of age-related multi-morbidity, or to treat dysfunction in a multi-cell type organ such as the kidney.
Nuclear factor erythroid 2-related factor 2
The NRF2 system may provide a targeted node for intervention that is common to the range of morbidities manifesting within the diseasome of aging (197). More specifically, NRF2 expression is low in several burden of lifestyle diseases, including autoimmune (e.g., multiple sclerosis), respiratory (e.g., smoking-related lung emphysema), gastrointestinal (e.g., primary biliary cholangitis), metabolic (e.g., insulin resistance and nonalcoholic steatohepatitis), and neurodegenerative (e.g., Huntington disease) (29, 197). NRF2 and its repressor KEAP1 are key regulators of cellular stress and damage defenses and, therefore, represent potential treatment options for a wide range of disease states. In CKD, single-nucleotide polymorphism studies of the NRF2-coding NFE2L2 gene have revealed a link with outcome (191). Further, in addition to its beneficial effects on components of metabolic syndrome (241), NRF2 is also associated with attenuated risk for progression of diabetic kidney disease (DKD) (89) and to be in the center of inflammation- and metabolism-related pathways for CKD (134).
Mechanistically, NRF2 can induce the generation of NADPH and the expression of antioxidant enzymes through the ARE in the promoter regions of target genes (29). Thus, an NRF2-mediated antioxidant response is activated in human CKD glomeruli that are under increased oxidative stress (89). Interestingly, higher levels of oxidative stress and damage, as assessed by 8-oxo-dG staining, were evident in glomeruli of NRF2-deficient mice compared with wild-type controls (89). Besides its antioxidative response, NRF2 exerts direct and indirect anti-inflammatory effects through the inhibition of NF-κB signaling (214) and pro-inflammatory cytokines, for example, IL-1β and IL-6 (99). Moreover, ER stress, which can activate a maladaptive unfolded protein response (UPR) potentially affecting different renal pathologies (84), is directly linked to NRF2. Thus, ER stress induces nuclear translocation and DNA-binding activity of NRF2, as well as NRF2-dependent gene expression in 3T3-L1 adipocytes in a protein kinase RNA-like ER kinase (PERK)-dependent fashion (30), that is, one of the major adaptive UPR pathways (83). Importantly, these findings were also validated in human kidney 2 (HK-2) tubular epithelial cells (20). NRF2 nuclear translocation is further mediated by ER-produced H2O2 during ER oxidative protein folding, which, in turn, is related to intracellular Ca++ homeostasis (66). As ER stress also directly causes premature senescence (125, 126), NRF2 hypothetically can link tissue dysfunction and premature aging in CKD, not only through antioxidative effects but also by affecting cell organelle stress/crosstalk. In addition, autophagy, as one of the key mechanisms for clearance of mis-folded proteins (32), is also bi-directionally linked to NRF2 through the adaptor protein p62 (88). Thus, the NRF2 inducer sulforaphane exerts beneficial renal effects in a model of obesity-related glomerulopathy with higher potency compared with the conventional antioxidant N-acetylcysteine (129). Lu et al. (129) have demonstrated that in addition to the well-established antioxidative effects, NRF2 enhances markers of autophagy in podocytes (129), a finding similar to data obtained in pancreatic islets (121) and cardiomyocytes (231). By way of contrast, when autophagy is diminished in adipocytes, the adaptor protein p62 accumulates and acts as an endogenous inducer of NRF2 (13) by competing for NRF2 binding to KEAP1 (101). It needs to be pointed out that chronic activation of autophagy can also have deleterious effects. Thus, induction of p62 expression and subsequent NRF2 might stimulate tumor growth (82, 235). Indeed, NRF2 is related to cancer progression, metastasis, as well as resistance to chemo- and radiotherapy (174).
Keeping the aforementioned issues in mind, NRF2-based treatment could be considered one option to reduce the chronic burden of lifestyle diseases and particularly CKD by attenuating persistent inflammation, oxidative stress, as well as ER stress and autophagy. Several targets for NRF2 activation have recently been summarized, including both pharmacologic and nutriceutical compounds (29). Bardoxolone is one of the most promising pharmacological candidates with positive renal outcome data in a phase 2 trial in patients with DKD (162). Using a closely related bardoxolone analog in rodents, NRF2 treatment has been shown to improve redox balance, mitochondrial function, and to suppress inflammation (144). However, previous treatment approaches for patients with DKD using bardoxolone have been terminated (250) due to an excess of heart failure hospitalizations in the bardoxolone group. Interestingly, post hoc analyses have recently demonstrated that bardoxolone users were significantly less likely to develop a composite renal endpoint (23). Thus, the question remains as to whether bardoxolone exerts positive effects on renal function in carefully selected patients without signs of heart failure, which is presently being investigated in randomized controlled trials (21).
