
Abstract
Significance:
The significant role of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) in signal transduction is mediated by the production of reactive oxygen species (ROS), especially in the central nervous system (CNS). The pathogenesis of some neurologic and psychiatric diseases is regulated by ROS, acting as a second messenger or pathogen.
Recent Advances:
In the CNS, the involvement of Nox-derived ROS has been implicated in the regulation of multiple signals, including cell survival/apoptosis, neuroinflammation, migration, differentiation, proliferation, and synaptic plasticity, as well as the integrity of the blood/brain barrier. In these processes, the intracellular signals mediated by the members of the Nox family vary among different tissues. The present review illuminates the regions and cellular, subcellular localization of Nox isoforms in the brain, the signal transduction, and the role of NOX enzymes in pathophysiology, respectively.
Critical Issues:
Different signal transduction cascades are coupled to ROS derived from various Nox homologues with varying degrees. Therefore, a critical issue worth noting is the varied role of the homologues of NOX enzymes in different signaling pathways and also they mediate different phenotypes in the diverse pathophysiological condition. This substantiates the effectiveness of selective Nox inhibitors in the CNS.
Future Directions:
Further investigation to elucidate the role of various homologues of NOX enzymes in acute and chronic brain diseases and signaling mechanisms, and the development of more specific NOX inhibitors for the treatment of CNS disease are urgently needed. Antioxid. Redox Signal. 35, 951–973.
Introduction
The pathological disorders of the central nervous system (CNS) include Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), traumatic brain injury (TBI), subarachnoid hemorrhage, and ischemic stroke, as well as psychiatric disorders, including autism spectrum disorder, excited delirium syndrome, and schizophrenia. All of these diseases are found to be associated with neuroinflammation and neurodegeneration.
In the past few decades, a large number of studies involving clinical patient samples and animal models have confirmed the presence of increased oxidative stress in brain diseases. Therefore, treatment strategies for many brain diseases have targeted oxidative stress and used antioxidants, such as vitamin C (244), vitamin E (162), and coenzyme Q10 (4) to scavenge the free radicals in the brain. Unfortunately, almost all of these treatments have failed in clinical trials. Later on, the enzymes involved in producing reactive oxygen species (ROS) attracted the attention of scientists.
The reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Noxes) are professional ROS producers (123), unlike other oxidoreductases, such as cyclooxygenase (118), lipoxygenase (118, 203), cytochrome P450 enzymes (183), xanthine oxidase (20), and mitochondria complex I (122), that produce ROS as by-products. The excessive activation of the NADPH oxidase (Nox) enzymes is well-documented in a variety of brain diseases (50, 60, 76, 147, 189, 204), especially the patient samples manifested overexpression of Noxes (133, 151, 215). Moreover, robust neuroprotective effects have been reported by the use of inhibitors and/or gene deletion of Noxes in a variety of diseases, including multiple neurodegenerative disorders (17, 93, 148, 193, 237), stroke (190, 254), TBI (67, 144), autism (5). Our group also confirmed greater improvement in the mouse model of stroke on the application of combined NADPH and the Nox inhibitor apocynin (187). Inhibition of the expression of proinflammatory proteins, including NLRP3, ASC, caspase-1, interleukin (IL)-1β, and IL-18, is the mechanism associated with the anti-inflammatory and neuroprotective effects. However, the different roles of Noxes in the CNS physiology and pathology, and in the signal transduction pathways engaged in mediating normal and abnormal actions of Noxes are yet to be fully elucidated. The seven members of the Nox family that are identified so far include Nox1, Nox2, Nox3, Nox4, Nox5, and dual oxidases 1 and 2 (DUOX1 and DUOX2) (23). Each isoform performs a varied role in the CNS dysfunction, except for DUOX1 and DUOX2, since DUOX1 and DUOX2 are barely reported in the CNS. In this review, we summarize current research on Nox-mediated modulation of signal transduction in CNS pathophysiology with a focus on Nox1–5 by referring to the literature on the PubMed database. All relevant articles reporting Nox1–5 in the CNS until December 2020, including various neuropsychiatric diseases, excluding peripheral nerve diseases, such as dorsal root ganglion, were taken into consideration for this article.
The Family of Noxes
The oxidative pentose phosphate pathway (PPP) primarily produces NADPH, called reduced coenzyme II, which accounts for 30%–50% of the cytoplasmic NADPH (68). Interestingly, NADPH exhibits bidirectional effects in vivo. On the one hand, participation of NADPH as a hydrogen donor has been documented in a variety of intracellular reductive reactions and ensures reductive capacity for the antioxidant systems to protect cells from oxidative stress (231). Moreover, studies in our laboratory have revealed a protective effect of NADPH in several disease models, including ischemic stroke (131), myocardial ischemia (284), kainic acid-induced excitotoxicity (137, 139), 1-metyl-4-fenyl-1,2,3,6-tetrahydropyridin (MPTP)-induced dopaminergic neurodegeneration, and neuroinflammation (282, 283). On the other hand, NADPH acts as a substrate for NOX enzymes delivering electrons for the production of ROS (17).
Available literature suggests that the multisubunit protein complex, NOX (23), comprises a family of seven major members, namely Nox1, Nox2 (gp91phox), Nox3, Nox4, Nox5, DUOX1, and DUOX2, which are expressed both outside and inside the brain (23). Structurally, all the seven isoforms of NOX enzymes are composed of at least six transmembrane domains and cytosolic binding sites for NADPH and FAD (193). Despite this similarity in structure, each isoform is activated in different ways, which requires association with other proteins as subunits (193). For example, activation of Nox1–3 necessitates the recruitment of multiple proteins to plasma membrane involved in p22phox transmembrane protein along with the cytosolic organizer subunits (p47phox/NoxO1 [Nox organizer 1]) and activator subunits (p67phox/NoxA1 [Nox activator 1] and p40phox), as well as the small GTPase Rac1 or Rac2. Nox4 requires p22phox only for activity, whereas Ca2+ specific binding is directly indispensable for the activation of Nox5, DUOX1, and DUOX2 (237). Accumulation and activation of these protein complexes in the plasma membrane are followed by the transfer of an electron from NADPH to oxygen to form the superoxide extracellularly or intracellularly from vesicles, which can directly indulge in oxidative damage, or react with nitric oxide (NO) to generate peroxynitrite (ONOO−) (23). These radicals are mostly unstable in the cell and are largely converted by superoxide dismutase (SOD) to hydrogen peroxide, which is highly permeable through cell membranes (155). Subsequently, superoxide and hydrogen peroxide act as second messengers and target cysteine residues or oxidize thiols of protein among cellular proteins and lipids, subsequently leading to a variety of downstream effects (228). Under physiological conditions, ROS generated by Nox enzymes function as signaling molecules and thereby contribute to the defense mechanisms against pathogens (202). The mouse model of chronic granulomatous disease (CGD) prominently substantiates the involvement of Nox in the N-methyl-
Nox1
Regional and cellular distribution in CNS
In the CNS, Nox1 has been reported in neurons (245), astrocytes (169), microglia (154), and neurovascular endothelial cells (120) (Table 1). The prevalence of Nox1 is also documented in neurons among different brain regions, including cerebral cortical neurons (208), hippocampal (44) and thalamic (192) neurons, cerebellar granule neurons (53), and in the dopaminergic neurons in the substantia nigra (SN) (40) and striatum (124), as well as in the ventral tegmental area (VTA) (102) (Table 1). The expression of Nox1 has been found to be enhanced in those neurons in response to various environmental toxins (55) or damage-associated molecular patterns (DAMPs) (160), and it is possibly responsible for neurodegeneration. Furthermore, Nox1 expression in activated microglia and astrocytes is considered a major cellular source of oxidative stress during chronic neuroinflammation (161).
