
Abstract
Aims:
Emerging evidence suggests that the pathogenesis of osteoporosis, characterized by impaired osteogenesis, is shifting from estrogen centric to oxidative stress. Our previous studies have shown that the zinc-finger transcription factor krüppel-like factor 5 (KLF5) plays a key role in the degeneration of nucleus pulposus and cartilage. However, its role in osteoporosis remains unknown. We aimed to investigate the effect and mechanism of KLF5 on osteogenesis under oxidative stress.
Results:
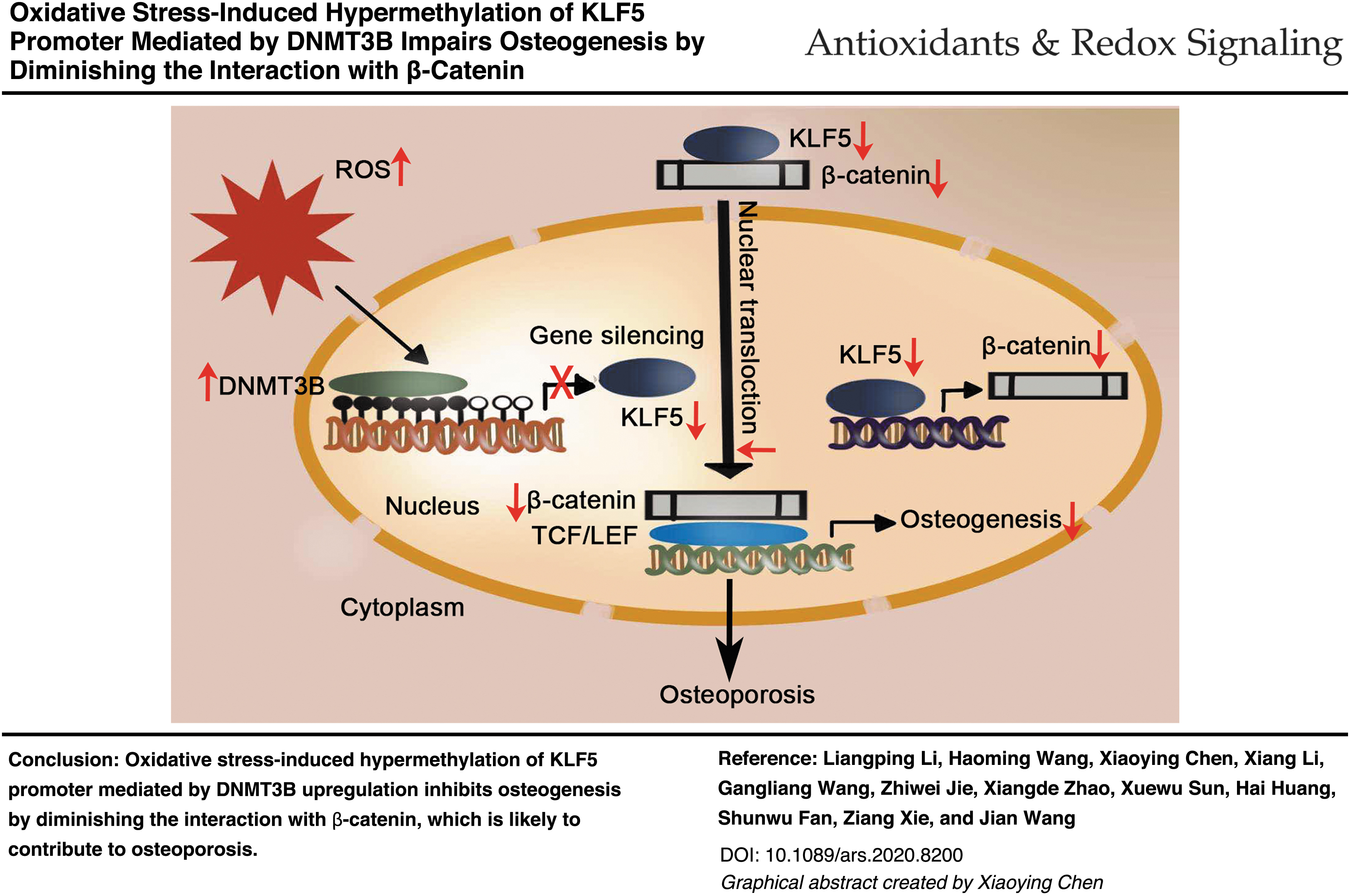
First, KLF5 was required for osteogenesis and stimulated osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs). KLF5 was hypermethylated and downregulated in ovariectomy-induced osteoporosis mice and in BMSCs treated with H2O2. Interestingly, DNA methyltransferases 3B (DNMT3B) upregulation mediated the hypermethylation of KLF5 induced by oxidative stress, thereby impairing osteogenic differentiation. The inhibition of KLF5 hypermethylation using DNMT3B siRNA or 5-AZA-2-deoxycytidine (5-AZA) protected osteogenic differentiation of BMSCs from oxidative stress. Regarding the downstream mechanism, KLF5 induced β-catenin expression. More importantly, KLF5 promoted the nuclear translocation of β-catenin, which was mediated by the armadillo repeat region of β-catenin. Consistently, oxidative stress-induced KLF5 hypermethylation inhibited osteogenic differentiation by reducing the expression and nuclear translocation of β-catenin.
Innovation:
We describe the novel effect and mechanism of KLF5 on osteogenesis under oxidative stress, which is linked to osteoporosis for the first time.
Conclusion:
Our results suggested that oxidative stress-induced hypermethylation of KLF5 mediated by DNMT3B impairs osteogenesis by diminishing the interaction with β-catenin, which is likely to contribute to osteoporosis. Targeting the hypermethylation of KLF5 might be a new strategy for the treatment of osteoporosis. Antioxid. Redox Signal. 35, 1–20.
Color images are available online.
Introduction
Oxidative stress is an accumulation that occurs due to an imbalance in the generation and scavenging of reactive oxygen species (ROS) (53). Formation of ROS is an inescapable consequence of life in oxygen-rich environments and occurs primarily in the mitochondria (36). Excessive ROS damage nucleic acids, proteins, and lipids. Drastic oxidative stress inhibits cell survival, proliferation, and differentiation (26). Previous studies by us and others have demonstrated that osteogenic differentiation is inhibited by oxidative stress and is rescued by antioxidants (15, 30). Oxidative stress can be initiated by a variety of factors, including aging and estrogen deficiency, and is associated with numerous diseases such as cancer and osteoporosis.
Osteoporosis, characterized by low bone mass and high susceptibility to fracture, is caused by impaired bone formation and/or excessive bone resorption (65, 72). ROS levels in the bone marrow increase with age in both female and male mice (37). The increases in the levels of oxidized metabolites and decreases in antioxidant enzyme activities in the blood and saliva of patients with osteoporosis suggest that they are under oxidative stress (67, 69, 74). In addition, lineage commitment of mesenchymal stem cells (MSCs) is controlled by oxidative stress (33). Poor bone formation mediated by insufficient osteoblastogenesis is an important etiology of osteoporosis (39). Thus, a better understanding of the molecular mechanisms of osteogenesis in the context of oxidative stress contributes to elucidating the pathogenesis of osteoporosis.
Krüppel-like factor 5 (KLF5), a member of the large KLF family of transcription factors, is structurally characterized by three zinc-finger domains at the C-terminus (50). KLF5 is ubiquitously expressed in various tissues, including the gut epithelium, cardiovascular and adipose tissue, muscle, bone, and cartilage (13). KLF5 regulates numerous cellular functions, including cell proliferation, differentiation, apoptosis, migration, and stemness in response to diverse environmental stimuli (2, 14, 54). KLF5 has been reported to regulate smooth muscle cell differentiation (1), adipocyte differentiation of 3T3-L1 preadipocytes (44), muscle differentiation of C2C12 myoblasts (21), odontoblastic differentiation of dental papilla mesenchymal cells (8), luminal differentiation of basal progenitors (70), and osteogenic differentiation of human periodontal ligament cells (17), suggesting that KLF5 regulates cellular differentiation in a context-dependent manner.
Several members of the KLF family, including KLF2, KLF4, and KLF15, regulate osteoblast differentiation and skeletal development (38, 41, 68). Moreover, a previous study demonstrated that KLF5 also induced cartilage matrix degradation by matrix metalloproteinase 9, contributing to endochondral ossification during skeletal development (51). The authors reported that the femoral and tibial limbs of KLF5+/− embryos were 10%–15% shorter than that of the wild-type littermates, and that bone formation around the primary ossification center was delayed in the KLF5+/− limb shaft. They also found that KLF5 was mainly expressed in primary osteoblasts, chondrocytes, and the osteoblastic (MC3T3E1) and chondrogenic (ATDC5 and OUMS27) cell lines, but not osteoclasts. These findings suggest that KLF5 might promote skeletal development through the control of bone formation.
