
Abstract
Significance:
During aging, excessive production of reactive species in the liver leads to redox imbalance with consequent oxidative damage and impaired organ homeostasis. Nevertheless, slight amounts of reactive species may modulate several transcription factors, acting as second messengers and regulating specific signaling pathways. These redox-dependent alterations may impact the age-associated decline in liver regeneration.
Recent Advances:
In the last few decades, relevant findings related to redox alterations in the aging liver were investigated. Consistently, recent research broadened understanding of redox modifications and signaling related to liver regeneration. Other than reporting the effect of oxidative stress, epigenetic and post-translational modifications, as well as modulation of specific redox-sensitive cellular signaling, were described. Among them, the present review focuses on Wnt/β-catenin, the nuclear factor (erythroid-derived 2)-like 2 (NRF2), members of the Forkhead box O (FoxO) family, and the p53 tumor suppressor.
Critical Issues:
Even though alteration in redox homeostasis occurs both in aging and in impaired liver regeneration, the associative mechanisms are not clearly defined. Of note, antioxidants are not effective in slowing hepatic senescence, and do not clearly improve liver repopulation after hepatectomy or transplant in humans.
Future Directions:
Further investigations are needed to define mutual redox-dependent molecular pathways involved both in aging and in the decline of liver regeneration. Preclinical studies aimed at the characterization of these pathways would define possible therapeutic targets for human trials. Antioxid. Redox Signal. 35, 832–847.
Introduction
According to the World Health Organization, aging is a biological process characterized by the progressive accumulation of a broad range of cellular and molecular injuries, which leads to structural and functional decline, altered conservation and repair systems, increased exposure to disease and death (184a). Mutual damage underlying this process was described as “hallmark of aging,” and includes genomic instability, mitochondrial impairment, telomere shortening, epigenetic modifications, proteostasis failure, defective nutrient sensing, cellular senescence, collapse of stem cell pool, and flawed intercellular network (95). These hallmarks may be triggered by (and may also cause) free radicals and related oxidants produced by endogenous metabolism and derived from the environment, supporting the Free Radical Theory of Aging (67, 178). Since this theory cannot completely explain the aging process, it has been recently integrated by a loss of adaptive homeostasis, which is characterized by the impaired protective capacity against environmental and metabolic stressors (132).
The liver is a unique organ designated to the maintenance of whole-body homeostasis by modulating energy metabolism, clearance of exogenous and endogenous toxic substances, and biosynthesis of molecules (139). Although several morphological alterations occur in old age, the liver shows a peculiar aging process characterized by a noticeable preservation of its functions (3, 86). Organ reserve in old people is impaired, leading to increased susceptibility to acute liver injury and liver fibrosis, with a more serious course of diseases with respect to young/adult patients (46). Indeed, aging increases both the risk of several liver diseases and liver-related mortality rate (51, 146, 157). Nevertheless, the most remarkable consequence of aging on the liver refers to its proliferative capacity (168). Despite several progresses in the field, the molecular mechanisms underlying the impairment of regenerative process in aging liver remain to be fully elucidated. Age-related phenotypic alterations potentially impacting liver regeneration include both genomic and epigenomic alterations, dysfunction of mitochondria and nutrient sensing, cell senescence, and low-grade inflammation (73).