Cellular Distribution and Subcellular Localization of Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase Isoforms in Various Regions of the Brain
DG, dentate gyrus; ER, endoplasmic reticulum; GCPs, cerebellar granule cell precursors; MAM, mitochondria-associated ER membrane; NE-LC, noradrenergic locus coeruleus; Nox, reduced nicotinamide adenine dinucleotide phosphate oxidase; NSC, neural stem cell; OL, oligodendrocyte; PVN, paraventricular nucleus; SFO, subfornical organ; SVZ, subventricular zone; VTA, ventral tegmental area.
The onset and progression of a variety of vascular diseases, including stroke (111), subarachnoid hemorrhage (114), and Alzheimer's dementia (44), are associated with ROS generation by Nox1 expressed in cerebral arteries (157). However, the distribution and function of Noxes in cerebral vascular are controversial. Recent studies suggest a significant elevation of Nox1 levels in neurovascular endothelial cells in blast-induced traumatic brain injury (bTBI) (120). Since the dorsal root ganglia (DRG) do not belong to the CNS, Nox1 expressed in DRG neurons regulating pain signaling (103) is not discussed.
Subcellular localization
Various subcellular localizations of Nox1 have been reported in the CNS (Table 1). In vitro studies confirmed the elevated levels of Nox1 localized in the nucleus in N27 (rat dopaminergic cells) after treatment with 6-hydroxydopamine (6-OHDA) (40). Similarly, mitochondria are found to be the area of subcellular localization of both Nox1 and p67phox proteins in untreated N27 DA cells (222). Following treatment with dieldrin and lindane, increased mitochondrial localization of Nox1 protein is considered to be a risk factor for the development of PD. Moreover, Nox1 gene silencing in microglia helped to determine the subcellular distribution and functions of Nox1. [Nox1-p22phox] dimers localized in intracellular vesicular compartments, including lysosomes, are recruited for phagosomal membranes during microglial phagocytosis triggered by zymosan and lipopolysaccharide (LPS) (38). However, more solid evidence is needed to confirm subcellular localization of Nox1 subunits with immune electromicroscopic examination and separation of organelles, such as mitochondria and lysosomes.
Signal transduction and function
Regulation of neuronal death
There are a number of studies that explored the involvement of Nox1 in neuronal death (75) (Fig. 1). Nondifferentiated PC12 cells manifested ∼10 times higher Nox1 messenger RNA (mRNA) level than Nox2 (gp91phox). Nerve growth factor (NGF)-induced neurite outgrowth was found to be suppressed by increased superoxide production triggered by Nox1 upregulation (101). Since ROS are considered a key risk factor in animal models of PD (66), accumulation of Nox1 in SN dopaminergic neurons of paraquat-treated rats was responsible for oxidative stress and the subsequent dopaminergic neuronal death (55). Use of a chemical inhibitor, rottlerin, or RNA interference (RNAi)-mediated protein kinase C (PKC) δ knockdown strategy clearly indicated that PKCδ was the key mediator of Nox1 expression in dopaminergic cells after exposure to paraquat (54). Moreover, Nox1-mediated oxidative stress could be a major player in α-synucleinopathy in paraquat-induced PD models (56). Nox1 knockdown-mediated significant reduction of both α-synuclein expression and aggregation, as well as the formation of A11 oligomers, largely attenuated dopaminergic neuronal loss in vivo. Furthermore, adeno-associated virus-mediated Nox1 knockdown (40) or inhibition of Nox1 expression (276) ameliorated 6-OHDA-induced dopaminergic neuronal degeneration. The mechanism was predicted to be alleviating oxidative DNA damage and reducing the DNA oxidative stress marker, 8-oxo-dG, subsequently suppressing nuclear phospho c-jun (p-c-jun) (41). Similar beneficial effects of AAV-mediated Nox1 knockdown were witnessed in the middle cerebral artery occlusion (MCAO) model (42). Induction of matrix metallopeptidase 3 (MMP-3) expression and activation mediated by 6-OHDA (43) further enhanced Nox1 expression and Rac1 activation through mitochondrial ROS production. Consequently, the accumulation of ROS produced by Nox1 results in neuronal apoptosis. Actually, ROS derived from mitochondria are essential but not enough to induce cell death. Therefore, this demands the sustained accumulation of ROS by the subsequent activation of Nox1 (126). The aforementioned signaling effects may be correlated with the subcellular localization of Nox1 in the nucleus and mitochondria.
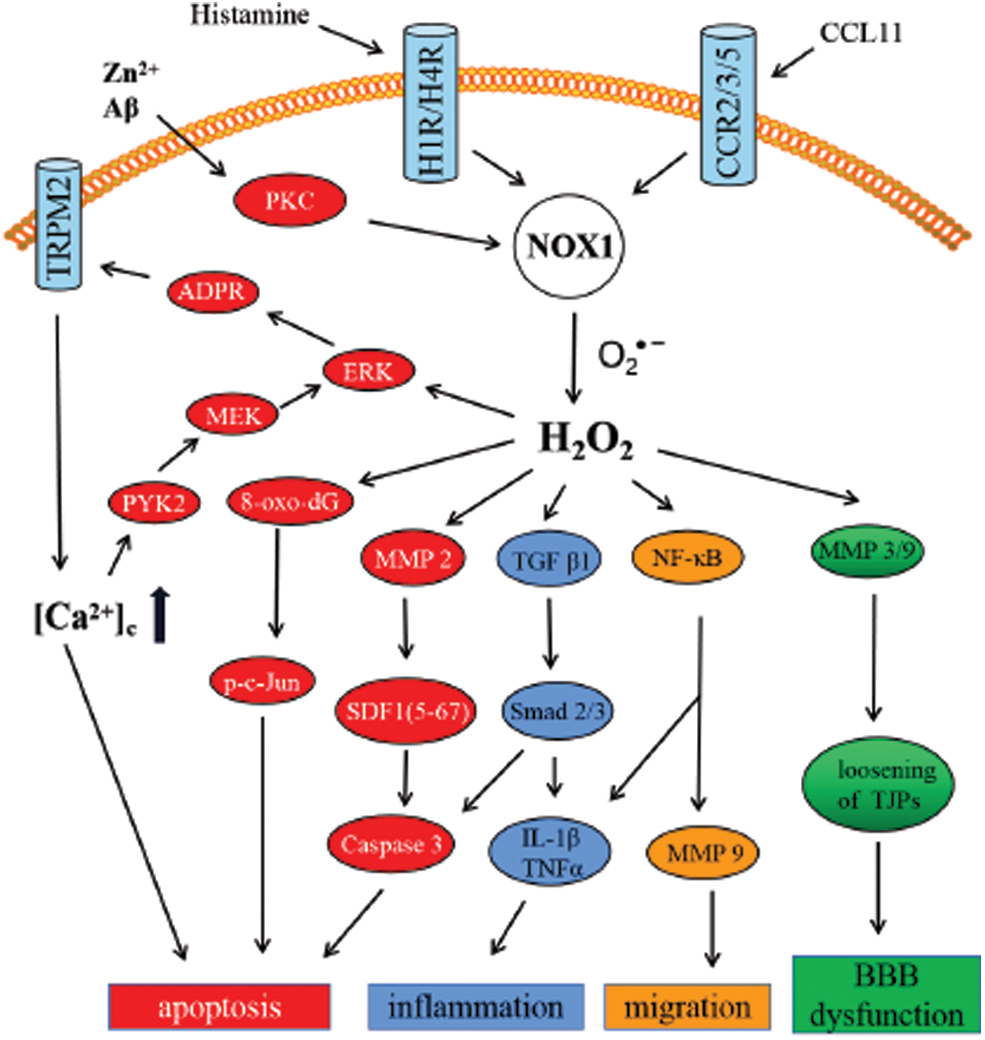
Regulation of glial cell death
Research further substantiated the involvement of Nox1 in the microglial apoptosis mediated by trace metal zinc ion (Zn2+), a damage-associated molecular pattern molecule (74). Excessive ROS are generated by Zn2+-stimulated PKC and Noxes. ROS promote poly(ADP-ribose) polymerase 1 (PARP-1) and poly(ADP-ribose) glycohydrolase (PARG) in the nucleus and lead to adenosine diphosphate ribose (ADPR) production, thereby activating transient receptor potential melastatin 2 (TRPM2)-dependent Ca2+ influx to increase the cytoplasmic Ca2+ concentrations ([Ca2+]c). A positive feedback mechanism ensued by elevated [Ca2+] concentration-mediated activation of the protein tyrosine kinase (PYK2)/MEK/extracellular signal-regulated kinase (ERK) signaling pathway amplifies PARP-1 activation, leading to TRPM2-mediated Ca2+ overload and cell death (160). Induction of the same signaling cascade by Nox2- and Nox4-derived ROS is also reported in response to amyloid beta (Aβ)42 (159). Mild traumatic brain injury (mTBI) reflected a significant increase in the Nox1 expression and the level of ROS to promote MMP2 activation. Activated MMP2, in turn, cleaved stromal cell-derived factor (SDF)-1α into the neurotoxic product SDF-1 (5 –67) to induce apoptosis in a caspase 3-dependent manner (2). On the contrary, transforming growth factor beta (TGF-β) 1 signaling was activated by increased ROS. Subsequently, Smad2 and Smad3 were phosphorylated to activate the caspase 3 signaling pathway, thereby promoting apoptosis (180).
