
Abstract
Significance:
Protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductase 1 (ERO1) are crucial for oxidative protein folding in the endoplasmic reticulum (ER). These enzymes are frequently overexpressed and secreted, and they contribute to the pathology of neurodegenerative, cardiovascular, and metabolic diseases.
Recent Advances:
Tissue-specific knockout mouse models and pharmacologic inhibitors have been developed to advance our understanding of the cell-specific functions of PDI and ERO1. In addition to their roles in protecting cells from the unfolded protein response and oxidative stress, recent studies have revealed that PDI and ERO1 also function outside of the cells.
Critical Issues:
Despite the well-known contributions of PDI and ERO1 to specific disease pathology, the detailed molecular and cellular mechanisms underlying these activities remain to be elucidated. Further, although PDI and ERO1 inhibitors have been identified, the results from previous studies require careful evaluation, as many of these agents are not selective and may have significant cytotoxicity.
Future Directions:
The functions of PDI and ERO1 in the ER have been extensively studied. Additional studies will be required to define their functions outside the ER.
Introduction
Oxidation and isomerization of intramolecular disulfide bonds are essential for appropriate protein folding and maturation in the endoplasmic reticulum (ER). Dysregulation of this process often results in cell and tissue pathology and may lead to neurodegenerative diseases and diabetes. Thiol isomerases, such as the prototypic enzyme, protein disulfide isomerase (PDI), are catalysts that have critical roles in facilitating disulfide bond modification. After PDI has catalyzed the oxidation of one or more disulfide bonds in a protein substrate, the reduced protein is enzymatically reoxidized by ER oxidoreductase 1 (ERO1). This interaction between ERO1 and PDI represents a catalytic redox cycle that is critical for oxidative protein folding in the ER.
Studies with conditional knockout (CKO) mice and pharmacological inhibitors have advanced our understanding of the physiological roles played by both PDI and ERO1. Both PDI and ERO1 are upregulated in various cell types in response to pathologic conditions, and small amounts of each enzyme are secreted extracellularly. These aspects highlight the rationale for developing novel therapies that target PDI and ERO1 and suggest that these enzymes might be developed as biomarkers to predict disease severity. In this review, we focus on the structure, function, and pathophysiological roles of PDI and ERO1 and the identification and development of their inhibitors as novel therapeutics.
Structure of PDI
PDI includes four major domains (a, b, b′, and a′). The a and a′ domains contain a TrpCysGlyHisCysLys active site. The b and b′ domains are catalytically inactive but can interact with substrates primarily via a hydrophobic region in the b′ domain (29). The b′ and a′ domains of PDI are linked with a flexible linker peptide containing 19 amino acids. This linker region regulates substrate binding by capping and uncapping a hydrophobic site on the b′ domain (119, 180). Many acidic residues in the C-terminal region maintain the functional conformation of PDI and prevent self-aggregation (170). PDI has an ER retrieval sequence (LysAspGluLeu) at its C-terminus that promotes binding a receptor in the cis-Golgi and facilitates its return to the ER (24).
Despite the ER retention signal, PDI can be released from cells and has been detected on the plasma membrane where it is bound to molecules on the cell surface (51, 62, 97). Previous studies suggested several molecular mechanisms by which ER-resident molecules can be exported and released; these include (1) masking of the LysAspGluLeu sequence by glycosylation (2, 210) saturation or inactivation of LysAspGluLeu receptors (3, 202) membrane translocation of LysAspGluLeu receptor 1 (10) and (4) alteration of cytosolic calcium levels (20, 173).
The crystal structures of yeast and human PDI were resolved at resolutions of 2.4 and 2.5 Å, respectively (169, 181). Human and yeast PDI share 31% amino acid sequence identity. The four domains (a, b, b′, and a′) are arranged in a “U” shape with the a and a′ active sites facing each other. In the reduced form of human PDI, the a, b, and b′ domains are in a linear configuration, with the a′ domain angled at ∼45° from the plane; in this configuration, the distance between the sulfur atoms of Cys53 and Cys397 is maintained at 27.6 Å (181). In contrast, the four domains are organized differently in the oxidized form of human PDI, with a 40.3 Å distance separating the two active sites in the same plane.
Based on the relative positions of the substrate-binding pocket and catalytic sites, reduced and oxidized forms of PDI are considered to be in a closed and open conformation, respectively. Although most proteins are capable of interacting with oxidized PDI, it is critical to note that some substrates can bind preferentially to the reduced form. For example, we showed that the β-switch region of the platelet-specific receptor, glycoprotein Ibα (GPIbα), interacts with the b′ domain of reduced PDI on the platelet surface (97). Further, we found that the Cys4–Cys17 disulfide bond in the β-finger region of GPIbα is closer to PDI Cys53 when complexed with the reduced rather than the oxidized form of the enzyme (16 Å vs. 28 Å, respectively). These results suggest that GPIbα may bind more favorably to the reduced form of PDI.
The existence of different conformations of the b′ domains (181) implies the possibility of substrate interactions that favor either the oxidized or the reduced forms of PDI. In addition, it was recently reported that Fam20C, a Golgi-associated secretory pathway kinase, phosphorylates Ser357 in the x-linker region of PDI in response to ER stress. Phosphorylation at this site induces an open conformation with a 70 Å distance between the a and a′ domains and results in a functional switch from an oxidoreductase to a molecular chaperone (203). Future studies are required to explore the impact of Ser357 phosphorylation on the interaction between PDI and ERO1α and ERO1α-mediated PDI oxidation.
Function of PDI
PDI was first identified in 1963 as a microsomal enzyme of rat liver and pigeon/chicken pancreas that was capable of oxidizing reduced bovine pancreatic ribonuclease (1, 47). Twenty-four years later, PDI was identified as the β-subunit of prolyl 4-hydroxylase subunit β (P4HB) (132). Since prolyl 4-hydroxylase forms an α2β2 tetramer and catalyzes the formation of 4-hydroxyproline in collagen, the PDI subunit could contribute to oxidoreductase and/or molecular chaperone activity as a monomer and as a part of the prolyl 4-hydroxylase tetramer together with the α-subunit.
A previous study revealed that PDI forms disulfide-independent dimers within cells and that dimerization impairs both the catalytic and chaperone function by blocking substrate-binding sites in the b and b′ domains (11). However, because the results of this study were based on PDI overexpression, it is not clear whether endogenous PDI can undergo dimerization under pathophysiological conditions or the nature of its functional role in this conformation.
PDI catalyzes disulfide bond oxidation and isomerization in unfolded substrates in the ER and thus plays an indispensable role in the process of protein folding (152). In a highly oxidizing environment, reduced Cys thiols in substrates form transient disulfide bonds with thiols with active site residues in PDI (CysGlyHisCys), thereby resulting in an oxidized, folded protein. Misfolded proteins are reduced and isomerized by PDI and converted to their appropriate native conformation. Reduced PDI is subsequently reoxidized by oxidases such as ERO1, a topic that will be discussed later in this review. To facilitate this process, the active site of PDI is maintained in a reduced state, and glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH) promote cleavage of the disulfide bonds (15, 111).
In addition to oxidoreductase activity, PDI-mediated chaperone activity prevents the accumulation of misfolded proteins that are formed in response to oxidative ER stress and facilitates the maturation and transport of secretory proteins. As described earlier, phosphorylation of Ser357 in PDI results in a significant decrease in its isomerase activity and a significant increase in its chaperone activity (203). This result suggests that the post-translational modification in PDI prevents aggregation of misfolded proteins under conditions of ER stress.
The expression and function of PDI in the ER are indispensable for survival in yeast, with no apparent compensation provided by any of the four additional members of the PDI enzyme family (53, 90). Consistently, our unpublished data suggest that deletion of the gene encoding PDI results in an embryonic lethal phenotype in mice, suggesting that none of the 21 PDI oxidoreductase family members can provide adequate compensation (16). Importantly, dysregulation of PDI function in the ER has been associated with numerous disease processes, including neurodegenerative disorders, diabetes, and cardiovascular diseases (CVDs). Specifically, these disorders have been characterized by accumulations of misfolded and immature proteins or by impaired function of cell surface molecules, which will be discussed later in this review.
Structure of ERO1
The yeast gene, Ero1, encoding Ero1p was discovered in 1998 (42, 134). Two human genes, ERO1-L and ERO1-Lβ, encoding ERO1α and ERO1β, respectively, were identified thereafter (23, 126). Although ERO1α is expressed in most cell types (126), ERO1β is mainly expressed in intestinal and pancreatic β cells (7, 36). ERO1α and ERO1β share 65% amino acid sequence identity. Inaba et al. reported the crystal structure of hyperactive and inactive human ERO1α at resolutions of 2.35 and 3.07 Å, respectively (58). Interestingly, one recent study showed that there are no substantial differences between the conformations of active and inactive forms of human ERO1α (209).
Consistent with findings from mass spectrometry (4), a crystallographic analysis revealed that hyperactive and inactive ERO1α form a single globular domain enriched in α-helices with five intramolecular disulfide bonds (Cys35–Cys48, Cys37–Cys46, Cys85–Cys391, Cys208–Cys241, and Cys394–Cys397) (58). Although the results of one earlier study suggested that the intramolecular Cys85–Cys391 disulfide bond regulates ERO1 activity (9), subsequent studies revealed a structural role for this linkage (6, 121, 208). Further, two essential Cys triads (Cys85–Cys94–Cys99 and Cys391–Cys394–Cys397) are highly conserved in yeast and human ERO1 and serve to control enzymatic activity (17).
The results from mutational studies revealed that ERO1α is activated by the formation of a Cys94–Cys99 disulfide bond, whereas it is inactivated on disulfide bond formation between the Cys94–Cys131 and Cys99–Cys104 (4, 6). A protruding β-hairpin that includes Trp272 interacts with the substrate-binding site of the b′ domain of reduced PDI, and the Cys94–Cys99 pair then transfers electrons from PDI to the Cys394–Cys397 disulfide bond, resulting in PDI reoxidation (104).
As a flavin adenine dinucleotide (FAD)-binding enzyme, ERO1 oxidizes reduced PDI during oxidative protein folding. Our docking study confirmed the FAD-binding site in human ERO1α and identified additional molecular interactions between FAD and ERO1α as compared with a previous report (58). Among these results, we found that FAD interacts with ERO1α via multiple hydrogen bonds with Arg187, Thr189, Trp197, Trp200, Ser252, Asn259, Arg287, and Arg300 and two π-π interactions with His255 and Tyr191 (Fig. 1A, B). One study reported that the carbon 4a in the isoalloxazine ring of bound FAD is only 3.3 Å away from Cys397 of ERO1α, implicating the possibility of an interaction between FAD and Cys397 that takes place after cleavage of the Cys394–Cys397 disulfide bond (58).