DNA methylation is associated with a variety of physiological activities, including gene silencing, chromatin modification, and embryonic development (47). DNA methylation is mainly mediated by maintenance enzyme DNA methyltransferases (DNMTs) DNMT1 and de novo enzymes DNMT3A/3B (45). Extensive studies have demonstrated that DNA methylation is closely associated with oxidative stress, aging, and cancerogenesis (22). Recent studies have shown that KLF5 silencing might be correlated with DNMT3A in acute myeloid leukemia (11, 33), and hypermethylation of KLF5 promoter is mediated by DNMT1 in renal cell carcinoma (16).
KLF5 is involved in various cancers, cardiovascular disorders, and age-associated diseases (10, 23, 24). Our previous studies have shown that KLF5 plays a key role in the degeneration of nucleus pulposus and cartilage (55, 64). However, the role of KLF5 in osteoporosis has not been defined. Here, we investigated the effect and underlying regulatory mechanism of KLF5 on osteogenesis under oxidative stress to further elucidate the pathogenesis of osteoporosis.
Results
KLF5 is required for osteogenesis
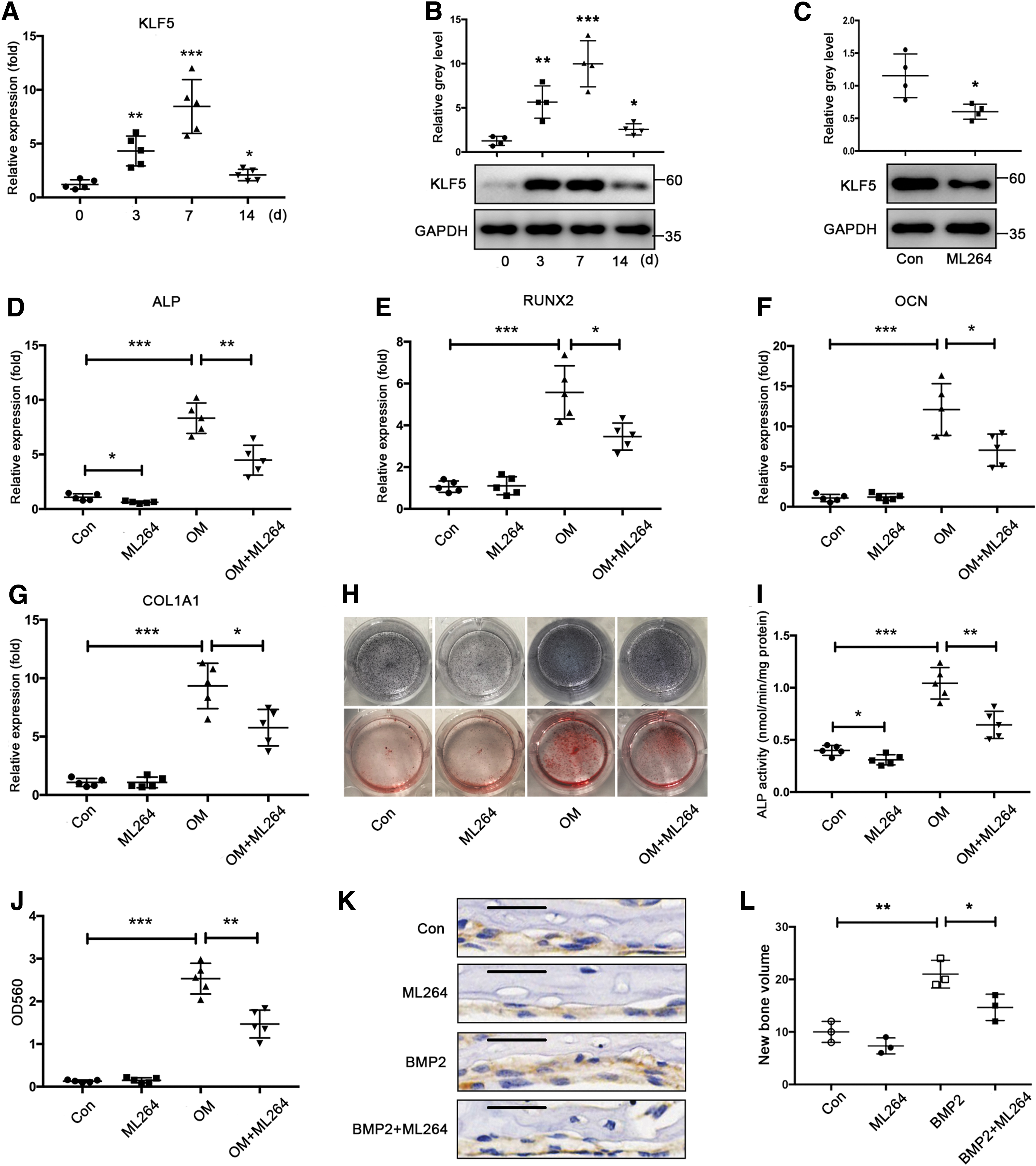
To determine whether KLF5 is involved in bone formation, we first determined KLF5 expression patterns during osteogenic differentiation. We observed that both mRNA and protein expression of KLF5 gradually increased in the early and middle stages, and declined in the later period during osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) (Fig. 1A, B). ML264, a KLF5 inhibitor, was used to investigate the effect of KLF5 on osteogenesis. ML264 significantly suppressed KLF5 expression, whereas it had no effect on the proliferation of BMSCs (Fig. 1C and Supplementary Fig. S1). Intriguingly, ML264 reduced the mRNA expression of osteogenic markers, including alkaline phosphatase (ALP), runt-related transcription factor 2 (RUNX2), osteocalcin (OCN), and collagen type I alpha 1 (COL1A1) at 7 days (Fig. 1D–G). It also inhibited ALP activity at 7 days and calcium deposition at 14 days, respectively, during osteogenic differentiation of BMSCs, as shown by ALP staining and Alizarin red staining (Fig. 1H–J). Subsequently, we performed calvarial organ culture to assess the effect of KLF5 on bone morphogenic protein-2 (BMP2)-induced bone formation. As expected, BMP2 induced new bone formation in subperiosteal calvarial. However, ML264 significantly repressed BMP2-induced new bone formation (Fig. 1K, L). Collectively, these in vitro and ex vivo findings indicated that KLF5 was induced during osteogenic differentiation and required for bone formation.