This review aims to epitomize the redox-dependent mechanisms, which impair the hepatic regeneration in aging. Discussion of basic aspects of redox biology is beyond the scope of this article, and the reader is referred to several outstanding reviews on this topic (50, 155, 158, 159). The term “reactive species,” referring to reactive oxygen and nitrogen species, will be used throughout this review. After a brief presentation of the redox reactions occurring in the liver and the age-related modifications in hepatic redox balance, we outline the recent knowledge of the role played by reactive species in the disruption of the adaptive regenerative homeostasis in old age.
Redox Homeostasis in the Liver
Even though every tissue and organ exert their own metabolic activity, the liver represents the metabolic hub of the human body for nutrients and xenobiotics. Indeed, the liver accounts for carbohydrate and lipid metabolism, ammonia metabolism, biotransformation of endogenous and exogenous by-products, and bile synthesis. To let these different metabolic pathways efficiently run in parallel, and to limit futile cycles, liver parenchyma is functionally specialized through metabolic zonation related to oxygen supply (Fig. 1). Indeed, periportal (zone 1) differ from perivenous hepatocytes (zone 3) in subcellular organelles, enzymes, translocators, receptors, exhibiting different functional capacities (80). Metabolic zonation can be considered as a dynamic process, since several factors (such as nutrients, hormones, but also oxygen and reactive species) may regulate gene expression patterns and enzyme distributions (85). Anabolic and catabolic pathways in liver cells are regulated by the following factors:
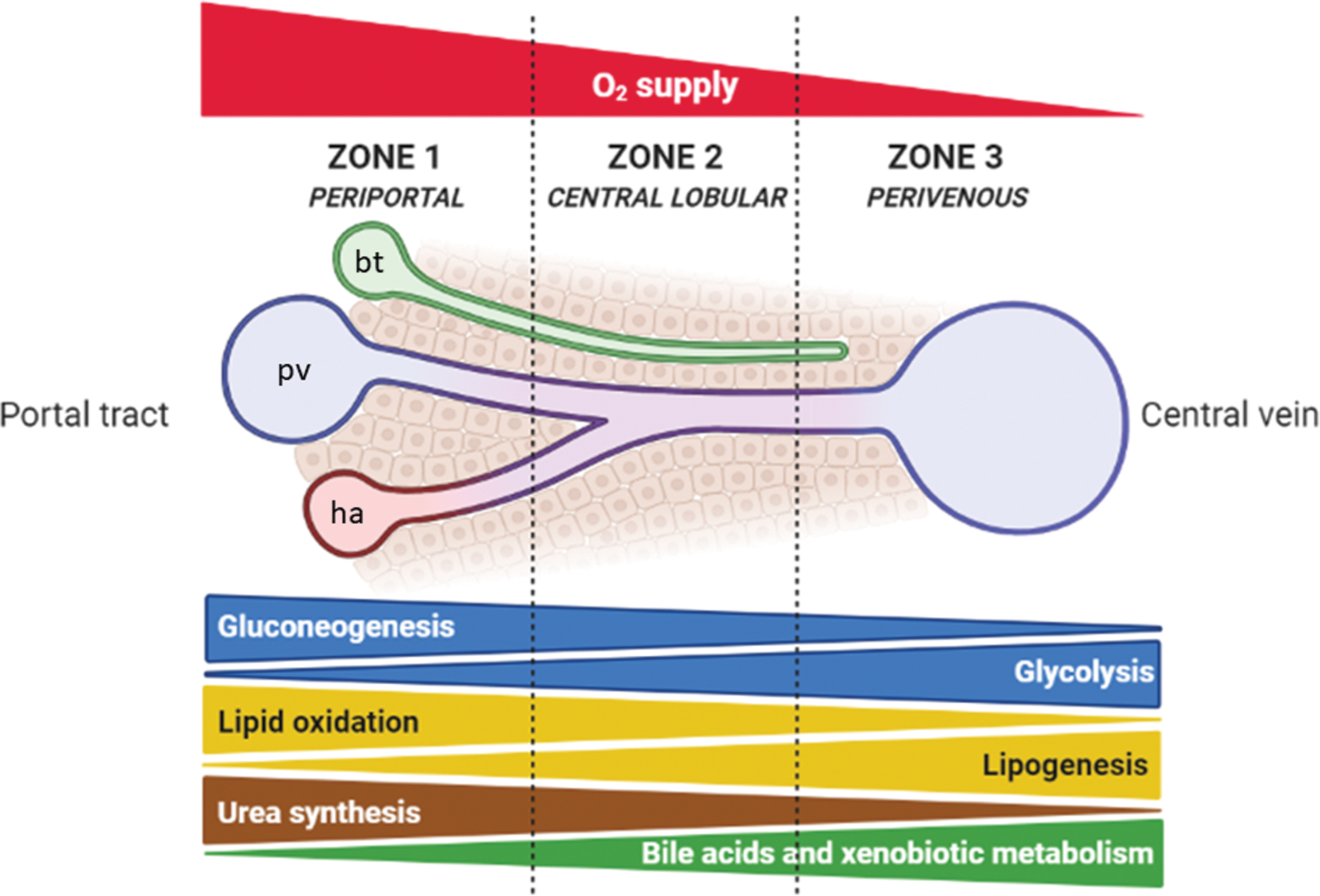
Cellular compartments—several oxidative reactions occur in mitochondria and peroxisomes, while reductive reactions take place mainly in the cytosol (69);
availability of coenzymes—the couple oxidized/reduced nicotinamide adenine dinucleotide (NAD+/NADH) for oxidation reactions in catabolism, and the couple oxidized/reduced nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) as reducing agents in anabolic reactions (186);
cellular AMP/ATP ratio—increased ATP consumption or reduced ATP production activates AMP-activated protein kinase (AMPK) to induce catabolic reactions, while inhibition of AMPK by high ATP availability switches metabolism toward anabolic pathways (52).
Hepatic sources of reactive species
Reactive species in the liver are produced by several metabolic reactions, occurring in different cell types and cellular compartments (Fig. 2).
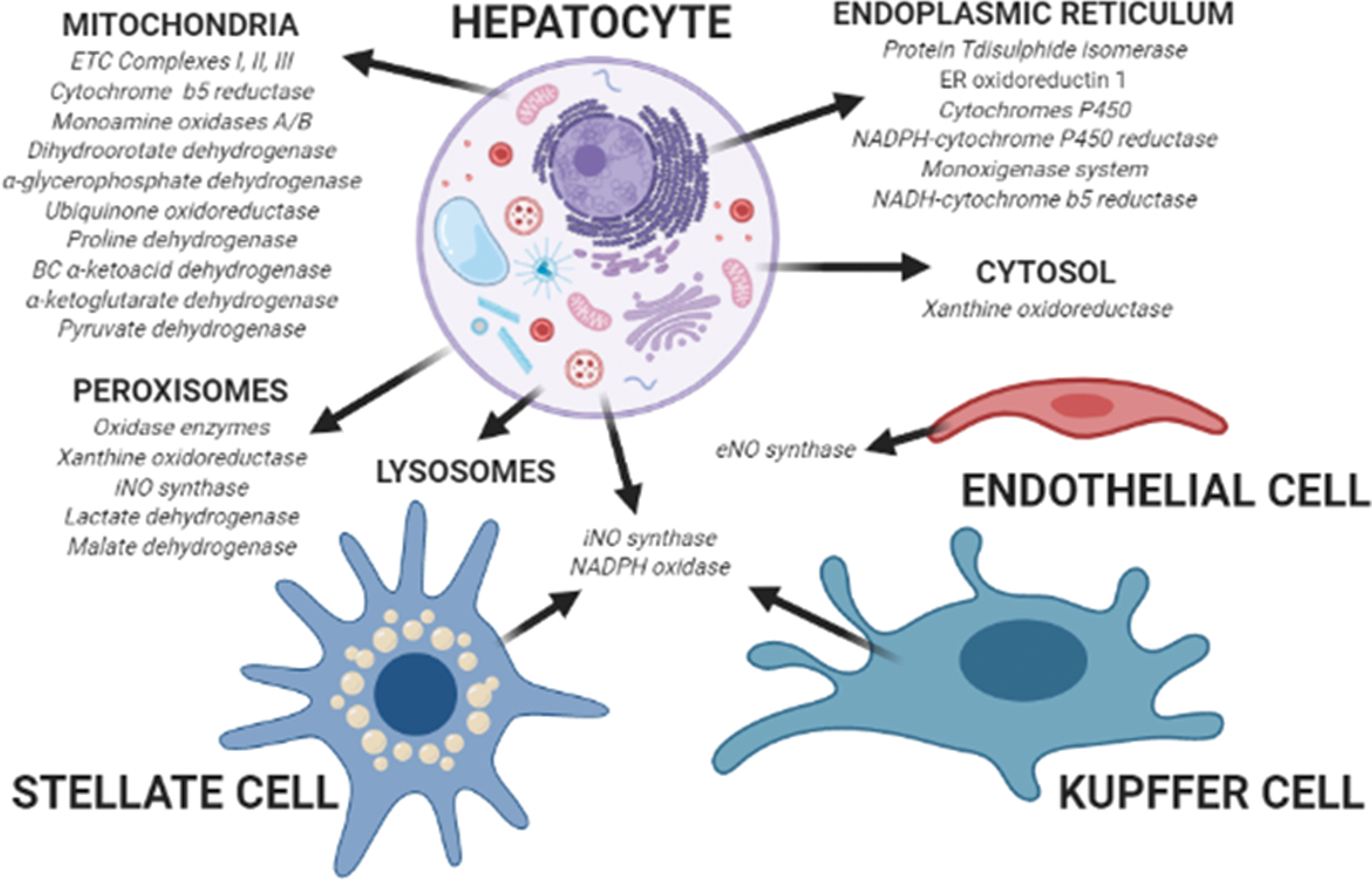
Among the different metabolic enzymes producing reactive species in the cytosol, xanthine oxidoreductase (XOR) plays a determinant role by catalyzing the last two steps of purine catabolism, which involve oxidation of hypoxanthine to xanthine, and the further oxidation of xanthine to uric acid (11). In the human liver, XOR is mostly present as a dehydrogenase, transferring electrons to NAD+; however, several stimuli can convert the enzyme into the oxidase isoform, transferring electrons to O2 producing reactive species (162).