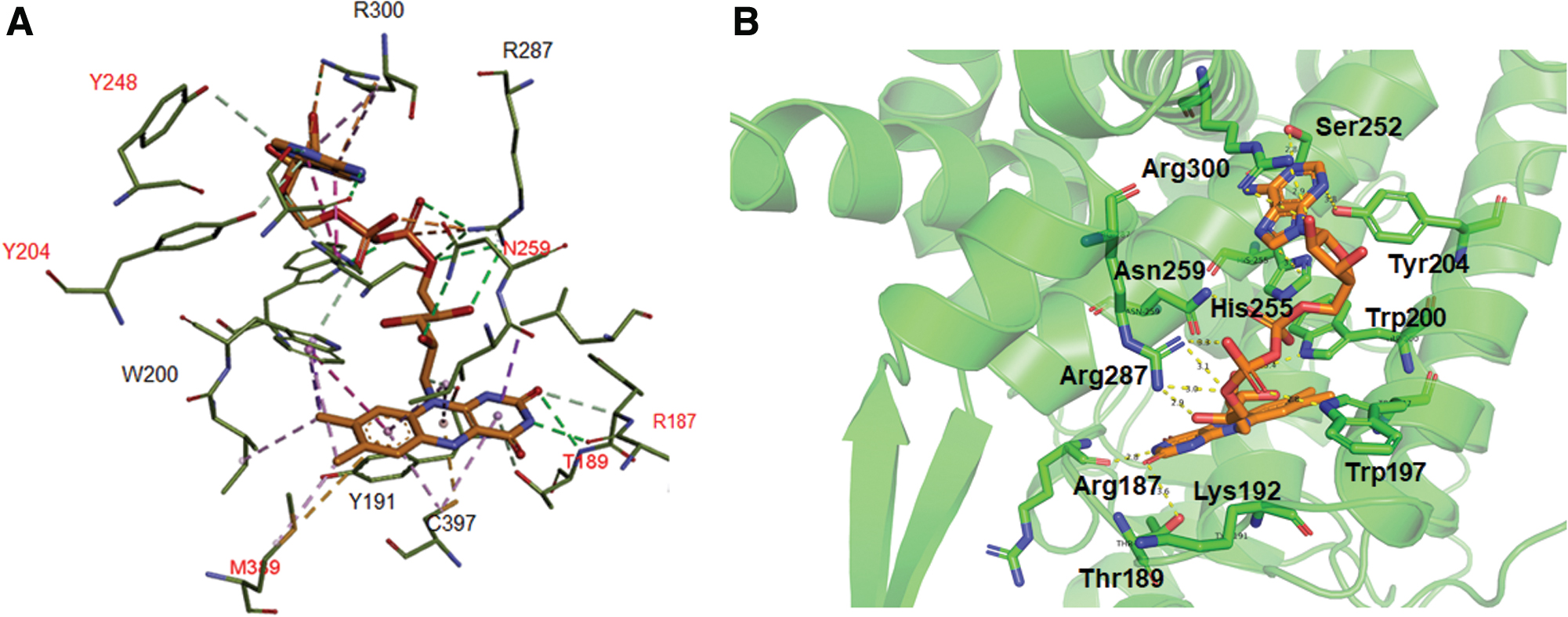
Function of ERO1
Deletion of Ero1 has a marked effect on oxidative protein folding and cell viability in yeast (42). In contrast, mice with loss-of-function mutations in Ero1-l and Ero1-lβ are viable and exhibit minimal defects (214). The Ero1-l and Ero1-lβ mutant mice are hypersensitive to β-adrenergic blockade, whereas an Ero1-l gene deletion alters calcium homeostasis in cardiomyocytes and protects against the progression of heart failure (27). Consistent with its abundant expression in pancreatic β-cells, deletion of Ero1-lβ results in compromised oxidative folding of proinsulin and promotes glucose intolerance (214). The different outcomes resulting from disruption of ERO1 function are largely due to the presence of compensatory mechanisms for ERO1-mediated thiol oxidation in mice but not in yeast.
The expression of ERO1α is upregulated via hypoxia-inducible factor-1α (HIF-1α) under hypoxia (105), whereas the expression of ERO1β is enhanced by the unfolded protein response (UPR) (126). ERO1α serves as the primary oxidase that restores reduced PDI and other PDI family thiol isomerases, which together constitute an electron transfer network of ER oxidoreductases. However, given the known dissociation constants for each oxidoreductase, ERO1α is likely to bind preferentially to PDI (K d = 1.7 μM), compared with ERp44 (K d = 21 μM), ERp5 (K d = 70 μM), ERp57 (K d = 180 μM), ERp72 (K d = 160 μM), or ERp46 (K d = 280 mM) (5).
One study revealed that peroxiredoxin 4 preferentially recognizes and oxidizes ERp5 and ERp46 and that peroxiredoxin 4-catalyzed oxidation of ERp5 and ERp46 is accelerated in combination with PDI (150). Interestingly, ERO1α-catalyzed PDI oxidation was impaired in the presence of a hyperactive mutant of ERp44 (104). Collectively, these results suggest that ER oxidases target specific thiol isomerases during protein folding and that thiol isomerases cooperate or compete with one another to promote protein reoxidation.
A previous study using full-length and mutant PDI revealed that the b′-x-a′ domains of PDI provide the structural basis for the preferential binding and oxidation of ERO1α (183). Figure 2 summarizes ERO1α-PDI redox activity and oxidative protein folding in the ER. A study using docking simulations and mutations suggested that Trp272 in ERO1α interacts with the hydrophobic residues (Phe240, Phe249, and Phe304) of the b′ domain of reduced PDI (104).
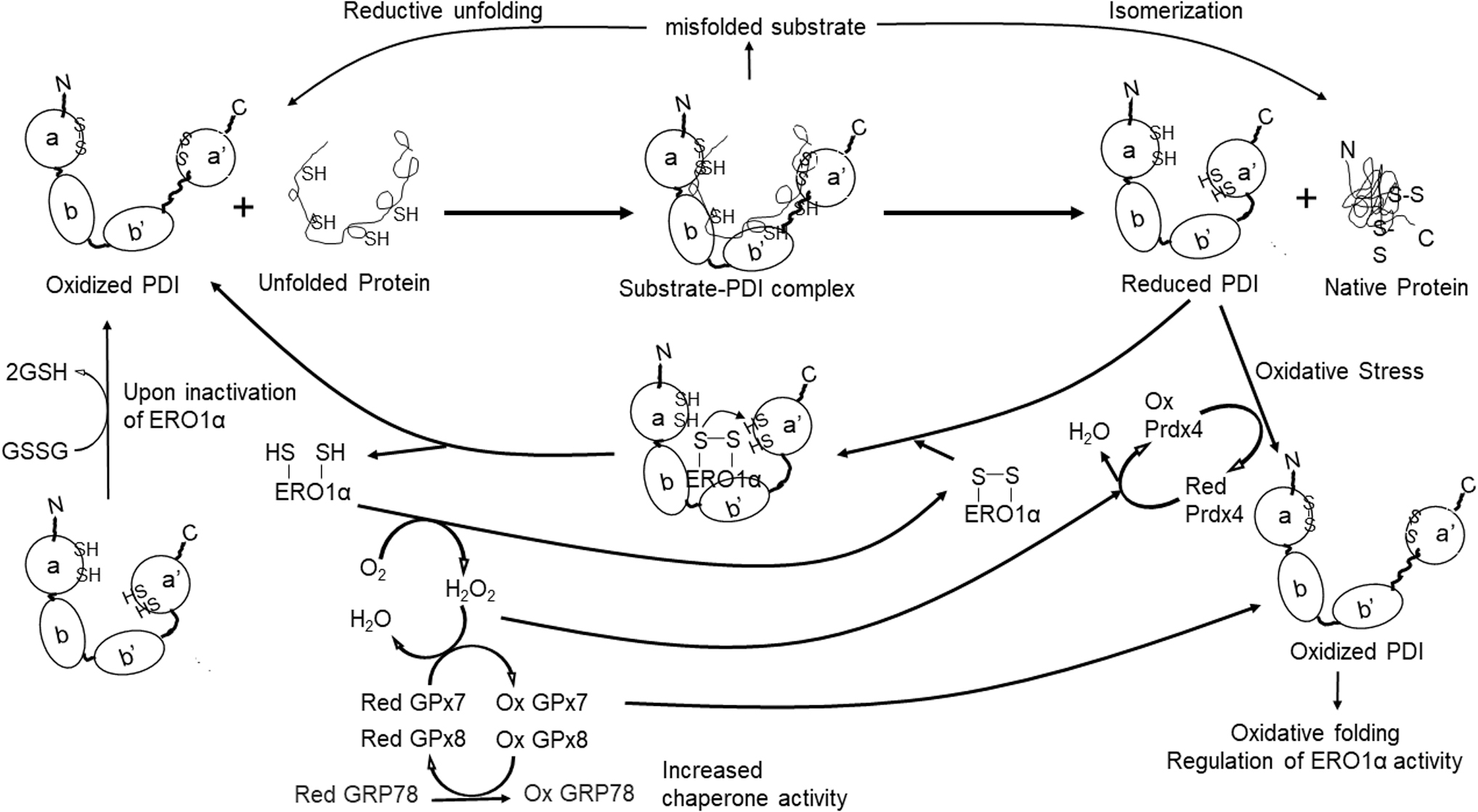
Preferential oxidation of the PDI a′ domain is catalyzed by nucleophilic attack initiated by the C-terminal active site in PDI targeting the Cys94–Cys99 shuttle disulfide bond in ERO1α. This results in a mixed-disulfide bond that forms a transient link between the C-terminal active site of PDI and Cys94 of ERO1α. Oxidized PDI shuttles a disulfide bond to an unfolded substrate bound to its hydrophobic pocket. ERO1 uses O2 as an electron acceptor in this reaction and produces H2O2 as a byproduct, inducing ER stress (183). Therefore, the ERO1-PDI redox cycle is tightly regulated under homeostatic conditions.
The ER-resident peroxidases, GSH peroxidase 7 (GPx7), and GPx8 utilize ERO1α-generated H2O2 to oxidize PDI, facilitating oxidative protein folding (118, 184). One recent study suggested that GPx7 reacts more efficiently with H2O2 and promotes higher levels of PDI oxidation than GPx8. The differential activity observed was attributed to the presence of Gln92 in the GPx7 catalytic tetrad; this residue stabilizes sulfenylated Cys57 that forms a hydrogen bond with ERO1α-generated H2O2 (70).
Mouse embryonic fibroblasts deficient in GPx7 exhibit increased production of reactive oxygen species (ROS) and, thus, oxidative stress (188). This study also showed that oxidized GPx7 generates disulfide bonds and activates the chaperone protein, glucose-regulated protein 78 (GRP78). However, a very high concentration of H2O2 (20 mM) was used in this study, which raises questions regarding physiological relevance. Deletion of GPx8 results in leakage of ERO1α-produced H2O2 from the ER to the cytosol and induces ER stress and cell death (139). Further, knockdown of peroxiredoxin 4 results in impaired cellular tolerance to ER stress (165). Taken together, these results suggest that cells are protected from H2O2-induced ER stress by the actions of GPx7, GPx8, and peroxiredoxin 4.
