
Abstract
Significance:
Numerous abnormalities in T cells have been described in patients with systemic lupus erythematosus (SLE), including lymphopenia, DNA demethylation, expression of endogenous retroviruses (ERVs), increased cell death, enlarged mitochondria, production of reactive oxygen species (ROS), and the appearance of unusual CD4−CD8− T cells. Our studies propose a model in which accelerated homeostatic proliferation of T cells promotes an epigenetic and metabolic program, leading to this cluster of abnormalities.
Recent Advances:
Growing knowledge of the innate immune disorders in SLE has included increased mitochondrial size and ROS production that induces oligomerization of the mitochondrial antiviral signaling (MAVS) protein and type I interferon production, as well as DNA demethylation, upregulation of inflammatory genes, and expression of certain ERVs in SLE peripheral blood mononuclear cells. All these events are part of the cellular program that occurs during homeostatic proliferation of T cells. Evidence from a murine model of SLE as well as in human SLE reveals that increased T cell homeostatic proliferation may be a driving factor in these processes.
Critical Issues:
Despite extensive knowledge of the myriad autoantibodies in SLE and other immune abnormalities, a cogent model has been lacking to link the numerous and seemingly disparate immune aberrations. This may partly explain the general lack of new drugs specifically for SLE in over 50 years. A more coherent model of SLE would not only unify the variety of immune abnormalities is SLE but would also suggest new therapies.
Future Directions:
The model of augmented homeostatic proliferation leading to increased mitochondrial mass, ROS, DNA demethylation, and upregulation of inflammatory genes suggests strategic new targets for SLE, including antioxidants and certain inhibitors of metabolism. Antioxid. Redox Signal. 36, 410–422.