Mitochondria consume ∼95% of O2 in the liver to oxidize different substrates by transferring electrons to carriers such as NAD+, flavin mononucleotide, and flavin adenine dinucleotide. In turn, the reduced forms of these carriers transfer electrons to the respiratory chain complexes, and finally to O2 in a multistep process associated with production of reactive species (198). In physiological conditions, liver mitochondria produce 13%–15% of the total H2O2 amount in the liver by using ∼2% of the total O2 consumed (22, 198). The main mitochondrial sources of reactive species are Complexes I and III, even though production by Complex II has also been described (108, 133). Other mitochondrial sources of reactive species are cytochrome b5 reductase (118) and monoamine oxidases A/B (130) in the outer membrane, dihydroorotate dehydrogenase (53), α-glycerophosphate dehydrogenase (109), the electron transfer flavoprotein ubiquinone oxidoreductase (184), proline dehydrogenase and the branched-chain α-ketoacid dehydrogenase complex (122) in the inner membrane, and α-ketoglutarate dehydrogenase and pyruvate dehydrogenase in the matrix (122).
Peroxisomes are organelles involved in several metabolic pathways, including fatty acid catabolism, metabolism of
The endoplasmic reticulum (ER) accomplishes several important functions in liver cells such as protein synthesis, folding and trafficking, lipid and steroid metabolism, calcium storage, and xenobiotic metabolism (93). Protein folding requires oxidation of sulfhydryl groups, a process favorized by a high oxidized glutathione (GSSG) to reduced glutathione (GSH) ratio in the ER lumen (30). The electron transport for protein folding involves protein disulfide isomerase (PDI) and ER oxidoreductin 1 (ERO1): while PDI directly accepts electrons, ERO1 transfers electrons to O2 as final acceptor, leading to the production of ∼25% of hepatocellular reactive species (38, 98). Xenobiotic metabolism includes phase I mono-oxygenation reactions involving cytochromes P450 and a flavoprotein (NADPH-cytochrome P450 reductase), and phase II conjugation reactions. The microsomal mono-oxygenase system, which further includes squalene mono-oxygenase, fatty acid desaturase, and 7-dehydrocholesterol reductase (involved in lipid and steroid metabolism), is one of the major sources of reactive species in the ER. Indeed, the electron transfer from NADPH to P450 during the mono-oxygenation reaction presents with a leakage that contributes to the generation of reactive species (194). A further leakage in the electron transfer process with consequent production of reactive species may occur through the NADH-cytochrome b5 reductase during fatty acid desaturation (141).
Lysosomes are fundamental for autophagy, an extremely conserved recycling process characterized by removal of cellular components. More than being merely devoted to final degradation of molecules, these organelles preserve energy balance in hepatocytes by modulating mitochondria quality, metabolic enzymes, and substrates availability (97). Thus, lysosomes protect against oxidative stress by removing damaged mitochondria, unfolded proteins, and toxic cellular waste compounds (131, 135). Nevertheless, lysosomes are provided of an electron transport chain including ubiquinone (UQ), which is reduced by the presence of cytosolic NADH with oxygen as final electron acceptor; acidification of the lysosomal matrix promotes the partial reduction of O2 with production of reactive species (62, 119).
Further sources of reactive species in the liver are NADPH oxidase and NO synthases (NOS). NADPH oxidase is found in both parenchymal and nonparenchymal liver cells, in phagocytic and nonphagocytic isoforms (40). Kupffer cells have a phagocytic form of NADPH oxidase, which produces high quantity of reactive species (82). Hepatic stellate cells express a nonphagocytic NADPH oxidase isoform, which produces mild amounts of reactive species but can be activated by different fibrogenic stimuli (2, 10, 195). NOS in the liver are present as constitutive in endothelial cells (endothelial NO synthase [eNOS]) and inducible (inducible NO synthase [iNOS]) in hepatocytes, Kupffer cells, endothelial and stellate cells. Production of NO by eNOS is crucial to preserve the hepatic blood flow (42). The role of iNOS in the liver is extremely intricate, since it can be modulated by different cytokines leading to the formation of several reactive species, with both protective and damaging effects (42, 166).