Besides pharmacological compounds (29), nutritional NRF2 modulators have been described (197), and distinct nutritional components can modulate NRF2, especially in CKD (46). Among others, the alkyl-catechols sulforaphane (inter alia derived from broccoli) and curcumin (inter alia derived from turmeric plant) appear to be the most attractive candidates (46, 197). A few studies have investigated the effects of nutritional NRF2 modulators in human burden of lifestyle diseases (111), such as CKD (17, 18). Importantly, curcumin's effects to decrease renal fibrosis, inflammation, and oxidative stress (2, 3) have been already validated in several small randomized controlled trials conducted in patients with CKD (2) and HD (3). Taken together, NRF2 is a promising candidate for the treatment of inflammation, oxidative stress, and related disturbances, including ER stress and autophagy, in numerous chronic burden of lifestyle diseases, including CKD, and several pharmacological and nonpharmacological treatment options are already available.
Klotho
Klotho is a single-pass transmembrane protein predominantly expressed in the kidneys that regulates aging and morbidity (114). Klotho-deficient mice display increased renal inflammation (124, 252), oxidative stress (96), a senescent phenotype (124), decreased autophagy (185), as well as premature aging (115)—a condition resembling the deleterious features of CKD. As patients and animals with CKD have decreased protein levels of klotho in the plasma, urine, and the kidneys (79), targeting klotho is a promising approach for the prevention of progression, complications, and the premature aging processes in renal dysfunction (78).
Similar to NRF2, klotho expression in CKD can be targeted by different approaches, by either increasing endogenous klotho expression or direct administration of exogenous klotho (253). From a practical perspective, this might be achieved from an epigenetic approach via demethylation of the klotho promoter and/or associated histone deacetylation to loosen the local chromatin configuration (149, 253). In this context, it is notable that IS and PCS directly induce hypermethylation of the klotho-coding KL gene (149, 204). Further, several drugs, already approved for the treatment of other diseases frequently observed in CKD, upregulate klotho expression (253). However, and in contrast to extensive studies in rodents, human data for klotho-based treatment approaches are lacking.
Although no human study to date has investigated distinct klotho therapies, including administration of exogenous klotho on CKD (254), several clinical trials on endogenous klotho inducers have been published. The peroxisome proliferator-activated receptor γ agonist pioglitazone, for example, increases renal klotho expression and reduces oxidative stress (242). Thus, thiazolidinedione treatment, that is, pioglitazone, in patients with CKD reduces markers of renal dysfunction possibly by affecting oxidative stress, inflammation, and other CKD-associated maladies [reviewed in Sarafidis and Bakris (176)]. Further, several vitamin D analogs induce the expression of klotho in distinct tissues, as well as increase urinary and serum klotho levels (117, 170). Conversely, vitamin D deficiency is associated with increased oxidative stress, inflammation, and cellular senescence [i.e., “inflammageing” (56)] (52) and vitamin D supplementation improves markers of oxidative stress according to a recent systematic review and meta-analysis (181). However, data from studies in patients with CKD show conflicting results of vitamin D receptor activators and their effects on markers of oxidative stress (86, 213, 216). Moreover, statins have been observed to increase klotho mRNA expression (146) and further data suggest that statins (246) reduce oxidative stress in experimental cyclosporine nephropathy via modulation of klotho. However, in patients with CKD, statin treatment, as part of an antioxidative therapy, does not induce circulating klotho compared with placebo (1) and atorvastatin treatment does not reduce oxidative stress (49). Randomized controlled trials indicate that the effects of statins on mortality in CKD are also independent of inflammation (108, 202), and statin treatment did not affect the association of lipid profile with mortality in a large observational dialysis cohort (42a). Collectively, these data suggest that the beneficial renal effects of statins on klotho observed in rodents do not translate in increased systemic klotho and oxidative stress in humans. In addition, activation of the nuclear androgen receptor (e.g., by testosterone) increases renal klotho gene expression (77).
