
Abstract
Significance:
Mitochondria-Associated Membranes (MAMs) are highly dynamic endoplasmic reticulum (ER)-mitochondria contact sites that, due to the transfer of lipids and Ca2+ between these organelles, modulate several physiologic processes, such as ER stress response, mitochondrial bioenergetics and fission/fusion events, autophagy, and inflammation. In addition, these contacts are implicated in the modulation of the cellular redox status since several MAMs-resident proteins are involved in the generation of reactive oxygen species (ROS), which can act as both signaling mediators and deleterious molecules, depending on their intracellular levels.
Recent Advances:
In the past few years, structural and functional alterations of MAMs have been associated with the pathophysiology of several neurodegenerative diseases that are closely associated with the impairment of several MAMs-associated events, including perturbation of the redox state on the accumulation of high ROS levels.
Critical Issues:
Inter-organelle contacts must be tightly regulated to preserve cellular functioning by maintaining Ca2+ and protein homeostasis, lipid metabolism, mitochondrial dynamics and energy production, as well as ROS signaling. Simultaneously, these contacts should avoid mitochondrial Ca2+ overload, which might lead to energetic deficits and deleterious ROS accumulation, culminating in oxidative stress-induced activation of apoptotic cell death pathways, which are common features of many neurodegenerative diseases.
Future Directions:
Given that Sig-1R is an ER resident chaperone that is highly enriched at the MAMs and that controls ER to mitochondria Ca2+ flux, as well as oxidative and ER stress responses, its potential as a therapeutic target for neurodegenerative diseases such as Amyotrophic Lateral Sclerosis, Alzheimer, Parkinson, and Huntington diseases should be further explored. Antioxid. Redox Signal. 37, 758–780.
Introduction
Proteinaceous contact sites between the endoplasmic reticulum (ER) and the mitochondria, called mitochondria-associated membranes (MAMs), modulate multiple cellular processes, including lipid exchange, calcium (Ca2+) homeostasis, mitochondrial and ER dynamics, ER stress response, autophagy, inflammation and apoptosis, as well as redox state (175). In the past few years, the physiological relevance of MAMs has been extensively studied and their structural and functional alterations have been described in neurodegenerative diseases, which are closely associated with ER stress, mitochondrial dysfunction, and increased ROS production, among other MAMs-related events (84).
Several neurodegenerative diseases, namely Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Amyotrophic Lateral Sclerosis (ALS), have been shown to be associated with alterations in Sigma-1R (Sig-1R) (181). The Sig-1R is an ER chaperone that translocates to MAMs during stress conditions to control ER-mitochondria Ca2+ flux through interaction with the ER inositol 1,4,5-trisphosphate receptor (IP3R), and it also modulates ER and oxidative stress responses (181).
Here, we present an overview of the major components of MAMs and the physiological processes that are regulated by these close ER-mitochondria contacts, and how their structural and functional alterations are implicated in the pathophysiology of neurodegenerative diseases, highlighting the regulation of reactive oxygen species (ROS). Further, evidence for the modulation of pathological events associated with these diseases by Sig-1R is provided and its potential as a therapeutic target is discussed.
Structure and Function of MAMs
The MAMs are specialized contact sites created by physical and biochemical interaction between the ER and the mitochondria that were first isolated from rat liver in 1990 by Jean Vance (9). Thereafter, MAMs were extensively investigated and, in the past few years, their involvement in several physiological and pathological conditions was investigated in-depth, especially in the context of neurodegenerative diseases (99).
It is estimated that 5%–20% of the mitochondrial surface is connected to the ER and the intermembrane distance between these organelles is about 10–30 nm (99). However, these values depend on the cell type, as well as on the level and duration of environmental alterations, such as exposure to ER stress or other insults (15). This contact site is a dynamic structure due to the presence of a set of specialized proteins, which works as an intracellular signaling platform that is able to determine cell fate. Indeed, MAMs regulate multiple cellular processes, including lipid exchange, Ca2+ homeostasis, mitochondrial and ER dynamics, ER stress response, autophagy, redox status, inflammation, and apoptosis (175).
The architecture of MAMs includes several ER-mitochondria tethering proteins inserted in the outer mitochondrial membrane (OMM) that interact with ER membrane-resident proteins: the ER IP3R linked to the mitochondrial voltage-dependent anion channel 1 (VDAC1) through the chaperone glucose-regulated protein 75 (GRP75); ER VAMP-associated protein B (VAPB) and mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51); B cell receptor-associated protein 31 (BAP31) in the ER that interacts with mitochondrial fission 1 (FIS1); and mitofusin 2 (MFN2), located at both ER and mitochondria, which homo-oligomerizes and hetero-oligomerizes with mitofusin 1 (MFN1) to tether both organelles (52, 141).
The ER-resident motile sperm domain containing 2 protein (MOSPD2) was recently proposed as a new ER-mitochondria tether that acts through its interaction with the mitochondrial PTPIP51 (110). In addition to inter-organelle tethering, other functions have been described for these proteins. The IP3R, which acts as a channel that mediates Ca2+ release from the ER, forms a complex with VDAC1 at the OMM to facilitate Ca2+ transfer from ER to the mitochondrial intermembrane space (160). FIS1, MFN1, and MFN2 are mediators of mitochondrial fission and fusion (100). MFN2 also modulates ER and mitochondria morphology, and it regulates Ca2+ and lipid transfer between both organelles (26, 33, 70).
The OMM-resident protein FIS1 interacts with ER BAP31 and promotes its cleavage into pro-apoptotic p20Bap31, which, in turn, enhances ER-mitochondria Ca2+ transfer, leading to apoptosis (82). Mitochondrial Ca2+ overload and apoptotic cell death are also modulated by PTPIP51 located at the OMM (178). VAPB regulates proteostasis, namely the ER Unfolded Protein Response (UPR) and autophagy (53, 57).
Several other proteins, which regulate Ca2+ homeostasis and act as chaperones, have been detected at MAMs, namely the ER-resident chaperone Sig-1R that interacts and stabilizes the IP3R modulating Ca2+ signaling (66). The chaperones calnexin and calreticulin regulate Ca2+ concentration in the ER lumen (117, 145), and the GRP78 chaperone regulates the UPR promoting the folding of nascent and misfolded proteins within the ER under stress conditions (73).
The ryanodine receptor (RYR) is another channel present at MAMs that mediates ER Ca2+ release, and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) that is responsible for ER Ca2+ uptake is also found at these ER-mitochondria contacts (32). The protein disulfide isomerase (PDI) and ER oxidoreductase 1 α (ERO1α) that are also enriched at MAMs catalyze isomerization, reduction, and oxidation of disulfide bonds in proteins synthesized de novo and regulate ER redox status (4,141).
Inositol-requiring enzyme 1 α (IRE1α) and protein kinase RNA-like ER kinase (PERK) are sensors of ER stress that trigger adaptive or pro-apoptotic responses of the UPR (73, 172). The MAMs also include proteins involved in autophagosome formation such as the PI3K complex, proteins involved in apoptotic cell death such as BCL2, BAX, and BAK, and components of the NLRP3 inflammasome (129, 179, 186) (Fig. 1). Enzymes involved in lipid synthesis and trafficking are also present in MAMs, namely acyl-CoA:cholesterol acyltransferase/sterol O-acyltransferase 1 (ACAT1/SOAT1), diacylglycerol O-acyltransferase 2 (DGAT2), phosphatidylserine synthases 1 and 2 (PSS1 and PSS2), phosphatidylethanolamine N-methyltransferase 2 (PEMT2), and fatty acid CoA ligase 4 (FACL4/ACS4) (Fig. 2) (161).
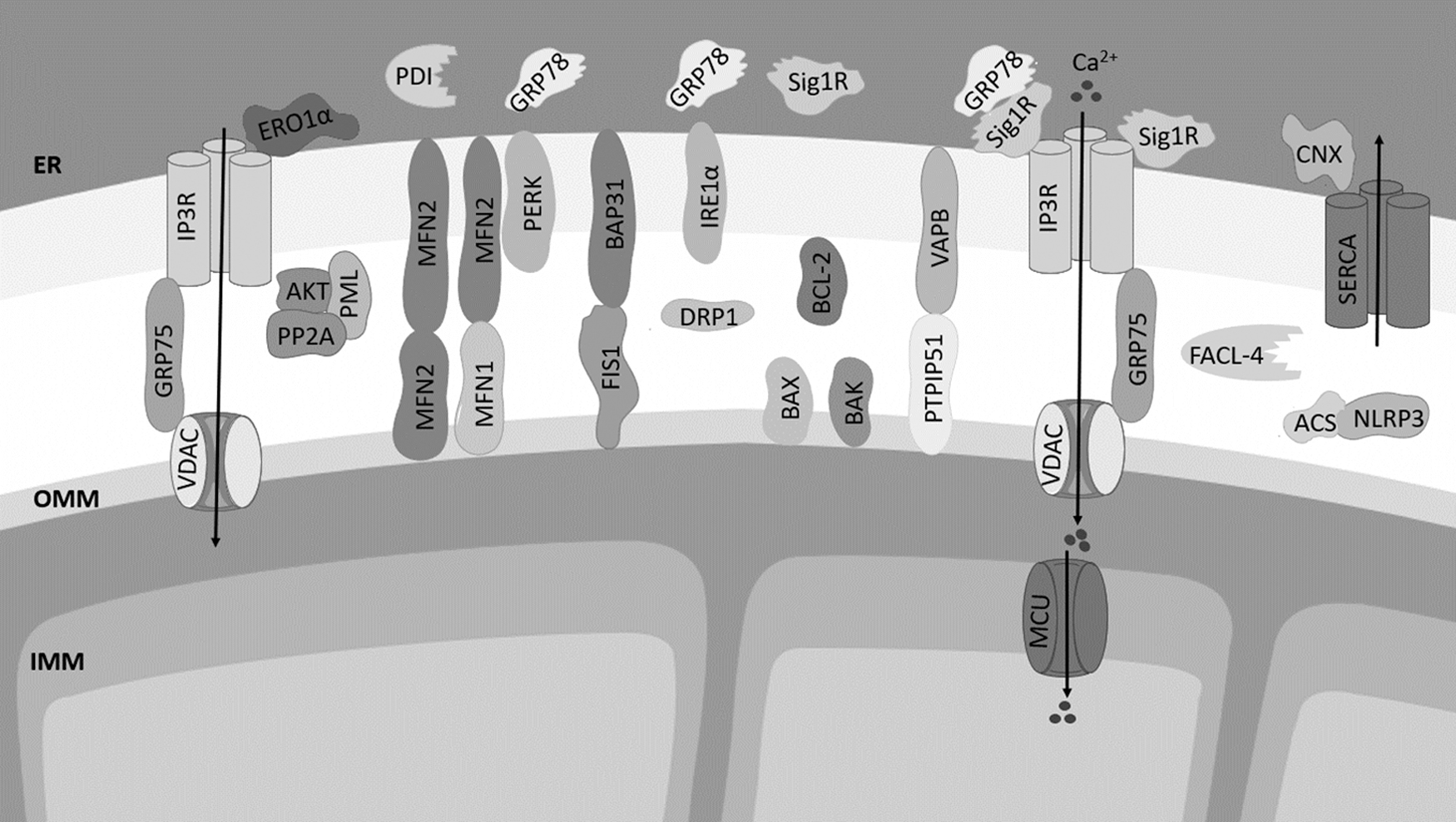
The architecture and functioning of MAMs is influenced by the silencing or overexpression of tethering proteins. For example, overexpression of VAPB or PTPIP51 increases, whereas their knockdown decreases, ER-mitochondria contacts followed by impairment of Ca2+ exchange between both organelles (56, 99). MFN2 ablation inhibits ER-mitochondria Ca2+ signaling, changes ER morphology, and prevents autophagosome formation (25, 61, 126). However, recent studies support the fact that MFN2 acts as a tethering antagonist that negatively modulates the number of close contacts between both organelles.