Because the reduced form of PDI is necessary for isomerization of thiol-disulfide bonds, H2O2-induced ER stress must be tightly controlled. Molteni et al. demonstrated the antagonistic roles of cytosolic GSH and ERO1α in regulating the ER redox state (111). GSH depletion accelerates the formation of protein disulfides most likely via enhanced ERO1 oxidase activity and the formation of high-molecular protein aggregates. The rate of protein oxidation is consistently higher in cells that overexpress ERO1α. These results suggest that pathological conditions that alter the ratio of oxidized to reduced glutathione (i.e., GSSG/GSH) would have an indirect effect on disulfide bond formation via its impact on the activity of ERO1 and/or thiol isomerases.
A study performed in yeast revealed that oxidative protein folding alters intracellular levels of GSSG but not GSH and thus influences the GSSG/GSH ratio (35). Increased expression of Pdip and Ero1p together reduces the cytosol redox state, whereas Pdip expression alone supports oxidation. Thus, maintaining the appropriate redox state in intracellular organelles can be a complex issue. Overall, oxidative protein folding and ER stress have an impact on the cytosolic redox balance and may be a key factor in the pathogenesis of protein folding-related diseases.
Pathological Roles of ERO1α and PDI
Neurodegenerative diseases
The accumulation of misfolded proteins triggers the UPR and induces ER stress (130). Of particular note, the accumulation and ultimately the aggregation of amyloid β, tau, and α-synuclein together with oxidative stress induced by ROS have been associated with the development of several neurodegenerative diseases (67, 151). Among the most common of these neurodegenerative diseases is Alzheimer's disease (AD), which can have a profound impact on memory and cognitive judgment (151). Similarly, Parkinson's disease (PD) impairs mobility and mental ability (67) and is a major cause of morbidity and mortality in the elderly. Figure 3 illustrates the roles of PDI and ERO1α in the pathogenesis of neurodegenerative diseases.
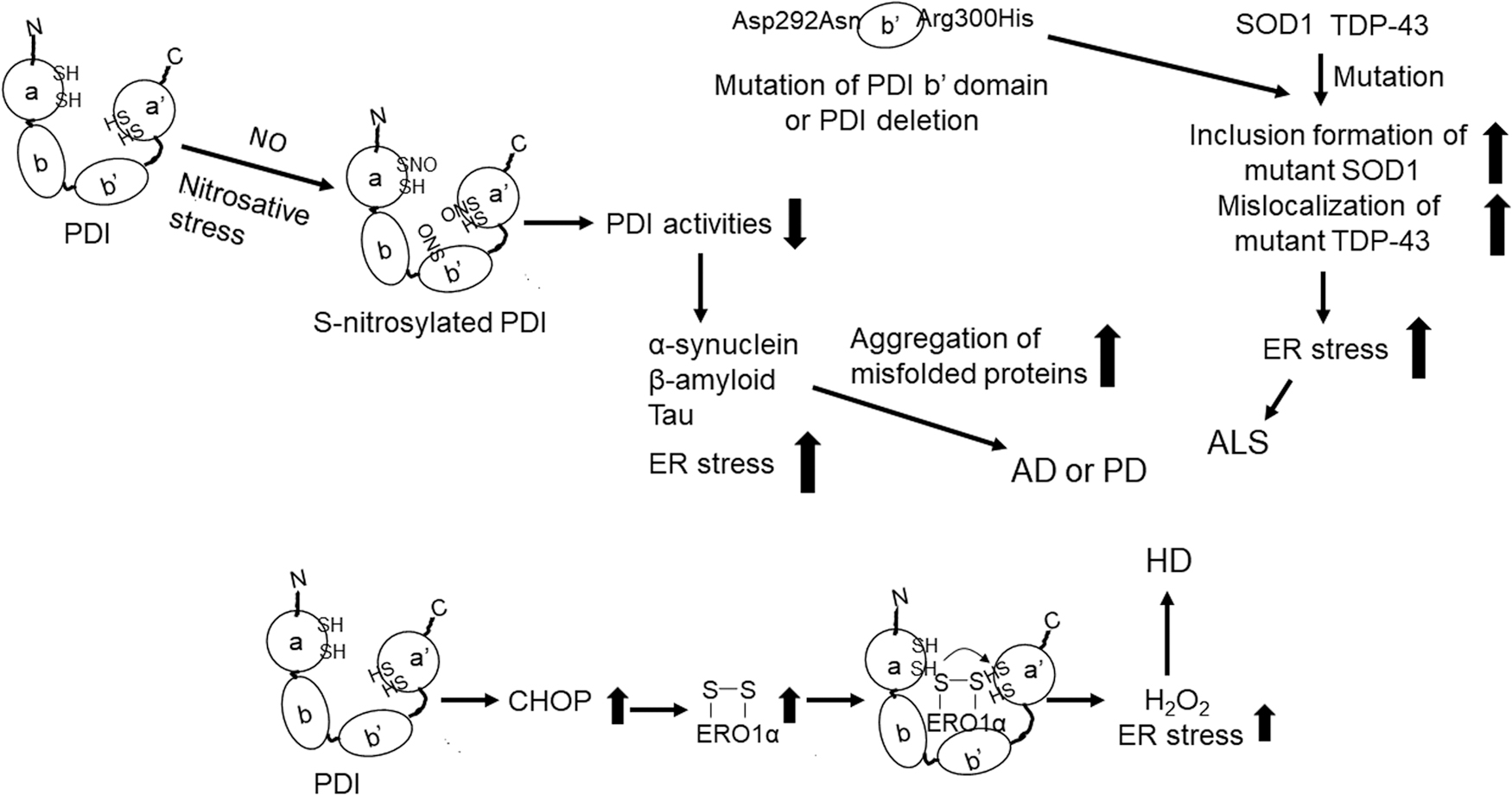
PDI has been found in S-nitrosylated form in the brains of patients with sporadic AD or PD (175). S-nitrosylation of PDI impairs both its isomerase and chaperone activities and thus induces protein misfolding and ER stress. The binding of PDI to tau protein prevents tau misfolding and fibrillization (198). One recent report documented that the chaperone activity of PDI inhibits the phosphorylation and abnormal aggregation of tau and thus protects cells from mitochondrial damage and tau-mediated cytotoxicity (182). PDI is recruited to tissues by tau protein in liquid droplets, inhibiting phase separation and stress granule formation at these sites. By contrast, PDI that is S-nitrosylated at Cys312 in the b′ domain is unable to recognize and undergo recruitment in response to tau protein (182). Likewise, intra-hippocampal injection of fibrillar amyloid β in rats increases the production of nitric oxide (NO) and decreases PDI activity; these responses result in the accumulation of misfolded protein in the ER lumen and ER stress (75).
Amyotrophic lateral sclerosis (ALS) is a disease in which degeneration of motor neurons in the brain and spinal cord leads to muscle weakness and paralysis (143). Many gene mutations have been associated with the familial form of ALS, including mutations in SOD1 (cytosolic superoxide dismutase 1) and TARDBP (TAR DNA-binding protein 43 [TDP-43]) (107). Similar to other neurodegenerative diseases, protein misfolding and inclusion formation are among the hallmarks of ALS.
Mutant SOD1 and TDP-43 proteins form aberrant, non-native disulfide bonds and ultimately undergo aggregation (21, 33). Alterations in nuclear localization or export of TDP-43 result in protein aggregation and recapitulate the biochemical profile of pathological TDP-43-associated findings in ALS (191). The findings from a recent study demonstrated that the isomerase activity of PDI protects neuronal cells from inclusion formation, protein unfolding, and aberrant cytoplasmic localization of mutant forms of TDP-43, as well as ER stress induced by mutant TDP-43 or SOD1 and ER-Golgi transport dysfunction (127).
Of particular note, both oxidoreductase and chaperone activities of PDI protect against apoptosis in neuronal cells that express mutant forms of TDP-43 or SOD1. Further, Asp292Asn and Arg300His mutations in the b′ domain of PDI have been linked to ALS; the expression of these PDI variants in zebrafish results in the disruption of motor neuron connectivity and impairs dendrite outgrowth (193).
Overexpression of PDI in the motor neuron-like cell line, NSC-34, results in reduced aggregation and inclusion formation due to mutant SOD1 as well as reductions in ER stress, whereas knockdown of PDI promotes mutant SOD1 inclusion formation (179). Similar to findings reported in the brains of patients with AD and PD, S-nitrosylated PDI has been detected in spinal cord tissues of patients with ALS as well as in transgenic mice that express the SOD1 mutation Gly93Ala (26). Collectively, these findings suggest that PDI protects against ER stress and protein misfolding in patients with AD, PD, or ALS and that this function is impaired by S-nitrosylation.
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by the degeneration and loss of neurons in the striatum (178). This genetic disorder is caused by the expansion of cytosine-adenine-guanine repeats within the huntingtin gene. This genetic aberration leads to the misfolding and aggregation of huntingtin protein and apoptotic cell death in the striatum and cortex. Currently, there is no effective therapy available to prevent or slow the progression of this disease.
A recent report documented upregulated expression of both PDI and ERp57 in the brains of patients with HD and a mouse model of HD; inhibition of PDI in this mouse model suppressed ER stress in the brain and results in improved survival (213). These results suggest that PDI might be a therapeutic target for the treatment of HD patients. Although it is not yet clear how PDI contributes to the pathology of HD, one group has suggested that this may result from the upregulation of CCAAT/enhancer-binding protein (C/EBP)-homologous protein (CHOP), which is an ER stress protein that modulates ERO1α expression after activation (93).
As the ERO1α-PDI redox cycle produces H2O2, accumulation of mutant huntingtin protein may lead to overactivation of the ERO1α-PDI cycle and the overproduction of H2O2, leading to ER stress in cells. Therefore, unlike its protective role vis-à-vis the pathogenesis of AD and ALS, PDI may aggravate the pathology associated with HD. It is interesting to note that PDI inhibitors were capable of attenuating ER stress, huntingtin-associated toxicity, and motor dysfunction in both in vitro and preclinical studies (55, 72, 213). The results of these studies will be discussed later in this review.
No published studies have focused on the contributions of ERO1α to the pathogenesis of neurodegenerative diseases. One study showed that inhibition of ERO1α or PDI reduces neurotoxicity and accumulation of α-synuclein in the ER of the human SH-SY5Y neuroblastoma cell line and improves cell survival after treatment with the PD-associated neurotoxin, 1-methyl-4-phenylpyridinium (92). However, it is not clear whether ERO1α modulates α-synuclein accumulation directly or indirectly via its actions on PDI. Future studies aimed at identifying the specific function of PDI and ERO1α in neurodegenerative diseases may provide insights into potential therapeutic inventions that address the mechanisms underlying ER stress and the UPR.