Regulation of inflammation
Numerous studies have confirmed that ROS produced by Nox not only affect the neurons but also modulate microglial activity (201). Nox1-induced TGF-β1 signaling phosphorylated Smad2 and Smad3, which, in turn, facilitated neuroinflammation and inflammatory cytokine secretion, including IL-1β and tumor necrosis factor alpha (TNF-α) (180). The microglial neurotransmitter receptors engaged in modulating the Nox activation of microglia included metabolic mGlu3 receptors, group III receptors, GABAA receptors, and purinergic P2X7 or P2Y2/4 receptors (154). Literature suggests the association of the Nox1 signaling pathway with both microglial phagocytosis via histamine receptor 1 (H1R) activation and ROS production via H1R and H4R activation induced by histamine (197). In microglia activated with LPS, Nox1 localized in phagosomal membranes generates ROS, which augments inducible nitric oxide synthase (iNOS) expression and IL-1β secretion (38). On the contrary, in rat brain astrocyte (RBA-1) stimulated with LPS, Nox1 could elicit ROS-dependent nuclear factor kappa B (NF-κB) activity to promote IL-1β production and neuroinflammation (265).
Regulation of glial cell migration
Along with Nox1/ROS-dependent NF-κB activation in RBA-1 cells treated with LPS, matrix metalloproteinase 9 (MMP-9) overexpression facilitated cell migration (265). Studies have validated that the predominant expression of the chemotactic cytokine (chemokine) eotaxin (CCL11) receptor (C-C motif chemokine receptor [CCR] 2, CCR3, and CCR5) in microglia was simulated by CCL11 released from activated astrocytes (174). Subsequently, the CCL11 receptor in microglia significantly promoted migration via Nox1 upregulation.
Regulation of endothelial permeability and blood/brain barrier
Studies have substantiated the pivotal role of blood/brain barrier (BBB) integrity in CNS pathology. Significant reduction of BBB permeability was attributed to phosphoinositide 3-kinase (PI3K) γ deficiency mediated by suppressing the expression of Noxes (Nox1, Nox2, and Nox4) and MMP-9 followed by reperfusion (109). In bTBI models (120), increased superoxide production both within and outside the endothelial cells was induced by ROS produced from Nox upregulation in neurovascular endothelial cells. Superoxide, in turn, elevates MMP-3/9 production, which dissolves the tight junction complexes connecting adjacent endothelial cells, ultimately leading to the disruption of the BBB. Furthermore, angiotensin II (Ang II)-stimulated increase in superoxide was about 70% smaller from Nox1-knock out versus wild type in cerebral blood circulation following ischemic stroke (105). It was also suggested that Nox1 plays a crucial role in Ang II-induced oxidative stress and endothelial dysfunction in the brain (47). More recently, by inhibiting Nox1 in brain vascular endothelial cells, some drugs have been suggested to protect the BBB integrity and alleviate the loss of tight junction proteins zonula occludens protein-1 (ZO-1) and occludin, which are essential for BBB integrity, such as cordycepin (270), GKT136901 (100), and phoenixin-14 (273).
Regulation of cognitive function and emotional behavior
In addition to the above physiological and pathological functions, the involvement of Nox1 has also been demonstrated in psychiatric disorders (214). A few studies have revealed that intraperitoneal administration of ketamine (30 mg/kg) at postnatal days 7, 9, and 11 in male C57/B16 pups resulted in early and persistent neurochemical imbalance and psychotic-like behavioral dysfunctions that could be improved by the Nox inhibitor, celastrol (211). Using a mouse model induced by corticosterone (CORT) (102), Ibi et al. found significantly upregulated Nox1 mRNA only in the VTA among brain areas responsible for emotional behaviors. Furthermore, increased ROS oxidated cysteine residues of NMDA receptor 1 (NR1), thereby attenuating NMDA receptor activity and reducing brain-derived neurotrophic factor (BDNF) expression via Nox1-dependent DNA methylation, leading to the development of depressive-like behaviors. Moreover, CORT-induced depressive-like behaviors were alleviated in mice deficiency of Nox1 (Nox1–/Y) or delivery of Nox1 microRNA (miRNA) to VTA.
Nox2
Regional and cellular distribution in CNS
Identification of Nox2 in phagocytes emphasized its role in host defense. Loss-of-function mutations in genes encoding gp91phox (major subunit of Nox2), p47phox, p67phox, or p22phox contribute to the prognosis of the CGD in human, a rare syndrome described in 1957 (202). CGD is an inherited immunodeficiency disease characterized by life-threatening bacterial and fungal infections mostly prevalent in peripheral organs (202). Interestingly, cognitive dysfunction in CGD patients and synaptic plasticity deficits recognized among gp91phox and p47phox mutant mouse models of human CGD (116, 173) indicate the importance of Nox2 in the development and physiological function of the CNS.