Direct administration of exogenous soluble klotho is another option to increase circulating klotho levels, which has been reported to exert renoprotective effects in rats with AKI (80) and mice progressing from AKI to CKD (185). In conclusion, animal and human data on endogenous klotho inducers show contradicting results for renal compared with systemic oxidative stress and inflammation in CKD.
SGLT2 inhibitors/other approaches
SGLT2 inhibitors were introduced as a new class of glucose-lowering medication, and the first members received their EMA- and/or FDA approval in 2012/2013. SGLT2 inhibitors induce glucosuria by different mechanisms, but particularly through reduced tubular reabsorption of glucose (37). With respect to effects in CKD, data from randomized controlled CV outcome trials have shown beneficial renal effects for empagliflozin (233), dapagliflozin (239), and canagliflozin (147). Since two recent renal outcome trials with a prespecified primary renal endpoint have confirmed these findings (69a, 163), SGLT2 inhibitors are considered as a novel therapy for CKD due to type 2 diabetes reducing the risk of dialysis, transplantation, or death and protecting against AKI (148).
Potential mechanisms through which SGLT2 inhibitors improve renal function include beneficial hemodynamic, vascular, metabolic, anti-oxidative, inflammatory, hypoxic, and diuretic effects (69). Although hemodynamic changes due to altered tubulo-glomerular feedback (95) are often believed to be a key mechanism of SGLT2 inhibitors, inflammation and oxidative stress, as well as renal hypoxia, could also play major roles. Indeed, recent data suggest that SGLT2 inhibitors protect kidney function through improvement of renal hypoxia (72, 179). As hyperglycemia causes tubular senescence via an SGLT2- and a p21-dependent pathway in the type 1 diabetic kidney (98), the effects of SGLT2 inhibitors on kidney senescence, SASP, and NRF2 expression need to be tested. Human studies suggest that SGLT2 inhibitors reduce circulating markers of inflammation and improve the adipokine profile (11). As several adipose tissue-secreted cytokines are associated with inflammatory markers in CKD (40, 75, 104, 105, 151), Bonnet and Scheen (11) have suggested that SGLT2 inhibitors act on adipose tissue, as well as on other distinct organs, to reduce systemic inflammation. Regarding renal inflammation, data are limited to rodent studies (11, 93), where the SGLT2 inhibitor empagliflozin reduced renal inflammation in mouse models of DKD (65, 152, 220). SGLT2 inhibitors have also improved renal oxidative stress in several rodent models of DKD. In more detail, different SGLT2 inhibitors reduced oxidative stress in diabetic rats (152, 155) or mice (68, 212) by improved antioxidant enzyme activities (155), suppression of the AGE/RAGE-axis (152), as well as decreased production of ROS and Nox4 expression (68, 212). It should be noted, however, that these effects of SGLT2 inhibitors on rodent DKD cannot specifically determine the cause of reduced inflammation/oxidative stress. Thus, a direct effect could still be possible. In contrast, they might also be the result of secondary improvements due to lower body weight, improved hyperglycemia, and other effects. It also has to be pointed out that there is a lack of rodent models of DKD replicating the main features of human DKD (5) and there is a huge difference in the renal phenotype, for example, on albuminuria, between standard diabetic mouse models compared with accelerated models (106, 215). This is congruent with a general feature of laboratory studies, where the rodents employed are metabolically morbid and lacking appropriate features of either human pathogenesis or normative aging (120, 133, 156). A biomimetic approach identifying wild animals that during evolution in extreme environments have developed mechanisms that protect them against aging, CKD and metabolic dysfunctions may yield important additive clues (135, 198). Therefore, carefully matched animal experiments are needed to verify the results excluding secondary effects. If such pleiotropic effects, that is, independent of metabolic and hemodynamic changes, can be confirmed, SGLT2 inhibitors can be a treatment target for other CKD causes than just DKD—a concept being currently investigated, for example, in the EMPA-KIDNEY study (70).