Cancer
Cancer is the second leading cause of death worldwide (205). Hypoxia is the most common characteristic of the tumor microenvironment. Hypoxia plays a critical role in the initiation and progression of solid tumors and leads to resistance to radiation and chemotherapeutic interventions (64). Numerous proteins, including growth factors, extracellular matrix proteins, and proteases, are involved in promoting tumor growth. Since O2 is required for disulfide bond formation, hypoxia significantly compromises this process. There is now substantial evidence for ERO1α and PDI overexpression in numerous cancers; these enzymes constitute a key pathway for oxidative protein folding in the disease state. Figure 4 summarizes our current knowledge regarding the roles of PDI and ERO1α in cancer.
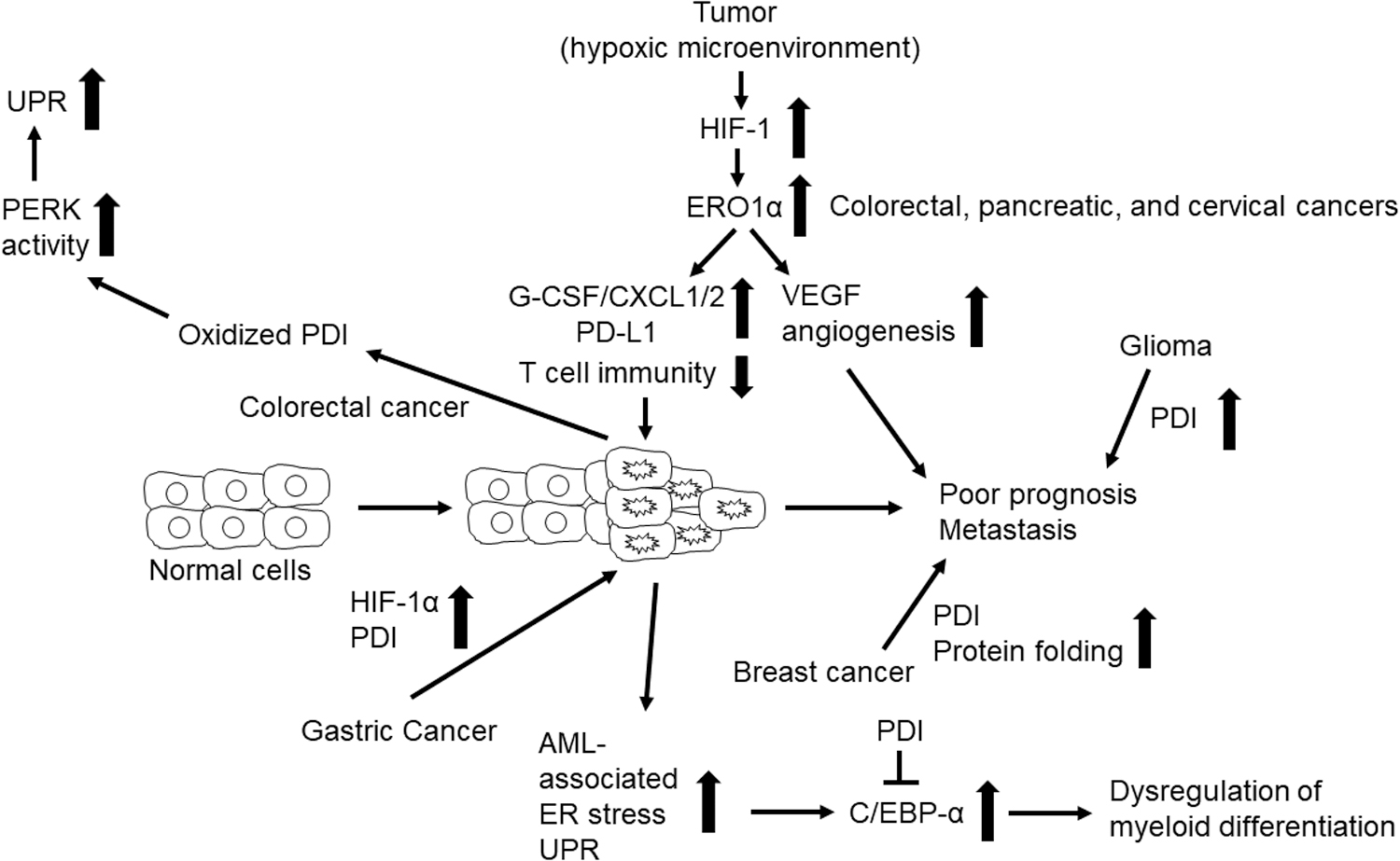
Studies using human cancer cell lines have demonstrated a role for ERO1α as an endogenous marker of hypoxia in the liver, pancreatic, colon, and breast cancer tissues (85, 161). ERO1α was upregulated in the mouse Hepa-1c1c7 hepatocellular carcinoma cell line in response to hypoxia in a manner that was dependent on HIF-1, but not p53 (105). Knockdown of ERO1α reduced the secretion of vascular endothelial growth factor (VEGF) and resulted in cell cycle arrest and apoptosis (105). In another study that featured both cell lines and patient-derived hepatocellular carcinoma tissue, cancer-associated ERO1α was found to promote tumor cell angiogenesis, migration, and invasion via upregulation of VEGF-A expression (200). Collectively, these findings suggest that ERO1α might be an actionable target for the development of strategies to control VEGF-driven angiogenesis and tumor growth.
Proteomic analysis revealed that ERO1α is highly upregulated in pancreatic cancer cells under hypoxia and that it serves as a potential biomarker predicting the survival of patients with pancreatic cancer (49). Kutomi et al. reported that deletion of ERO1α in murine 4T1 breast cancer cells markedly reduces tumor growth and lung metastasis after their injection into mice (86). Importantly, these authors found that patients with ERO1α-positive breast cancer exhibit poorer survival than those who are negative for this marker. These results suggest that ERO1α might be developed as a novel biomarker to predict the survival of patients with breast cancer.
Mechanistically, ERO1α promotes increased production of granulocyte colony-stimulating factor and chemokine CXC motif ligand 1/2 (CXCL1/2) by facilitating the formation of intramolecular disulfide bonds, recruiting polymorphonuclear myeloid-derived suppressor cells and inhibiting T cell-mediated immune responses (163).
Further, ERO1α facilitates oxidative protein folding and enhances the expression of programmed death-ligand 1 (PD-L1) in MDA-MB-231 breast cancer cells, impairing the functional activity of cytotoxic T cells (164). By contrast, other studies revealed that hypoxia-induced expression of ERO1α in a human SW480 colorectal cancer cell line promotes the oxidative folding and expression of MHC class I molecules via the actions of oxidized PDI, increasing their susceptibility to cytotoxic T cells (66, 85). Future studies will be needed to determine whether ERO1α interacts with and alters responses to immunotherapy that are currently in use as treatments for various cancers.
It was reported that PDI plays a role in the pathology of breast cancer (3, 192), gastric cancer (206), gliomas (215), acute myeloid leukemia (AML) (50), and pancreatic adenocarcinoma (43). The growth and ongoing survival of rapidly growing, highly invasive metastatic breast cancer cells would most likely require more active oxidative protein folding machinery than that found in nonmetastatic cells. One recent study revealed that PDI is overexpressed in nonadherent breast cancer cells and that PDI knockdown inhibits anchorage-independent cell proliferation and mammosphere growth (192). These results suggest that cancer-associated PDI may contribute to breast cancer cell metastasis.
Bioinformatics and biochemical studies revealed that PDI is a target gene of HIF-1α and that both proteins are upregulated in human gastric cancer cell lines (206). Knockdown of HIF-1α impairs invasion and metastasis of gastric cancer cells, an effect that was partially reversed by overexpression of PDI. Although the detailed underlying mechanism remains to be determined, these findings suggest the importance of the HIF-1α-PDI pathway in promoting gastric cancer metastasis.
Analyses of Gene Expression Omnibus and the Human Protein Atlas showed upregulated expression of PDI in gliomas; elevated PDI expression has been recognized as a novel predictor for poor prognosis in patients diagnosed with glioma (215). Inhibition of cell surface PDI with an anti-PDI antibody blocks the adhesive function of glioma cells (48). Collectively, these results suggest that PDI might be a useful prognostic biomarker and therapeutic target for gliomas. Further, a recent study demonstrated that ERO1α expression is upregulated in human cervical cancer and correlates with the grade of malignancy and poor prognosis and that blocking ERO1α-PDI signaling by mutating the Val101 residue in ERO1α reduces the migration, invasion, and growth of a human cervical carcinoma cell line (HeLa) (209).
Many cancers are associated with increased ER stress and activation of the UPR, which have a significant effect on protein expression (155). The transcription factor C/EBP-α plays a crucial role in the production of granulocyte/monocyte progenitors from common myeloid progenitors (145). Dysregulation of C/EBP-α function is a common finding in AML (145). PDI interacts with calreticulin binding to the stem-loop region of the C/EBP-α mRNA and blocks C/EBP-α translation (50). Further, other studies revealed that PDI expression is enhanced in cells from patients with AML on activation of the UPR. Collectively, these findings suggest that PDI suppresses C/EBP-α function and perturbs myeloid differentiation in AML.
Activation of protein kinase R-like endoplasmic reticulum kinase (PERK) is a critical step in the initiation of the UPR (144). A study using the HCT116 human colorectal carcinoma cell line suggested that oxidized PDI acts to activate PERK and that deletion or inhibition of PDI reduces PERK-mediated signaling and sensitizes cancer cells to hypoxia and ER stress (84). These results provide evidence suggesting that PDI might be a therapeutic target for the treatment of patients with colon cancer. Overall, ERO1α and PDI expression is upregulated by tumor hypoxia, and these enzymes function to promote tumor growth and metastasis.
Cardiovascular diseases
The CVDs are a leading cause of death and contribute to substantial disability worldwide (1, 65). In addition to inherited genetic risk factors, hypoxia, cell death, and ongoing activation of intravascular cells and the coagulation cascade are crucial for the initiation and progression of CVDs (59, 153, 157, 166).
Hypoxia activates the UPR in cardiomyocytes and induces apoptosis (167). Figure 5 summarizes the roles of ERO1α and PDI in myocardial infarction. A study using the ERO1 inhibitor, QM295, and mice with Ero1-l loss-of-function mutations showed that ERO1α is upregulated in fibroblasts under hypoxic conditions and that the enzyme plays an important role in calcium homeostasis in cardiomyocytes and progressive heart failure (27).
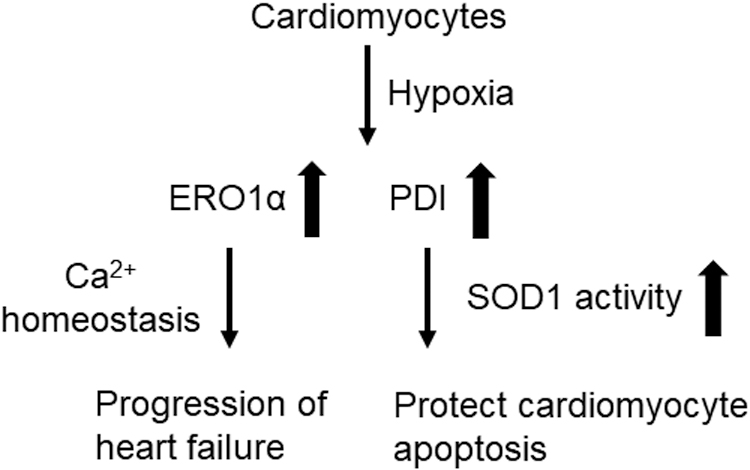
The PDI upregulation was detected in the infarcted area after ligation of the left anterior descending artery (81). The expression of both ERO1 and PDI was enhanced in the murine HL1 cardiomyocyte cell line in response to acute hypoxia. The PDI protects these cells from undergoing apoptosis via an activity-dependent mechanism (153). Postmortem analysis revealed that PDI is a key factor underlying cardiomyocyte survival in patients with ischemic cardiomyopathy (153). Although an injection of adenoviral vectors expressing PDI into heart tissue results in reduced infarct size and cardiac remodeling in a mouse model of myocardial infarction (153), it is not clear whether the expression of ERO1α results in similar effects.