Hepatic antioxidants
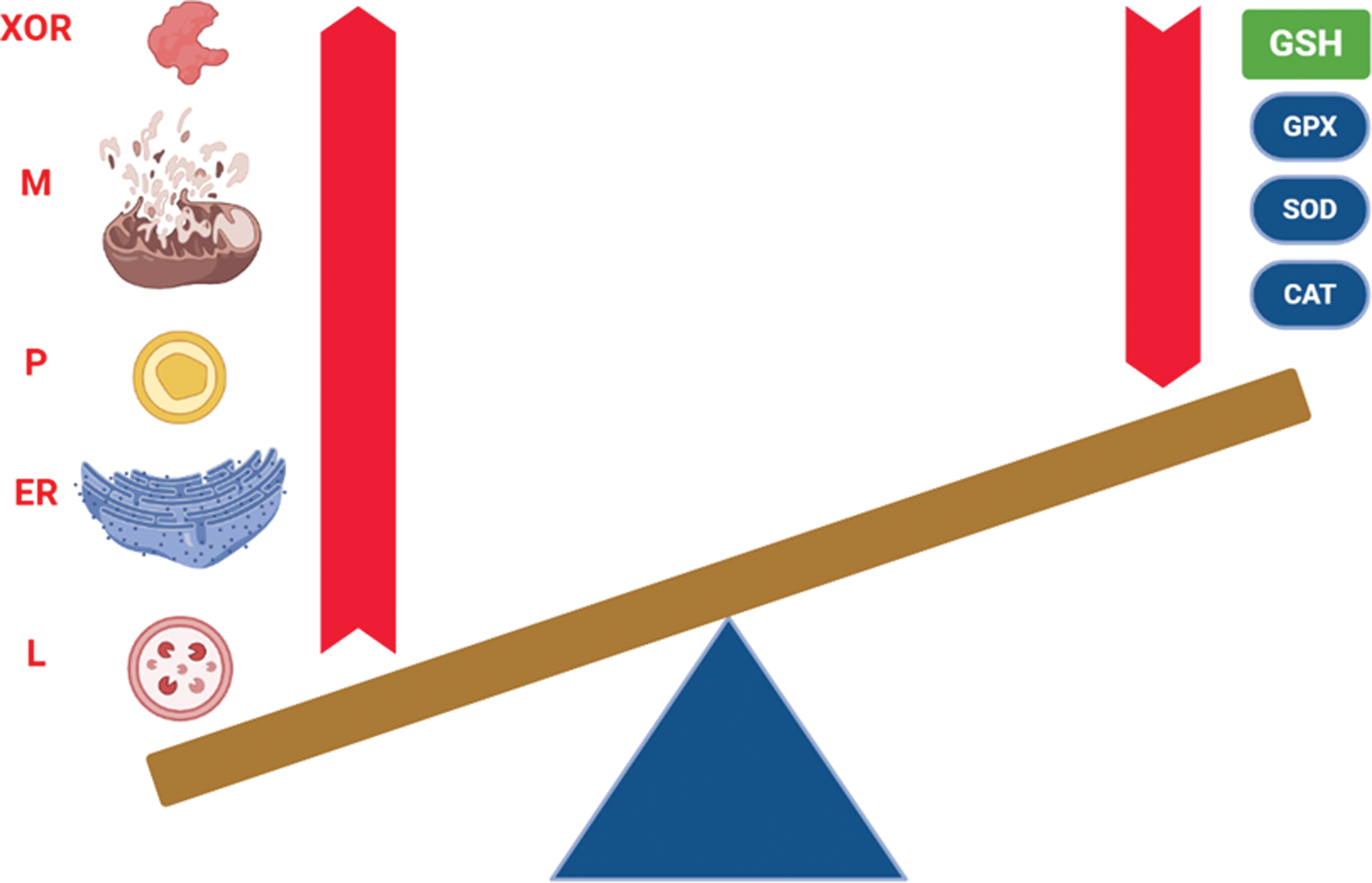
The liver is supplied of several antioxidants to counteract the production of reactive species and maintain redox homeostasis. Hepatic endogenous antioxidants may be grouped into nonenzymatic and enzymatic. Endogenous antioxidants form a huge system with interconnected redox reactions, which occur in the cytosol and within subcellular organelles (Fig. 3).

Nonenzymatic antioxidants include GSH, thioredoxin (TRX), and UQ. Being the most concentrated intracellular antioxidant in the liver, GSH is a tripeptide including a sulfhydryl group in a cysteine residue, allowing its reductant activity for several oxidized enzymes and antioxidants (88). TRX is a tetrapeptide characterized by two sulfhydryl groups in two cysteine residues subjected to reversible redox reactions by the NADPH-dependent thioredoxin reductase (TRXR). Playing a fundamental role in the hepatic redox homeostasis, the reduced form of TRX is necessary to reduce oxidized peroxiredoxin (PRX) (121). UQ (or coenzyme Q) is present in all liver cells and, because of its lipophilic properties, it is located within several cell membranes, acting as antioxidant in its reduced form (ubiquinol, UQH2). Nevertheless, it can be also partially reduced (ubisemiquinone, UQ•−), and the capacity to undergo between three redox states allows UQ to act as an electron carrier from Complexes I and II to Complex III in the mitochondrial respiratory chain (182). In rat liver, UQ is mostly concentrated in the Golgi vesicles, followed by mitochondria and lysosomes (81).
Enzymatic antioxidants include isoforms of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPX), glutathione reductase (GR), TRXR, and PRX. SOD accounts for the dismutation of superoxide anion; in humans, there are three different SOD isoforms, containing Cu/Zn (SOD1 and SOD3) or Mn (SOD2) in their active site (57). In rat liver, Cu/Zn-SOD is mostly sited in the cytosol and lysosomes, while Mn-SOD is mainly located in mitochondria, even though Cu/Zn-SOD is also found in the intermembrane space (120). The liver expresses the highest amount of both CuZn-SOD and Mn-SOD among human tissues (100). CAT acts as an iron-dependent peroxidase, which transforms two H2O2 into two H2O and one O2. In humans, CAT activity is highest in the liver and erythrocytes (63). GPX isoforms are selenium-dependent peroxidases, which become oxidized by transforming H2O2 into H2O or organic hydroperoxide (ROOH) to corresponding alcohol (ROH) and are reduced again by GSH. There are eight GPX isoforms in humans, but only GPX1, GPX2, GPX4 (phospholipid hydroperoxidase), and GPX7 are located in the liver (171). GPX1, GPX2, and GPX7 target H2O2 in the cytosol (GPX1), extracellular space (GPX2, GPX7), ER (GPX7), mitochondria (GPX1), while GPX4 targets ROOH in the cytosol, mitochondria, and nucleus (171). GR, together with cytosolic and mitochondrial TRXRs (TRXR1 and TRXR2, respectively), uses NADPH to reduce disulfides to dithiols in the liver. In the reaction catalyzed by GR, the dithiol reduces GSSG; being selenoproteins, TRXRs form selenothiol pairs, which reduce TRX (106). Similar to GPX, the six isoforms of PRX (thiol hydrolases) can be oxidized by H2O2 or ROOH, being reduced again by TRX. The human liver expresses all PRX isoforms, targeting both H2O2 and ROOH in the cytosol (PRX1, PRX2, PRX5, PRX6), mitochondria (PRX3, PRX5), extracellular space (PRX4), nucleus (PRX5), and endosomes (PRX3, PRX6) (136).