KLF5 stimulates osteogenesis
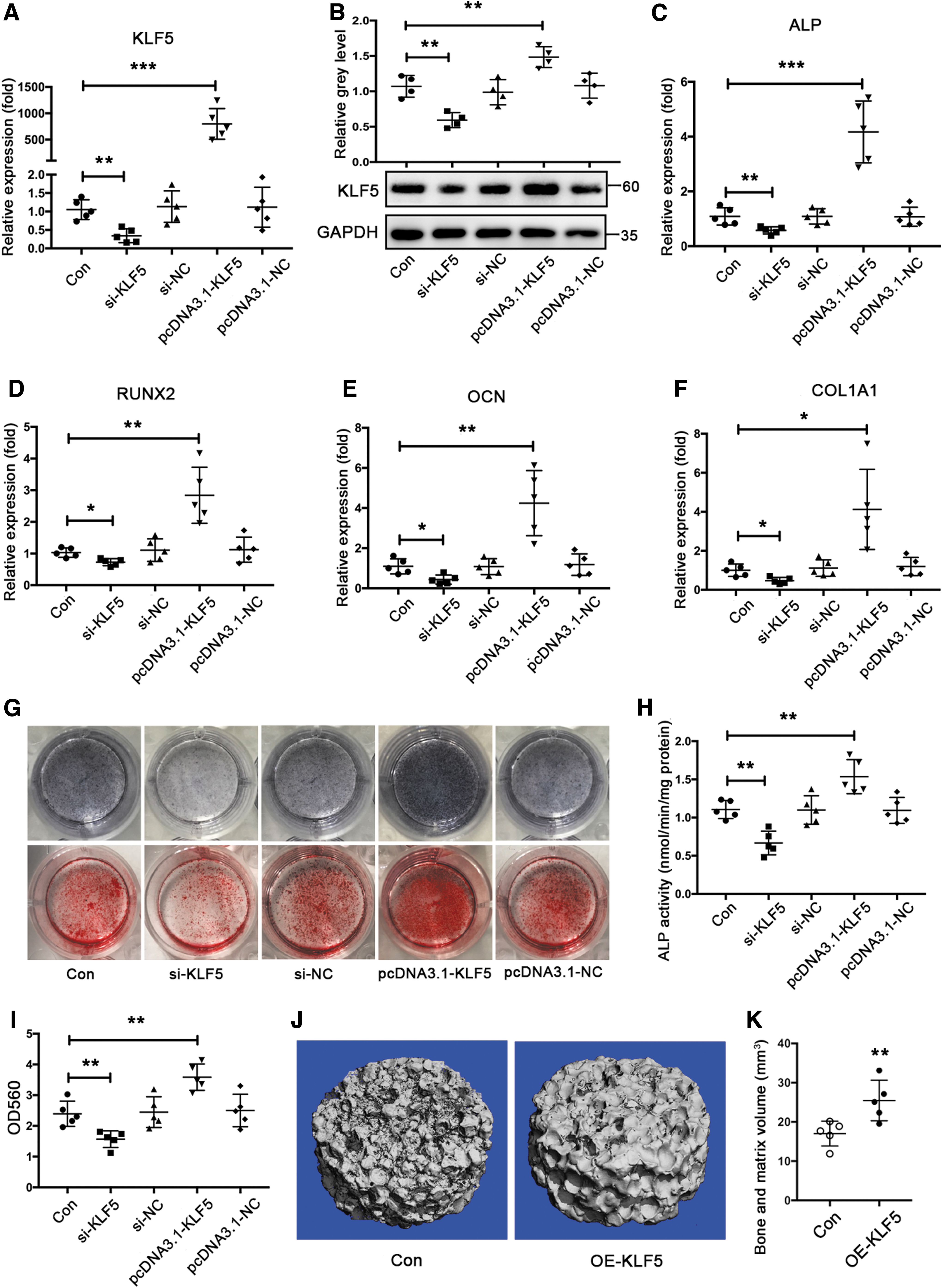
To test whether KLF5 regulates osteogenic differentiation, BMSCs were incubated with OM for osteogenic induction after transfection of si-KLF5 or pcDNA3.1-KLF5. Initially, the transfection of si-KLF5 decreased KLF5 mRNA and protein expression, whereas the transfection of pcDNA3.1-KLF5 had the opposite effect (Fig. 2A, B). Thereafter, we found that the mRNA levels of osteogenic markers, including ALP, RUNX2, OCN, and COL1A1, were diminished in response to KLF5 knockdown and elevated in response to KLF5 overexpression in differentiated BMSCs (Fig. 2C–F). The ALP activity shown by ALP staining and calcium deposition shown by Alizarin red staining changed in a similar manner during osteogenic differentiation of BMSCs (Fig. 2G–I). Finally, we used a model of subcutaneous heterotopic bone formation with β-tricalciumphosphate (β-TCP) scaffolds to confirm the in vivo effect of KLF5 on osteogenesis. Compared with that of the control group, KLF5 overexpression significantly enhanced the new bone formation in vivo (Fig. 2J, K). Taken together, these findings suggested that KLF5 stimulated osteogenic differentiation and enhanced bone formation.
Oxidative stress-induced KLF5 downregulation impairs osteogenic differentiation
To investigate the role of KLF5 in osteoporosis, we determined the expression of KLF5 in osteoporotic mice using an ovariectomized (OVX) model. As expected, immunohistochemistry (IHC) showed that OCN expression was significantly reduced in the tibias of OVX mice. Moreover, both mRNA and protein levels of KLF5 were downregulated in the OVX group compared with that of the control group (Fig. 3A, B). Thereafter, BMSCs incubated with H2O2 were used to induce oxidative stress and simulate the microenvironment of BMSCs in patients with osteoporosis. Cell viability was not significantly reduced when BMSCs were incubated with H2O2 at concentrations up to 200 μM (Fig. 3C). Accordingly, BMSCs incubated with 200 μM H2O2 were used to establish a cell model of oxidative stress. Subsequently, KLF5 expression was downregulated in BMSCs upon H2O2 treatment, while the antioxidant N-acetyl L-cysteine (NAC, 2 mM) abolished the inhibitory effect of H2O2 on KLF5 expression (Fig. 3D, E). Similarly, cell immunofluorescence also demonstrated that H2O2 inhibited KLF5 protein expression, whereas NAC abolished this inhibitory effect (Fig. 3F).
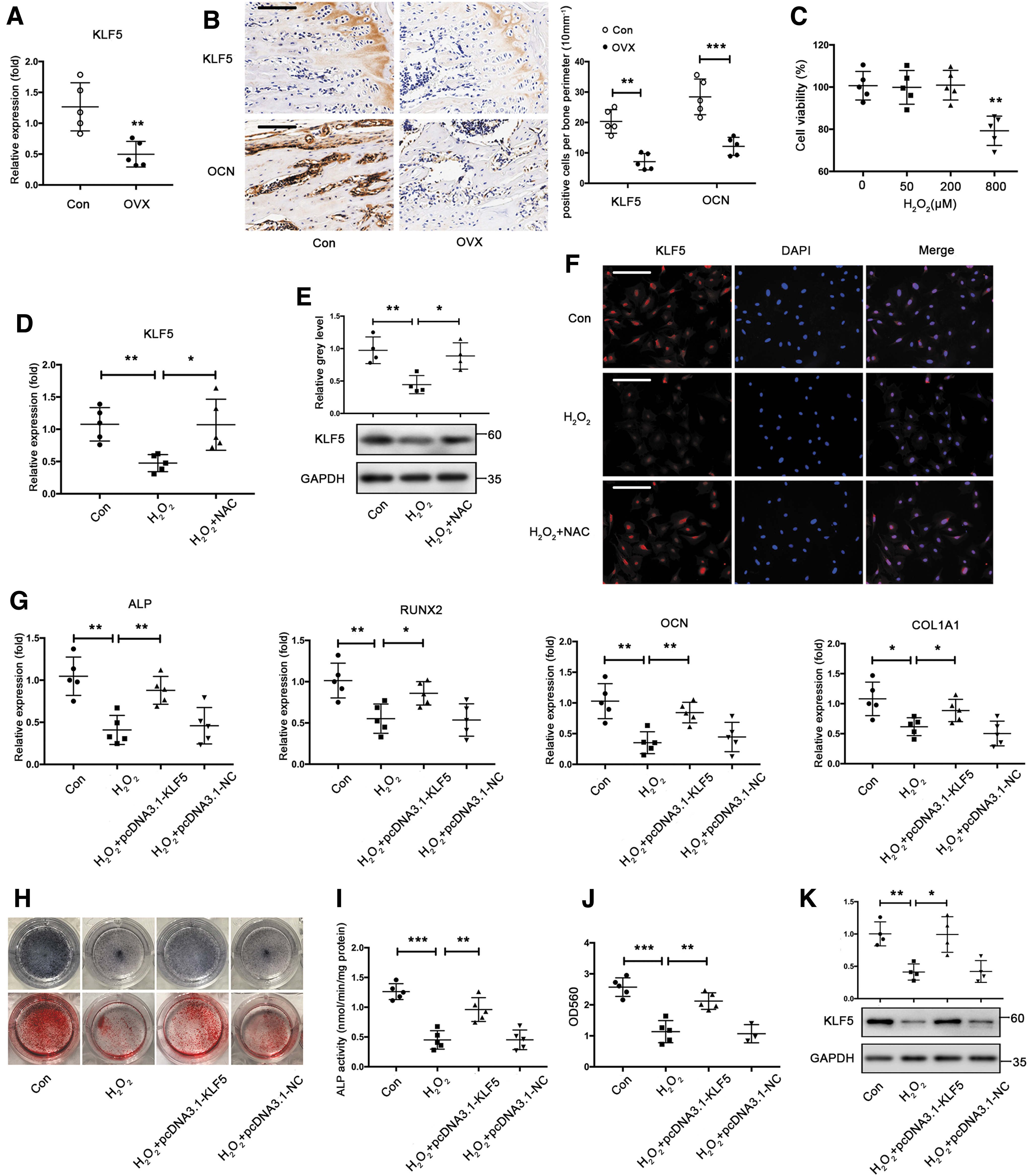
Furthermore, we investigated whether KLF5 downregulation induced by oxidative stress resulted in compromised osteogenic differentiation. H2O2 treatment resulted in decreased mRNA expression of osteogenic markers, ALP activity, calcium deposition, and downregulated KLF5 expression in differentiated BMSCs (Fig. 3G–K). However, KLF5 overexpression, at least in part, rescued the inhibitory effect of H2O2 on the mRNA expression of osteogenic markers, ALP activity, and calcium deposition (Fig. 3G–J). In addition, transfection of pcDNA3.1-KLF5 reversed KLF5 downregulation in differentiated BMSCs exposed to H2O2 (Fig. 3K). Taken together, these results suggested that KLF5 was downregulated in osteoporotic mice and in BMSCs treated with H2O2, thereby impairing osteogenic differentiation under oxidative stress.