MFN2 ablation or silencing increases ER-mitochondria tethering that promotes excessive Ca2+ transfer between these organelles, leading to cell death due to mitochondrial Ca2+ overload (44). Genetic depletion of MFN2 was also shown to activate ER stress-induced UPR, suggesting the role of this MAMs-resident protein in stress responses (127). In fact, MFN2 directly interacts and inactivates the stress sensor PERK that is known to potentiate the formation of deleterious ER-mitochondria contacts, leading to apoptosis (124).
Due to their composition, MAMs regulate multiple cellular processes, including lipid exchange, Ca2+ homeostasis, mitochondrial and ER dynamics, ER stress response, autophagy, redox status, inflammation, and apoptosis (138, 174). Therefore, ER-mitochondria tethering and crosstalk have to be tightly regulated to promote an inter-organelle homeostatic communication that is able to preserve cell survival (179).
Cellular Events Regulated by MAMs
Lipid metabolism
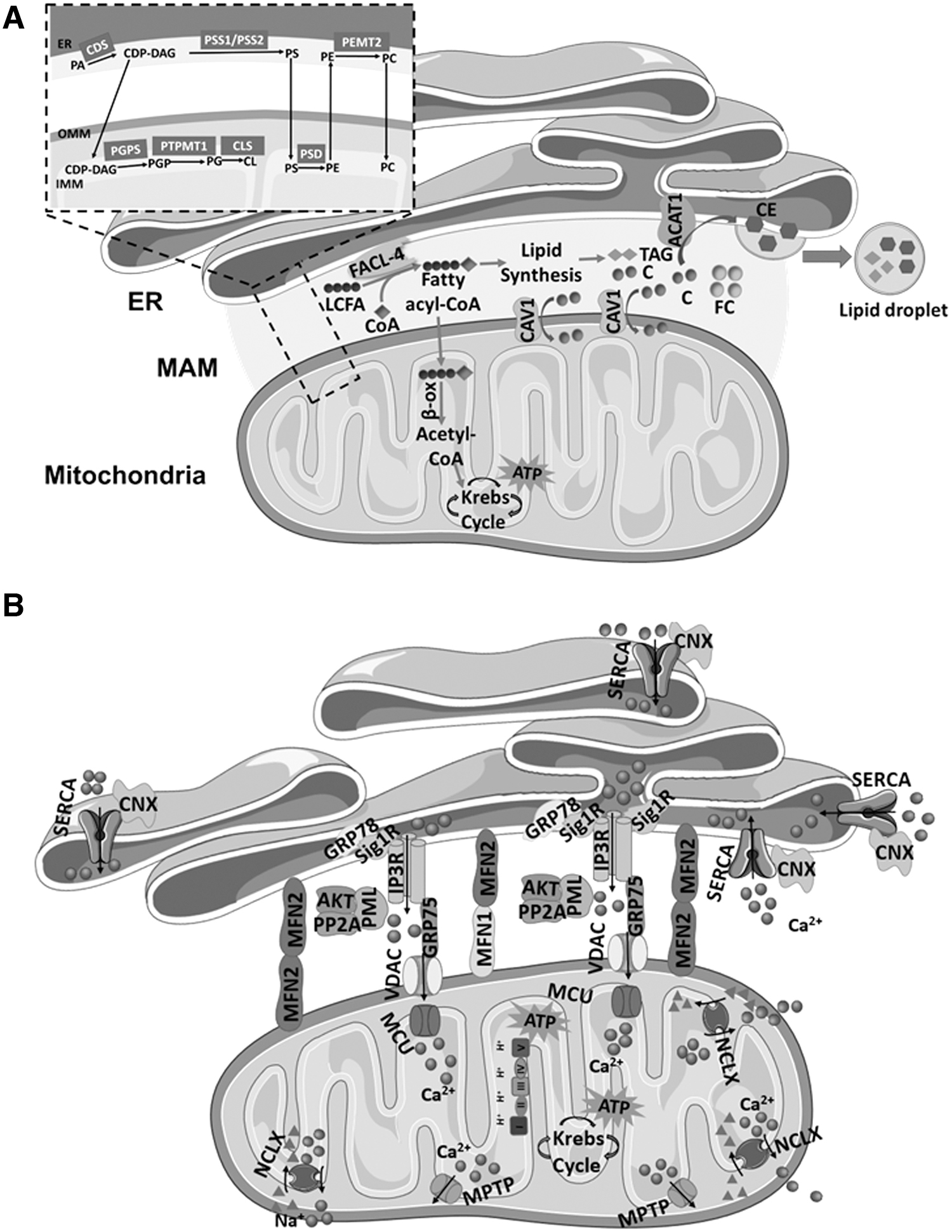
Biochemical studies show that the synthesis of triacylglycerol, phosphatidylcholine (PC), and phosphatidylethanolamine (PE) requires the activity of enzymes present at both ER and mitochondria (85, 141). These findings demonstrate the relevance of MAMs in lipid metabolism and transfer between both organelles. The MAMs involvement in lipid synthesis and exchange results from the accumulation of GM1-ganglioside enzymes in the glycosphingolipid-enriched microdomain of MAMs (151) and the presence of lipid rafts domains associated with Sig-1R receptors and erlin-1 and erlin-2 (27).
The MAMs are enriched in phospholipid-synthesizing enzymes, such as FACL4, which is involved in the ligation of fatty acids to coenzyme A (CoA) and in triacylglycerol synthesis; PSS1/2, which catalyzes phosphatidylserine (PS) synthesis; PEMT2, which is involved in PC synthesis; and ACAT1/SOAT1, which catalyzes the production of cholesterol esters, which are incorporated into lipid droplets (161, 171). PS is synthesized in the ER by PSS1 and PSS2 and translocated to the inner mitochondrial membrane (IMM) where it is decarboxylated by phosphatidylserine decarboxylase into PE, which is rapidly exported to other organelles, such as the ER, where it is converted to PC by PEMT2 (161, 171) (Fig. 2).
Enzymes involved in cholesterol biosynthesis are also accommodated at MAMs, namely ACAT1 that catalyzes the intracellular esterification of free cholesterol, allowing a dynamic equilibrium between membrane-bound and cytoplasm-stored cholesterol in resting state (84), and cholesterol-binding caveolin-1 (CAV1) that promotes cholesterol transfer into mitochondria (150). Cholesterol was found to negatively regulate MAMs-mitochondria association. Using an in vitro membrane association assay, it has been observed that depletion of cholesterol in isolated MAMs with methyl-β-cyclodextrin significantly increased the association between MAMs and mitochondrial fractions.
These results suggest the existence of two different mechanisms regulating MAMs-mitochondria association: one that promotes the membrane association via specific membrane-tethering proteins, such as Mfn2, and another that restricts the association wherein cholesterol plays a pivotal role (Fig. 2).
Calcium homeostasis
The ER, mitochondrial, and cytosolic Ca2+ homeostasis are regulated at MAMs through SERCA transport of Ca2+ from the cytoplasm into the ER lumen, IP3R-mediated ER Ca2+ release, and mitochondria uptake by VDAC at the OMM (84, 161) (Fig. 2). The MAMs form a Ca2+ signaling hub regulating ER chaperone-assisted folding of de novo synthesized proteins and activity of mitochondria-resident Ca2+-dependent dehydrogenases that are required for mitochondrial bioenergetics, as well as proteins involved in cell death such as BAX and BCL-2 (135, 142).
Therefore, fine-tuned Ca2+ transfer between the ER and the mitochondria is essential to determine cell fate. The MAMs form a microdomain for Ca2+ transfer, and physical proximity of ER and mitochondria allows a direct and selective transmission of Ca2+ signals (135, 161). Agonists of plasma membrane Ca2+ permeable ionotropic receptors activate phospholipase C that catalyzes the formation of IP3, which binds to IP3R and stimulates ER Ca2+ release (135). Due to the proximity to mitochondria, Ca2+ crosses the freely permeable OMM through VDAC channels and the impermeable IMM by the low-affinity channel of the mitochondrial Ca2+ uniporter (MCU), finally accumulating in the mitochondrial matrix (135, 161).
The MAM chaperone GRP75 mediates the interaction between the IP3R and VDAC, acting as a bridge between these membrane proteins and therefore regulating Ca2+ transfer to the mitochondrial matrix via MCU (135). Usually, mitochondria take up Ca2+ released from ER in a very short time frame after its release; however, the amplitude of ER-associated Ca2+ puffs decreases in the presence of MCU inhibitors. Low or very high cytosolic Ca2+ concentrations reduce the IP3R opening and consequently diminish ER Ca2+ release (141), which might negatively impact mitochondrial function.