Apart from the high expression of Nox2 in phagocytes, Nox2 expressions have been detected in the CNS. Nox2 expression is generally identified in several regions of the brain, such as the cortex [including the prefrontal cortex (134), cingulate cortex (216), motor cortex (194), and visual cortex (256)], hippocampus (261), striatum (89), SN of midbrain (220), cerebellum (84, 170), amygdale (219), paraventricular nucleus (PVN), and supraoptic nucleus of the hypothalamus (96), noradrenergic locus coeruleus (NE-LC) in the brain stem (251), and the subventricular zone (SVZ) (33) (Table 1). At the cellular level (Table 1), Nox2 is observed in neurons (210), astrocytes (50), microglia (223), neurovascular endothelial cells (69), and oligodendrocytes (OLs) (32). In CNS, upregulation of Nox2 in microglia (223), particularly after injury, is presumed to be associated with neuroinflammation and neuronal apoptosis (76).
Subcellular localization
In phagocytic cells such as microglia, activation of Nox2 indulges in the translocation of the cytosolic subunits p40phox, p47phox, and p67phox to the plasma membrane where they assemble with gp91phox and p22phox to form a membrane-bound complex (227). Thus, Nox2, generally expressed in the microglia plasma membrane, may participate in the inflammatory responses in microglia (227). Colocalization of Nox2 with IL-1R1 and mutant Huntingtin (Htt) was documented in plasma membrane lipid rafts (167, 246). Nox2 localized in endosomes (called redoxosomes) further regulates endosomal ROS and proinflammatory signals (132). Gp91phox (the major Nox2 subunits) and p22phox are associated with TSPO (translocator protein 18 kDa) in microglia (85). Meredith and his colleagues used coimmunoprecipitation, confocal immunofluorescence imaging, and proximity ligation assay and observed that TSPO interacts with gp91phox and p22phox at the mitochondria-associated endoplasmic reticulum (ER) membrane (MAM) to modulate redox homeostasis in microglia (140) (Table 1).
Signal transduction and function
Extensive research has substantiated the role of Nox2 in various CNS pathologies, such as AD (189), PD (124), ALS (26, 150), MS (194), TBI (147), subarachnoid hemorrhage (277), and ischemic stroke (110), even psychiatric disorders, including autism spectrum disorder (5), excited delirium syndrome (212), and schizophrenia (15). The pathological role of Nox2 in CNS has also been summarized in detail in previous reviews (165). Interestingly, Nox2 manifested significant activation in all of these diseases. Clinical studies validated Nox2 activation in whole blood of PD patients compared with healthy controls (151).
Regulation of glutamate receptors and excitotoxicity
In cerebral ischemia/reperfusion injury, activation of receptor-like kinase (ALK5) resulted in TGF-β signaling, phosphorylation of SMAD2/3, which further regulated the expression and activity of Nox2 and Nox4 (141, 142) (Fig. 2). The neuronal damage via calpain is also mediated by Nox2 activation via the metabolic glutamate receptors (mGluR1 and mGluR5) (83). In vivo experimental stroke model has confirmed the contribution of Nox2-derived ROS to oxygen/glucose deprivation-induced glutamate release via complexin II/SNARE signaling (254). ROS produced by Nox2 are also responsible for the NMDA receptor-dependent glutamate release in the prefrontal cortex (229). Nonetheless, Nox2-induced NMDA receptor activation triggers superoxide production and extracellular release, which enforces apoptosis of neurons, even neighboring neurons, and astrocytes (196). This process is involved in Ca2+ influx through NMDAR channels, further triggering PI3K/PIP3 signaling to activate PKC (22, 158). HIF-1α also acts as an upstream signal molecule to mediate Nox2 expression and activation. Chromatin immunoprecipitation assay reveals the binding of HIF-1α to the promoter of Nox2 in CoCl2-induced apoptosis of PC12 cells (82). Endogenous CHOP (ER stress protein) plays a crucial role in the upregulation of HIF-1α and Nox2 in long-term intermittent hypoxia (LTIH) to promote apoptosis (46). Extracellular glutamate-induced OL differentiation is mediated by the activation of the NMDA receptor, which subsequently activates the PKC/Nox2 cascade to generate ROS and activate the PI3K/Akt/mTOR (mammalian target of rapamycin) and/or ERK pathway (32). In accordance with this, several studies have reflected the importance of NMDA receptor-dependent Nox2 activation for synaptic plasticity induced by the long-term potentiation and depression in the hippocampal cornu ammonis (hippocampal CA1) area (117, 267) and visual cortex (61, 73).
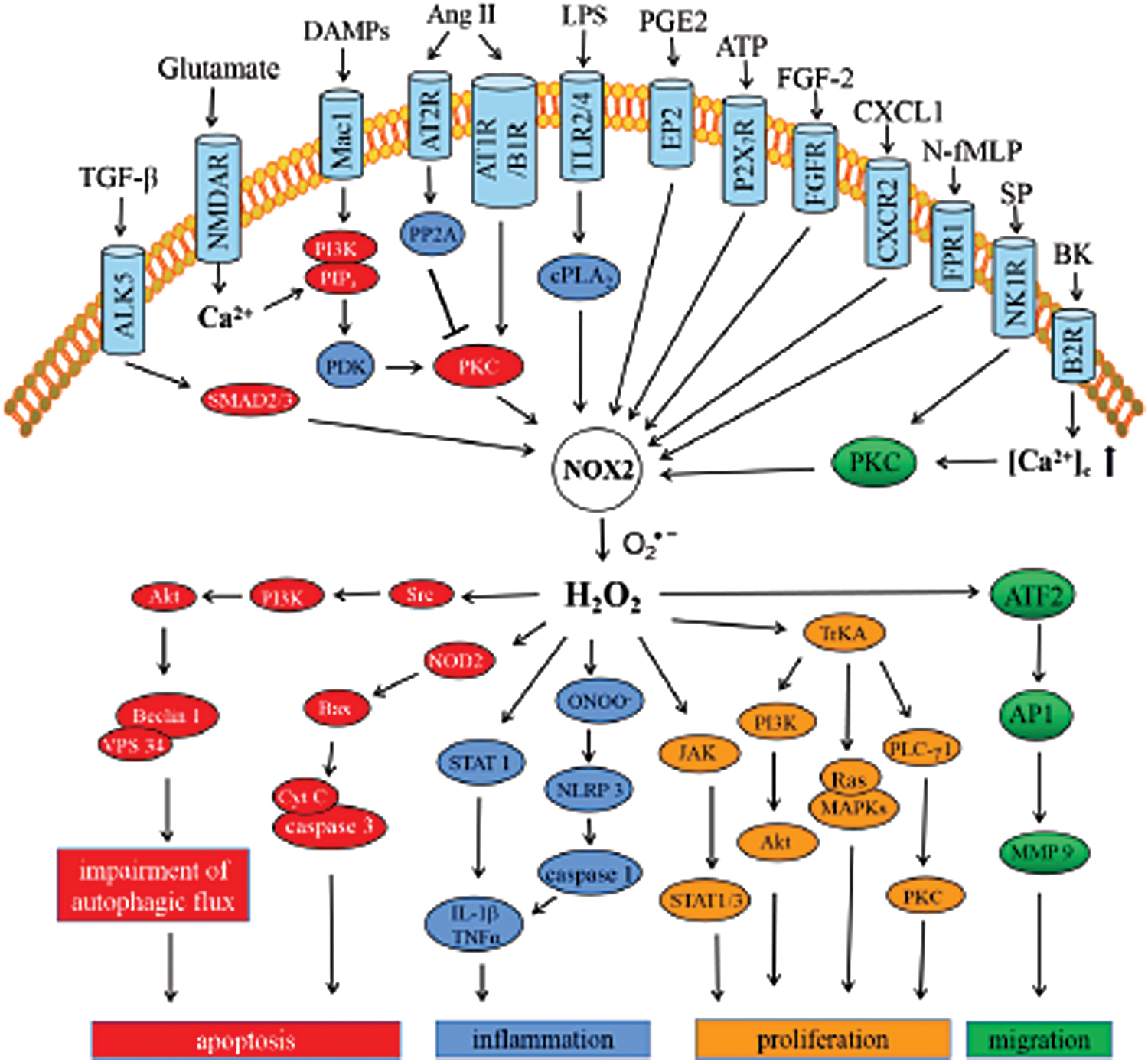
Regulation of autophagy and cell death
Furthermore, ROS-induced apoptosis is also involved in autophagy. In fact, research has shown that the autophagic process is regulated by the oxidation of cysteine residue near the Atg4 catalytic site by ROS, specifically H2O2 (209). The TBI rat model corroborated significantly decreased expression of LC3 and Beclin1 in the hippocampus following apocynin (Nox inhibitor) treatment (71). In a rotenone-inducible cellular model of PD, activated Nox2 was attributed to impair the interaction between Beclin1 and VPS34 through Src/PI3K/Akt-dependent activation to induce the disruption of autophagic flux, ultimately instigating apoptosis (172). The 6-OHDA-induced PD model determined that Nox2-mediated oxidative stress facilitated the expression of nucleotide-binding oligomerization domain-containing protein (NOD2) to promote apoptosis via Bax- and caspase 3-dependent pathway (37). In different neuronal injury models, activated Nox2 produces ROS, which either inactivates protein phosphatase 5 (PP5) or activates the JNK (c-Jun N-terminal kinase) pathway (263) or NLRP1 inflammasome/caspase-1 signaling (233, 253, 264) and induces neuronal apoptosis. Studies have validated the involvement of Nox2 in the loss of parvalbumin-immunoreactive interneurons (PV-IN) (188, 213, 271). However, the molecular mechanisms have not been elaborated in CNS injury.