The Aging Liver and Redox Balance
Liver structure changes in aging
Age-related modifications occurring in the liver include both anatomical and physiological features. With respect to adults, reduction in liver volume of ∼20%–40% and decreased blood flow of 35%–50% are the most reported alterations in aged subjects (76, 91, 185, 193). Lower blood flow can be considered as the main cause for reduced liver volume, even though the sinusoidal perfusion rate does not change with aging (179). However, alterations in hepatic microcirculation characterized by thickening of endothelium and reduction of endothelial cell fenestrations occur in aging (36, 103). Even though these reports would suggest a reduced oxygen delivery to liver cells, no significant changes in hepatic oxygenation arise in aging liver (33). Another macroscopic alteration of the aging liver is referred to its color, which is described as “brown” because of an accumulation of lipofuscin (147). Of interest, lipofuscin is derived by the oxidation of unsaturated fatty acids and proteins, thus representing a sign of chronic oxidative stress (79).
Age-related microscopic liver alterations include increased number of hepatocytes with polyploidy, reduced number of mitochondria but increased mitochondrial volume (143, 146, 183). Furthermore, studies report a greater volume and number of secondary lysosomes and residual bodies (dense bodies) (145, 147). The surface of both the smooth and the rough ER is decreased in old hepatocytes, and associated with a reduced synthesis of enzymes and microsomal proteins (3).
Changes in hepatic morphology, structure, and function during aging may be underpinned by alterations in redox homeostasis determined by both an enhancement of reactive species production and defects in the endogenous antioxidant systems (Fig. 4). However, it is worth to note that, despite resulting effective in experimental models, no clinical intervention aimed at restoring this homeostasis—such as antioxidant supplementation—proved to be beneficial in human aging and age-related liver diseases (18, 37, 58).
Production of reactive species in the aging liver
Age-related changes in the liver are consistent with increased levels of reactive species, which may derive from almost all the sources previously described.
Hepatic XOR expression and activity (mostly as oxidase) are increased, and associated with lipid peroxidation in old mice, sustaining a determinant role for XOR-induced oxidative stress in the aging liver (175, 176).
Mitochondrial dysfunction represents a major hallmark of liver aging. Early evidence on hepatocytes showed that age was associated with decreased mitochondrial membrane potential, but an increase in mitochondrial size and peroxide production (143). Our experiments on old rats confirmed impairment in hepatic mitochondrial bioenergetics, characterized by dissipation of membrane potential and alteration of ATP homeostasis; these findings are associated with altered composition of mitochondrial membranes and high production of reactive species (13, 153). Both animal and human studies also demonstrate that respiratory chain complexes present with defective expression and activity in old liver, acting as major sources of reactive species (110, 111, 116).
Aging greatly influences both metabolism and biogenesis of peroxisomes in the liver (191). In particular, the amount of peroxisomal reactive species is tightly controlled during young and adult age; redox balance is lost in the aging liver, characterized by excess of peroxides and related oxidative damage sustained by a decline of scavenging activity rather than enhanced oxidation reactions (127, 170). Peroxisomal homeostasis is further impaired in aging by both accumulation of damaged organelles due to defective autophagy, and dysfunction of peroxisomal proliferation (29).
The aging liver exhibits markers of ER stress, particularly related to the unfolded protein response (UPR) (61). Components of the UPR pathway are oxidatively damaged in the liver of old rodents (48, 134). Even though the possible source of reactive species accounting for such oxidative injury remains to be fully elucidated, it is conceivable that defective cytochromes P450 could play a role in triggering hepatic ER stress (174, 188). Reduced activity of NADH-cytochrome b5 reductase also occurs in liver microsome of aged people, which may also contribute to the impairment of redox homeostasis in the ER (60, 101).
Lysosomal activity in the aging liver is reduced, impacting autophagy and removal of damaged organelles, as well as availability of metabolites (148). The accumulation of peroxide within lysosomes leads to the formation of lipofuscin in aged hepatocytes, which in turn results in a malfunction of lysosomal enzymes in autophagosomes, causing accumulation of dysfunctional mitochondria and proteins (89, 187).
Reactive species derived by NADPH oxidases and NOS may be implicated in the aging process of several tissues and organs, such as central nervous system, cardiovascular system, and kidneys (12, 25, 43, 149). Future research needs to investigate the role of NADPH oxidase- and NOS-derived reactive species in the aging liver.
Endogenous antioxidants in the aging liver
A decline in endogenous antioxidants occurs in old age, promoting a disruption of redox balance. Glutathione and its associated enzymes show important alterations in the aging liver: hepatic GSH progressively decreased in rats with advancing age, associated with the reduction of GPX (105, 161). This observation was not confirmed in the liver of old mice, where GSH levels are stable but an increase in GSSG with respect to young animals suggests a pro-oxidant hepatic environment (78, 172). Moreover, hepatic GSH decreased during time in strains of mice prone or resistant to senescence; this time-dependent decrease also occurred for the activities of hepatic GPX and SOD (68).
Even though studies on the hepatic TRX level in the aging liver are lacking, an increase in TRXR activity, associated with high mitochondrial TRXR expression, arises in three different long-lived mutant mice models (129). Maintenance of TRX in a reduced state may be of extreme importance to counteract reactive species in senescent tissues and organs (190).
Despite several studies using exogenous supplementations of coenzyme Q10 to counteract oxidative stress in old age, there is lack of consistent data related to UQ concentration in the aging liver. Endogenous UQ content seems not to change in the liver of old mice, even though there is a shift to oxidized form in centenarians, suggesting a severe tissue shortage of Q10 (113, 160).
The expression and activity of Mn-SOD did not change with age in rat liver, while there was a decrease in CuZn-SOD; no difference was reported in the liver of young and old mice (84, 116, 142). Of interest, the cytoplasmic CuZn-SOD isoform resulted as the most oxidized protein in the liver of old rats (31). Other than SOD, even CAT and GPX activities are reduced in the liver of old rats (26, 151). With respect to young and adults, activities of GPX and SOD were reduced while GR activity did not change, and CAT activity increased in the liver of old rhesus monkeys (28). Moreover, old hepatocytes fail to respond to oxidant stimulation for the induction of GPX1 (140). Among other antioxidant enzymes, an overoxidation of PRX3 occurs in the liver of old rats, a condition associated with a loss of enzymatic activity (112).
A further antioxidant effect may be exerted by uncoupling proteins (UCPs), a family of mitochondrial proton transporters, which uncouple respiration from ATP production, playing an important role in the aging process and life span (138). UCPs expression is tissue specific, so that their impact on senescence may be variable in different organs. UCP2 is the most expressed isoform in the liver, even though in physiological conditions it is mainly found in nonparenchymal cells (90). UCP2 expression in hepatocytes is triggered by overload of energetic substrates and increased oxidative pressure, as it happens in liver steatosis and steatohepatitis (32, 154). Age-related changes in hepatic UCP2 expression need further investigation, since controversial results (increased levels or no modifications) are reported in different animal models (9, 13).