Sig-1R binds to IP3R, preventing the ubiquitin-mediated degradation of IP3R-associated Ca2+ channels, increasing Ca2+ transfer from the ER to the mitochondria. By doing this, Sig-1R stimulates ATP production due to activation of Ca2+-dependent metabolic enzymes in the Krebs cycle, and consequently, preserves cell viability (141, 161). On the other hand, SERCA overexpression leads to ER Ca2+ overload, which stimulates ER Ca2+ release, namely through the IP3Rs. As a consequence, ER-mitochondria Ca2+ transfer is exacerbated, promoting mitochondrial Ca2+ overload and dysfunction (105, 161).
Several proteins recruited to MAMs are involved in Ca2+ modulation, namely AKT/PKB and cytochrome c. In response to survival signals, the anti-apoptotic protein AKT/PKB translocates to MAMs and inactivates IP3R reducing ER Ca2+ release. Cytochrome c, which is released from mitochondria by apoptotic stimuli, can bind IP3Rs at MAMs, increasing ER Ca2+ release and subsequent mitochondrial Ca2+ overload, as well as cytochrome c release, thus enhancing pro-apoptotic signaling (135). Ca2+ is taken up into ER through Ca2+ pumps of the SERCA family, after release from mitochondria through the mitochondrial Na+/Ca+ exchanger (NCLX). This closes the cycle of intracellular Ca2+ signaling between the ER and the mitochondria, which determines the correct functioning of these organelles (141).
In addition to the regulation of Ca2+ and lipid homeostasis, MAMs are also involved in other physiologic events, namely the modulation of energy metabolism, ER stress response, autophagy, apoptosis, and innate immunity (Fig. 3).
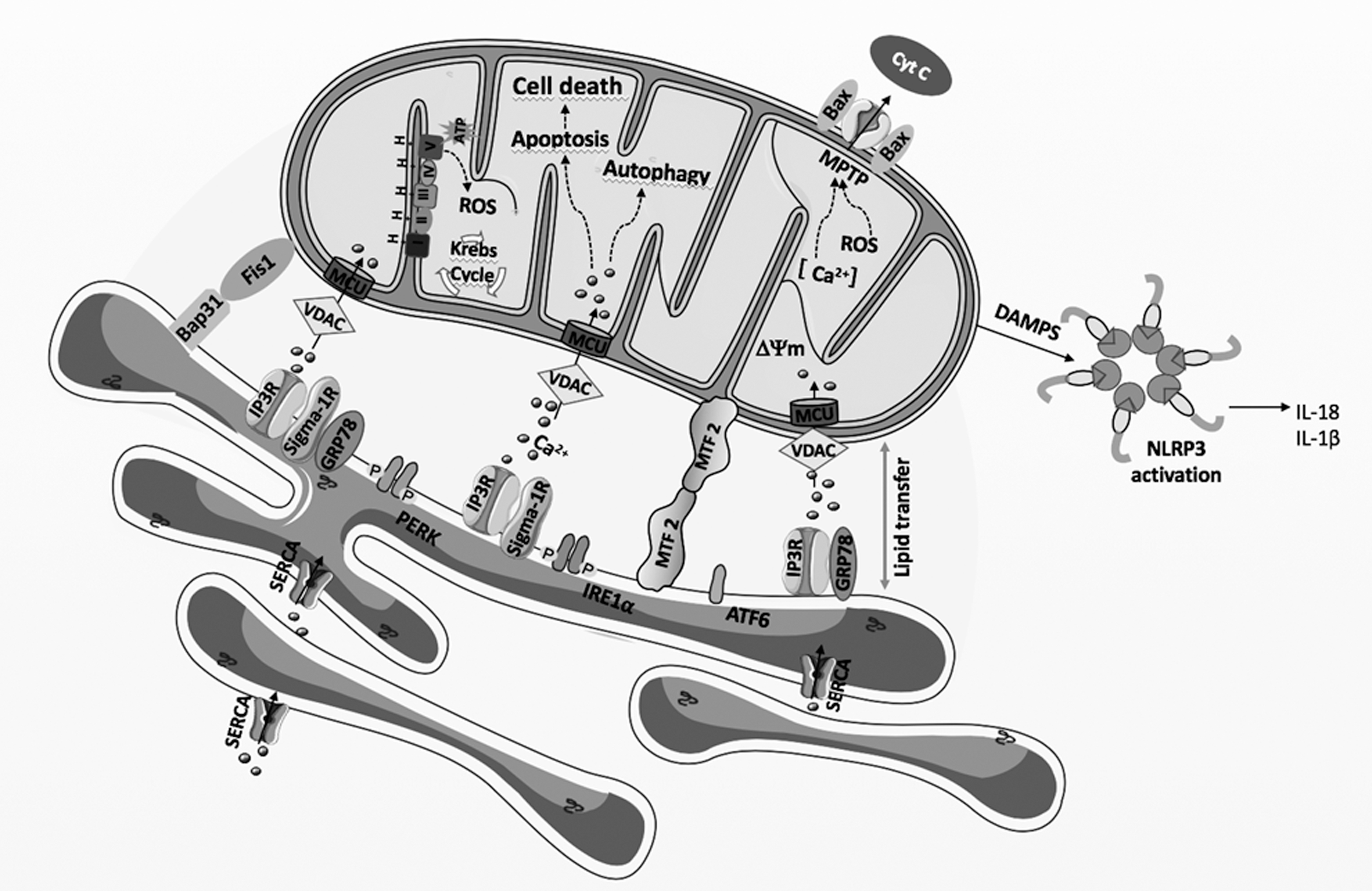
Energy metabolism
Mitochondria play a central role in cell metabolism since they are responsible for ATP synthesis, which is the main cellular energetic source. Mitochondrial uptake of Ca2+ released by the ER is required to activate the tricarboxylic acid cycle (TCA), which provides reducing equivalents required for ATP production at the electron transport chain (ETC) (31). On the other hand, the ATPase activity of the ER resident GRP78 (32) depends on ATP produced by mitochondria (79). Disruption of ER-mitochondria tethers, such as VAPB-PTP1P51, perturbs Ca2+ flux between these organelles and, consequently, impairs mitochondrial ATP production (130).
IMM and OMM are strongly enriched in lipids. Although mitochondria can synthesize lipids such as cardiolipin (CL), the structural and functional maintenance of these organelles is highly dependent on lipids imported from the ER (108). CL is synthesized in mitochondria from ER-derived phosphatidic acid (PA) (87). CL deficiency inhibits the ETC complex IV, supporting the fact that CL plays a key role in mitochondrial metabolism (115). CL modulates OPA1 functions and therefore regulates mitochondrial morphology and quality control (13), which is fundamental to adjust ATP synthesis to metabolic needs (55). PC, the most abundant mitochondrial phospholipid, is only synthesized in the ER and then exported to the mitochondria (77) and decreased mitochondrial PC/PE molecular ratio is associated with decreased oxygen consumption and ATP depletion (166).
The earlier findings indicate that lipid transfer between the ER and the mitochondria is crucial to maintain normal cell functioning, namely bioenergetic metabolism. Therefore, the impairment of ER-mitochondria contacts negatively affects mitochondrial metabolism and cell survival.
ER stress response
One of the major ER functions is the folding of proteins synthesized de novo. Several insults, such as nutrient deprivation, hypoxia, loss of Ca2+ homeostasis, as well as accumulation of mutant proteins, can compromise the efficiency of protein folding, leading to the accumulation of unfolded/misfolded proteins in its lumen, a state known as ER stress that affects cell homeostasis. In response to ER stress, the adaptive UPR signaling pathway, which is mediated by activation of the ER transmembrane protein sensors PERK, IRE1α, and activating transcription factor 6 (ATF6), is induced to reestablish ER homeostasis and promote cell survival (72).
On ER stress, PERK phosphorylates eukaryotic translation initiation factor 2 subunit-α (eif2-α), attenuating global protein synthesis (64), which limits the loading of the ER lumen with misfolded proteins. Phosphorylated eif2-α paradoxically increases translation of the ATF4 (102) that upregulates the genes involved in redox homeostasis, amino acid metabolism, protein synthesis and degradation by macroautophagy or ER-associated protein degradation (ERAD), and also apoptosis (11, 62, 65).
IRE1α oligomerizes and autophosphorylates on ER stress to promote its RNase activity (195). IRE1α cuts off a small 26-nucleotide intron from the mRNA encoding the transcription factor X-box-binding protein 1 (XBP1) (190), increasing the levels of a spliced active form of XBP1 (XBP1s), which, in turn, upregulates the genes involved in ER protein translocation, folding, and secretion, as well as degradation of misfolded proteins (95). ATF6 is translocated to the Golgi apparatus when the ER is under stress; it is cleaved by Site 1 and Site 2 proteases (S1P and S2P) into an active ATF fragment (ATF6p50) that migrates to the nucleus and stimulates the transcription of UPR target genes involved in protein folding and degradation (185, 188).
Numerous evidence demonstrate that the ER stress response is intimately associated with ER-mitochondria contacts. Activation of the PERK-ATF4 arm of UPR was found to prevent ER stress-related events through parkin upregulation, which, in turn, avoids mitochondrial fragmentation and ATP depletion (22). Accordingly, stress-induced parkin overexpression has been associated with enhanced ER-mitochondria coupling and mitochondrial Ca2+ uptake to preserve energetic metabolism (29).
Moreover, Verfaillie et al. demonstrated that PERK is enriched at MAMs and that its ablation disturbs ER morphology and Ca2+ signaling, as well as diminishes ER-mitochondria contact sites (172). Remodeling of these contacts on ER stress has been shown to be mediated by protein kinase A (PKA) that enhances Ca2+ transfer toward mitochondria to avoid cell dysfunction and death. These protective effects were abolished in the presence of the MAMs-located CAV1, and cells become unresponsive to ER stress and more sensitive to Ca2+-mediated cell death (23).
Calnexin is a chaperone located at MAMs that interacts with SERCA regulating ER-mitochondria Ca2+ exchange. Under short-term ER stress, it is preferentially located at the ER and shifts its function from Ca2+ signaling regulation toward modulation of protein folding and quality control in the ER lumen (104). During the early adaptive phases of ER stress, there is a temporary increase in ATP production in an attempt to preserve cell survival (174). Increased mitochondrial energetic metabolism results from enhanced ER-mitochondria coupling and Ca2+ transfer (27).