Another study demonstrated that the effects of PDI on preventing cardiomyocyte apoptosis during acute myocardial infarction disappear in patients with diabetes and mice, in which most of the PDI was in reduced form (171). Although the detailed mechanism is unknown, the beneficial effects of PDI may result from the increased activity of SOD1 (172, 185). In contrast to acute hypoxia, chronic hypoxia (3 weeks) before myocardial infarction protects cardiac function in mice (168). This discrepancy may relate to the upregulation of myocardial endothelial cell (EC) PDI, which induces angiogenesis and protects cardiomyocytes against myocardial infarction. These results suggest that each of the known cellular functions of PDI has a distinct role in mitigating the progression of myocardial infarction.
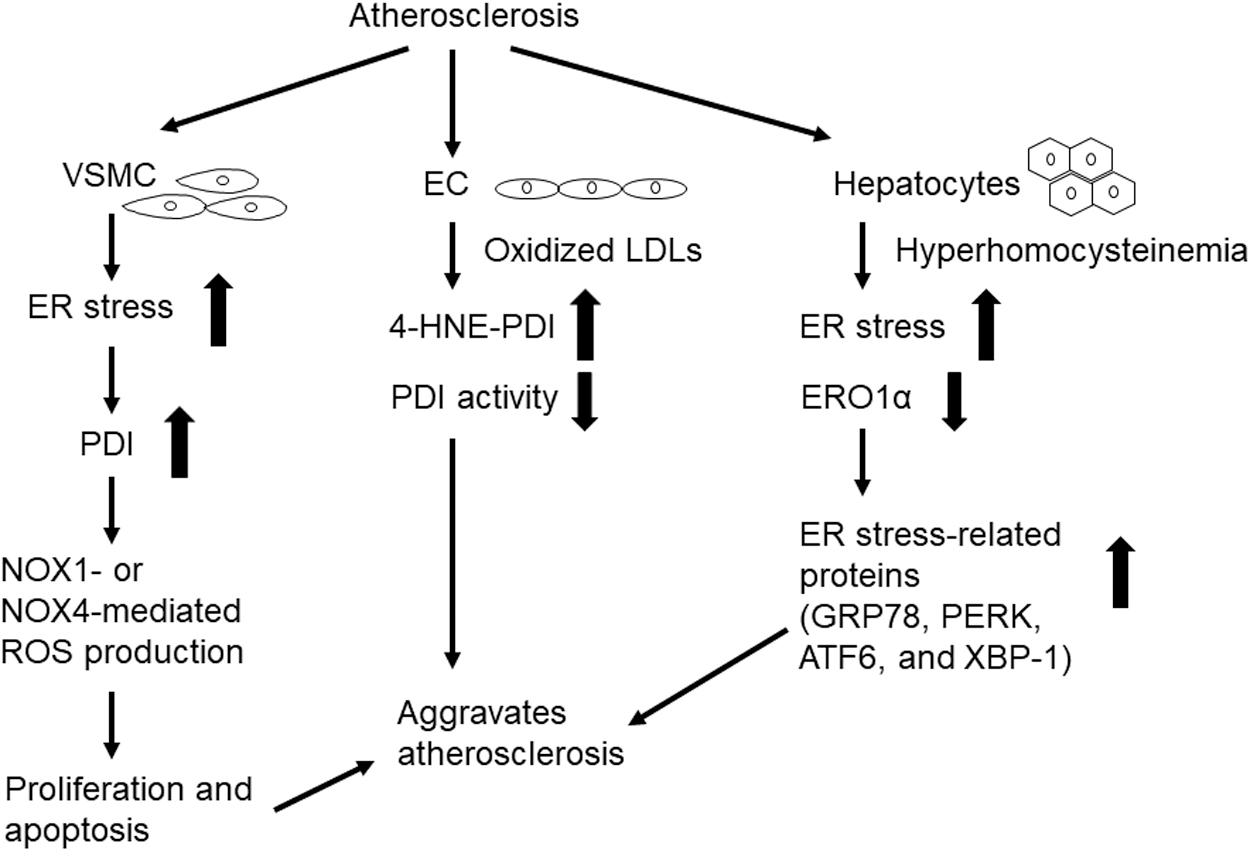
Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries, and it is associated with elevated levels of low-density lipoproteins (LDLs) (99). During the disease process, ER stress induces phenotypic switching and apoptosis of vascular smooth muscle cells (VSMCs) (60, 160) and it promotes foam cell formation, pro-inflammatory cytokine production in macrophages (122), and EC apoptosis (2). Elevated LDL levels and cytokines can further activate ER stress.
Proteomic analysis revealed PDI upregulation in rat aortic VSMCs after stimulation with growth factors (128). The ROS produced via the actions of NADPH oxidase 1 (NOX1) are crucial factors underlying the proliferation and migration of VSMCs in atherosclerosis (187). One study using an siRNA approach suggested that PDI enhances the expression of NOX1 but not NOX4 and ROS production in VSMCs in response to stimulation with platelet-derived growth factor and that PDI positively regulates the activities of both Rac1 and RhoA, thereby promoting VSMC migration (131). Stretch stress and advanced glycosylation end products also upregulate PDI and NOX1 in VSMCs; the PDI produced by this mechanism enhances proliferation and apoptosis of VSMCs, thereby accelerating the atherosclerosis of diabetic vein grafts (133).
One recent paper using biochemical analysis and proximity ligation assays showed that PDI interacts with p47phox through Cys residues found in its two active sites and that PDI interacts with p47phox in thrombin-stimulated VSMCs and the carotid artery after wire-mediated injury in mice (46).
Fernandes et al. used an inducible PDI overexpression system in VSMCs to show that PDI overexpression modulates the expression of both NOX1 and NOX4 (41). At an early time point (24–48 h), PDI overexpression resulted in upregulated expression of NOX1, elevated levels of H2O2, and VSMC migration; however, at a later time point (72–96 h), PDI overexpression resulted in upregulated expression of NOX4 and VSMC differentiation, which was associated with sustained high levels of NOX1. Further, the authors demonstrated that NOX1 expression is downregulated whereas NOX4 expression is enhanced in the carotid arteries of PDI-overexpressing transgenic mice. This result suggests that PDI can regulate both NOX1 and NOX4 activity and VSMC function.
Elevation plasma LDLs are strongly associated with the increased risk of atherosclerosis (113). Oxidized LDLs inhibit PDI activity in human microvascular ECs by inducing the formation of 4-hydroxynonenal (4-HNE)-PDI adducts, which were detected in advanced atherosclerotic plaques obtained from patients undergoing carotid endarterectomy (115). However, these results relied on the use of dieosin glutathione disulfide (Di-E-GSSG) in cell-based assays, which raises questions about the specificity of these assays for the assessment of PDI activity.
Treatment of human umbilical vein ECs with oxidized LDLs induces the binding of the RNA-binding protein, heterogeneous nuclear ribonucleoprotein E1, to the PDI 5′UTR, upregulating PDI expression (108). Hyperhomocysteinemia, another risk factor for atherosclerosis (106), increases ER stress in hepatocytes of ApoE KO mice and downregulates both mRNA and protein levels of ERO1α (201). Overexpression of ERO1α in hepatocytes reduces the expressions of ER stress-related proteins, including GRP78, PERK, activating transcription factor 6 (ATF6), and X-box binding protein 1 (XBP-1). These results imply that ERO1α limits the impact of hyperhomocysteinemia-induced ER stress during atherosclerosis. Figure 6 presents the roles of PDI and ERO1α in the pathology of atherosclerosis.
Ischemia is an underlying factor in ∼80% of all strokes. Ischemia results from occlusion of the cerebral artery, which blocks blood flow and O2 delivery to the brain, leading to the damage and death of brain cells (156). Figure 7 summarizes the role of PDI and ERO1α in ischemic stroke. One study using a rat model of ischemic stroke showed that both CHOP and ERO1α are upregulated in the hippocampus in response to ischemic injury (135). Although CHOP regulates ERO1α expression in ER-stressed cells (93), hypothermia results in downregulated CHOP expression and augmented ERO1α expression in hypoxic cells. These results suggest that HIF-1α may induce the upregulation of ERO1α. PDI expression is also enhanced in rat primary astrocytes in response to hypoxia (2% O2 for 48 h) and subsequent reoxygenation (20% O2 for 48 h) (162).
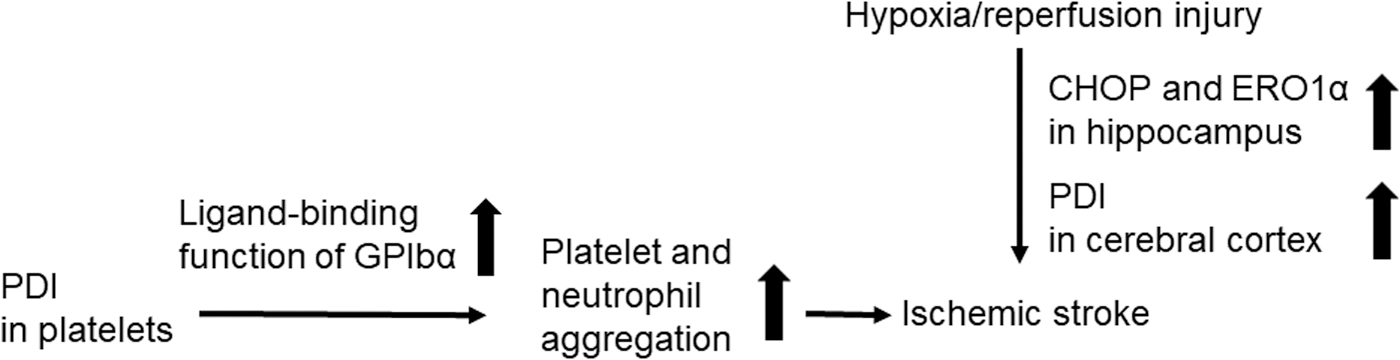
Another study revealed that PDI is upregulated in the cerebral cortex of the ischemic brain and that its oxidoreductase activity is a critical factor in promoting protection against hypoxia-induced cell death (162). Although the molecular mechanism remains elusive, the cytoprotective effects of p-hydroxy benzyl alcohol and tanshinone IIA in ischemic stroke may be derived from their capacity to upregulate PDI expression in brain tissue (68, 190).