Regulation of inflammation
In CNS, neuroinflammation characterized by microglial activation is closely related to neuronal death. Indeed, a number of studies have established the role of microglial Nox2 in brain inflammation (34, 45, 48, 67, 88, 97, 98, 119, 215, 252, 280). The receptors on microglia cell membranes involved in the initiation of Nox2-mediated inflammatory responses include integrin receptor Mac1 [also known as CD11b/CD18 (92), CR3 (35), or αMβ2 (274)] stimulated by DAMPs, such as α-synuclein, β-amyloid, HMGB-1 (76); angiotensin II type 2 receptor (AT2R) (21); angiotensin II type I receptor (AT1R) and kinin B1 receptor (B1R) stimulated or upregulated by Ang II (175, 198); toll-like receptor (TLR) 2 and TLR4 stimulated by LPS (186) or DAMPs (138, 184); E-type prostaglandin receptor 2 (EP2) stimulated by prostaglandin E2 (PGE2) (77); and purinergic receptors (P2X7R) stimulated by ATP or BzATP (179). The previous review by Vilhardt and colleagues (87) vividly described the receptor-mediated Nox activation in microglia. Just like NOD2 mediating Nox2/ROS-induced apoptosis as aforementioned, NOD2-deficient (NOD2−/− ) mice attenuated the inflammatory responses mediated by NF-κB, p38 mitogen-activated protein kinases (MAPKs), and JNK signaling (136). Moreover, NOD2 deficiency inhibits the upregulation of Nox2 and ROS generation following cerebral ischemia/reperfusion injury, which may be induced through the NF-κB and MAPK pathway (136). After the activation of the receptor, Nox2 activation is either directly induced (8, 77) or regulated by other signaling molecules. Aβ-activated Mac1 receptor activated the PI3K-PIP3-PDK-AKT/PKC signaling cascade and promoted the assembly of Nox2 subunits (39, 275). However, the AT2 receptor-mediated suppression of the PKC activation via PP2A restrained Nox2 activation and ROS generation (21). Nox2 is activated by TLRs via the activation of cytosolic phospholipase A2 (cPLA2α) (149). In TLR-induced neuroinflammation, activated Nox2 induces downstream inflammatory pathways, such as the activation of signal transducer and activator of transcription (STAT) 1 and NF-κB and CD40 protein expression (10, 149). After ATP release-mediated P2X7 receptor activation, the Nox2-derived superoxide anion quickly reacts with NO to form peroxynitrite (ONOO−). Thereafter, this peroxynitrite triggers NLRP3 inflammasome assembly and caspase1 activation (70, 146). Most studies have reported regulation of NF-κB and MAPK inflammatory signals by Nox2 (112, 185, 199, 226). A few studies have also indicated the association of gp91phox transcription with NF-κB regulation (7). Moreover, overexpression of MKP-1 (mitogen-activated protein kinase phosphatase-1) or inhibition of MAPKs downregulated the expression of Nox-2 (99).
Regulation of cell proliferation
In addition to inflammation, Nox2 has also been implicated in cell proliferation. Amyloid-β-induced microgliosis and astrogliosis exhibited a significant association with Nox2-mediated redox signaling (78, 262). Astrogliosis via the activation of JAK (Janus kinase) and transcription factor STAT1/3 can be triggered by the extracellular release of Nox2-derived H2O2 in microglia (94). In APP/PS1 mice, instigated by the chemokine (C-X-C motif) ligand 1 (CXCL1), C-X-C motif chemokine receptor 2 (CXCR2) triggered Nox2-PI3K-Akt signaling cascade and promoted the proliferation of neural stem cells (NSCs) in the SVZ (221). AKT signaling maintains cell proliferation, which is inhibited by PTEN. However, the mitogen fibroblast growth factor 2 (FGF-2) signal promotes the Nox2 activation and production of H2O2 in the adult hippocampal stem/progenitor cells (AHPs), which can oxidize and deactivate PTEN, ultimately resulting in proliferation (64). Furthermore, in SH-SY5Y cells, formyl-peptide receptor 1 (FPR1) stimulation by n-formyl methionyl-leucyl-phenylalanine (N-fMLP) facilitates cell proliferation by inducing the neurotrophin receptor TrkA transactivation through Nox2-dependent ROS. In this process, TrkA transactivation initiates intracellular PI3K-Akt, Ras-MAPK, and PLCγ1-PKC cascades (31).
Regulation of cell migration
In microglia and astrocytes, Nox2 signaling has also been implicated with migration mediated by various receptors, such as VEGFR1/CSF-1R (128), neurokinin-1 receptor (NK1R) stimulated by substance P (SP) (250), TLR2 stimulated by lipoteichoic acid (LTA) (95), and B2R stimulated by bradykinin (BK) (135). PKC is a key molecule that orchestrates the activation of Nox2 induced by SP, LTA, and BK. Subsequently, the ATF2-AP1 signaling pathway may trigger sustained expression of MMP-9, which is required for astrocytic migration (95, 135).
Others
In addition to the above signaling pathway, Nox2 is also involved in a variety of signal transduction pathways in CNS, including differentiation (33), synaptic plasticity (1, 204), and cerebrovascular dysfunction (69). Moreover, LPS-induced acute microglial inflammation failed to drive the axon initial segment (AIS) structural plasticity in Nox2−/− mice (18). A more integrated description regarding the role of Noxes in neuronal development has been highlighted in several recent reviews (238, 260). The Ang II- and amyloid peptide-induced cerebrovascular dysregulation is also mediated by radicals produced by endothelial Nox2 (47, 79, 113, 177, 178). Indeed, during NMDA receptor activation, ROS produced by Nox2 activation are attributed to the modulation of cerebral blood flow (80). An in-depth presentation of Nox2-mediated endothelial dysfunction can be found in a recent review (24).