Autophagy
Macroautophagy is a catabolic mechanism that is involved in several physiological processes, namely quality control strategies to eliminate dysfunctional organelles and intracellular misfolded proteins (121). Macroautophagy is initiated by the isolation of specialized membranes from compartments of the ER enriched in phosphatidylinositol 3-phosphate (PI3P), called omegasomes, to form autophagosomes (10).
Several findings support the hypothesis that autophagosomes are formed at ER-mitochondria contact sites. For example, the pre-autophagosome/autophagosome marker ATG14 and the autophagosome-formation marker ATG5 are recruited to the ER-mitochondria platform on starvation. Moreover, the disruption of coupling between organelles avoids the formation of ATG14 puncta, preventing autophagosome formation (61). The MAMs have also been proposed as platforms for autophagy based on their role in modulating lipid composition of ER-mitochondria contacts. For instance, PE upregulation at MAMs improves autophagic flux (144).
In addition, the molecular interaction between the components of MAM's lipid-rafts microdomains, such as the ganglioside GD3, and core-initiator autophagic proteins, including AMBRA1 and WIPI1, is required to initiate autophagy (50). The stearoyl-CoA desaturase 1 (SCD1), an enzyme enriched in MAMs that regulates membrane content of fatty acids, promotes autophagy and its inhibition depletes membrane phospholipids, resulting in the impairment of autophagosome formation and autophagic cargo degradation (83).
During mitophagy, which is the autophagic process that selectively eliminates damaged mitochondria, the ER-mitochondria tether Mfn2 is rapidly degraded and ER-mitochondria contacts are destroyed. This process involves Mfn2 phospho-ubiquitination by the PINK1/parkin system, which is a signal to mark damaged mitochondria and initiate their lysosomal degradation. Moreover, ER-mitochondria contacts themselves are negative mitophagy regulators, through the Mfn2 antagonist effect (116).
Apoptosis
The activation of the intrinsic apoptotic pathway when cells are exposed to prolonged stress implicates the communication between the ER and the mitochondria. Ca2+ released from the ER IP3R- or RyR-associated Ca2+ channels located at MAMs increases cytosolic Ca2+ levels in the vicinity of the mitochondria. Excessive Ca2+ entry through VDAC and MCU leads to mitochondrial Ca2+ overload that is noxious for cells, leading to permanent opening of the high conductance mitochondrial permeability transition pore (mPTP), which is considered as a non-return point of apoptosis.
The permanent pore opening is involved in cytochrome c release and dissipation of the mitochondrial membrane potential, with concomitant cell death associated with the release into the cytosol of mitochondrial proteins such as cytochrome c, apoptosis-inducible factor, and Smac/DIABLO proteins. Once released, these proteins trigger both caspase-dependent and caspase-independent apoptosis (30).
Some BCL-2 family proteins are located at MAMs and modulate the amount of Ca2+ transfer between the ER and the mitochondria (20). For instance, BCL-2 inhibits pro-apoptotic signals by reducing Ca2+ release through its interaction with the IP3R at MAMs (146). In addition, the interaction of BCL-xL with the IP3R promotes a pro-survival Ca2+ transfer to mitochondria (182). On the other hand, during severe stress conditions, the IP3R-interacting protein IRBIT inhibits the anti-apoptotic BCL2l10 and promotes ER-mitochondria contact and Ca2+ transfer, which, in turn, induces mitochondrial Ca2+ overload, culminating in apoptosis (19).
Apoptotic cell death is induced under chronic or severe ER stress conditions on UPR activation (62). The role of ER-mitochondria coupling in apoptosis is further supported by data showing that the ER stress sensor PERK, which is mainly localized at MAMs, is required for ER stress-induced apoptosis triggered by ROS that promote the transfer of Ca2+ from the ER to the mitochondria (172). Under stress conditions, Mfn2-deficient cells, which present alterations in ER-mitochondria contact sites, were shown to present reduced apoptosis markers and PERK deficiency rescued these cells from apoptosis triggered by ER stress (124). Further, IRE1α ubiquitination at MAMs by the mitochondrial ubiquitin ligase (MITOL/MARCH5) inhibits apoptosis under ER stress conditions (164).
Innate immunity
The MAMs play a critical role in innate immunity, providing a molecular platform for NLRP3 inflammasome assembly (120). Inflammasomes are multiprotein complexes that are activated in the presence of pathogen-associated molecular patterns and damage-associated molecular patterns (DAMPs) (60). They are composed of a sensor protein, an adaptor protein called apoptosis-associated speck-like (ASC) protein containing a caspase activation and recruitment domain (CARD) and pro-caspase 1. Once assembled, caspase 1 is activated by auto-cleavage from pro-caspase 1 and induces the maturation and secretion of the pro-inflammatory cytokines interleukin-1β/18 (IL-1β/18) (60).
During NLRP3 inflammasome formation, de novo synthesized NLRP3 and ASC are translocated to MAMs, resulting in the apposition of ASC on the mitochondria to NLRP3 on the ER (196). At MAMs, inflammasome senses DAMPs that emanate from damaged mitochondria such as ROS, DNA, and CL externalization, membrane depolarization, and Ca2+ deregulation, among others (120). Cellular processes that are modulated by ER-mitochondria contacts at MAMs such as Ca2+ and lipid transfer play a crucial role in NLRP3 activation. In the presence of NLRP3 agonists, blocking mitochondrial Ca2+ uptake by silencing VDAC isoforms 1 and 2 prevents NLRP3 inflammasome activation, as well as mitochondrial ROS production (196). Moreover, cholesterol trafficking to the ER is required for NLRP3 inflammasome activation (92).
ROS generation
The ROS are biological molecules with at least one oxygen atom and with higher reactivity than molecular oxygen (O2). These molecules include non-radical and free radical (with at least one free electron) species such as hydrogen peroxide (H2O2) and the superoxide anion radical (O2 .−), respectively, which are key redox signaling agents (154).
Apart from transmembrane NADPH oxidases (NOXs) and the mitochondrial ETC, which are the major endogenous sources of H2O2 and O2−•, H2O2 is also produced by other oxidases present in the ER, peroxisomes and plasma membrane, as well as by superoxide dismutases (SOD) (154). Mitochondria produces O2 −• as a by-product of the inefficient transfer of electrons by the ETC during oxidative phosphorylation (OXPHOS).
This ROS is rapidly converted to H2O2 by the copper-zinc SOD (SOD1) in the intermembrane space and by the manganese-dependent SOD (SOD2) in the mitochondrial matrix space and then reduced to water by catalase (CAT) or glutathione peroxidase (74, 153). H2O2 can react with metal ions, such as Fe2+, via the Fenton reaction, to generate highly reactive hydroxyl radicals (88). In the ER, H2O2 is produced during the protein folding process mediated by the PDI and Ero1α chaperones (194).
Although the oxidative environment is conducive to protein folding, excessive H2O2 accumulation can deregulate the ER redox balance, leading to the accumulation of misfolded proteins causing ER stress (194). Similarly, the flavoprotein quiescin sulfhydryl oxidase catalyzes disulfide formation by coupling disulfide oxidation to the reduction of O2 generating H2O2 (90). NOX4 located at the ER membrane also produces H2O2, which may be involved in apoptotic cell death activation after prolonged ER stress (191). To maintain the ER redox balance, some oxidoreductases such as peroxiredoxin 4 (PRDX4), glutathione peroxidase 7/8 (GPx7/8), and ascorbate peroxidase scavenge H2O2. In addition, reduced glutathione (GSH) also contributes to the removal of excessive H2O2 to avoid oxidative stress (6).
The ER-mitochondria interface hosts dynamic H2O2 nanodomains, which originate from the mitochondrial cristae and are induced by and influence Ca2+ signals. These structures are compressed on Ca2+ uptake to the matrix that stimulates the influx of K+ through Ca2+-activated mitochondrial K+ channels, causing rapid osmotic changes in the matrix, compensated by H2O influx (21). Sig-1R, which is a MAMs-resident chaperone that modulates Ca2+ exchange between the ER and the mitochondria by interacting with IP3Rs, is also involved in ROS generation.
In physiological conditions, Sig-1R agonists were found to generate moderate oxidative stress in mouse brain mitochondria stimulating ROS production at ETC complex I level. Under pathological conditions, Sig-1R activity may contribute toward restoring mitochondrial function (54). Ero1α, a key controller of oxidative folding and ER redox homeostasis, also localizes at MAMs and regulates Ca2+ fluxes. Downregulation of Ero1α inhibits mitochondrial Ca2+ fluxes and modifies the activity of MCUs. On the other hand, overexpression of redox active Ero1α increases passive Ca2+ efflux from the ER, lowering ER Ca2+ concentration and fluxes to mitochondria in response to IP3R agonists (4).
Ero1α activity results in H2O2 formation in the ER (197). During oxidative protein folding, PDI chaperones (e.g., ERp44) oxidatively fold proteins before Ero1α oxidase transfers electrons from reduced PDI to the terminal acceptor, which is usually O2 that is reduced to H2O2 (197) (Fig. 4). The interplay between Ero1α and ERp44, which is an ER luminal chaperone also present in the MAMs fraction, is responsible for the regulation of ER Ca2+ release via IP3R. Ero1α interacts with IP3R1 and oxidizes it, potentiating the Ca2+ release during ER stress (4).