Using megakaryocyte-specific Pdi CKO mice, we demonstrated that platelet-derived PDI promotes platelet-neutrophil aggregation and contributes to the pathogenesis of ischemic stroke (97). Platelet–neutrophil interactions are mainly mediated by the binding of P-selectin and GPIbα, which are found on the platelet surface to neutrophil P-selectin glycoprotein ligand-1 and αMβ2 integrin, respectively (94, 95).
Adherent platelet–neutrophil aggregates can form thrombi in the inflamed endothelium, which will ultimately result in microvessel occlusion (79, 96). We found that PDI cleaves two allosteric disulfide bonds in GPIbα, thereby enhancing its ligand-binding function. Importantly, although the inhibition of extracellular PDI or platelet GPIbα results in decreased infarct volume and protects against the development of neurological deficits in a mouse model of ischemic stroke, no additive effects were observed (97). These results suggest that a specific blockade of PDI-GPIbα signaling might be an attractive strategy for the treatment of thromboinflammatory diseases.
Platelets adhere to extracellular matrix proteins, such as collagen and von Willebrand factor, at the site of arterial injury where they undergo receptor-mediated activation and aggregation (120). Although this process is essential for hemostasis, excessive accumulation of platelets will result in thrombosis and vascular occlusion. Since current antiplatelet therapies that target signaling molecules or receptor–ligand interactions increase the risk of major bleeding (13), many efforts have been made to develop potent but safer antithrombotic agents that do not impair hemostasis.
Modification of thiol-disulfide bonds identified in platelet receptors induces conformational changes and modulates their ligand-binding activity (22). Extracellular PDI has been identified as a target for novel treatments of thrombotic disease. Figure 8 illustrates the role of extracellular PDI in the pathology of thrombosis. Several studies that used thiol-reacting agents and PDI inhibitors suggested that platelet-derived PDI enhances platelet adhesive function (39, 40, 88, 89).
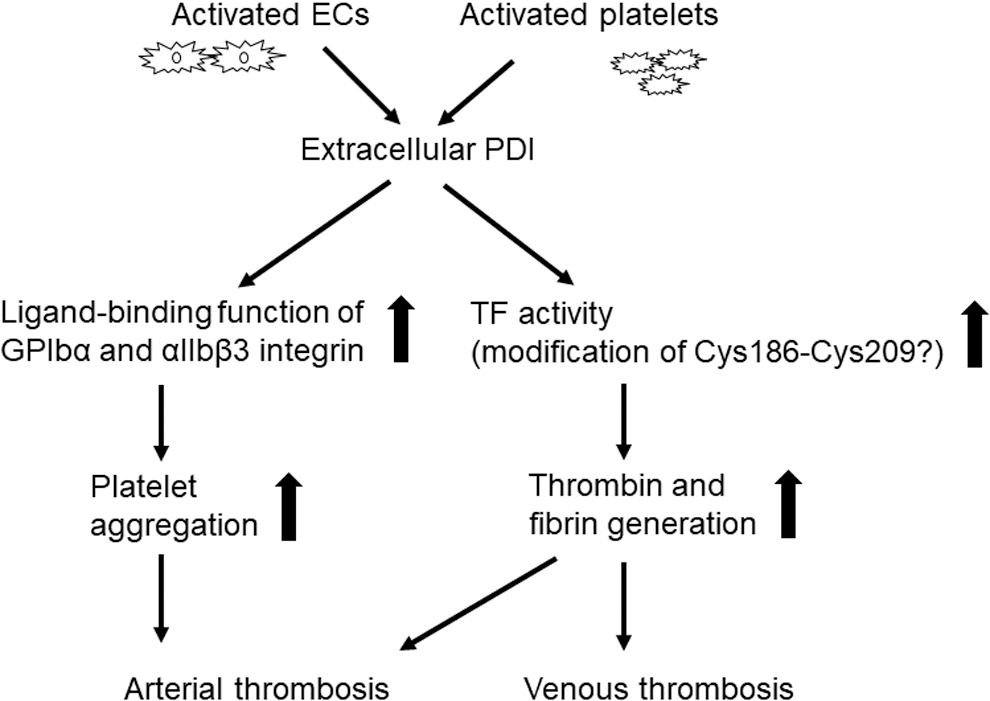
Circulating PDI detected on the surface of platelet-derived microparticles also promotes aggregation (140). Reinhardt et al. demonstrated that extracellular PDI plays a crucial role in regulating the activity of tissue factor (TF) and fibrin generation in a mouse model of FeCl3-induced arterial thrombosis (141). Further, the inhibition of extracellular PDI with a blocking antibody perturbs arterial thrombus formation and prolongs bleeding times at the site of vascular injury (30), suggesting that extracellular PDI plays an important role in both thrombosis and hemostasis.
Studies using megakaryocyte-specific Pdi CKO and β3 KO mice demonstrated that platelet-derived PDI binds directly to the αIIbβ3 integrin and is required for its full activation (31, 78, 212). This interaction promoted platelet accumulation, with only a minimal effect on fibrin generation at the site of vascular injury. Although EC-specific Pdi CKO mice have not yet been generated, one study using an αIIbβ3 antagonist suggested that EC PDI contributes to the generation of fibrin (62).
The EC microparticles isolated from diabetic mice contain PDI, and the treatment of mouse platelets with the microparticles activates the αIIbβ3 integrin (137). Further, EC-derived PDI exposes an ArgGlyAsp sequence in thrombospondin-1, thereby promoting its binding to αvβ3 integrin (56), which may also contribute to thrombosis.
An in vitro study suggested that platelet ERO1α may regulate the function of PDI and αIIbβ3 and platelet aggregation (159). However, detailed molecular mechanisms and the function of ERO1α in thrombosis in vivo remain elusive.
Venous thromboembolism is the third most common CVD. This condition is the direct result of blood stasis, hypercoagulability, and vascular wall injury (189). TF-initiated coagulation is a critical feature of this disease process. Mass spectrometric analysis using purified TF and PDI revealed that the Cys209 residue in TF is constitutively conjugated with GSH and that PDI deglutathionylates TF and catalyzes oxidation of the Cys186-Cys209 disulfide bond (141). In contrast, PDI is found to enhance the pro-coagulant activity of TF detected on EC-derived microparticles via a mechanism that is independent of its oxidoreductase activity (177). These results imply that both oxidoreductase and chaperone activities of PDI may be required to regulate TF activity.
Another study revealed that ATP-induced P2X7 receptor signaling induces the release of TF-positive microparticles via thiol- and PDI-dependent mechanisms (44). Of the two blocking anti-PDI antibodies (RL90 and BD34), only RL90 effectively reduced the release of these microparticles. Another study revealed that the inhibition of extracellular PDI with high concentrations of RL90 (50 μg/mL) or quercetin-3-rutinoside (rutin; 100 μM) interferes with the pro-coagulant activity of TF in the human THP-1 monocytic cell line treated with antithymocyte globulin (91). However, since RL90 and rutin inhibit the activity of both PDI and ERp57 (197, 212), these results may implicate the contribution of ERp57 to microparticle release and TF activity.
The TF activation in myelomonocytic cells induced by antiphospholipid antibodies is inhibited by treatment with the PDI inhibitor, 16F16, or by blocking TF-PDI binding (116). A study featuring a rat model of venous thrombosis documented upregulation and colocalization of PDI and TF in leukocytes and ECs in the inferior vena cava (211). Similarly, Subramaniam et al. demonstrated that blocking PDI with PACMA 31 attenuates platelet accumulation and TF-dependent fibrin formation in a mouse model involving partial stenosis of the inferior vena cava (158). Although the aforementioned studies provide evidence for PDI-mediated modulation of TF activity, several published reports have challenged this concept (8, 83, 129).
Diabetes
Diabetes is a metabolic disease that results in high blood sugar. Glucose homeostasis is tightly regulated by insulin, which is a hormone secreted from β cells found in pancreatic islets (52). The production and secretion of insulin depend on the synthesis and appropriate folding of its proinsulin precursor in the ER. Chronic elevation in proinsulin synthesis results in the accumulation of misfolded protein due to incorrect pairing of the intramolecular disulfide bonds (101). Increased ER stress and UPR result in the dysfunction and apoptosis of β cells, which can cause diabetes. There are two main subtypes of this disorder. Type 1 diabetes results from a defect in insulin secretion, and type 2 diabetes is characterized by insulin resistance and defective insulin secretion (149). Figure 9 illustrates the role of PDI and ERO1α in diabetes.
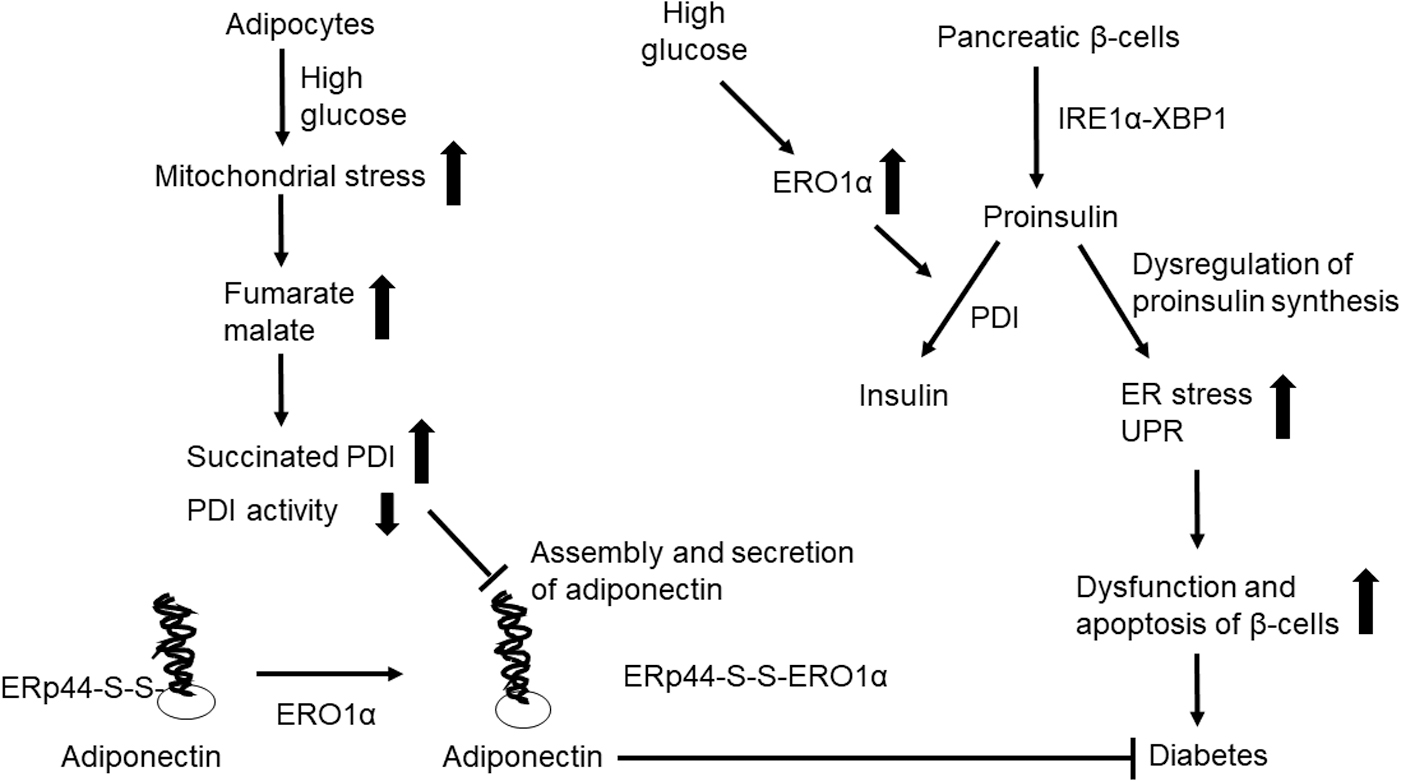
Higher levels of PDI-bearing platelet microparticles are detected in patients with diabetes compared with healthy control subjects (140). One study revealed that both the redox states and enzymatic activities of PDI and ERO1α are altered in liver microsomes isolated from streptozotocin-treated rats (117). Proteomic analysis revealed that insulin- and TNF-α-induced oxidative stress in differentiated 3T3-L1 adipocyte-like cells induces PDI upregulation (25). Overexpression of wild-type but not an activity-null mutant form of ERO1α prevented proinsulin misfolding and reduced ER stress; co-expression of PDI impaired the beneficial effects observed in response to ERO1α overexpression (196).