Nox3
Regional and cellular distribution in CNS
Together with Nox4 and Nox5, Nox3 in nonphagocytic cells was first discovered in 2001 (36), which is expressed primarily in fetal tissues (36) and the inner ear (9). Overexpression of Nox3 in specific portions of the inner ear is confirmed by several studies, and also its role is found to be indispensable for otoconia morphogenesis to the vestibular system (171). The low availability of the Nox3 gene in the brain justifies the minimal accessibility of information about Nox3 in the CNS (9). The earliest study about Nox3 in CNS substantiated its increased expression in the AD brain (60). Nox3 expression in neurons has been reported only after brain injury (49). High expression of Nox3 is also noted in the cerebrum of 10-week-old stroke-prone spontaneously hypertensive rats (SHRSP) (156). Tight regulation of the differentiation of precursor cells with the intracellular redox state in OL (224) and the Nox3 enzyme seems to be the key modulator of ROS homeostasis in OL. Nox3 is expressed effectively in the human OL precursor cells MO3–13 (3) and phorbol-12-myristate-13-acetate (PMA) and H2O2 significantly increased the mRNA level of Nox3 for 1 and 4 days. Specific Nox3 small interfering RNA (siRNA) used in knockdown experiments prevented PMA effects on the mRNA levels of MBP (myelin basic protein) and Olig-2 (the oligodendrocyte lineage genes). In the Nox3eqlb mutant mouse (152), Nox3 expression in cerebellar granule cell precursors (GCPs) and NSCs plays a vital role in cerebellar development (Table 1).
Subcellular localization
The only study in HEK-293 cells (human embryonic kidney cells) on Nox3 subcellular localization reveals that the Nox regulatory subunits NoxO1 and NoxA1 activated by transfecting, functional murine Nox3 coexpressed with p22phox are located to the plasma membrane (163).
Signal transduction and function
Although there is minimal knowledge about the functions of Nox3 expressed in cortical neurons after brain injury (49) and the model of SHRSP (156), ROS generated by Nox3 and Nox5 contribute to the MO3–13 cell differentiation induced by PMA or hydrogen peroxide via ERK1/2- and cyclic adenosine monophosphate (cAMP)-responsive element-binding protein (CREB)-dependent mechanism in OLs (3). PKC and redox state seem to be the central modulators of differentiation of glial precursor cells (224). Nox3-derived ROS amplified PMA-induced PKC activation and the subsequent phosphorylation of ERK1/2 and CREB1. Thereafter, translocating into the nucleus, the phosphorylated CREB increases the expression of specific OL differentiation markers (207): the Olig-2 (143, 205) and MBP (14), ultimately leading to OL differentiation (Fig. 3).
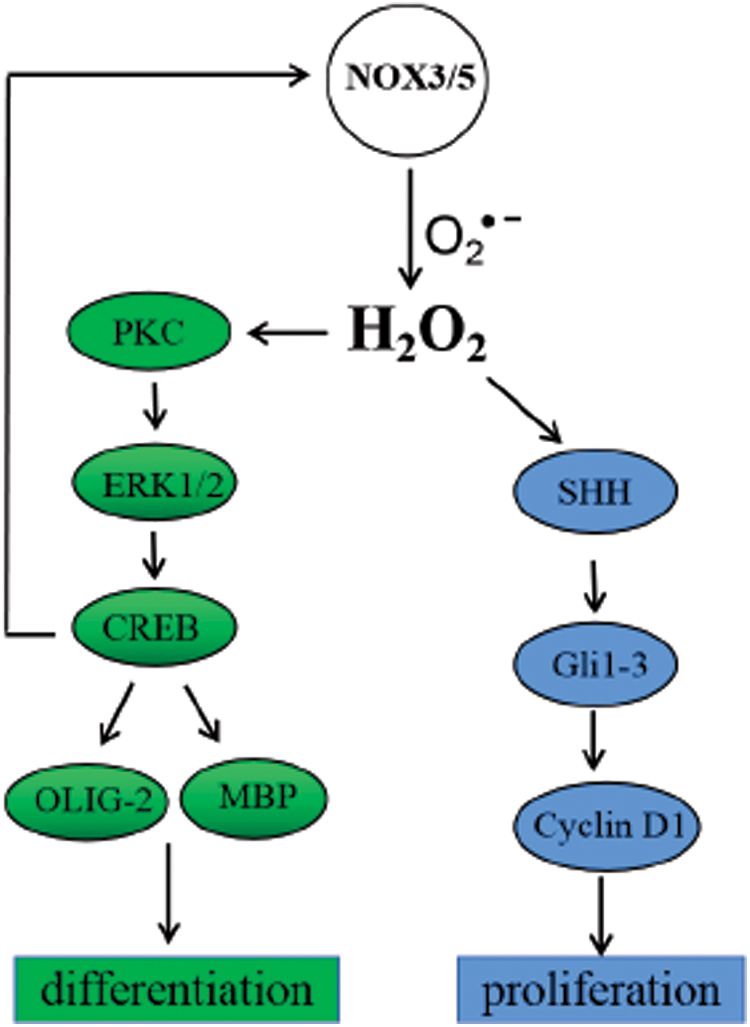
A study performed with the Nox3eqlb mutant mouse model confirmed proliferation in GCPs and NSCs promoted by ROS derived from Nox3 (152). Indeed, high endogenous ROS levels in proliferative NSCs rationalize the importance in regulating self-renewal and neurogenesis in a PI3K/Akt-dependant manner (125). Sonic hedgehog (Shh) signaling plays a vital role in the development and patterning of the mouse cerebellum (129, 259). Nox3eqlb mouse GCPs and NSCs produce higher amounts of ROS, which activate the Shh intracellular signaling pathway by upregulating the expression of the GLI family of transcription factors Gli1, 2, and 3 (51, 57) and cyclin D1 (a main target of the Shh pathway) (181) to promote proliferation.
Nox4
Regional and cellular distribution in CNS
Like Nox1 and Nox2, Nox4 expression is manifested in many cell types among various brain regions. In situ hybridization demonstrated strong expression of Nox4 in neurons of all six layers of the cortex, in the pyramidal cells of the Ammon's horn of the hippocampus, and Purkinje cells of the cerebellum (247). The distribution of Nox4 in the brain is also witnessed in the dopamine neurons of the SN (272), granule cells of the cerebellum (258), the subfornical organ (SFO) (30), and the hippocampal dentate gyrus (DG) (40). The abundance of Nox4 expressions is more prominent in cells, including neural stem/precursor cells (268), astrocytes (169), microglia (130), pericytes (166), and endothelial cells (81) (Table 1).
Subcellular localization
Several studies have addressed the subcellular localization of Nox4. In HEK-293 cells, Nox4 has been identified in the plasma membrane, the perinuclear regions, and the ER (127, 182, 278). Interaction of Nox4 with TLR4 is documented at neuronal plasma membranes in CNS (145, 234). Moreover, mitochondrial and intranuclear localization of Nox4 has been recognized in neurons (30, 272). Nox4 expression is also obtained in the cytoplasm of endothelial cells (81) (Table 1). The cytoplasmic localization of Nox4 could be resulted from detection of unassembled Nox4 subunits by anti-Nox4 antibodies.