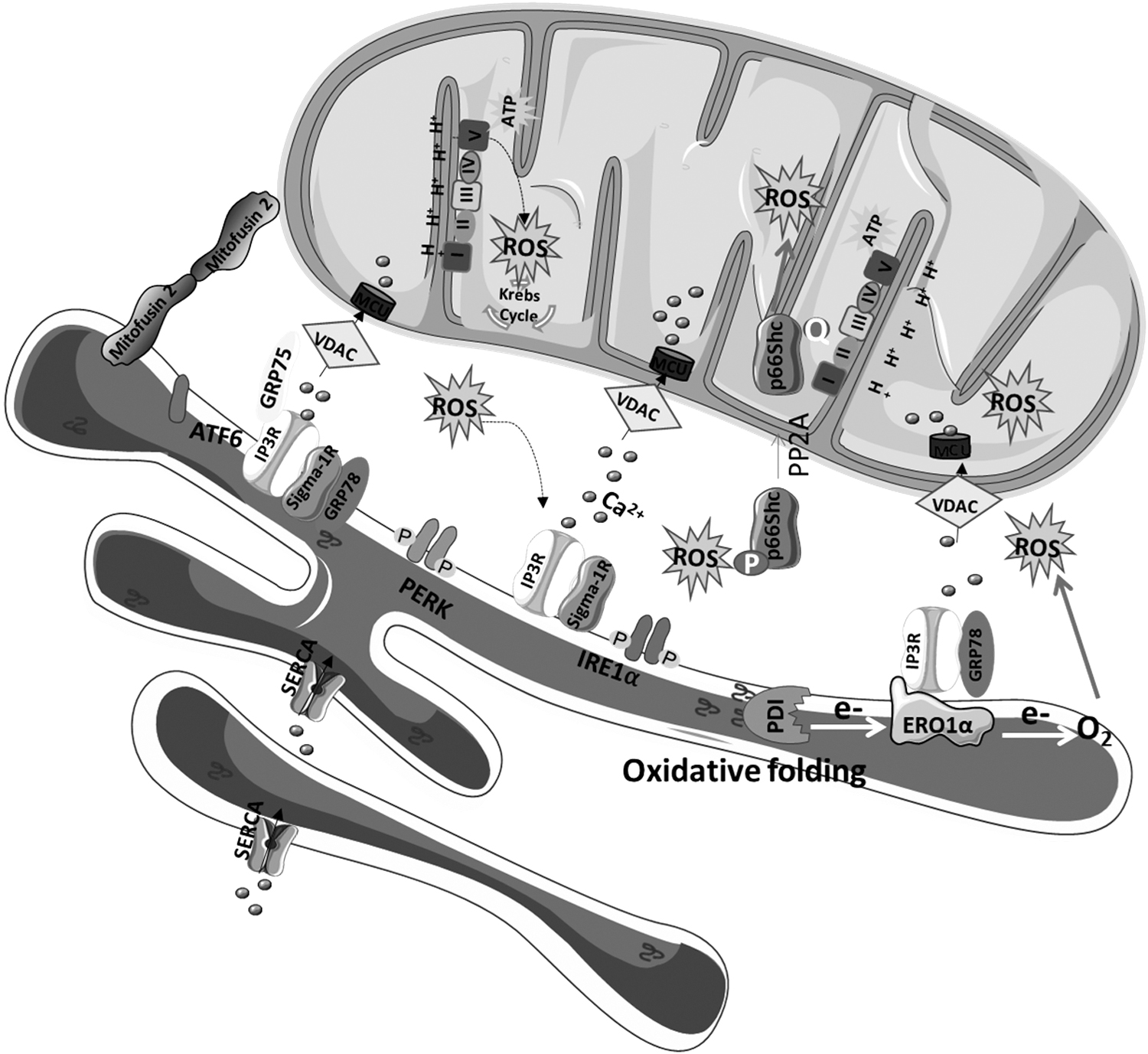
Thioredoxin-related transmembrane protein 1 (TMX1), another member of the PDI family of ER proteins that catalyze protein folding and thiol-disulfide interchange reactions, also regulates Ca2+ flux between the ER and the mitochondria. At MAMs, TMX1 inhibits SERCA2b, increasing cytoplasmic Ca2+ levels that activate Ca2+ import in mitochondria to stimulate OXPHOS (59). TMX downregulation alters mitochondrial morphology, repositions mitochondria close to the plasma membrane, enhances bioenergetics, and increases mitochondrial- and NOX-derived ROS generation (193).
GRP75 is essential to maintain the physical contact between the ER and the mitochondria by promoting the interaction between IP3R and VDAC1, and it also plays a major role in redox homeostasis regulation. GRP75 silencing was demonstrated to attenuate both cytosolic and mitochondrial Ca2+ overload in conditions of oxidative stress, block ROS formation, and preserve mitochondrial respiration (76). The canonical ER stress sensor PERK also serves as a structural tether at the ER-mitochondria interface that regulates inter-organelles crosstalk during ROS-induced cell death. It was observed that weak ER-mitochondria contacts are present in PERK−/− cells, preventing the propagation of ROS signaling to the apoptotic machinery (172).
Another MAMs-resident protein, the p66Shc, is activated by oxidative stress and translocates to the mitochondria where it participates in ROS formation. p66Shc protein is a growth factor adaptor protein belonging to the ShcA family involved in Ras signaling under physiologic conditions (136). Under oxidative stress, p66Shc can be phosphorylated at Ser36 by JNK (89), protein kinase (PK)Cβ (184), or ERK (78), leading to a cascade of events including isomerization of Ser36-phosphorylated p66Shc by the prolyl isomerase Pin1, dephosphorylation of Ser36-P-p66Shc by phosphatase A2, and translocation of p66Shc to the mitochondria and/or MAMs fraction, where it participates in ROS production (52) (Fig. 4).
A positive correlation between increased p66Shc phosphorylation at Ser36 and oxidative damage of DNA and proteins was found in fibroblasts expressing mutations in complex I subunits. PKCβ inhibition decreased oxidative stress levels and restored complex I activity under these conditions (184, 187). Accordingly, knockdown of p66Shc alleviated ROS levels and apoptosis in an in vitro oxidative stress model (63), further supporting a role of p66Shc in ROS production. Sirt1-regulated acetylation modulates the capacity of p66Shc to induce ROS formation. Lysine acetylation of p66Shc facilitates its phosphorylation on Ser36 and translocation to the mitochondria where it promotes H2O2 production (91).
Modulation of Intracellular Signaling Pathways by ROS
The deleterious roles of high ROS levels in cells and organisms have been the focus of a huge amount of work in the past, but physiologic levels of ROS support crucial cellular processes, namely neuronal development, synaptic plasticity, metabolic processes such as proteasome function, autophagy, and general inflammatory signaling (46, 183, 198).
H2O2 is recognized as one of the main molecules in the sensing, modulating, and signaling of redox metabolism, by acting as a messenger molecule in signal transduction pathways mediated by NF-κB, MAPKs, or PI3K-Akt (39, 109, 192). The transcription factor NF-κB is crucial for cellular processes, including immunity, inflammatory response, cellular adhesion, differentiation, proliferation, autophagy, senescence, and apoptosis (18). Cumulative evidence suggests an interrelation between ROS and NF-κB. H2O2 influences the activation of the NF-κB pathway mainly by inhibition of the IκBα phosphorylation (163). H2O2 can also disturb the ubiquitination and degradation of IκB and, therefore, the NF-κB activation (97).
On the other hand, NF-κB can influence ROS levels by increasing the expression of antioxidant enzymes such as SOD1, SOD2, and GPx (192). H2O2 can also activate the ERK1/2 MAP kinase, c-Jun N-terminal kinases (JNK), p38 kinase (p38), and the big MAP kinase 1 (BMK1/ERK5) pathways that are also intracellular signal transduction pathways playing an important role in processes such as cell growth, differentiation and development, cell cycle, cell survival, and cell death (192).
The PI3K/Akt signaling pathway regulates many cellular functions, namely, cell proliferation, migration, differentiation, and apoptosis through activation or inhibition of downstream proteins. Phosphatase and tensin homolog (PTEN) transforms PI-3,4,5-P3 into PI-4,5-P2; therefore, Akt and the downstream signaling pathway is activated when PTEN activity is inhibited (96). H2O2 activates PI3K either directly or concurrently, inactivating PTEN and thus amplifying Akt downstream signaling (192). H2O2 also regulates the Keap1 (Kelch-like ECH-associated protein 1)-Nrf2 signaling pathway, which plays a critical role in the maintenance of cellular redox balance and metabolism and in the induction of an adaptive response to oxidative stress.
Keap1 is physiologically associated with Nrf2, promoting its ubiquitination and thus suppressing its transcriptional activity (67). On H2O2-induced oxidative stress, Keap1 loses its ability to ubiquitinate Nrf2, allowing it to accumulate in the nucleus and induce the expression of its target genes, namely, genes encoding proteins involved in the antioxidant response such as glutathione synthetase, SOD1, and CAT that are essential for protection against oxidative insults and, therefore, of many diseases that have oxidative stress as underlying pathological features (67).
Cytosolic Ca2+ concentration is determined by a dynamic balance between mechanisms that bring Ca2+ into the cytosol, including Ca2+ influx from the extracellular medium and the release from intracellular stores, and processes that remove Ca2+ from the cell, including Ca2+ efflux across the plasma membrane and sequestration into the ER and the mitochondria (143). Intracellular Ca2+ modulates both ROS generation and clearance processes and thereby shifts the redox state to either a more oxidized or reduced state.
Mitochondrial Ca2+ promotes ATP synthesis and ROS generation via the stimulation of Krebs cycle enzymes (pyruvate dehydrogenase, isocitrate dehydrogenase, and oxoglutarate dehydrogenase) and OXPHOS (39). In turn, H2O2 directly and indirectly affects Ca2+ transport proteins located in the plasma membrane, ER, and mitochondria such as plasma membrane Ca2+ ATPases (PMCA), Na+/Ca+ exchanger (NCX), IP3R, RyR, SERCA, and MCU (69, 106). RyR/IP3R that are responsible for Ca2+ release from the ER and NCX that elicits Ca2+ extrusion from the cell are activated in response to thiol oxidation by H2O2.
The oxidation of PMCA and of SERCA pumps, which extrudes Ca2+ from the cell and transports cytosolic Ca2+ to the ER, respectively, is largely inhibitory (69). STIM and ORAI proteins that mediate the store-operated Ca2+ entry activated by ER Ca2+ depletion are also regulated by oxidative modifications in Cys residues. These redox modifications of Cys residues alter STIM1 Ca2+ sensing and reduce ORAI1 channel function (17). The oxidation of MCU, the main Ca2+ channel in the IMM, induces persistent MCU channel activation, increasing mitochondrial Ca2+ uptake and ROS generation (41). On the other hand, Ca2+ modulates ROS clearance processes, directly activating antioxidant enzymes (CAT and GSH reductase), promoting SOD upregulation, or inducing the release of mitochondrial GSH in response to Ca2+-induced mPTP opening (192).
The ROS also react with unsaturated lipids (lipid peroxidation), producing a wide variety of oxidation products. Lipid aldehydes, such as malondialdehyde, 4-hydroxy-nonenal, 4-oxo-nonenal, methylglyoxal, and isolevuglandins, are products of lipid peroxidation that can react with cellular nucleophiles, such as proteins, nucleic acids, and aminophospholipids at MAMs (e.g., PEs and PCs) to form stable adducts that change cellular function (38). When the redox equilibrium is disturbed, due to the excessive generation or impaired clearance of ROS, many cellular signaling pathways are compromised, leading to cell dysfunction that influences the development of various pathologies, including neurodegenerative disorders such as AD, PD, HD, and ALS (34).