Overall, studies suggest that the altered redox homeostasis in different compartments of the aging liver can be characterized by a combination of increased production of reactive species counteracted by relatively stable antioxidants, plus decreased antioxidants facing normal amounts of reactive species.
Regeneration in the Aging Liver
The liver is characterized by unique features of regeneration after various injuries (genetic, metabolic, viral, toxic, or immunologic). In the healthy liver, remaining functional parenchymal cells replace the loss of hepatocytes (104). However, when a continual or acute liver injury overcomes the replicative capacity of mature hepatocytes, activation and differentiation of hepatic progenitor cells (HPCs, also termed oval cells in rodents) through ductular reaction will help replace damaged parenchyma (49).
Failure in regenerative capacity is the most striking alteration in the aging liver. With respect to young and adults, regeneration after partial hepatectomy in old rats is reduced because of a lag in cell cycle entry and lower number of replicating hepatocytes (Fig. 5) (23, 56, 75, 169). Dysfunctional liver regeneration in aging may be detrimental for cell or organ transplantation. Indeed, transplantation of hepatocytes from old rats leads to the formation of smaller clusters as compared with young donors, suggesting a drop in regenerative capacity (152). In humans, liver regeneration rate among old donors is lower as compared with young ones, suggestive of a decrease in HPC population (124). Molecular mechanisms underlying the reduced regeneration capacity are complex, but include changes in signal transduction pathways at translational and post-translational levels (168).
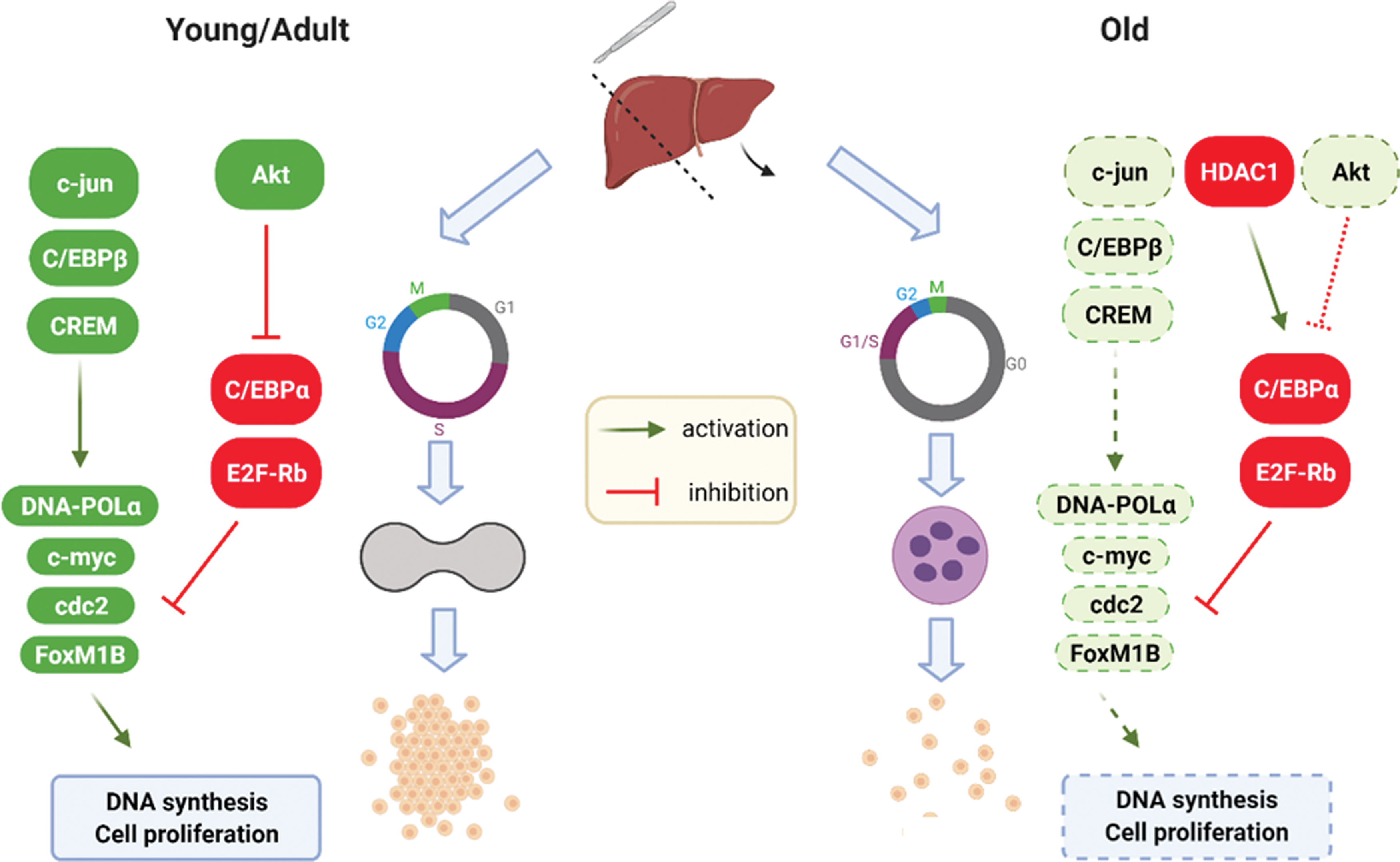
In healthy liver, the CCAAT-enhancer-binding protein α (C/EBPα) reduces proliferation of parenchymal cells through direct inhibition of cyclins (181). In the aging liver, C/EBPα is no more able to inhibit cyclins but represses the E2F transcription factor, impairing hepatic regeneration after partial hepatectomy (75). This repression occurs by an epigenetic modification determined by the binding of C/EBPα with the chromatin remodeling protein Brm, and this complex also inhibits the transcription factor Forkhead box M1b (FoxM1b) (180). Furthermore, the aging liver shows increased expression of histone deacetylase 1 (HDAC1), which interacts with the C/EBPα–Brm complex through a cyclin D3-dependent mechanism (180). An additional contribution to the activation of C/EBPα may derive from the impaired insulin signaling and reduced Akt activity described in the old liver (27, 156). Decreased regeneration in the aging liver also depends on further epigenetic modifications, relying on decreased acetylation and increased phosphorylation of H3 histones, leading to chromatin impairment (83).