DNMT3B mediates the hypermethylation and downregulation of KLF5 induced by oxidative stress
We investigated the mechanism by which KLF5 was downregulated by oxidative stress. Recent studies have shown that the downregulation of KLF5 expression is associated with the hypermethylation of its promoter in tumors, which is also closely associated with oxidative stress (11, 12, 16). Thus, we examined whether decreased KLF5 expression was accompanied by increased DNA methylation levels under oxidative stress. Bisulfite sequencing demonstrated higher methylation levels of the KLF5 promoter in the OVX group than in the control group (Fig. 4A). H2O2 treatment increased methylation levels of the KLF5 promoter in BMSCs, whereas NAC attenuated the hypermethylation effect (Fig. 4B). Accordingly, BMSCs were transfected with siRNAs to investigate the effect of DNMTs on KLF5 expression, including DNMT1, DNMT3A, and DNMT3B. The silencing of DNMT3A or DNMT3B increased the mRNA and protein expression of KLF5, while the silencing of DNMT1 had no effect (Fig. 4C, D). Subsequently, we determined DNMT expression under oxidative stress. Among the three DNMTs, only DNMT3B mRNA was upregulated in BMSCs upon H2O2 treatment and was attenuated by NAC (Fig. 4E). The changes of DNMT3A mRNA were contrasted with those of DNMT3B mRNA in response to H2O2 and NAC. Western blot also confirmed that DNMT3B expression was upregulated in BMSCs under oxidative stress (Fig. 4F). Thereafter, transfection of siDNMT3B reduced DNMT3B expression and abrogated increased methylation levels of the KLF5 promoter in BMSCs exposed to H2O2 (Supplementary Fig. S2 and Fig. 4G). Accordingly, DNMT3B silencing increased the mRNA and protein expression of KLF5 compared with that of the negative control (Fig. 4H, I). Immunofluorescence also confirmed that DNMT3B silencing increased KLF5 expression (Fig. 4J). Collectively, these findings indicated that DNMT3B upregulation mediated the increased methylation levels and decreased expression of KLF5 under oxidative stress.

DNMT3B-mediated KLF5 hypermethylation impairs osteogenic differentiation under oxidative stress
We further investigated whether DNMT3B-mediated KLF5 downregulation impaired osteogenic differentiation under oxidative stress. We found that DNMT3B silencing significantly attenuated the inhibitory effect of H2O2 on osteogenic markers, ALP activity, and calcium deposition (Fig. 5A–D). Moreover, ML264 abolished the protective effect of DNMT3B silencing on osteogenic differentiation of BMSCs under oxidative stress (Fig. 5A–D). Likewise, DNMT3B silencing significantly attenuated the inhibitory effect of H2O2 on KLF5 expression during osteogenic differentiation of BMSCs, whereas ML264 abolished the protective effect of DNMT3B silencing on KLF5 expression (Fig. 5E). Subsequently, we evaluated DNMT3B expression in the tibias of osteoporotic mice. Compared with the control group, the OVX group had significantly higher protein expression of DNMT3B (Fig. 5F).

Inhibition of KLF5 hypermethylation by DNMT inhibitors protects osteogenic differentiation under oxidative stress
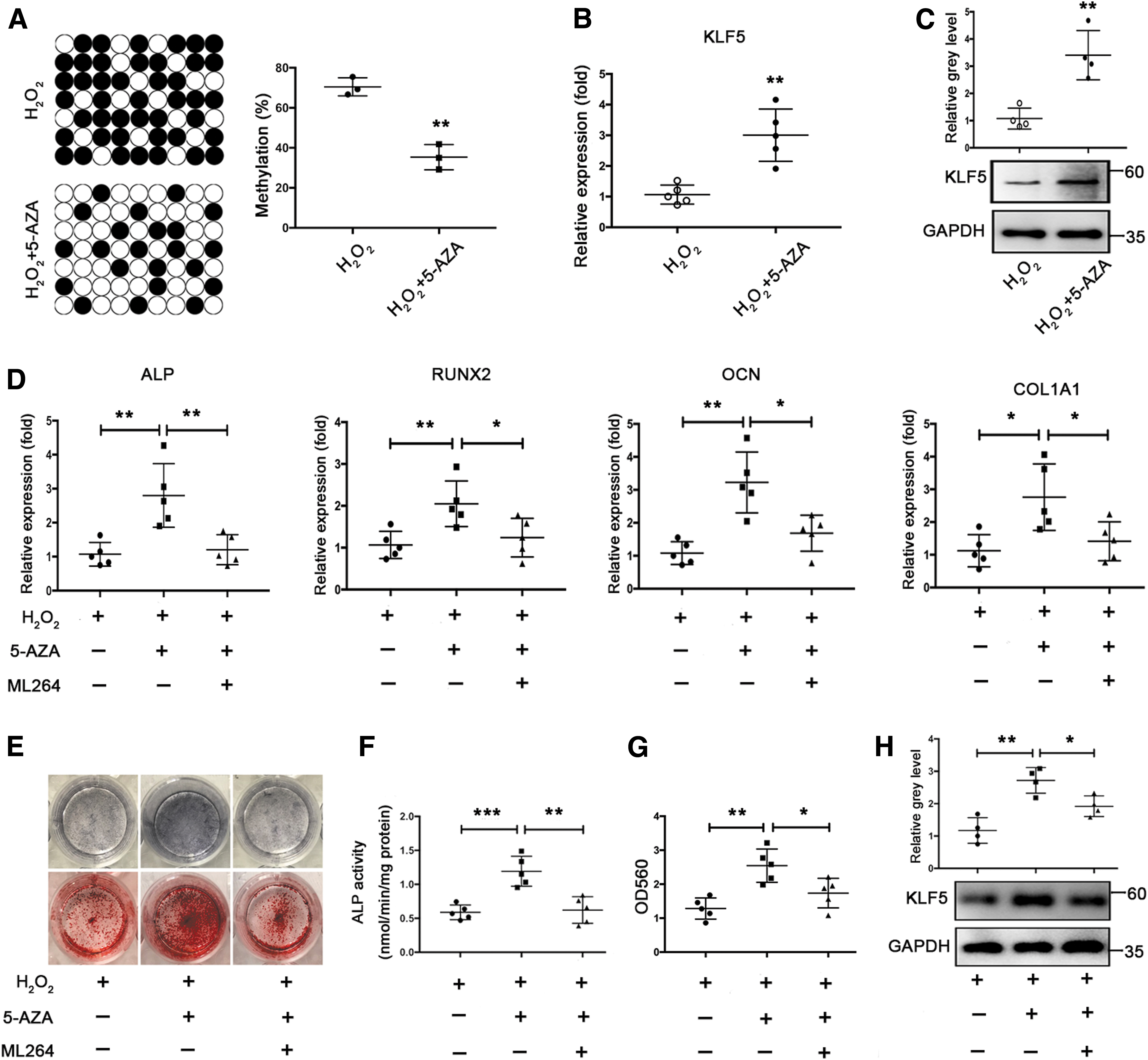
We first examined whether 5-AZA-2-deoxycytidine (5-AZA), a commonly used DNMT inhibitor, reduced KLF5 hypermethylation and increased KLF5 expression under oxidative stress. Administration of 5-AZA resulted in decreased methylation levels of the KLF5 promoter from 70.5% to 35.3% in BMSCs incubated with H2O2 (Fig. 6A). Nanaomycin A is a new DNMT3B-selective small molecule inhibitor (28). Both 5-AZA and Nanaomycin A significantly increased KLF5 mRNA and protein expression (Fig. 6B, C and Supplementary Fig. S3A, B). We further investigated whether inhibition of KLF5 hypermethylation using DNMT inhibitors protected against the impaired osteogenic differentiation under oxidative stress. Both 5-AZA and Nanaomycin A resulted in increased mRNA expression of osteogenic markers, ALP activity, and calcium deposition during osteogenic differentiation of BMSCs in the presence of H2O2. However, ML264 abrogated the stimulatory effect of 5-AZA and Nanaomycin A on osteogenic differentiation of BMSCs under oxidative stress (Fig. 6D–G and Supplementary Fig. S3C–I). Moreover, 5-AZA increased KLF5 expression during osteogenic differentiation of BMSCs in the presence of H2O2. ML264 also abrogated the stimulatory effect of 5-AZA on KLF5 expression in differentiated BMSCs exposed to H2O2 (Fig. 6H). Taken together, these results suggested that inhibition of KLF5 hypermethylation using DNMT inhibitors, resulting in upregulated KLF5 expression, and thus protected the impaired osteogenic differentiation of BMSCs under oxidative stress.