These diseases share several common features, namely oxidative stress, mitochondrial dysfunction, and accumulation of abnormal aggregated proteins such as amyloid-beta (Aβ), tau, α-synuclein and huntingtin, which, in turn, induce mitochondrial dysfunction and oxidative stress (1, 35, 49).
Disease-Associated Pathological Alterations of MAMs
Alzheimer's disease
A growing body of evidence has revealed the impact of altered communication at MAMs in the pathophysiology of neurodegenerative disorders (175) (Fig. 5). Some of them are associated with increased ER-mitochondria contacts (7, 8, 140, 165), whereas in others a decrease of these structures has been described (43).
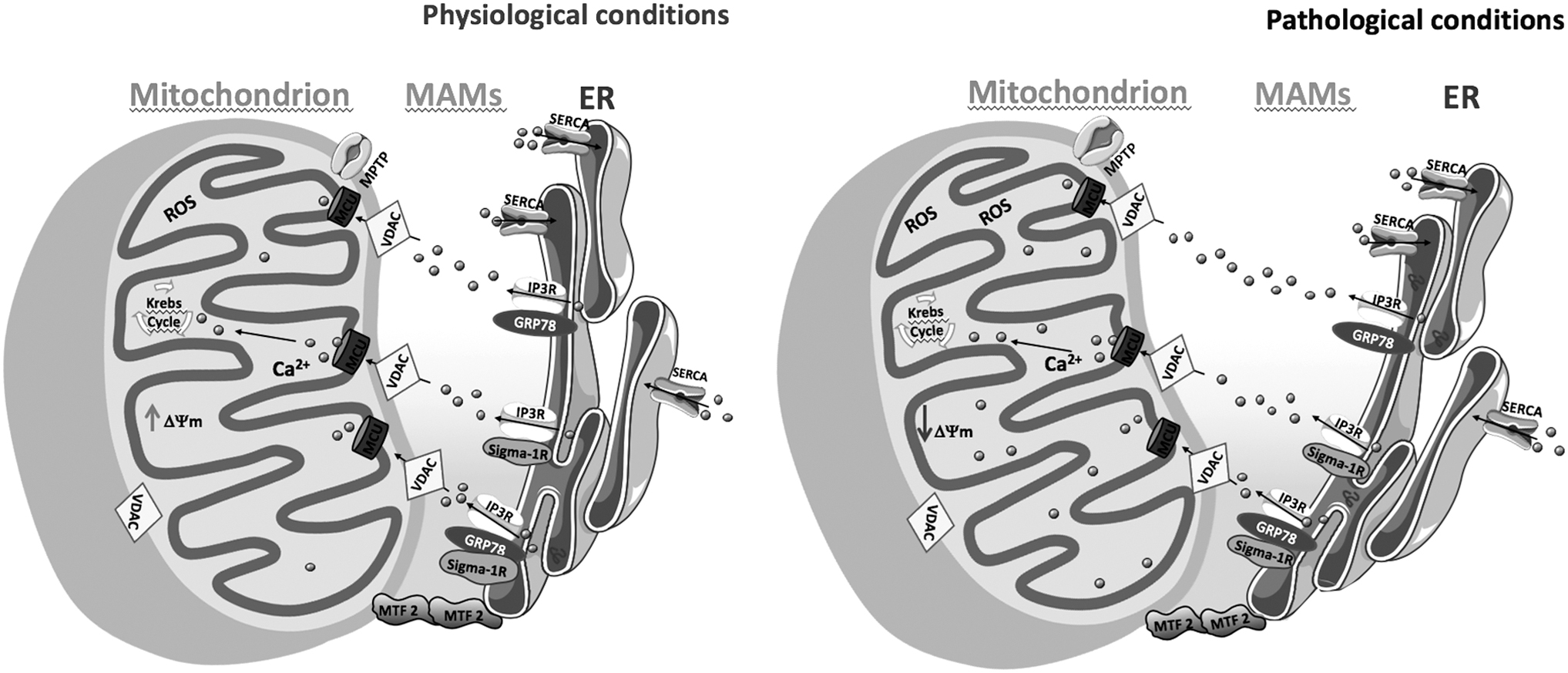
Area-Gomez et al. proposed the “MAM hypothesis” for AD based on evidence of mitochondria toxicity that arises from enhanced ER-mitochondria contacts caused by increased levels of C99, a product of amyloid-β precursor protein (APP) cleavage by β-secretase (8). Accordingly, ER-mitochondria communication was shown to be increased in fibroblasts from both familial and sporadic AD patients (7) and several MAM proteins were found to be overexpressed in human post mortem AD brain samples (68).
Interestingly, the activation of MAMs has been associated with the presence of the ɛ4 variant of apolipoprotein E (ApoE4), the major risk factor for AD (165). Del Prete et al. reported increased ER-mitochondria contact sites in SH-SY-5Y cells overexpressing the APP Swedish mutation (APPswe) (140). Using in vitro and in vivo AD models, these authors also found that both APP and its proteolytic products, namely the Aβ peptide, are located at MAMs where they modulate the ER and mitochondria functions, namely the activities of the E3 ubiquitin-protein ligase and complex III or cytochrome b-c1 complex, respectively (140). These data highlight the role of APP processing in AD-associated MAMs deregulation.
However, it is not clear how α- and β-secretases may function in the ER, mitochondria, and MAMs compartments in intact cells. Recent studies by Fernandes et al. that observed a decrease of close ER-mitochondria contacts together with activation of the ER UPR, reduced ER-mitochondria Ca2+ transfer, and impaired mitochondrial function in a mouse neuroblastoma cell line (N2A) overexpressing human APPswe (43) further support MAMs alterations in AD.
Using the MAMs fraction isolated from the cerebral cortex of the APP/PS1 mouse model of AD, Völgyi et al. detected an increase in the levels of markers of protein synthesis, ERAD, oxidative stress response, and evidence of decreased mitochondrial protein transport and ATP production. These changes precede the onset of dementia-like symptoms in the APP/PS1 model that is characterized by cerebral accumulation of the Aβ peptide, suggesting their importance in AD progression (176).
Mutant presenilin-2 (PS2) that causes familial AD and also induces Aβ deposition was demonstrated to decrease mitochondrial ATP production by reducing ER Ca2+ content and thus ER-mitochondria Ca2+ transfer, which impairs the ETC (147). The ATP depletion directly affects cell homeostasis and could explain changes in protein degradation, synaptic function, neuritic growth, neurotransmission, and long-term potentiation involved in memory formation, which are closely associated with AD (170).
There is strong evidence that p66Shc is implicated in numerous oxidative stress-associated pathologies, such as AD and other neurodegenerative disorders (159). The expression and activation of p66Shc renders central nervous system (CNS) cells more sensitive to the toxicity of Aβ by promoting mitochondrial OXPHOS and ROS production (101). In brain mitochondria isolated from p66Shc−/− aged mice, redox imbalance was demonstrated to be attenuated and both ROS generation and clearance were decreased (137). Moreover, p66Shc ablation in PS/APP transgenic mice rescued Aβ-induced cognitive deficits accompanied by improvement of brain mitochondrial respiration, reversal of mitochondrial complex I dysfunction, restoration of ATP production, and reduction of ROS levels (40).
These results support a role for p66Shc in Aβ-related mitochondrial dysfunction and oxidative damage in vivo, and they suggest that p66Shc silencing may be a promising novel therapeutic strategy against AD.
Parkinson's disease
The MAMs also play a crucial role in the pathogenesis of PD mediating PD-related events, including proteostasis loss, namely compromised autophagy (56). Besides that, several proteins implicated in PD are involved in ER-mitochondria signaling. For instance, α-synuclein regulates mitochondrial Ca2+ homeostasis by stimulating ER-mitochondria contacts (28). Recently, Basso et al. demonstrated that parkin plays a key role as regulator of ER-mitochondria communication by showing decreased inter-organelle interaction in parkin-deficient cells and in human fibroblasts expressing the familial PD-related mutant parkin (14).
In addition, it was demonstrated that MFN2 mutants that are not ubiquitinated by parkin do not promote ER-mitochondria interactions. These findings were strengthened by using an in vivo drosophila PD model in which the reestablishment of ER-mitochondria tethering using a synthetic linker was able to rescue PD-related motor deficits (14). Mitochondrial impairment and altered ER-mitochondria contacts are closely associated with other familial PD-related genes such as PINK1 and DJ-1 (134). Decreased ER-mitochondria contacts and impaired mitochondrial motility in neurites have been observed in PINK1 knockdown cells (134).
DJ-1 physically interacts with and is an essential component of the IP3R3-Grp75-VDAC1 complexes at MAMs. Depletion of DJ-1 levels, decreased IP3R3-DJ-1 interaction, as well as compromised ER-mitochondria association were found in the substantia nigra of sporadic PD patients (98).
Amyotrophic lateral sclerosis
Several strands of evidence support the role of MAMs disruption in ALS pathophysiology. Stoica et al. demonstrated that the ALS-related pathologic TDP-43 protein disrupts ER-mitochondria contacts, specifically targeting the interaction between the ER-resident protein VAPB and the mitochondrial protein PTPIP51, which is associated with impaired Ca2+ homeostasis (156). Loss-of-function mutations in Sig-1R are also strongly implicated in the familial forms of this disease (2, 180) and are closely associated with decreased ER-mitochondria contacts (16, 58). The ALS-linked Sig-1R variants are not able to bind IP3Rs and thus, to control Ca2+ homeostasis at MAMs, leading to calpain activation, mitochondrial dysfunction, and neurodegeneration.
Further, MAMs collapse was observed in motor neurons isolated from Sig-1R-deficient mice (180). SOD1 mutations are one of the most common causes of inherited ALS, and they are also linked with MAMs disruption and Sig-1R aggregation (180), which highlights MAMs dysfunction as a common mechanism underlying both Sig-1R- and SOD1-linked ALS forms.