As ERO1β is the dominant isoform expressed in pancreatic β cells (7, 36), Zito et al. generated homozygous Ero1β mutant mice and demonstrated a key role for this protein in oxidative folding of proinsulin and glucose tolerance (214). ERO1β is upregulated in mouse islets treated with high glucose, and pancreatic and duodenal homeobox 1 promote transcriptional regulation of ERO1-Lβ expression, thereby facilitating oxidative protein folding (77). The knockdown of ERO1β reduces insulin content and secretion in the mouse MIN6 insulinoma cell line and promotes tunicamycin-induced cell death (77). In contrast to ERO1β, knockdown of PDI augments proinsulin folding in the rat INS-1 β-cell line (138). The PDI can interact directly with proinsulin via the formation of a mixed disulfide bond, which results in its retention in the ER.
One study performed in β cell-specific Pdi CKO mice on a high-fat diet demonstrated that PDI is crucial for optimal insulin production and glucose tolerance under conditions of metabolic stress (61). The IRE1α-XBP1 signaling pathway is constitutively activated in pancreatic β cells under homeostatic conditions (174). Tsuchiya et al. (174) showed that β cell-specific deletion of Ire1α results in reduced insulin secretion, decreased insulin and proinsulin content, and downregulated expression of PDI family isomerases, including PDI, ERp5, and ERp44. Taken together, PDI is a downstream effector of IRE1α-XBP1 signaling and it plays a critical role in oxidative folding of proinsulin. Overall, these results suggest that expression and/or activity levels of PDI and ERO1 are altered under diabetic conditions and that these enzymes facilitate appropriate proinsulin folding and insulin secretion and thus are critical regulators of glucose homeostasis.
The plasma level of adiponectin, an antidiabetic and antiatherogenic hormone secreted from adipocytes, is decreased in patients diagnosed with diabetes (194). ERO1α regulates the assembly and secretion of adiponectin by releasing Erp44 thiol-mediated retention (186).
Mitochondrial stress in adipocytes due to high glucose levels results in increased levels of both fumarate and malate in diabetes; this leads to succinylation of the PDI active site Cys residues and impaired assembly and secretion of adiponectin (103). This article also noted that succinylation perturbs the oxidoreductase activity of PDI in adipose tissue of diabetic mice. Restoring glucose levels from high to normal concentrations also restores PDI reductase activity and reduces ER stress in adipocytes. Although it remains to be determined whether PDI regulates adiponectin function, these results suggest that PDI succinylation could be a novel regulatory mechanism by which altered mitochondrial metabolism induces ER stress in adipocytes in patients with diabetes.
The deposition of islet amyloid polypeptide (IAPP) is a common feature of type 2 diabetes (18). Misfolding and aggregation of IAPP lead to β cell dysfunction and death. The administration of adenovirus encoding P4HB to transgenic mice expressing human IAPP specifically in β cells resulted in amelioration of insulin secretion and inhibition of β cell apoptosis by PDI under glucolipotoxic conditions (112).
Inhibitors
ERO1 inhibitors
A high-throughput screening study identified two compounds, EN460 and QM295 as ERO1 inhibitors, both with an IC50 value of 1.9 μM (Fig. 10A, B) (19). Both inhibitors activate the UPR in 293T cells and protect hypersensitive PERK-null fibroblasts against severe ER stress. However, EN460 and QM295 are weak inhibitors when evaluated in cell-based assays, and they also exhibit cellular toxicity, potentially due to poor solubility and cell permeability, nonspecific interactions with free thiols, and/or inhibition of other FAD-binding enzymes.

As ERO1α is upregulated in response to tumor hypoxia, the inhibitors have been tested for their efficacy in numerous cancer models. Among these findings, the treatment of multiple myeloid cells with EN460 reduced cell proliferation and induced apoptosis (54). Treatment with EN460 inhibited the proliferation of pancreatic ductal adenocarcinoma cells in vitro and also impeded tumor growth in vivo (207).
Costanzo et al. evaluated a network of genetic interaction profiles and reported that erodoxin interacts with yeast genes involved in protein folding, glycosylation, and cell wall biosynthesis, and it also inhibits ERO1 activity (Fig. 10C) (34). However, it is unknown whether erodoxin has in vivo efficacy in animal models. Due to the high (65%) amino acid sequence identity shared between ERO1α and ERO1β, no isoform-specific inhibitor has been identified.
PDI inhibitors
Given its contributions to pathology associated with a variety of diseases, PDI has received considerable attention as a promising druggable target. Studies using a high-throughput screen identified rutin and the bepristats as potential PDI inhibitors (Fig. 11A–E) (14, 63). As flavonoid-based antioxidant compounds, both quercetin and rutin are known to inhibit thrombosis (12, 32, 114), inflammation (37, 146), neurodegenerative diseases (38, 102), and cancer (57, 73). Rutin binds to the b′ domain of PDI and exerts an inhibitory effect on platelet aggregation and thrombus formation at the site of arteriolar injury (63, 100). Nevertheless, several studies have addressed the nonspecific effects of rutin at the same concentration as that used to inhibit PDI activity (78, 97, 212).
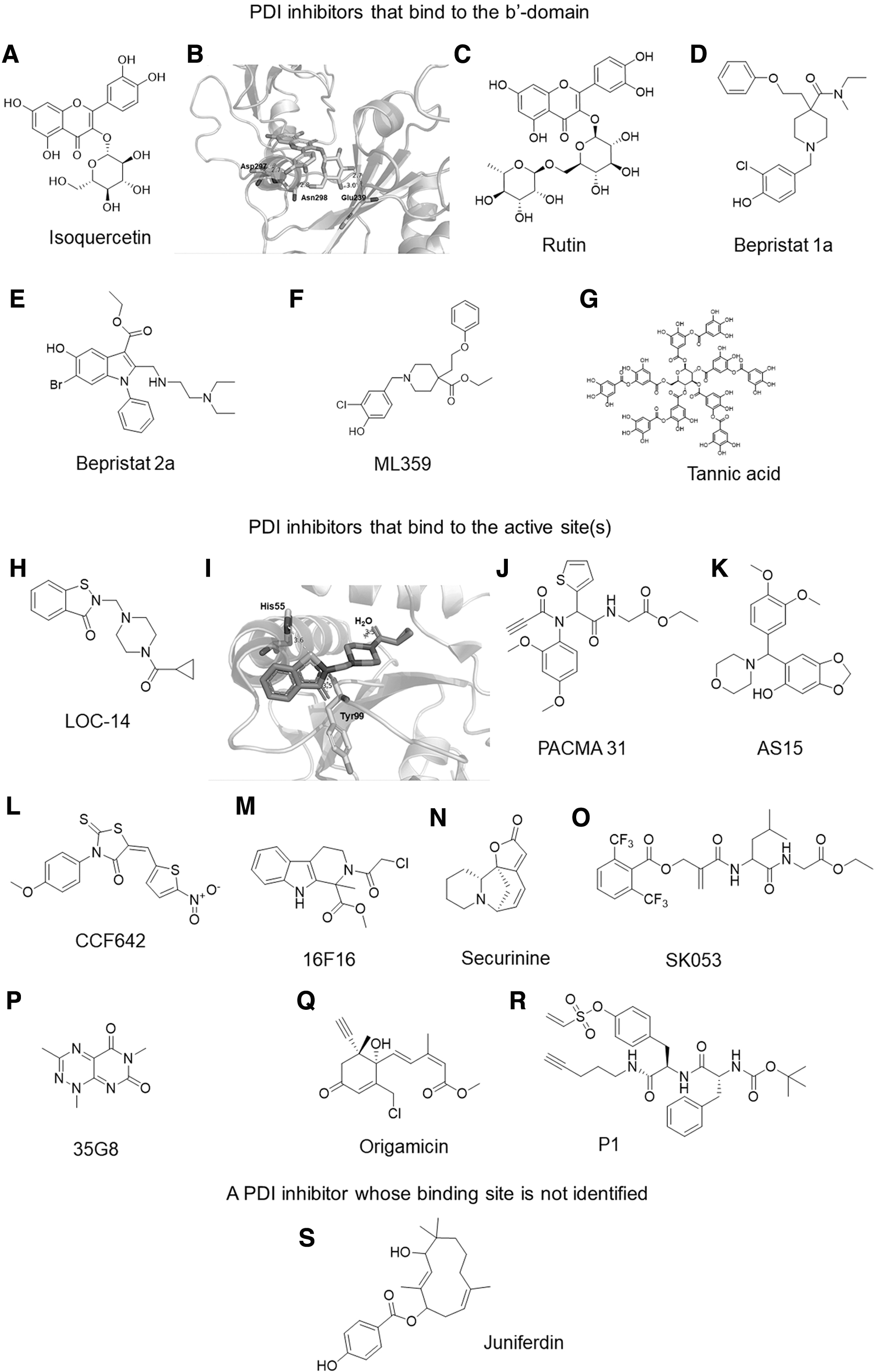
The results of a Phase II clinical trial revealed that quercetin-3-glucoside (isoquercetin) reduces hypercoagulability in patients with advanced cancer (216). Simialr to rutin, isoquercetin interacts with the PDI b′ domain via the formation of hydrogen bonds with Glu239, Asp297, and Asn298 (Fig. 11B). The bepristats also bind to the substrate-binding pocket of the PDI b′ domain, blocking substrate binding and inhibiting platelet aggregation and thrombus formation (15). Although PDI-null platelets exhibit a moderate defect (∼50% inhibition) in agonist-induced aggregation (78, 97, 212), treatment with bepristats abrogates platelet aggregation (14), implicating their off-target effects.