Classified as HDACs, sirtuins are NAD+-dependent deacetylases involved in the modulation of aging, metabolism, and genome preservation (35). In particular, Sirtuin-1 (SIRT1) is implicated in the control of aging and regulation of circadian clock in the liver (66, 114). A determinant role for SIRT1 in regeneration of the aging liver comes from evidence that liver-specific knockout of SIRT1 in mouse impairs response to partial hepatectomy, and decreased expression of SIRT1 occurs in the aging liver (15, 144).
Other than epigenetic alterations, impaired regeneration in old liver is associated with increased levels of genes/proteins regulating autophagy and apoptosis, such as microtubule-associated proteins 1A/1B light chain 3B (LC3), autophagy-related protein 5 (Atg5), and caspase-3 (47). Of interest, knockdown of Atg5 in the mouse liver impairs regeneration after partial hepatectomy, inducing hepatocyte senescence and mitochondrial dysfunction, suggesting that preservation of autophagy would be beneficial for maintaining liver regeneration in aging (173).
Impaired regulation of mitosis may be crucial to the decline of regeneration in the aging liver. Budding uninhibited by benzimidazole-related 1 (BubR1) prevents chromosome unequal segregation during mitosis, and its decreased expression is associated with cell senescence (6 –8). BubR1 insufficiency in mice leads to failure in liver regeneration by altering desmocollin 1 (DSC1), a desmosome transmembrane cell adhesion protein greatly expressed in the liver (77). BubR1 is a transcriptional target of Yes-associated protein (YAP), whose expression increases in old as compared with young mice undergoing partial hepatectomy (128). Macrophage-stimulating proteins 1/2 (MST1/2) are inhibitory kinases, which may phosphorylate YAP in the aging liver, contributing to impaired regeneration (94).
Other than affecting the regenerative potential of hepatocytes, aging may weaken the activation and proliferation of HPCs. The importance of HPCs in regenerating the aging liver is highlighted by evidence that induction of hepatocyte senescence through specific knockout of Mouse double minute 2 homolog (Mdm2) gene promotes rapid ductular reaction and activation of oval cells (96). Furthermore, a mouse model of β-catenin knockout shows hepatocyte senescence and an inflammatory microenvironment that promotes HPC activation via C1q (70). Nevertheless, an age-related impairment in HPC-dependent regeneration occurs in a rodent model of ductular reaction, suggesting that the stem cell exhaustion is a further hallmark of the aging liver (34).
Impaired Signaling in the Aging Liver: Redox Deregulation of Hepatic Regeneration
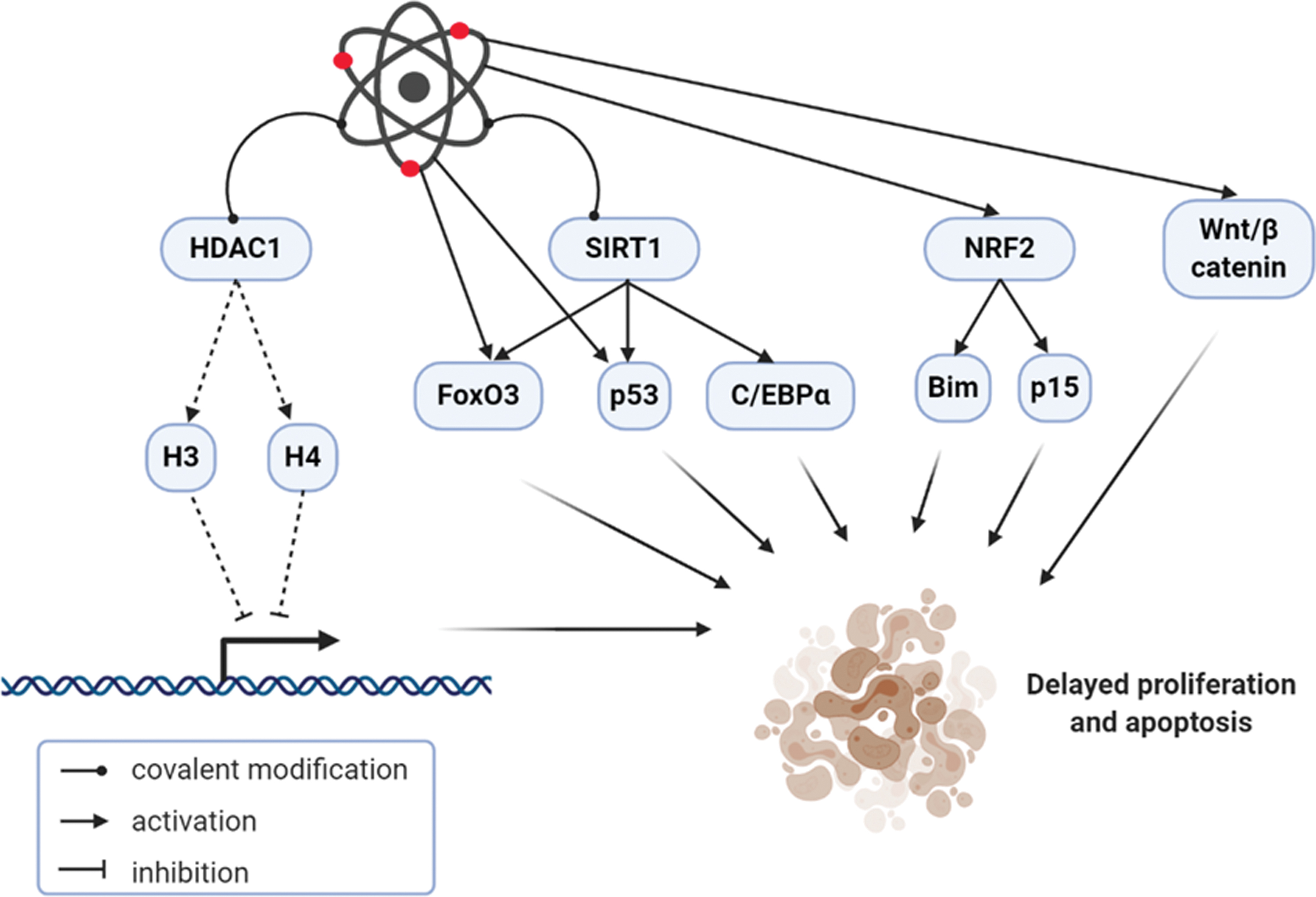
Several investigations suggest that oxidants, rather than being always dangerous, could exert a positive effect, since small quantities of reactive species may induce protective pathways against widespread stress (71). Thus, the free radical theory of aging may be replaced by the “cell signaling disruption theory of aging,” proposing that reactive species contribute to aging by altering the cellular signaling network (177). In the liver, oxidants modulate age-related processes, which may impact liver regeneration. These consist in epigenetic and post-translational modifications, but also activation/repression of transcription factors, which include (but are not limited to) Wnt/β-catenin, the nuclear factor (erythroid-derived 2)-like 2 (NRF2), members of the Forkhead box O (FoxO) family, the p53 tumor suppressor (41). Furthermore, reactive species may reduce HPC activation and proliferation because the regenerative niche is infiltrated by neutrophils recruited by chemokines from activated hepatic stellate cells in the aging liver (34).