Huntington's disease
Cherubini et al. were pioneers in implicating ER-mitochondria miscommunication in HD. These authors reported reduced ER-mitochondria contact sites in striatal neurons from R6/1 HD mice (36). Moreover, they demonstrated that HD-related defects on mitochondrial dynamics, caused by increased Drp1-mediated mitochondrial fragmentation, disrupt ER-mitochondria communication, affecting Ca2+ homeostasis and leading to excessive ROS production (36). Further, it was reported that a significant depletion of ER-mitochondria tethering proteins, namely IP3R, Grp75, and Mfn2, occurs in the knock-in mutant HdhQ7/Q111 and R6/1 HD mice models (36).
Finally, a significant decrease of MAMs proteins in the putamen of HD patients compared with controls was observed. These findings suggest that the disruption of ER-mitochondria contacts, caused by either upregulated mitochondria fission or loss of MAM-resident proteins, is a relevant mechanism implicated in striatal dysfunction and neurodegeneration in HD (36).
Sigma-1 Receptor
The Sig-1R is an ER chaperone protein that is abundantly expressed in the CNS, namely in neurons, astrocytes, microglia, and oligodendrocytes (3, 86). Sig-1R is highly enriched at MAMs and controls, among other mechanisms, IP3R-mediated Ca2+ flux from the ER to the mitochondria, lipid dynamics, and oxidative and ER stress responses (181) (Fig. 6). These processes are regulated by MAMs (141, 161, 172), which are increasingly recognized to play a role in the pathophysiology of many neurodegenerative diseases, including AD, PD, HD, and ALS (8, 36, 131, 155), as described earlier. Further, recent evidence supports the fact that Sig-1R agonists exhibit strong neuroprotective effects in preclinical models, which highlight the importance of Sig-1R as a potential therapeutic target for AD, PD, HD, and ALS (149).
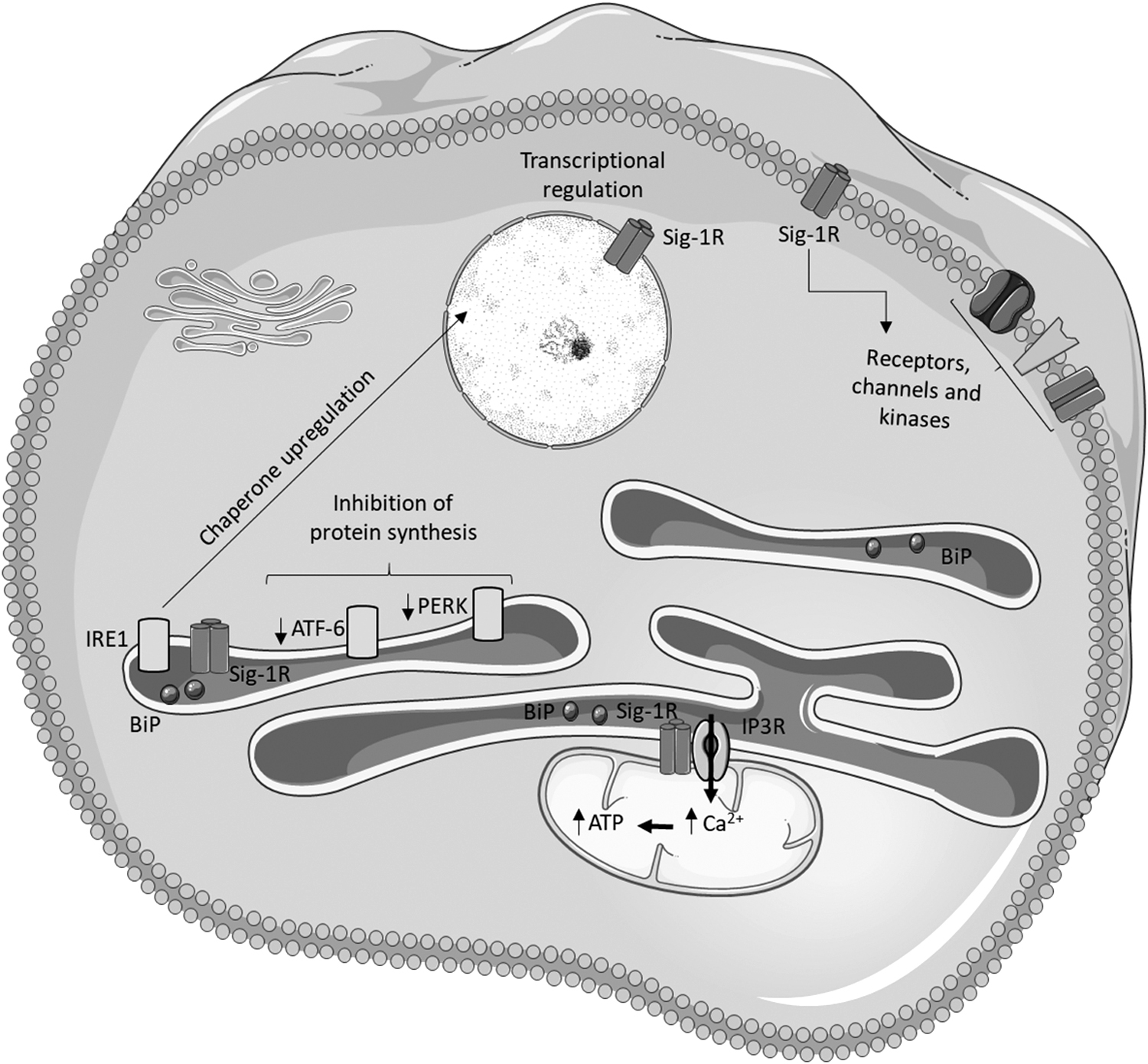
Regulation of mitochondrial and ER functions by Sig-1R
In physiological conditions, Sig-1R forms a complex with the ER-resident chaperone GRP78 at MAMs. When activated by agonists or under ER stress conditions, Sig-1R dissociates from GRP78 and binds to IP3Rs. The chaperone activity of free Sig-1Rs attenuates aggregation and degradation of IP3R, enhancing Ca2+ uptake by the mitochondria (66). As a result, redox reactions and ATP production are facilitated due to the regulation of Ca2+-dependent enzymes in the TCA cycle and of cell death pathways that can be initiated due to mitochondrial Ca2+ overload, namely, necrosis and apoptosis (143).
Under stress conditions, Sig-1R also stabilizes IRE-1α at MAMs and enhances cell survival by prolonging the activation of the IRE1-XBP1 signaling pathway. Accordingly, Sig-1R is upregulated by ER stress to preserve cell homeostasis (122) and is translocated to the plasma membrane to interact with and regulate many different membrane proteins, including receptors, ion channels, and kinases (158). Sig-1R also translocates from MAMs to the nuclear membrane on ER stress, interacting with chromatin remodeling factors that, in turn, regulate the activity of transcription factors and thereby gene expression (169) (Fig. 6).
Role of Sig-1R in oxidative stress
Sig-1R promotes cell survival by protecting against oxidative stress in several tissues, including the brain tissue (54), through activation of the antioxidant response element (ARE) and expression of genes encoding the antioxidant enzymes NAD(P)H quinone oxidoreductase 1 (NQO1) and SOD1 (132). Sig-1R knockout (KO) mice showed elevated endogenous ROS levels and glutathione depletion in Müller cells compared with wild-type mice. In addition, protein levels of the antioxidant enzymes SOD1, CAT, NQO1, and GPX1 were found to be decreased in Sig-1R KO Müller cells.
The Sig-1R agonist dehydroepiandrosterone (DHEA) and its sulfated analog (DHEAS) exhibited a neuroprotective role against H2O2-induced toxicity in mouse neuroblastoma cells. Moreover, the antioxidant enzymes peroxiredoxin 3 (Prx3) and SOD2 have been shown to be reduced in the hippocampi of mice deficient of zinc finger protein 179 (Znf179) that was identified as a Sig-1R downstream effector (157). Inhibition of Sig-1R by RNAi or antagonists significantly blocked the increase of Ferritin heavy chain 1 (FTH1) and transferrin receptor protein 1 (TFR1) levels induced by erastin and sorafenib, indicating that Sig-1R prevents ROS accumulation by negatively regulating iron metabolism (12).
Metal ions such as iron, copper, and zinc are important in several metabolic processes; therefore, their homeostasis is tightly regulated, particularly in the brain. Dysregulation of redox-active metal ions may cause uncontrolled free radical generation and contribute to the pathophysiology of neurodegenerative diseases (170).
Sig-1R Modulation as a Therapeutic Target
Oxidative stress and mitochondrial dysfunction, in which the Sig-1R has been shown to have a positive impact, are common features of neurodegenerative diseases such as AD, PD, HD, and ALS (34, 36). Therefore, Sig-1R is a putative target to prevent or delay the progression of neurodegenerative disorders.
Alzheimer's disease
Sig-1R has been associated with AD pathophysiology. In fact, Sig-1R density is significantly lower in AD patients' brains (68, 119, 167) and genetic studies demonstrated an association between SIGMAR1 polymorphisms and the risk for developing AD (42).
Sig-1R knockdown causes degeneration in mouse hippocampal neurons (68). Moreover, impaired neurogenesis in the dentate gyrus of adult hippocampus due to the dysfunction of glutamate NMDA receptors has been observed in Sig-1R KO mice (152). Accordingly, the protective effect of Sig-1R agonists in in vitro and in vivo models of AD has been reported. The Sig-1R agonists PRE-084, SA4503, ANAVEX1-41, and ANAVEX2-73 prevented learning and memory impairments in mice injected with Aβ25–35 (111, 112, 173).
Similarly, anti-amnesic and neuroprotective effects induced by the Sig-1R agonist (-)-methyl(1S,2R)-2-{[1-adamantyl(methyl)amino]methyl}-1-phenylcyclopropane-carboxylate have been observed in Aβ-treated rats (5). Improved cognitive function, as well as decreased synapse loss, BACE1 and GSK3β activity, p25/CDK5, neuroinflammation, soluble and insoluble Aβ levels, and plaque and tau pathologies have been reported in 3xTg-AD mice treated with AF710B, a muscarinic and Sig-1R agonist (45). Further, 2-73-(tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanmethanamine hydrochloride) blocked the accumulation of Aβ1–42 and C99 in the hippocampus of Aβ-injected mice, a sporadic model of AD (94).
ANAVEX2-73 and PRE-084 prevented dysfunction of the mitochondrial ETC and subsequent oxidative stress and apoptosis in Aβ25-35-injected mice (93). It has been observed that PRE-084, ANAVEX1-41, ANAVEX3-71, and DHEA induce mild oxidative stress in mitochondria and activate complex I in physiological conditions but protect against pathological oxidative stress, reducing Aβ1-42-induced ROS formation (54). These results highlight the role of Sig-1R in modulating mitochondrial function and ROS generation in both physiological and pathological conditions.