Signal transduction and function
Rapid conversion of Nox4-derived superoxide to hydrogen peroxide suggested that, unlike the other Nox isoforms, hydrogen peroxide is presumed to be the only molecule that mediates the downstream effects of Nox4 (217). Clinical brain sample studies indicated the significantly increased Nox4 expression at an early stage after TBI (133). Clinical studies also validated the restoration of neuronal injury and neurodegeneration by inhibition of Nox4 (153).
Regulation of cell survival and apoptosis
Specially, cell survival in cerebral microvascular endothelial cells (CMVECs) was promoted by Nox4-derived H2O2 (13). ROS induced by the inflammatory mediator TNF-α increase carbon monoxide (CO, a gaseous antioxidant mediator) production by heme oxygenase-2 (HO-2), subsequently inhibiting ERK1/2 and p38 MAPK phosphorylation through an Akt-dependent manner, in turn leading to the inhibition of Nox4 (13). On the contrary, TNF-α-induced Nox4 activation results in apoptosis through caspase 3-dependent DNA fragmentation in CMVECs (12) (Fig. 4). It is suggested that TNF-α not only induces apoptosis by directly activating Nox4, but also concurrently triggers endogenous cytoprotection mechanisms indirectly. The reason why Nox4 can mediate two opposite signaling pathways may be related to the quantity and speed in the production of ROS. Helena et al. emphasized that the key role of ROS is to cause cell death, even though Nox4-derived ROS also stimulate a prosurvival pathway through activation of constitutive HO-2. On the contrary, there is no obvious difference in the two opposite effects in TNF-α-induced ROS, and the direction of cell fate would be determined by other cellular conditions. Yet, the stress-induced upregulation of Nox4 at the ER-mitochondria contact sites (MAMs) provides a prosurvival mechanism that inhibits InsP3 receptor-dependent Ca2+ release into mitochondria (19). As supportive evidence, Nrf2 plays a neuroprotective role by hindering the Nox4/ROS/NF-κB pathway (58).

Regulation of neuronal apoptosis
Nox4 activation mediates neuronal apoptosis by Ca2+ influx via NMDA and AMPA receptors (86) and angiotensin 1 (AT1) receptor activated by AngII (272). Subsequently, intranuclear oxidized guanosine (8-OHdG) and caspase-3 activation were evident during apoptosis (272). In PC12 cells, Nox4 expression upregulated by Aβ instigated the autophagy flux and caused excessive autophagy, finally leading to apoptosis (108). The role of Nox4 in autophagy is documented in the cardiovascular system (72). During heart ischemia/reperfusion injury, Nox4 ensures cardioprotective effects by binding to the serine/threonine protein phosphatase 1 (PP1) subunit GADD34 at the ER, which subsequently leads to autophagy activation via inhibition of PP1 activity to increase eIF2α phosphorylation and activating transcription factor 4 (ATF4) levels (206).
Regulation of inflammation
Meaningfully, the interaction between TLR4 and Nox4 indulges in both neuronal apoptosis (234) and inflammation through NF-κB activation induced by LPS (176) and Aβ25–35 (239). Moreover, Nox4 conferred an inflammatory response by activating the JAK2/STAT3 pathway and NLRP3 inflammasome (269, 279). However, siRNA against Nox4 failed to alter the expression of major histocompatibility complex class II (MHCII), CD68, CD11b, iNOS, VEGF, and TGF-β, but markedly decreased IL-6 mRNA in the human microglia cell line clone 3 (HMC3) (130).
Regulation of cell proliferation and differentiation
Nox4 has also been implicated in NSC proliferation and astrocytic differentiation. Ang II- and bFGF-induced proliferation of neural stem/precursor cells witnessed increased Nox4 expression to activate the AKT/mTOR pathway (240, 268). During astrocytic differentiation, Nox4-induced ROS, which can be inhibited by PARP3, contribute to the reduction in the mTORC2 activity through degradation of Rictor to control the process of differentiation (200). Furthermore, pericyte Nox4 upregulated in peri-infarct areas following acute brain ischemia destroyed BBB through NF-κB activation and the expression of MMP-9 (166). Interestingly, Nox4 mediates ischemic damage in the brain but not in myocardial infarction and hind limb ischemia, which may be explained through Nox4-dependent BBB breakdown (27).
Nox5
Regional and cellular distribution in CNS
The infrequent occurrence of Nox5 in primary cells contributes to the major reason for the limited studies on Nox5 expression in the CNS. The only research has corroborated that together with Nox1, Nox2, and Duox, Nox5 expression during the early nervous system development may validate the relevance of neurodevelopment and regeneration in the developing zebrafish (255). Accetta et al. (3) reported that Nox5 is expressed in OLs, together with Nox3 (Table 1).
Subcellular localization
Similar to Nox3, none of the studies has reported the subcellular localization of Nox5 in the CNS. However, numerous studies have reported the abundance of Nox5 primarily within the intracellular membranes, including the plasma membrane (218), perinuclear regions, and ER (243) in HEK-293 cells.
Signal transduction and function
As above, ROS produced by Nox5 upregulated Nox3 expression, further accumulates the ROS in OLs, which promotes differentiation via ERK1/2-CREB-Olig-2/MBP signal transduction (3) (Fig. 3). The crucial role of Nox5 in the peripheral cardiovascular system (242) is well-documented, following its expression in vascular smooth muscle cells (107) and endothelial cells (16). Interestingly, a humanized knockin mouse model has been established harboring human Nox5 driven by the Tie2 promoter, which predominantly governs endothelial cell expression, and aggravation of the neurological dysfunction following ischemia and reperfusion was evident compared with wild type mice (25). In terms of its mechanism, activation of Nox5 following cerebral ischemia results in excessive amounts of ROS to lead to the breakdown of the BBB (28). This finding proposes the involvement of Nox5 in the maintenance of BBB integrity.
Nox Inhibitors and Their Potential Usage
Thus, it has been summarized above that Nox isoforms exert an essential role in various CNS disorders and signal transduction pathways. Simultaneously, methods for the detection of Nox activity and in vivo expression of Nox have been developed (65, 230). Thus, Nox inhibition contributes a potential antioxidant target for the treatment of neurological diseases. To date, a limited number of Nox inhibitors have been studied for the management of a variety of CNS diseases (Table 2). However, the low specificity and selectivity of these inhibitors restricted their clinical application. Therefore, the specificity and selectivity of Nox inhibitors may be the core problem to be resolved.