The KLF5–β-catenin interaction is mediated by the armadillo repeat region of β-catenin
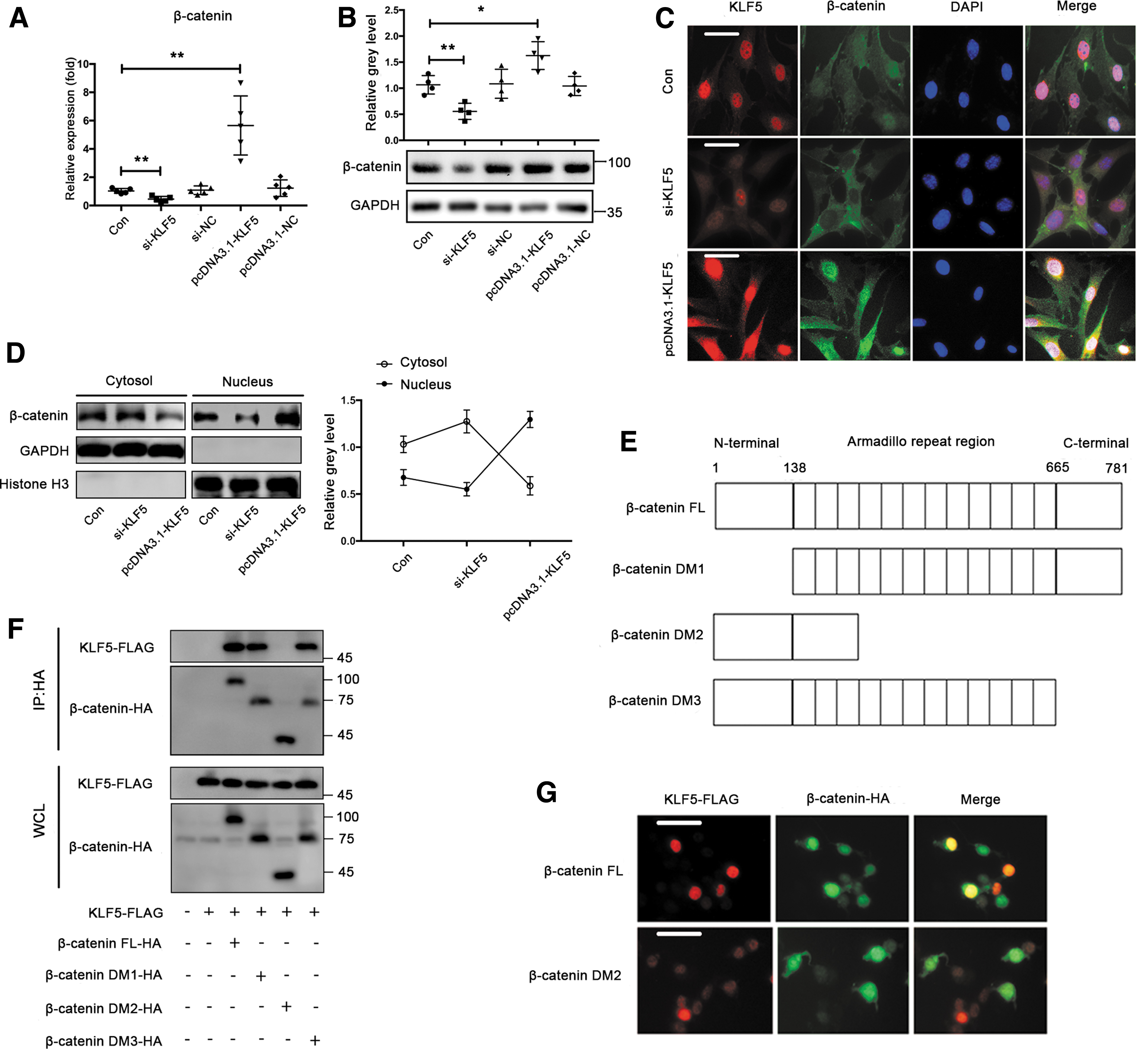
Next, we investigated the downstream mechanism by which KLF5 regulated osteogenic differentiation. Previous studies have demonstrated that KLF5 interacts with β-catenin in several cancer cells (19, 40, 57). Therefore, we examined whether β-catenin was regulated by KLF5 in BMSCs. We observed that KLF5 knockdown resulted in decreased mRNA and protein expression of β-catenin, whereas KLF5 overexpression resulted in opposite changes (Fig. 7A, B). More importantly, immunofluorescence showed that KLF5 knockdown significantly inhibited the nuclear translocation of β-catenin, whereas KLF5 overexpression enhanced its nuclear translocation (Fig. 7C). In addition, Western blot demonstrated that KLF5 silencing significantly decreased nuclear β-catenin but increased cytosolic β-catenin, whereas KLF5 overexpression led to the opposite effects (Fig. 7D). Given that the armadillo repeat region is critical for biological function of β-catenin, we further investigated whether KLF5 regulated β-catenin through the armadillo repeat region. One plasmid with the β-catenin full-length (FL) and three plasmids with β-catenin deletion mutants (DMs) were constructed to examine which region of β-catenin bound to KLF5 (Fig. 7E). Interestingly, coimmunoprecipitation experiments revealed that β-catenin DM2, spanning the armadillo repeat region, dramatically abolished the β-catenin–KLF5 interaction in 293T cells (Fig. 7F). Furthermore, immunofluorescence showed that β-catenin DM2 resulted in less colocalization of KLF5 and β-catenin in 293T cells compared with β-catenin FL (Fig. 7G). These findings indicated that KLF5 promoted the nuclear translocation of β-catenin, which was mediated by the armadillo repeat region.