Although the studies cited earlier support the neuroprotective effect of Sig-1R activation, there are studies showing contradictory results. For example, Sig-1R deficiency was shown to reduce Aβ25-35-induced hippocampal neuronal cell death and cognitive deficits in mice (189).
Parkinson's disease
Sig-1R has been reported to be decreased in the brain of PD patients (123, 167). In a 6-hydroxydopamine (6-OHDA)-induced PD mouse model, pharmacological modulation of Sig-1R with PRE-084 improved motor deficits, which was accompanied by an increase in dopaminergic fiber density and dopamine levels, neurotrophic factors (BDNF and GDNF), and survival signaling mediators (ERK1/2 and Akt) (48). In another study, afobazole and PRE-084 have been demonstrated to normalize motor function and prevent dopamine depletion in the striatum of the 6-OHDA-induced parkinsonism model (177).
Moreover, it has been observed that physiologic concentrations of dopamine induce apoptosis in Sig-1R knockdown CHO cells by an NF-κB/BCL-2-dependent mechanism (123). These results highlight the potential protective effect of Sig-1R in PD. Clinical benefits have also been reported, namely the attenuation of levodopa-induced dyskinesia in PD patients treated with Sig-1R agonists (47, 133). On the contrary, another study demonstrated that Sig-1R deficiency and pharmacological inactivation reduces 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced motor deficits and loss of dopaminergic neurons, suggesting that Sig-1R deficiency is neuroprotective (75).
Huntington's disease
Sig-1R levels are reduced in the PC6.3 cell line, a neuronal HD model overexpressing the mutant huntingtin protein (80). In this in vitro HD model, PRE084 upregulates antioxidants and inhibits NF-κB signaling, decreasing oxidative stress (80). SIGMAR1 siRNA significantly promoted the cytoplasmatic and nuclear accumulation of aggregates in HeLa cells transfected with N-terminal mutant huntingtin, whereas SIGMAR1 overexpression enhanced the degradation of intranuclear inclusions (118).
Pridopidine was originally described as a dopamine stabilizer. However, its mode of action includes Sig-1R activation, which is required for its effect on BDNF signaling pathways (51). Pridopidine stabilizes striatal medium spiny neurons spines, normalizes ER Ca2+ content and release in aged corticostriatal co-cultures from YAC128 transgenic HD mice, and the deletion of neuronal Sig-1R prevented spine protection (125, 148).
Amyotrophic lateral sclerosis
Sig-1R is enriched in motor neurons in the CNS (113). Further, Sig-1R accumulates in postsynaptic densities associated with cholinergic synapses and is enriched at ER structures of alpha motor neurons. A mutation in the gene encoding Sig-1R causes FTLD-ALS and juvenile ALS (2, 103, 180) and its protein levels are reduced in lumbar ALS patient spinal cord. Pharmacological Sig-1R activation induced the clearance of mutant protein aggregates in mice-cultured motor neurons (139). In mutant SOD1-overexpressing mice, Sig-1R deficiency accelerated the onset of ALS manifestations and induced MAMs disruption, calpain activation, and mitochondrial dysfunction (180).
Further, SIGMAR1 knock-out in an ALS mice model potentiated motor performance impairment and decreased longevity (114). Recently, Couly et al. provided evidence that mutant Sig-1R overexpression recapitulates ALS pathology in vivo and that Sig-1R rescues locomotor activity and ATP levels in flies overexpressing TDP-43, the key ALS protein (37).
The benefits of Sig-1R activation in ALS were obvious. Administration of the Sig-1R agonist SA4503 to an ALS mouse model prevented motor neuron degeneration and symptom progression, together with extended survival. Moreover, SA4503 reduced SOD1G93A mutation and serum free-induced cell death in NSC34 cells (128). It has also been reported that SA4503 can normalize bradykinin-sensitive intracellular Ca2+ stores and avoid cytosolic Ca2+ overload in spinal neurons isolated from embryonic mice overexpressing human SOD1 with the ALS-causing mutation G93A (162). In this mouse model, improved spinal motor neuron function, locomotor performance, preservation of neuromuscular junctions and of motor neurons in the lumbar spinal cord have been described on treatment with the agonist PRE-084 (107).
Further, pridopidine attenuated deficits in Sig-1R axonal transport, disruption of neuromuscular junction, and cell death in SOD1G93A motor neurons and in neuromuscular co-cultures (81). Recently, Couly et al. provided evidence that mutant Sig-1R recapitulates ALS pathology in vivo whereas Sig-1R rescues locomotor activity and ATP levels of flies overexpressing TDP-43 (37).
All the studies cited earlier support the involvement of Sig-1R in the pathophysiology of neurodegenerative diseases, and the effects of its modulation pinpoint Sig-1R as a potential therapeutic target for these disorders.
Conclusion
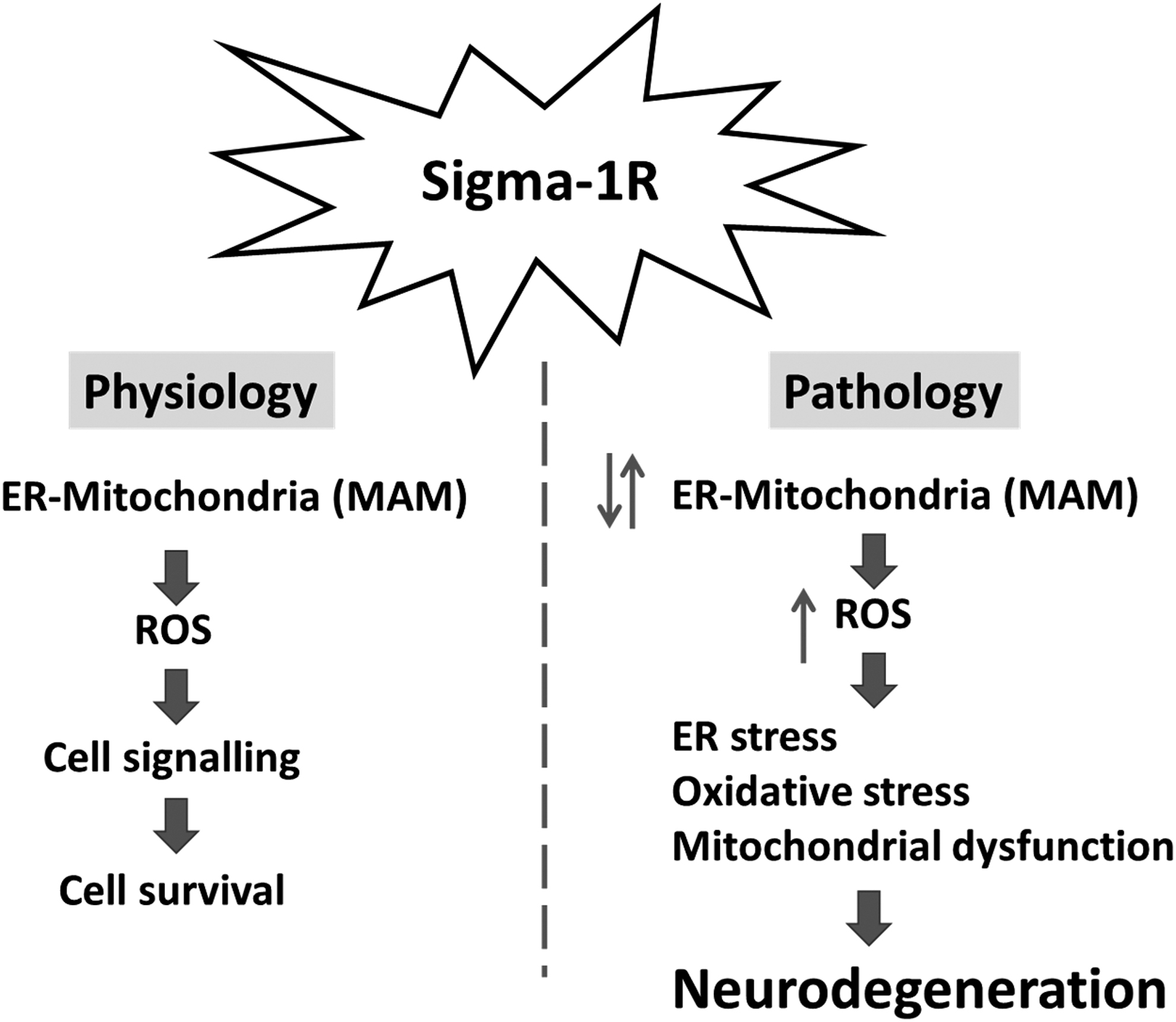
The key role of impaired communication between the ER and the mitochondria at MAMs in the pathogenesis of age-dependent neurodegenerative disorders is highlighted. It can be hypothesized that changes in ER-mitochondria contact sites in the asymptomatic stage can represent a stress response mechanism to promote ROS signaling, preserve cell metabolism and survival, whereas chronic alterations might exacerbate Ca2+ transfer and ROS generation, leading to deregulation of proteostasis and innate immunity, finally activating apoptotic cell death pathways.
The Sig-1R chaperone is highly enriched at MAMs and controls ER to mitochondria Ca2+ fluxes, lipid dynamics, as well as oxidative and ER stress responses, which are regulated by MAMs. Sig-1R modulation in preclinical models highlights its potential as a therapeutic target for neurodegenerative diseases, including AD, PD, ALS, and HD (Fig. 7).
Authors' Contribution
Conceptualization, C.F.P.; writing—original draft, R.R., T.F., A.C.P., and A.P. M.; writing—review and editing, R.R. and C.F.P. All authors have read and agreed to the published version of the article.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Programme under project CENTRO-01-0145-FEDER-000012 (HealthyAging2020), through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation, and Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, under projects POCI-01-0145-FEDER-028214 and UIDB/04539/2020, UIDP/04539/2020 and LA/P/0058/2020. European Social Fund (Post-Doctoral Researcher Contract SFRH/BPD/101028/2014 to Rosa Resende). Tânia Fernandes and Ana Catarina Pereira are recipients of FCT PhD fellowships (SFRH/BD/148801/2019 and SFRH/BD/148653/2019, respectively).