Live biotherapeutics
Multiple studies have linked gut dysbiosis and CKD (102, 194). Patients with CKD often display reduced microbial diversity, though this finding is not always consistent between studies, accompanied by a decrease in bacteria producing short-chain fatty acids (SCFA) from fiber and an increase in bacteria that produce uremic toxins such as TMAO, IS, and PCS (139, 194). Possible biotherapeutic treatment strategies for CKD include dietary therapy, prebiotics (ingredients that stimulate the growth of a desired microorganism), probiotics (live microorganisms), synbiotics (combining both prebiotics and probiotics), as well as host and bacterial enzyme inhibition (48). However, given the dietary restrictions, therapies, and frequent comorbidities found in patients with CKD, further studies may be warranted to identify specific causal links and therapeutic options (116, 194). For instance, potassium and phosphorus intake are restricted in patients with CKD due to the increased risk of adverse outcomes, but these restrictions also limit fruit and vegetable intake and affect the gut microbiome (116). Nevertheless, a number of studies have found beneficial effects in patients with CKD for a range of diets, most notably a Mediterranean or a vegetarian diet (139).
Perhaps in part due to the limits of current knowledge, studies with probiotics and prebiotics have produced inconsistent results (194). Increasing microbial SCFA production has been proposed as a possible therapeutic intervention (81). SCFA regulate immunity, blood pressure, and glucose metabolism; they have been shown to be involved in the health of colonic epithelial cells, signal transduction, epigenetic maintenance, and autophagy; and to ameliorate kidney dysfunction (81, 116, 238). Alternatively, decreasing the intestinal production of uremic toxins can also be a valid approach (194), and studies have shown some efficacy using synbiotics or prebiotics (139, 238).
One enzymatic approach has focused on acarbose, an inhibitor of α-glucosidase that limits the hydrolysis of polysaccharides and oligosaccharides in the intestine, stimulating the growth of saccharolytic bacteria, reducing the production of p-cresol (145). Indeed, studies in healthy volunteers indicate that acarbose may shift gut microbial metabolism away from proteolytic fermentation and toward saccharolytic fermentation (47). Importantly, acarbose therapy is also associated with experimental longevity (203). Another strategy under investigation is the use of 3, 3-dimethyl-1-butanol (DMB), an inhibitor of microbial TMA production that has led to decreased levels of TMAO in the blood and reduced atherosclerotic lesion development (145).
Conclusions
Increasing health care costs in patients with CKD, overall detrimental outcomes, and deficiency of optimal treatments, including organ donation, place a tremendous pressure on health care providers, scientists, and patients and their families per se. Innovative therapeutics based on the identification of selective injury targets might serve to develop precision medicine to optimize health care costs, disease monitoring, targeted interventions, as well as outcomes, including patients' quality of life. In this review, we view CKD as a burden of lifestyle disease, underpinned by a dysregulated aging process that is common to the “diseaseome of ageing.” A systemic approach to tackle CKD, based on attenuating the associated inflammatory and oxidative cell stress, mitochondrial dysfunction, and damage processes, has the potential to mitigate the effects of CKD, but also preempt the development and progression of associated morbidities. In effect, this will enhance health span and compress the period of morbidity. Pharmacological as well as nutritional and potentially lifestyle-based interventions are promising therapeutic avenues to achieve such a goal.
Footnotes
Authors' Contributions
T.E., O.N., P.S., and P.G.S. wrote the article and researched literature. A.W. and K.K. contributed to the discussion and reviewed/edited the article.
Author Disclosure Statement
P.S. serves on the scientific advisory boards of Baxter, Reata, and Astra Zeneca. O.N. is funded by a research award to Paul Shiels from Constant Pharma (Massachusetts). T.E. has served as a consultant for Sanofi-Aventis, as well as on a scientific advisory board of Boehringer Ingelheim Pharma. The other authors of this article have nothing to declare.
Funding Information
T.E. was supported by a Novo Nordisk postdoctoral fellowship run in partnership with Karolinska Institutet, Stockholm, Sweden, a Karolinska Institutet Research Foundation grant, a grant by the Stig och Gunborg Westmans stiftelse (Stig and Gunborg Westmans-Foundation), as well as by the EFSD Mentorship Programme supported by AstraZeneca. A.W., K.K., P.S., and T.E. were further supported by Njurfonden (Swedish Kidney Foundation). P.S. was supported by the Swedish Heart and Lung Foundation (No. 20160384), as well as by the Strategic Research Programme in Diabetes at Karolinska Institutet (Swedish Research Council grant No. 2009–1068). K.K. received other grants from the Swedish Research Council (No. 2018–00932). P.G.S. was supported by awards from 4D Pharma and Constant Pharma.