Effects of Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase Inhibitors in Central Nervous System Diseases
Aβ, amyloid beta; AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; BBB, blood/brain barrier; DPI, diphenyleneiodonium; EAE, experimental autoimmune encephalomyelitis; HD, Huntington's disease; HT, hemorrhagic transformation; ICH, intracerebral hemorrhage; LTIH, long-term intermittent hypoxia; MPTP, 1-metyl-4-fenyl-1,2,3,6-tetrahydropyridin; MS, multiple sclerosis; NMDA, N-methyl-
Nonspecific Nox inhibitors
The compounds that nonspecifically inhibit Nox enzymes, including celastrol, ebselen [2-phenyl-1,2-benzo-isoselenazole-3(2H)-ketone], as well as the widely used diphenyleneiodonium (DPI), and apocynin (4-hydroxy-3-methoxy-acetophenone), have been widely studied (11, 115). The effectiveness of celastrol isolated from Tripterygium wilfordi (a Traditional Chinese Medicine) as an Nox inhibitor with superior inhibition of Nox1 with an IC50 of 0.41 μM and Nox2 with an IC50 of 0.59 μM in enzyme activity analysis has been extensively investigated (106). Attributed to its ability to react with peroxynitrite, ebselen, in addition to being an inhibitor of Nox, has been found to inhibit a variety of enzymes, such as lipoxygenases, NO synthases, PKC, and H+,K+-ATPase (52). In the TBI model, ebselen-mediated amelioration of neurological injury is mediated by the modulation of NO levels and the TLR4-mediated p38 MAPK signaling pathway (257). Like ebselen, DPI can also inhibit various protease systems, including Nox, NO synthase, xanthine oxidase, NADH coenzyme Q reductase, and possibly others (11). The mechanism behind its inhibitory effect is found to be closely associated with the irreversible inhibition of flavoprotein (an electron transporter) (168). Furthermore, research has established the promising antioxidant and anti-inflammatory effects of apocynin, the most widely used Nox inhibitor in experimental stroke models, among phagocytic and nonphagocytic cells (121, 281). Research on myeloperoxidase (MPO)-deficient cells confirmed the necessity of MPO and hydrogen peroxide for the inhibitory function of apocynin (90, 232). MPO and hydrogen peroxide obstruct the p47phox translocation to the membrane by apocynin dimerization and thereby inhibit the assembly of the Nox complex (241). Interestingly, MPO-deficient cells observed that apocynin also plays a role in improving oxidative stress involving nonspecific oxidative scavenger function without inhibiting Nox (90). Although in vitro and in vivo experimentations validated the efficacy of these compounds in inhibiting Nox, the interpretation of results obtained with these inhibitors requires very careful analysis and controlled trials.
Selective Nox inhibitors
The identification of members of the Nox family and the gradual elucidation of their roles in different tissues further facilitate the development of specific Nox inhibitors as well (29, 59). So far, the most selective Nox inhibitor, chimeric peptide Nox2ds-tat (originally known as gp91ds-tat), revealed effective inhibition of Nox2 with an IC50 of 0.74 μM in vitro and in vivo (195). Nox2ds-tat consists of two components. The core inhibition region was designed based on the Nox2 catalytic core sequence peptide (62), and there are nine amino acid peptides derived from the HIV viral coat (HIV-tat) before the C-terminal, which facilitated the delivery of the peptide into the cell (248). Thus, this peptide binds specifically to p47phox blocking the assembly of the Nox2 complex (62). Furthermore, another peptide inhibitor, NoxA1ds, developed in a similar way to Nox2ds-tat, was characterized to effectively inhibit Nox1 activity, but not Nox2, Nox 4, Nox 5, or xanthine oxidase (191). With an 11-amino-acid sequence, NoxA1ds directly binds to Nox1 and displaces NoxA1, impeding the enzymatic activity with an IC50 of 0.02 μM (191). Both of these peptide inhibitors interact with the activation domain of Nox to regulate biological functions. Therefore, the researchers targeted the p47phox/p22phox protein interaction to develop inhibitors, exploiting the possibility of drug discovery and achieved some positive results (225). Recently, a small-molecule inhibitor, GSK2795039, has received attention (63, 249, 266). Although GSK2795039 inhibits multiple Nox isoforms, it demonstrates higher selectivity for Nox2 (91). WST-1 assay substantiates that GSK2795039 inhibits Nox2 with pIC50 of 5.54 ± 0.25 μM, while it has no activity against other Nox isoforms up to 100 μM (91). Besides, some other Nox inhibitors also display higher selectivity for certain Nox isoforms. For example, VAS2870 exhibited better selectivity to Nox2 with an IC50 of 0.77 μM (27, 153, 235). GKT137831 and its close analog, GKT136901, portrayed its Nox1/4 inhibitory activity (29). The IC50 of GKT137831 for Nox1 and Nox4 was 0.14 and 0.11 μM, respectively, while the IC50 of GKT137831 for Nox2 and Nox5 was 1750 and 0.41 μM, respectively (6). The IC50 of GKT136901 for Nox1 and Nox4 was 0.16 and 0.17 μM, while the IC50 of GKT136901 for Nox2 and Nox5 was 1530 and 0.45 μM, respectively (6). The IC50 values of Nox inhibitors for different Nox isoforms can directly reflect their efficacy, and more summaries of inhibitors' selectivity are detailed in previous reviews (6). Despite recent advances, there is still a need to explore novel compounds that specifically target Noxes and their pathological effects in CNS diseases.
Conclusions and Perspectives
Although signal transduction of Nox isoforms in the CNS has been only partially elucidated, one consensus observation is ultimately proposed. All Nox isoforms mediate downstream signal transduction by the ROS production, but the signaling pathways mediated by each Nox isoform are not completely consistent or even opposite. For example, Nox1 and Nox2 mediate apoptosis through different downstream pathways, while Nox4, on the one hand, plays a critical role in mediating apoptosis pathways, on the other hand, also participates in cell survival. So what is the key to the specific role of intracellular Nox isoforms? The explanations may be involved in several aspects. (i) Expression of various Nox isoforms in varying degrees in several cell types among different brain regions; (ii) differential functions of cells expressing Nox isoforms in varied brain regions; (iii) variation in the subcellular localization of Nox isoforms in the same or different cell types; (iv) differences in the functions of various cell organelles with Nox isoform; and (v) differential supply of NADPH, the substrate of Nox, in different subcellular environments. For instance, quantitative real-time PCR detected basal levels of Nox1, Nox2, and Nox4 expressions in different regions of adult C57BL/6 mouse brain (104). Nox2 and Nox4 are significantly expressed in the forebrain, midbrain, and hindbrain of mice, whereas Nox1 is detectable but at lower levels (104). Besides, higher levels of Nox2 are observed in the forebrain compared with the Nox1 and Nox4, whereas the abundance of Nox4 mRNA is more prevalent in the midbrain and hindbrain (104). In NSCs, Nox2 is stimulated by CXCL1 (221) to mediate cell proliferation, while Ang II induces Nox4 (240) to promote proliferation. Ang II activates Nox2 via AT1 receptors to promote inflammation in the microglia (198), while Ang II-induced Nox2 activation indulges in cerebral endothelial dysfunction in the endothelium (47). Furthermore, apoptosis in CMVECs is triggered by TNF-α-mediated activation of Nox4 (12); simultaneously, TNF-α stimulates Nox4 to initiate cell survival mechanism by increasing CO production (13). In addition, genetically encoded fluorescent sensors demonstrated a marked difference in NADPH level between the cytoplasm and the mitochondrial matrix, ∼3.1 ± 0.3 μM in the cytosol and 37 ± 2 μM in the mitochondrial matrix (236). Taking all these into consideration, the specific brain regions are worthy of attention while investigating the expression of different subtypes of Nox in CNS.
The past decade has been consistently working on the linkage between oxidative stress and brain dysfunction. Plenty of evidence reveals the upsurge in the activity and expression of Noxes in brain dysfunction. At the second messenger level, Nox-derived ROS has been directly or indirectly engaged in the downstream signal transduction pathway. However, the coupling of various Nox subtypes to tissue-specific signal transduction pathways is yet to be fully elucidated. Antibodies, detection systems of Nox activity, and NOX inhibitors are often nonspecific, making the relevant research very challenging. In the future, we are targeting further in-depth research focusing on Noxes and specific Nox inhibitors in the CNS.
Footnotes
Authors' Contributions
J.F. conceived and wrote the article, and was a major contributor to the article. R.S. and Z.-H.Q. revised the article. All authors read and approved the final article.
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
This work is supported by the Natural Science Foundation of China (Nos. 81730092 and 81973315).