Introduction
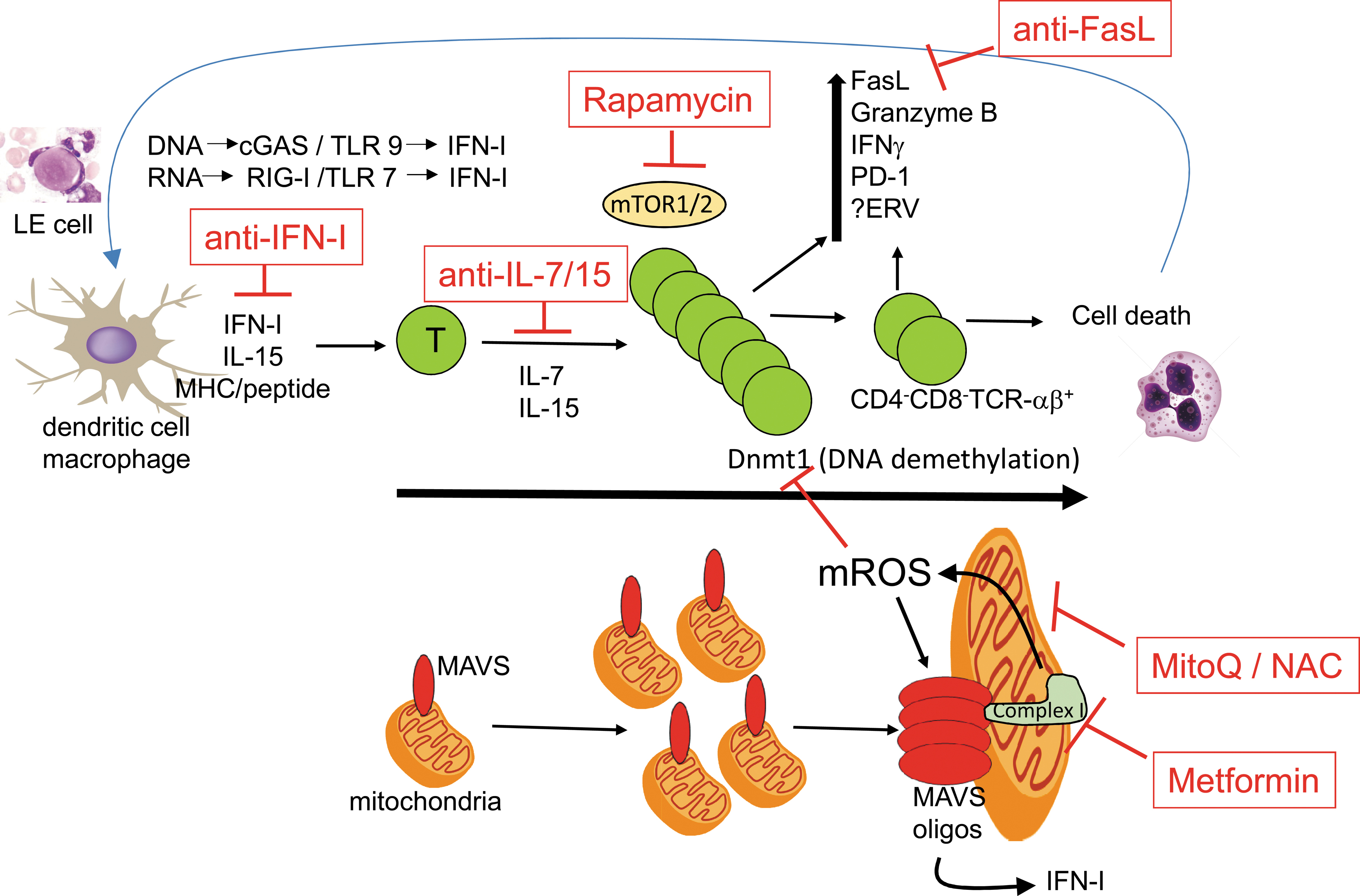
Multiple T cell abnormalities have been described by numerous investigators in systemic lupus erythematosus (SLE). These include lymphopenia, increased cell death (91, 99), enlarged mitochondria bearing increased reactive oxygen species (ROS) (13, 29, 30), oligomerization of mitochondrial antiviral signaling (MAVS) protein, increased type I interferon (IFN-I) (10, 88), hypomethylated DNA (49, 77, 105), and the appearance of an unusual population of polyclonal CD4−CD8− TCRαβ+ cells (19, 20, 78, 90). To date, no cellular process has been proposed to link these seemingly disparate aberrations of T cells. We propose a unifying model driven by increased T cell homeostatic proliferation in SLE. Homeostatic proliferation of T cells is the daily low-level cell cycling driven by self-peptide/major histocompatibility complex and the cytokines, IL-7 and IL-15. Recent studies show that progressive rounds of T cell homeostatic proliferation drive two parallel programs: (i) a metabolic program leading to enlarged mitochondria and increased ROS, which drives MAVS oligomerization and IFN-I production, and (ii) an epigenetic program consisting of DNA demethylation at discrete loci that correspond to the upregulation of numerous immune genes (25 –27) (Fig. 1). Linking these two processes may be the redox state of homeostatically expanded T cells. These programs are most manifest in the CD4−CD8− TCRαβ+ cell subset, which represents the extreme end of CD8+ T cell homeostatic proliferation (20, 48, 62, 70). This review will draw upon striking parallels in T cells between human SLE and the lupus-prone lymphoproliferative (lpr) mouse.