KLF5 hypermethylation impairs osteogenic differentiation by reducing the interaction with β-catenin under oxidative stress
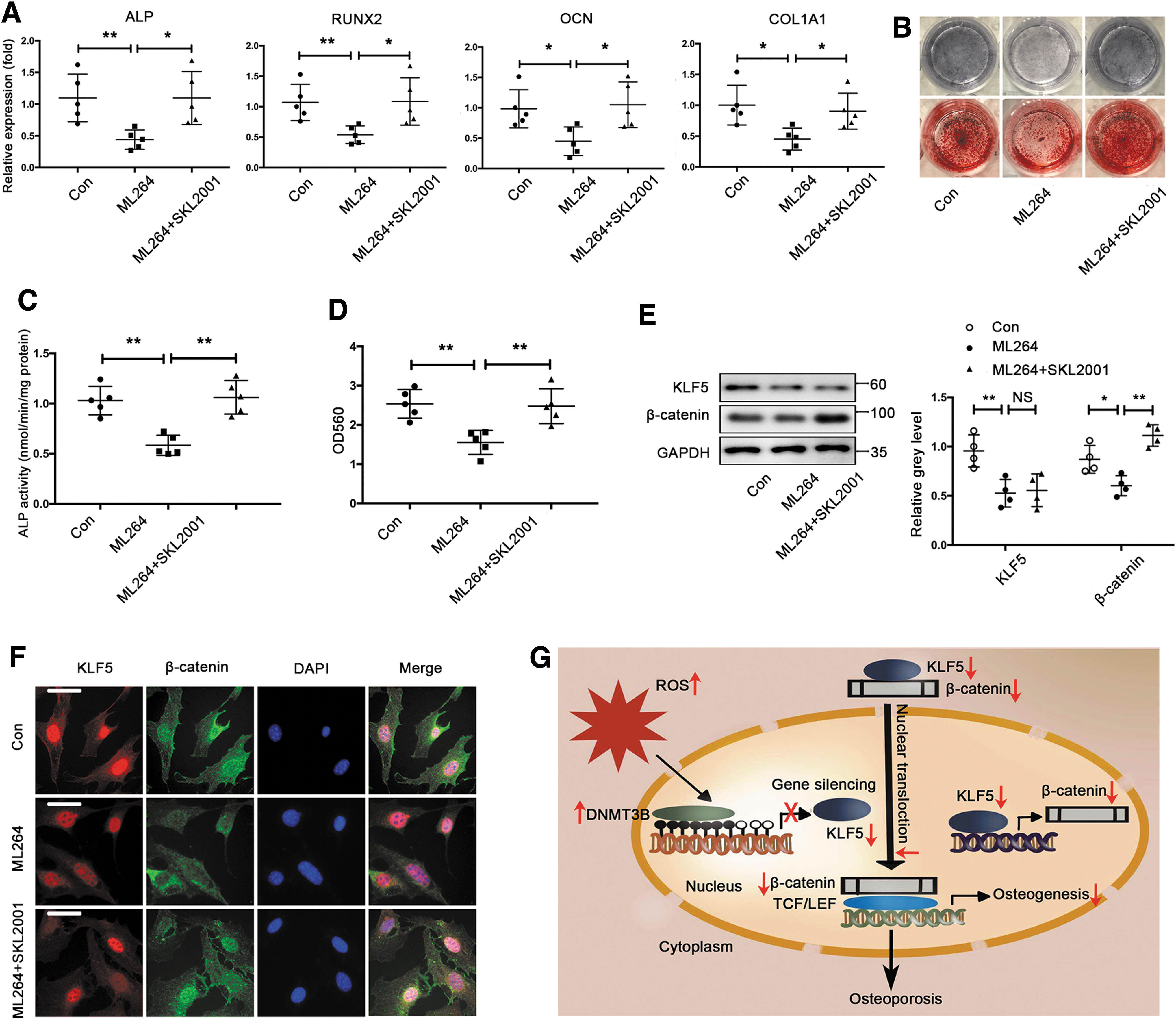
We first investigated whether KLF5 regulated osteogenic differentiation through β-catenin under normal condition. The Wnt/β-catenin pathway agonist SKL2001 that stabilizes intracellular β-catenin reversed the inhibitory effect of ML264 on osteogenic differentiation of BMSCs, as evidenced by the rescue of osteogenic marker genes, ALP activity, and calcium deposition (Fig. 8A–D). Meanwhile, ML264 suppressed β-catenin expression during osteogenic differentiation of BMSCs, whereas SKL2001 reserved the inhibitory effect of ML264 on β-catenin expression (Fig. 8E). Moreover, ML264 inhibited the nuclear translocation of β-catenin in differentiated BMSCs. However, SKL2001 abolished the inhibitory effect of ML264 on β-catenin nuclear translocation (Fig. 8F).
Next, we further examined whether KLF5 regulated osteogenic differentiation through β-catenin under oxidative stress. SKL2001 reserved the decreased mRNA expression of osteogenic markers, ALP activity, and calcium deposition during osteogenic differentiation of BMSCs upon H2O2 treatment (Supplementary Fig. S4A–D). Accordingly, SKL2001 abolished the inhibitory effect of KLF5 downregulation induced by H2O2 treatment on β-catenin expression in differentiated BMSCs (Supplementary Fig. S4E). Moreover, SKL2001 abolished the inhibitory effect of KLF5 downregulation on the nuclear translocation of β-catenin in differentiated BMSCs under oxidative stress (Supplementary Fig. S4F). Taken together, these results suggested that KLF5 hypermethylation impaired osteogenic differentiation by reducing the interaction with β-catenin under oxidative stress.
Discussion
Estrogen deficiency has been considered the seminal mechanism of osteoporosis, but emerging epidemiological surveys in humans and mechanistic studies in rodents suggest that aging and oxidative stress are the proximal culprits (36). Although the elevated ability of osteoclastic bone resorption is the leading causative factor of osteoporosis, the impaired capacity of osteogenic differentiation of BMSCs is another important factor (39). However, the molecular mechanisms underlying impaired osteogenesis under oxidative stress remain elusive. Herein, we describe the effect of the zinc-finger transcription factor KLF5 on osteogenesis under oxidative stress and the upstream and downstream mechanisms, which is linked to osteoporosis for the first time (Fig. 8G).
Although KLF5 has been identified to control adipogenesis (44), myogenesis (21), and odontogenesis (8), its effect on osteogenesis has not been defined yet. Several members of the KLF family have been identified to regulate osteoblast differentiation. For instance, KLF2 stimulates osteoblast differentiation of dental pulp-derived stem cells (38), whereas KLF4 represses osteogenic differentiation of primary calvarial osteoblasts (56). Several pieces of evidence gathered from the study demonstrated that KLF5 positively regulated in vitro osteoblast differentiation and in vivo bone formation. First, KLF5 expression was upregulated during osteogenic differentiation of BMSCs. In addition, ML264, a KLF5 inhibitor, significantly inhibited osteoblast differentiation of BMSCs, as evidenced by decreased mRNA expression of osteogenic markers, ALP activity, and calcium deposition. Second, KLF5 overexpression stimulated osteoblast differentiation of BMSCs, whereas KLF5 knockdown suppressed it. Third, enforced KLF5 enhanced bone formation in a nude mouse model of subcutaneous heterotopic osteogenesis, whereas ML264 attenuated the BMP2-induced bone formation in a neonatal mouse model of calvarial organ culture. Our findings were consistent with observations by Shin et al. that KLF5 overexpression induced intestinal ALP promoter–reporter activity and expression, and KLF5 knockdown attenuated intestinal ALP expression in colon cancer cells (49). Our results also provide another possible explanation of findings from Shinoda et al. that the femoral and tibial limbs of KLF5+/− embryos were 10%–15% shorter than those of the wild-type littermates, although they considered that the main reason is impaired cartilage degradation and endochondral ossification mediated by insufficient KLF5 (51). Given that KLF5 was mainly expressed in osteoblasts and chondrocytes, but not osteoclasts (51), the study focused the effect of KLF5 on osteoblasts and did not explored its role in osteoclasts.
The expression of multiple KLF family members is increased by H2O2 treatment in neonatal rat cardiac myocytes (9), and the KLF5 expression is significantly increased in endothelial cells from a mouse model of diabetes mellitus that is highly related to oxidative stress (62). In contrast with these studies, we found that mRNA and protein levels of KLF5 were downregulated in osteoporotic mice and in BMSCs treated with H2O2, which indicated that KLF5 expression was regulated at the transcriptional level. Therefore, we then investigated the reason KLF5 was downregulated under oxidative stress.
Genomic DNA methylation and the expression and activity of DNMTs are regulated by oxidative stress. For instance, DNMT1 and SIRT1 become tightly bound to chromatin in H2O2-treated human embryonic carcinoma cells, contributing to cancer-specific aberrant DNA methylation and transcriptional silencing (43). Oxidative stress induced by H2O2 or ONOO− decreases DNMT-1 levels, and causes hypomethylation and overexpression of genes in CD4+ T cells (32). In addition, oxidative stress induced by antimycin A increases the activity of DNMT3B, but not DNMT1 or DNMT3A, in mouse embryonic stem cells (63). Recently, hypermethylation of KLF5 promoter was found to be caused by DNMT1 in renal cell carcinoma (16). In this study, we found that the methylation levels of KLF5 promoter were elevated by H2O2 treatment and attenuated by NAC in BMSCs. The hypermethylation of KLF5 promoter induced by oxidative stress was also abolished by 5-AZA treatment. Interestingly, we found that DNMT3B, rather than DNMT1 and DNMT3A, mediated the hypermethylation of KLF5 promoter in BMSCs under oxidative stress in the study. Moreover, the silencing of DNMT3B or 5-AZA treatment decreased KLF5 methylation levels and increased KLF5 expression under oxidative stress, further confirming that the hypermethylation and downregulation of KLF5 were mediated by DNMT3B upregulation under oxidative stress.
Growing evidence suggests that DNA methylation and DNMTs play an important role in the regulation of osteogenic differentiation as well as bone metabolism (42, 71). A prior study by Ha et al. (20) reveals that nanohydroxyapatite modulates the osteoblastic differentiation by altering the methylation levels of ALP promoter. Our recent study has demonstrated that DNMT3A downregulation mediates the hypomethylation of ALP and RUNX2, thereby stimulating osteogenic differentiation of human MSCs cultivated in the scaffolds under appropriate oxidative stress (30). DNMT3A was also shown to be involved in the regulation of bone resorption and formation in myeloma (34). Although DNMT3A and DNMT3B are highly homologous and have overlapping biological functions (5), DNMT3B may play a more important role in osteogenic differentiation (48) and bone regeneration (60). Moreover, the evidence that DNMT3B mutation mice show abnormally shaped frontal bone and shorter nasal bone highlights its role in bone formation and skeletal development (59). In particular, a recent study reported that DNMT3B upregulation mediated the hypermethylation of sonic hedgehog and impaired osteogenic differentiation under mechanical unloading condition, whereas DNMT3B downregulation promoted osteoblastogenesis through hypomethylation of sonic hedgehog under mechanical loading condition (61). Consistent with these studies, we found that DNMT3B-mediated KLF5 hypermethylation impaired osteogenic differentiation under oxidative stress in the study. Accordingly, the inhibition of KLF5 hypermethylation by DNMT3B silencing rescued the impaired osteogenic differentiation of BMSCs under oxidative stress.