Reactive species may dose- and time-dependently induce covalent modification of HDAC1, reducing acetylation of histones H3 and H4 with consequent derepression of gene transcription (44). Similarly, oxidants may covalently modify the deacetylase SIRT1, leading to impaired regulation of target proteins such as FoxO3 and p53 (24, 74). Of note, high amounts of NAD+ may activate SIRT1, which can in turn deacetylate C/EBPα (192). Further research needs to define whether this mechanism might occur in the aging liver, with possible impact on organ regeneration.
The Wnt/β-catenin signaling is implicated in multiple biological pathways, ranging from embryonic and adult tissues development to stem cell fate, cancer, and aging (65). Antagonism of Wnt/β-catenin signaling by age-related oxidative stress could contribute to the development of the most prevalent diseases in the elderly (99). Wnt/β-catenin pathway is among the first signals to be activated by partial hepatectomy or acetaminophen overdose, promoting liver regeneration in rodents (5, 17, 107, 115). Studies using mice with specific hepatic deletion of β-catenin confirm the importance of this pathway in liver repair, since they show delayed hepatocyte proliferation after partial hepatectomy (150, 164). On the contrary, hepatocellular overexpression of β-catenin accelerates liver regeneration (117). Of interest, activation of Wnt/β-catenin and inhibition of p53 signaling are necessary for human hepatocyte proliferation (123). Wnt/β-catenin signaling is also determinant in the regulation of HPC biology. In rodent models of ductular reaction, HPCs exhibit the activation of this pathway, enter cell cycle after stimulation with Wnt3a, and may repopulate the liver in case of impaired hepatocyte proliferation (72, 189). Furthermore, macrophage Wnt3a expression facilitates hepatocyte regeneration through HPC activation and differentiation (21). Future research will clarify how the age-related redox perturbations may interfere with the Wnt/β-catenin pathway to modulate liver regeneration.
NRF2 is the most important controller of the adaptive response to oxidative stress by regulating the expression of antioxidant enzymes; indeed, the age-dependent defective antioxidant response relies on a reduced effectiveness of NRF2 signaling (197). The role of NRF2 in liver regeneration is unique and needs further clarification in the aging liver. In NRF2-knockout mice, hepatocyte proliferation is significantly delayed after partial hepatectomy, suggesting that NRF2 is necessary to the process of liver repair (16). The augmenter of liver regeneration, a regulator of hepatic tissue repair, is a target of NRF2 and exerts an antioxidant effect, connecting impaired redox homeostasis to hepatocellular proliferation (39). Nevertheless, constitutive activation of NRF2 impairs liver regeneration by delaying hepatocyte proliferation and enhancing apoptosis via p15 and Bim, respectively (87). However, chemical activation of NRF2 pathway limits age-related changes in hepatic morphology, function, and fibrosis in rats (196). The different results obtained from diverse approaches of NRF2 stimulation suggest that constitutive—rather than chemical—activation of this inducible transcription factor might be detrimental instead of beneficial for the aging liver. The role of NRF2 in HPC modulation in the process of liver regeneration is intriguing and worth of investigation.
The FoxO transcription factors are pivotal in the modulation of cell adaptation to age-related oxidative stress (102). Several FoxO-dependent pathways are regulated in the liver to adapt to metabolism and to respond to different stressors: FoxO1 is involved in the regulation of gluconeogenesis and lipid metabolism, while FoxO3 is essential for antioxidant response and autophagy (167). Liver regeneration after partial hepatectomy requires the inhibition of Akt-mediated FoxO1 signaling, whose liver-specific deletion in Akt1/2-deficient mice rescues the proliferative capacity of hepatocytes (126). Similarly, inhibition of FoxO3 via Wnt/β-catenin protects against oxidative stress-induced apoptosis in hepatocytes (165). It is conceivable that the age-related excess of reactive species in the liver would trigger FoxOs, with a negative impact on regeneration; further studies will clarify such a redox-dependent mechanism and provide evidence for potential future therapeutic strategies.
The tumor suppressor p53 is triggered by reactive species and may show antioxidant features to remove low quantities of oxidants; nevertheless, oxidative stress may stimulate a specific p53 transcriptional response, which modulates cell senescence and aging (59, 92). Deletion of p53 induces proliferation in the liver of mice subjected to acute injury by acetaminophen overdose: even though initiation of liver regeneration is delayed, cell cycle is fast and maintained by Akt and mammalian target of rapamycin signaling in p53-knockout mice (19, 20). Nevertheless, the deletion of p53 from HPC lines could contribute to immortalization and transformation to hepatocellular carcinoma cells (45, 125).
Conclusion
With advancing age, several changes related to structure and function occur in the liver, which may impact organ regeneration. Alteration of redox homeostasis is a main event characterizing the aging liver. Knowledge of several redox-dependent factors and pathways involved in the aging process of the liver has importantly advanced during the last few decades. Correspondingly, latest investigation broadened understanding of redox modifications and signaling related to liver regeneration. The associative mechanism between aging and altered regeneration is not clearly established. Despite redox imbalance being a common event of both aging and impaired organ repair, antioxidants neither demonstrated to be efficacious in slowing hepatic senescence, nor definitively improved liver repopulation after hepatectomy or transplant in humans. Whether molecular pathways modulated by redox homeostasis might be mutual to the process of aging and the decline of liver regeneration represents the fascinating aim of future research (Fig. 6). Preclinical studies aimed at the characterization of these pathways would define possible therapeutic targets for human trials.
Footnotes
Authors Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this work.