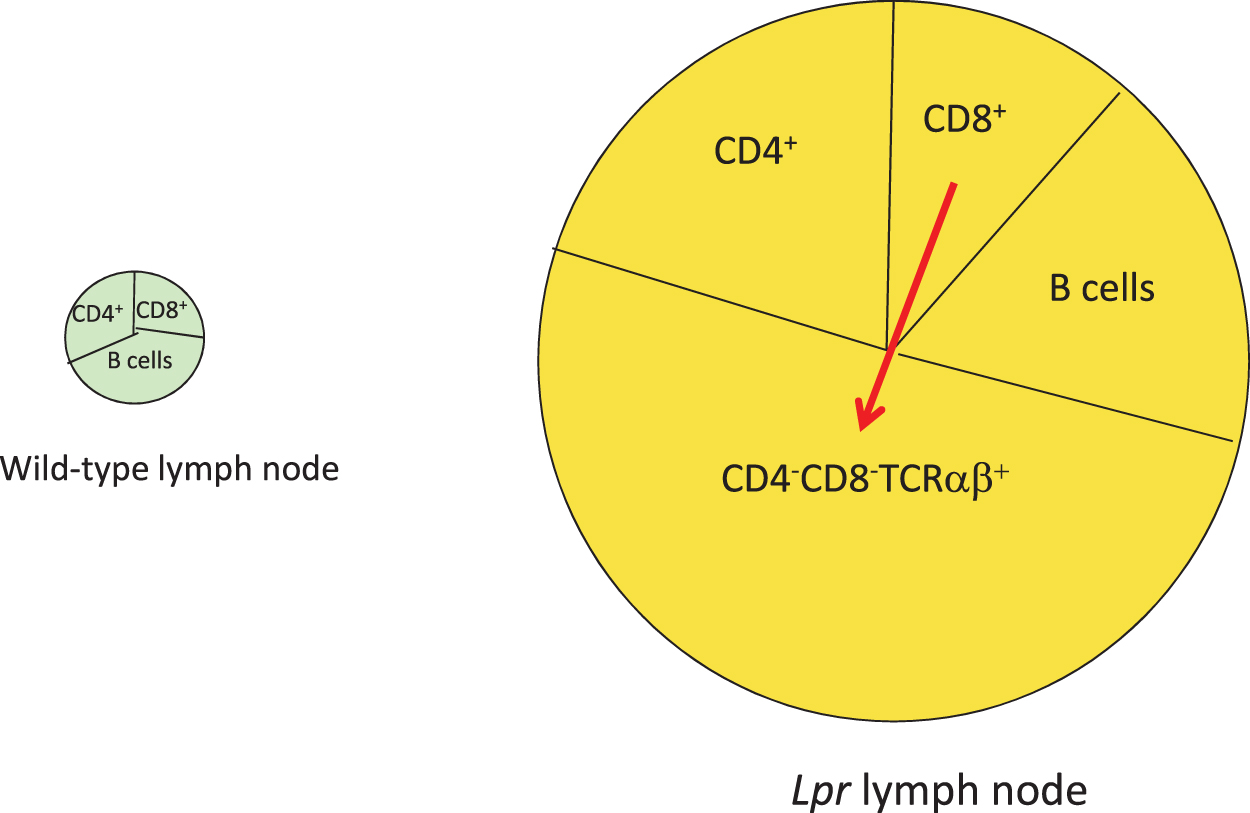
Increased T Cell Homeostatic Proliferation in Lupus Mice
During intrathymic T cell development, autoreactive T cells are largely deleted, a process known as central tolerance. Disappointingly, early investigations to demonstrate a defect in central tolerance in various lupus mouse models, either to deletion of endogenous superantigen-recognizing T cells or to conventional self-peptide-recognizing CD4+ or CD8+ T cell receptor transgenic mice, were unsuccessful (95). This was true even in lupus mice bearing defects in the proapoptotic Fas pathway (9). Lpr mice bear a retroposon insertion in the gene for fas, disrupting its expression (1). As a result, these mice accumulate a massive number of T and B lymphocytes with age, including an unusual subset of polyclonal CD4−CD8− TCRαβ+ cells (Fig. 2), which is also observed in human SLE (19, 20, 90, 99). Genetic studies demonstrated that these CD4−CD8− T cells arise from CD8+ precursors, but the mechanism is uncertain (Fig. 2) (20, 48, 53, 62, 70). As lpr mice demonstrated no abnormality in cell death during thymic negative selection (9) or following acute infections (41), the origin of their adenopathy remained a mystery for several years. We determined that the source of the accumulating T cells was dysregulated homeostatic proliferation (Fig. 3). A standard model to monitor homeostatic proliferation is to adoptively transfer T cells from one mouse into recipient mice lacking the recombinase protein Rag1, which are devoid of T and B lymphocytes (27). The transferred T cells then expand homeostatically to fill the void. Thus, when wild-type T cells were transferred into Rag1−/− mice, their expansion plateaued after about 2 weeks due to the balance of cell death, whereas lpr T cell numbers continued to expand (27). In addition, the transfer of purified CD8+ T cells, but not CD4+ T cells, gave rise to CD4−CD8− T cells (27). Thus, lack of T cell death during homeostatic proliferation explained both the adenopathy and the origin of CD4−CD8− T cells in lpr mice.