The aberration of genomic DNA methylation has been identified to be associated with numerous diseases, including cancers and osteoporosis (6, 22, 46). Decitabine (Dacogen), the clinical form of 5-AZA, has been approved by the US Food and Drug Administration (FDA) for the treatment of hematological malignancies, including myelodysplastic syndrome (18). Recently, a study demonstrated that the hypermethylation of lncRNA-H19 mediated by upregulated DNMT1 was associated with disuse osteoporosis (29). They found that 5-AZA abrogated the inhibitory effect of DNMT1 overexpression on osteogenic differentiation of UMR106 cells, and that DNMT1 silencing dramatically alleviated the development of disuse osteoporosis in hindlimb unloading rats. Moreover, 5-AZA not only enhanced osteogenic differentiation of MSCs under normal condition (52, 73) but also rescued the osteogenic capacity of human periodontal ligament stem cells under high-glucose condition (35). In line with these studies, our study demonstrated that 5-AZA-suppressed KLF5 hypermethylation increased KLF5 expression and protected the impaired osteogenic differentiation of BMSCs under oxidative stress. These findings by us and others indicated that the inhibition of KLF5 hypermethylation using 5-AZA under oxidative stress might be a novel strategy for the treatment of osteoporosis by enhancing bone formation. Given that both methylation changes and osteoporosis are closely associated with aging as well, we are intended to further study the role of KLF5 in aging and its relation to osteoporosis.
With regard to the downstream mechanism, KLF5 regulates a number of target genes in the regulation of cell proliferation and differentiation (13, 37, 44). Several previous studies have demonstrated the potential interactions between KLF5 and β-catenin in colorectal cancer and breast cancer cells (19, 40, 57). In detail, KLF5 not only binds to the promoter of β-catenin to regulate its transcription (57) but also enhances the nuclear localization and transcriptional activity of β-catenin (40). However, the relationship between KLF5 and β-catenin in MSCs remains unclear. Consistent with these studies, we observed that KLF5 not only upregulated mRNA and protein expression of β-catenin but also regulated its nuclear localization in BMSCs in this study. Furthermore, our data demonstrated that KLF5 was bound to the armadillo repeat region of β-catenin, as confirmed by coimmunoprecipitation assay and immunofluorescence. The primary structure of β-catenin consists of an NH2-terminal (130 amino acids), an armadillo repeat region (∼550 amino acids), and a COOH-terminal (100 amino acids). The armadillo repeat region, composed of 12 copies of a 42 amino acid sequence motif, is in the central region of β-catenin (66). The 12 repeats form a superhelix of helices and have a long, positively charged groove that mediates the interactions of β-catenin with cadherins and TCF-family transcription factors, critical for cell adhesion and Wnt signaling (25).
β-catenin is a crucial mediator of Wnt signaling, which plays a pivotal role in the differentiation of MSCs, including osteogenesis. Accordingly, we found that the Wnt/β-catenin pathway agonist SKL2001 abolished the inhibitory effect of ML264 on osteogenic differentiation of BMSCs, suggesting that KLF5 stimulated osteogenic differentiation through the regulation of β-catenin. Previous studies have shown that Wnt/β-catenin signaling is antagonized by oxidative stress, and that the activation of Wnt/β-catenin signaling protects against oxidative stress-induced inhibition of osteogenesis (3, 7). Moreover, Wnt/β-catenin signaling is inactivated in osteoporosis, and β-catenin expression is downregulated in osteoporotic bone samples (4, 31). β-catenin expression is also regulated by oxidative stress (58). Our findings revealed that both protein expression and nuclear translocation of β-catenin were decreased in response to KLF5 downregulation induced by H2O2 during osteogenic differentiation of BMSCs. Moreover, SKL2001 treatment abrogated the inhibitory effect of KLF5 downregulation induced by H2O2 on osteogenic differentiation of BMSCs. Collectively, these results suggest that KLF5 hypermethylation impairs osteogenesis by reducing the expression and nuclear translocation of β-catenin, mediated by the armadillo repeat region, under oxidative stress (Fig. 8G).
Conclusion
KLF5 hypermethylation, accompanied with reduced expression, is mediated by DNMT3B under oxidative stress that is closely associated with osteoporosis. Thereafter, KLF5 downregulation resulted in decreased transcription expression nuclear translocation of β-catenin. KLF5 regulates osteogenic differentiation through the interaction with β-catenin mediated by the armadillo repeat region. Collectively, oxidative stress-induced KLF5 hypermethylation impairs osteogenic differentiation diminishing the interaction with β-catenin, which is likely to contribute to osteoporosis.
Materials and Methods (Electronic Laboratory Notebook Was Not Used)
Chemicals and reagents
Dulbecco's modified Eagle's medium (DMEM), penicillin/streptomycin, and fetal bovine serum (FBS) were purchased from Gibco-BRL (Gaithersburg, MD). ML264 and SKL2001 were purchased from Selleck (Shanghai, China). 5-AZA-2-deoxycytidine (5-AZA) and Nanaomycin A were purchased from MCE (MedChemExpress, NJ) and APExBIO (Houston), respectively. Other chemicals (H2O2, NAC, dexamethasone, β-glycerophosphate, ascorbic acid, and Triton X-100) were purchased from Sigma-Aldrich (Mainland, China). Primary antibodies specific for KLF5, β-catenin, DNMT3B, and OCN were purchased from Abcam (Cambridge, MA). All other chemicals used were of analytical grade.
Cell culture and osteogenic induction
The BMSCs derived from C57BL/6 mice were purchased from Cyagen Biosciences (MUBMX-90011; Santa Clara). Cells were cultivated in an incubator (37°C, 5% CO2, and 95% humidity). DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin was used as the growth medium. Fresh osteogenic medium (OM, 0.1 μM dexamethasone, 50 mM ascorbic acid, and 10 mM β-glycerophosphate) was used for the osteogenic induction after the cells reached 70%–80% confluence and changed twice every week.
Cellular viability and proliferation
Cell viability and proliferation were probed using the cell counting kit-8 (CCK-8) assay (Kumamoto, Japan) according to the manufacturer's instructions. Cell viability was evaluated after BMSCs were treated with different concentrations of H2O2 (0, 50, 200, and 800 μM) for 24 h. Cell proliferation was evaluated after the cells were pretreated with 10 μM ML264. The absorbance was measured at 450 nm wavelength on an ELX800 absorbance microplate reader (BioTek Instruments, Winooski, VT).
Cell transfection
The si-DNMT1, si-DNMT3A, si-DNMT3B, and si-KLF5 were commercially constructed by Ribobio (Guangzhou, China), and si-NC (negative control) was used as the control. pcDNA3.1-KLF5 was purchased from Hanbio (Shanghai, China), and an empty pcDNA 3.1 vector (pcDNA3.1-NC) was used as the control. The cells were transfected with si-DNMTs and pcDNA3.1-KLF5 in Opti-MEM™ I reduced serum medium using Lipofectamine™ 3000 (Invitrogen) according to the manufacturer's instructions.
ALP staining and ALP activity assay
After osteogenic induction for 7 days, ALP staining was performed using a commercial kit according to the manufacturer's instructions. For the ALP activity assay, the cells were lysed with 0.5% Triton X-100 for 1 h at 4°C. After centrifugation, ALP activity in the supernatant was determined colorimetrically using an ALP assay kit (Nanjing Jiancheng Bioengineering Institute). Total protein was quantified using a BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). The ALP activity was normalized to the total protein and expressed as nmol/min/mg protein.
Alizarin red staining and quantitative detection
After osteogenic induction for 14 days, the mineralization of BMSCs was evaluated using Alizarin red staining. The cells were fixed with 4% paraformaldehyde for 30 min and then stained with 40 mM Alizarin red S (pH 4.0) for 15 min at room temperature. After rinsing with distilled water to completely remove the unbound stain, the cells were visualized and imaged using a light microscope and digital camera. After drying, staining was eluted with 10% hexadecylpyridinium chloride for 15 min, and the absorbance was measured at 560 nm (OD560).