ML359 (Fig. 11F) was identified as a potent PDI inhibitor with an IC50 value of 250 nM, but treatment with 30 μM of ML359 had only a minimal inhibitory effect on platelet aggregation (76). Although its binding site has not been reported, given its structural similarity with bepristat 1a (Fig. 11D), ML359 is likely to bind to the b′ domain.
A recent study using in silico molecular docking reported that tannic acid (Fig. 11G) binds to both active sites and the b′ domain of PDI. In contrast to cell-permeable small-molecule inhibitors, given its molecular mass of 1.7 kDa, tannic acid is likely to inhibit the activity of extracellular but not intracellular PDI (142). When injected intraperitoneally, tannic acid reduced thrombus formation without prolonging bleeding times in vivo. Another study revealed that the hexamer, LysPheTrpTrpPheSer, interacts with the substrate-binding site of PDI and eliminates ERO1α activity (109). This result suggests that ERO1α binding to the substrate-binding pocket in the PDI b′ domain is a critical step that promotes both ERO1α activation and the ERO1α-PDI redox cycle.
Most PDI inhibitors bind to the N- or C-terminal active site or both. As these interactions are typically thiol mediated, inhibition associated with molecules in this category is likely to be irreversible. Among those inhibitors, LOC14 (Fig. 11H) is the most potent PDI inhibitor with a Kd value of 62 nM (71). This compound was reported as binding to reduced PDI only, suggesting that the free thiols in the PDI active site are required for LOC14 binding. Using glide docking, we found that LOC14 fits well within the a domain of reduced PDI and interacts with His55 and Tyr99 (Fig. 11I). The compound is stable in mouse liver microsomes; blood plasma undergoes low rates of clearance while within microsomes, exhibits low binding affinity to plasma proteins, and penetrates the blood–brain barrier (71). Treatment with LOC14 improves the viability of PC12 cells expressing the mutant huntingtin protein (71) and suppresses ER stress in a mouse model of HD (213).
PACMA 31 (Fig. 11J) was identified as an orally available PDI inhibitor (199). Since PDI is overexpressed in ovarian tumors, it has been associated with poor prognosis in patients with ovarian cancer (148). PACMA 31 covalently binds to the Cys397 and Cys400 residues in the PDI a′ domain and irreversibly inhibits its activity with an IC50 value of 10 μM (199). This compound is cytotoxic and can suppress the growth of multiresistant ovarian cancer cells. PACMA 31 also exhibited a significant inhibitory effect on human ovarian cancer cell growth in a mouse xenograft model while displaying no substantial toxicity to normal tissues (199). Further, in vitro and in vivo studies revealed the beneficial effects of PACMA 31 in multiple myeloma (176), hepatocellular carcinoma (195), and breast cancer (136).
It is recently reported that similar to PACMA 31, the irreversible PDI inhibitor, AS15 (IC50 value of 300 nM; Fig. 11K), forms a covalent bond to the C-terminal active site of PDI (154). AS15 and its derivatives synergistically inhibit the growth of human glioblastoma cells (A-172 and U-118 MG lines) when treated in combination with a GSH synthesis inhibitor.
Unbiased screening experiments identified CCF642 (Fig. 11L) as a PDI inhibitor (176). Computational modeling suggested that the inhibitory effect is based on covalent binding between the carbonyl group of CCF642 and the Lys residue found at the two active sites of PDI (176). Compared with bortezomib, which is a U.S. Food and Drug Administration-approved drug for myeloma, CCF642 exhibits similar inhibitory effects on multiple myeloma in studies carried out in vivo. Another study showed that CCF642 reduces the expression of ER stress markers and neuronal apoptosis in a mouse model of experimental autoimmune encephalomyelitis, resulting in neuroprotection (69).
Hoffstrom et al. identified the compound 16F16 (Fig. 11M) as an irreversible inhibitor for both PDI and ERp57 (55). PDI accumulates at mitochondria-associated ER membranes in PC12 cells that express polyglutamine-expanded huntingtin exon 1; treatment with 16F16 suppressed apoptosis of the PC12 cells (55), suggesting mechanisms underlying PDI-mediated protection against apoptotic cell death in HD. In addition, 16F16 inhibits the migration of breast cancer cells (136) and the replication of influenza viruses (80). However, this inhibitor did not affect the pro-coagulant activity of TF (204). Although the sites of the 16F16-PDI interaction remain unclear, cystamine, which is a simple organic disulfide, competes with 16F16A for binding to PDI (55), suggesting that 16F16 is likely to bind to the active site of PDI.
One recent study in which the reactive group in 16F16 was replaced with an NO donor suggested that 16F16-NO S-nitrosylates PDI by releasing NO, resulting in the reversible inhibition of PDI activity (98). Securinine (Fig. 11N) was identified as another irreversible PDI inhibitor (72). Similar to the effects of phenylarsine oxide, which is a compound that reacts with vicinal thiols, securinine binds irreversibly to the N-terminal active site of PDI. Treatment with securinine results in improved viability of PC12 cells that express the mutant form of huntingtin (72). In addition, securinine inhibits SH-SY5Y cell viability with an IC50 value of 37 μM (123).
Several other PDI inhibitors have been characterized. SK053 (Fig. 11O) is an inhibitor of the thioredoxin–thioredoxin reductase system, and it also binds to the C-terminal active site of PDI and inhibits its enzymatic activity with an IC50 value of 10 μM (28, 82). Treatment with SK053 results in elevated C/EBP-α levels in the human HL-60 promyelocytic leukemia cell line and impairs cell growth and differentiation (28). A computational docking study revealed that compound 35G8 (Fig. 11P) binds to Cys397 in the C-terminal active site of PDI (87). This compound inhibits PDI activity with an IC50 value of 170 nM and is cytotoxic to glioblastoma cell lines at concentrations of 1–5 μM.
Origamicin (Fig. 11Q) was also identified as a PDI inhibitor that covalently binds to Cys residues in the active sites (124). Microarray analysis revealed that treatment with origamicin alters the gene expression profiles of both SH-SY5Y and Huh7 cells infected with the hepatitis C virus (124, 125). Origamicin inhibits hepatitis C virus replication and modulates the viability of SH-SY5Y cells at concentrations of 25–50 μM. Screening of the human MCF-7 breast cancer cell line identified a phenyl vinyl sulfonate-containing small compound (P1) (Fig. 11R) as a cell-permeable PDI inhibitor with an IC50 value of 1.7 μM, although it cross-reacts with both ERp72 and ERp5 (45). Treatment with P1 inhibits the proliferation of various cancer cells. The phenyl vinyl sulfonate group of P1 probably binds to Cys 397 in the C-terminal active site of PDI (45).
Juniferdin (Fig. 11S) is a natural compound that inhibits PDI reductase activity with an IC50 value of 156 nM and has no effect on the activities of ERp57 and ERp72 (74). The binding site in PDI has not been reported. Juniferdin perturbs PDI-catalyzed reduction of the human immunodeficiency virus type 1 protein, gp120, which is crucial for virus entry into cells (74, 147). Treatment with juniferdin also reduces the rates of intracellular replication of influenza A and B viruses (80). Further studies are required to determine whether this inhibitor interferes with other viral infections.
Concluding Remarks
ERO1 and PDI are upregulated in response to hypoxia and the ER stress-activated UPR that has been associated with numerous diseases. In particular, ERO1α has an emerging role in the hypoxic tumor microenvironment. Along with its critical function in the ER, PDI released from cells in the peripheral vasculature also regulates cell adhesive functions associated with thrombosis and inflammation.
Although the ERO1α-PDI redox cycle facilitates oxidative protein folding and disulfide bond modification, the target molecules and regulatory mechanisms might vary between individual cell types. Thus, future studies will be required to determine the answers to additional remaining questions. Among these issues, although absolute PDI deficiency would be lethal in humans, PDI that has undergone posttranslational modification (e.g., S-nitrosylation) has been found in patients diagnosed with neurodegenerative diseases. Therefore, it will be important to determine which posttranslational modifications or mutations occur in PDI and ERO1 under specific disease conditions.
Clinical and research data associated with altered expression or mutation of PDI and ERO1-L genes will greatly advance our understanding of their pathophysiological roles. Likewise, although most of the protein is retained in the ER, a small fraction of the total PDI is found in other organelles. The role and function of PDI detected outside the ER should be elucidated. Upregulated expression of both PDI and ERO1 might lead to their release under specific disease conditions. It would be important to understand whether and how PDI and ERO1 are released from cells as well as their individual roles in specific disease states.
Finally, as described earlier, PDI and ERO1 synthesized by and released from individual cell types might subserve unique functions in health and disease. The molecular and cellular mechanisms underlying the contributions of ERO1α and PDI to the initiation and progression of various diseases remain to be determined. Both specific inhibitors and tissue-specific CKO mice will be critical resources to answer the aforementioned questions.
There are several issues related to the identification and development of ERO1α and PDI inhibitors that remain of concern. First, due to the high amino acid sequence identity and the need to target the FAD-binding pocket, it may be challenging to identify ERO1α-specific inhibitors. Similarly, the selectivity and physicochemical properties (e.g., membrane-permeability, solubility, and others) of most PDI inhibitors have not been fully evaluated. For example, given that extracellular PDI activity is a critical feature contributing to the pathology of thrombotic diseases, cell-permeable inhibitors should be evaluated with caution as they might impair the indispensable function of PDI in the ER.
PDI is upregulated in areas of myocardial infarction in mouse models of left anterior descending artery ligation. The expression of ERO1 and PDI is enhanced by acute hypoxia in cells of the murine HL1 cardiomyocyte line, and PDI plays a protective role in limiting apoptosis via its activity-dependent mechanism (150). Further, postmortem analysis revealed that PDI is a key factor promoting cardiomyocyte survival in patients with ischemic cardiomyopathy.
Second, many PDI inhibitors are capable of forming covalent bonds with Cys residues in the active sites, thereby enabling them to inhibit PDI activity irreversibly. Since PDI inhibitors are likely to be administered for long periods of time, any adverse effects associated with these compounds should be carefully examined. Third, the administration of specific inhibitors of either PDI or ERO1α may not exhibit potent inhibitory effects in vivo, as other isoforms may compensate for diminished function. Thus, it would be interesting to test the combined effects of both sets of inhibitors in diseases such as cancer in which both PDI and ERO1α are upregulated.
Footnotes
Authors' Contributions
V.J., T.K., V.M., and J-K.M. wrote a draft; Z.A. and K.L.O. provided important comments; and J.C. wrote the article.
Author Disclosure Statement
Z.A. and K.L.O. are employees of Cayman Chemical Company Inc. The other authors declare no competing financial interests.
Funding Information
This work was supported by grants from the National Institutes of Health (HL153047, HL130028, HL148280, HL146559, and HL154107 to J.C.).