RNA isolation, complementary DNA synthesis, and quantitative real-time polymerase chain reaction
The total RNA was extracted using a TRIzol reagent following the manufacturer's protocol (Invitrogen). The RNA concentration was quantified using a spectrophotometer (Nanodrop; Thermo Scientific), and complementary DNA (cDNA) was synthesized using PrimeScript RT Master Mix (Takara Bio, Otsu, Japan). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using an ABI Prism 7500 system (Applied Biosystems, Foster City, CA) with a SYBR Green QPCR Master Mix (Takara Bio). Total cDNA (2 μL) was amplified in a 10 μL reaction system containing 5 μL of SYBR green (Takara Bio). The expression levels of each mRNA were normalized to that of glyceraldehyde-3-phosphate dehydrogenase, and the relative expression levels were calculated using the 2−ΔΔCt method. The primers used for amplification of the target mRNA are listed in Supplementary Table S1.
Western blot
In brief, the cells were harvested for protein extraction using a radioimmunoprecipitation assay buffer supplemented with phenylmethanesulfonyl fluoride. The protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto a 0.45 μm polyvinylidene fluoride membrane. The membranes were blocked, and then incubated with the primary antibodies KLF5 (Abcam; ab137676, 1:1000), DNMT3B (Abcam; ab79822, 1:1000), and β-catenin (Proteintech; 51067-2-AP, 1:1000) overnight at 4°C. After washing, the membranes were incubated with the secondary antibodies (1:1000) for 1 h at room temperature (RT). Protein bands were visualized using the LAS-4000 Science Imaging System (Fujifilm, Tokyo, Japan), and the obtained images were analyzed with ImageJ software.
Coimmunoprecipitation assay
One plasmid with the β-catenin FL and three plasmids with β-catenin DMs were constructed by Hanbio. The 293T cells were cotransfected with KLF5-FLAG and various β-catenin DMs-HA, and cultivated for 48 h. Cell extracts were precleared with protein A/G-agarose (50% v/v), and then the supernatants were immunoprecipitated with 2 mg of anti-β-catenin antibodies overnight at 4°C. Protein A/G–agarose–antigen–antibody complexes were collected by centrifugation at 12,000 rpm for 60 s at 4°C. Bound proteins were resolved by SDS-PAGE, followed by Western blotting with anti-FLAG (KLF5) and anti-HA (β-catenin) antibodies.
Cell immunofluorescence
The cells were fixed with 4% paraformaldehyde for 20 min and then rinsed with phosphate-buffered saline. For permeabilization, cells were incubated with 1% Triton X-100 for 20 min at RT. Unspecific binding sites were blocked with 5% goat serum for 1 h at RT after rinsing. The cells were incubated with primary antibodies KLF5 (Abcam; ab137676, 1:100) and β-catenin (Abcam; ab22656, 1:100) overnight at 4°C. After washing, the cells were incubated with the secondary antibodies DyLight 488 (Hangzhou fude; FD0150, 1:100) and DyLight 549 (Hangzhou fude; FD0132, 1:100) for 1 h in the dark at RT. Nuclei were counterstained by incubation with DAPI for 30 min at RT. Images of the staining were taken with a fluorescence microscope (BX51TRF; Olympus, Tokyo, Japan) and analyzed with ImageJ software.
Bisulfite sequencing
Bisulfite sequencing was performed as described in our previous study (5). In brief, total genomic DNA was isolated using a Wizard® Genomic DNA Purification Kit (Promega, Madison, WI), and then bisulfite conversion was carried out using a commercial EpiTectTM Bisulfite Kit (Qiagen, Hilden, Germany). To determine the methylation patterns of KLF5 promoter, PCR was carried out using Takara EpiTaq HS (Takara, Japan) after primers were designed to amplify the fragment with 591 bp including 63 CpGs. The PCR products were cloned into the T-vector pMD19-T after purification using a DNA gel recovery kit (Dongsheng Biotech, Guangzhou, China). Thereafter, the recombinant plasmid was transformed into the Escherichia coli strain DH5α. Eight clones per sample were sequenced by GENEWIZ (Suzhou, China). The number of total CpG sites was used as the denominator to identify the percentage of KLF5 promoter methylation in each sample.
BMP2-induced bone formation and immunohistochemistry
For calvarial organ culture, calvariae harvested from 5-day-old C57BL/6 pups were cultured in BGJb medium (Life Technologies). There were four groups: Con, ML264, BMP2 (100 ng/mL, n = 3), and BMP2+ML264 (10 μM, n = 3). After ex vivo culture for 7 days, calvariae were fixed in 4% paraformaldehyde and decalcified with 10% ethylene diamine tetraacetic acid for 2 weeks. For IHC, samples were paraffin embedded and sectioned (5 μm). Slides were subjected to sodium citrate buffer at 99°C for 20 min for antigen retrieval, and then incubated with primary antibodies overnight at 4°C.
Ovariectomy-induced osteoporosis model
All animal experiments were performed in accordance with the principles and procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the guidelines for the animal treatment of Sir Run Run Shaw Hospital. In brief, 10 healthy 8-week-old female C57BL/J6 mice purchased from the Laboratory Animal Center of Zhejiang University were subjected to either a sham operation (Con, n = 5) or bilateral ovariectomy (OVX, n = 5). Five mice were housed per cage with free access to water and food, and all mice were sacrificed 6 weeks after the ovariectomy. Left tibias were fixed in 4% paraformaldehyde for IHC, and right tibias were collected for DNA (Bisulfite sequencing) and RNA extraction (qRT-PCR).
Subcutaneous heterotopic bone formation
BMSCs were infected with lentivirus to stably overexpress KLF5 and then incubated with OM for 7 days. The differentiated BMSCs (1 × 106 cells/scaffold) were seeded into β-TCP scaffolds before they were subcutaneously implanted into adult male nude mice. Under general anesthesia, two subcutaneous pockets were created bluntly through one separate incision on the back. Each nude mouse was implanted one scaffold with BMSCs overexpressing KLF5 (OE-KLF5, n = 5) and another scaffold with BMSCs without overexpressing KLF5 (Con, n = 5). Five mice were housed per cage with free access to water and food. All mice were sacrificed 8 weeks after the operation.
Microcomputer tomography
The implanted scaffolds were analyzed using a high-resolution microcomputer tomography instrument (Skyscan 1072; Aartselaar, Belgium). The scanning protocol was set at an isometric resolution of 9 mm, with X-ray energy settings of 70 kV and 80 mA. The image matrix was 2048 × 2048 pixels. A high-resolution protocol (slice thickness 10 μm, feed 10 μm, pixel size 10 μm) was applied. The scaffold showed a mean gray level of 700 ± 50, and the newly formed bone showed a mean gray level of 500 ± 150. As bones were indistinguishable from the β-TCP sample (27), the threshold for distinction between connective tissue and calcified tissue was set at 358. To evaluate the amount of newly formed bone, unimplanted β-TCP control scaffolds (n = 5) were scanned and used as controls. Quantification of bone and matrix volume covered by bone and β-TCP was performed after reconstruction.
Statistical analysis
The data are presented as the mean ± standard deviation. Statistical differences were assessed by Student's t test or one-way analysis of variance (ANOVA), followed by Tukey's post hoc analysis where appropriate. p Values <0.05 were considered to be statistically significant. Each experiment was repeated independently at least three times.
Footnotes
Authors' Contributions
S.F., Z.X., and J.W. conceived the idea and designed the experiments. L.L., H.W., X.L., G.W., Z.J., and H.H. conducted the experiments. L.L., X.C., H.W., X.Z., and X.S. analyzed the results. L.L., Z.X., and X.C. wrote the article. S.F. and J.W. supervised and supported the study. All authors reviewed the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key R&D Program of China (2018YFC1105200), the National Nature Science Fund of China (81972089, 81871796), the China Postdoctoral Science Foundation (2019M652113), the Key Research and Development Plan of Zhejiang Province (2020C03043), the Natural Science Fund of Zhejiang Province (Q20H060042, Q20H060043), the Medicine and Health Technology Plan of Zhejiang Province (2019KY050), the Traditional Chinese Medicine Science and Technology Plan of Zhejiang Province (2019ZA026), the Scientific Research Fund of Zhejiang Provincial Education Department (Y201941402), and the Medical Health Science and Technology Project of Zhejiang Province (2A21904). No benefits in any form have been, or will be received from a commercial party related directly or indirectly to the subject of this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.