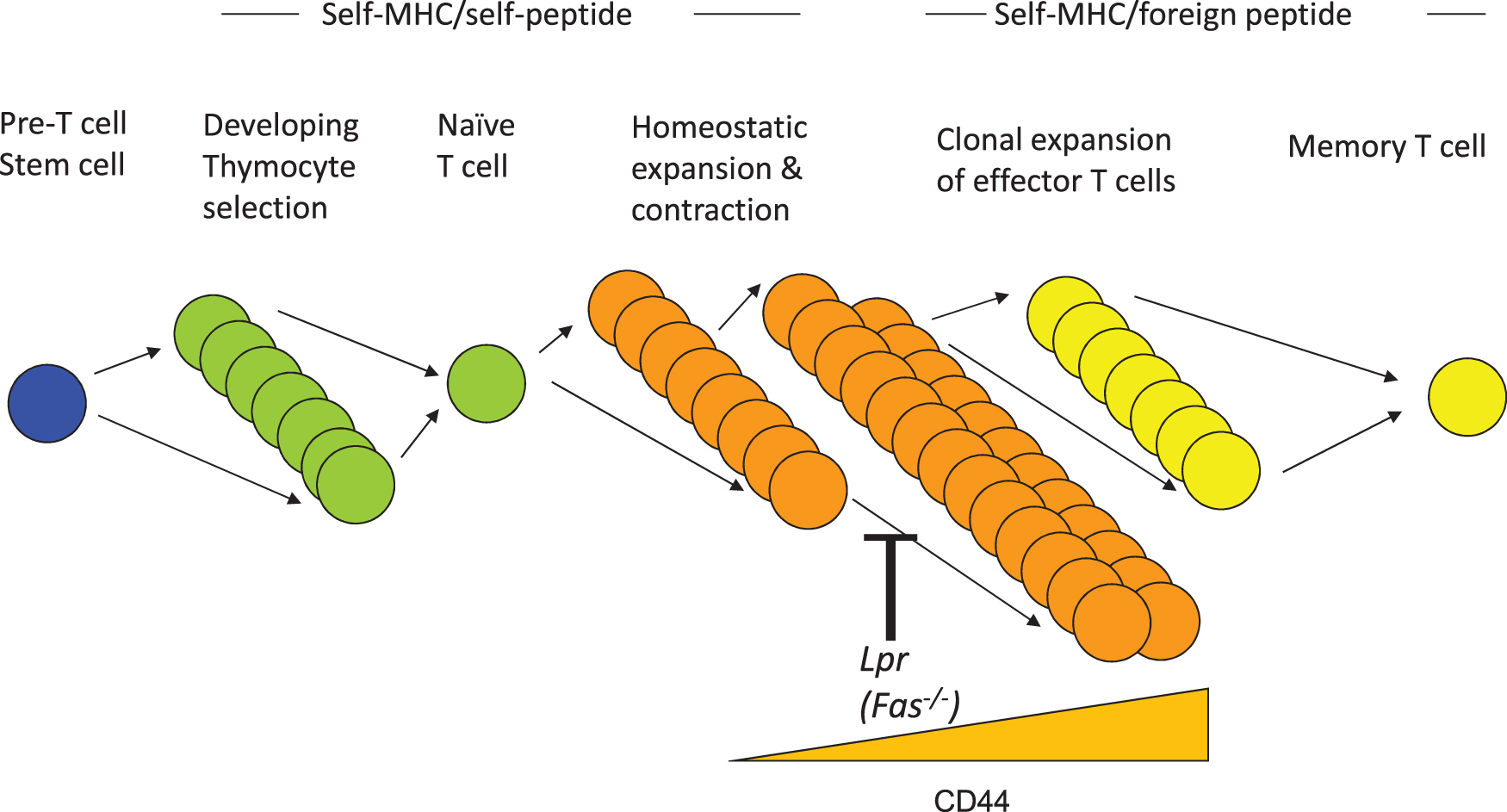
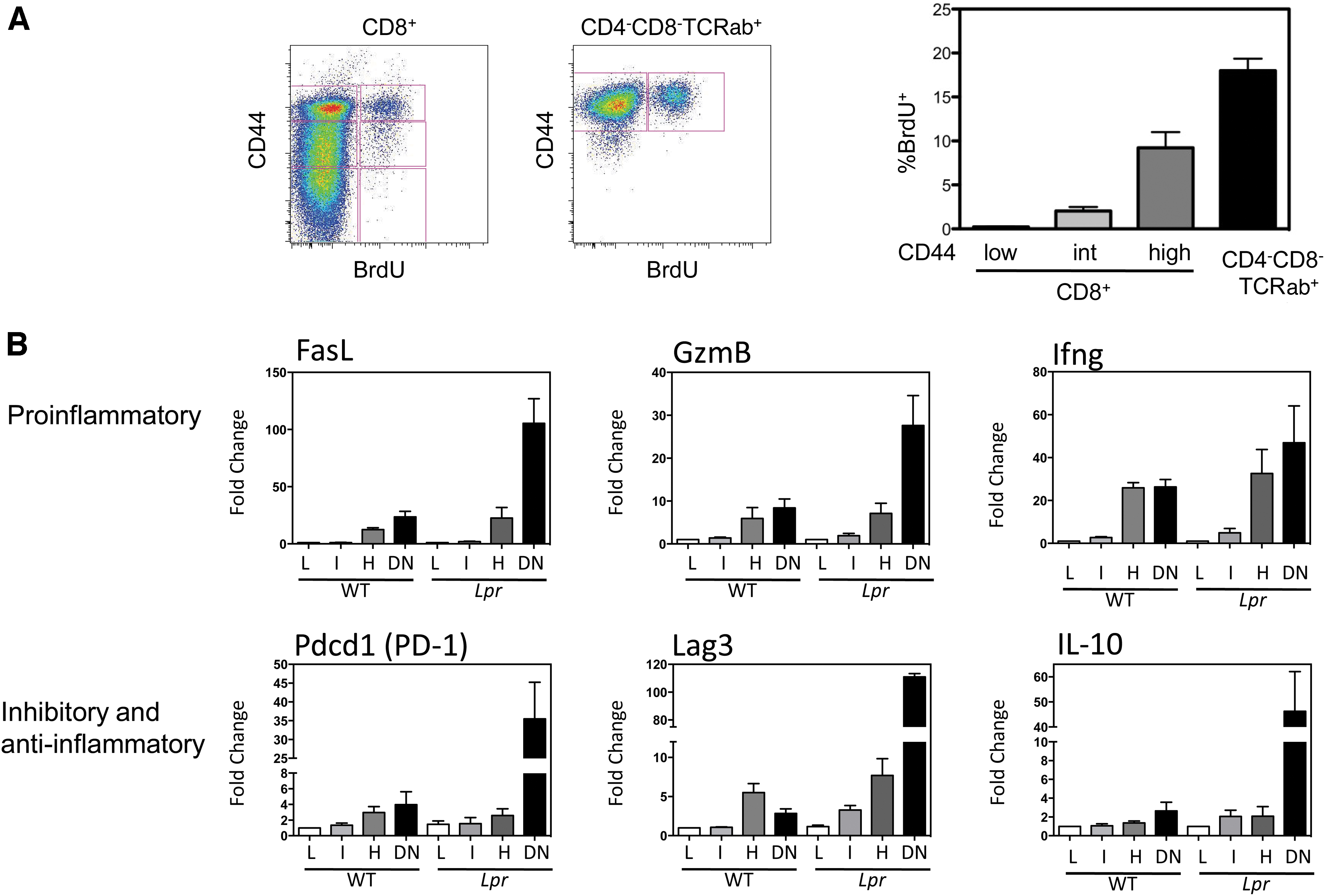
Accompanying homeostatic proliferation was the gradual upregulation of surface CD44 with each cycle of proliferation (26). In agreement with this, measurement of T cell proliferation in vivo in wild-type and lpr mice using a 24-h pulse of BrdU revealed that increasing levels of CD44 paralleled increased BrdU incorporation, with the CD4−CD8− CD44high T cell subset manifesting a remarkable 18% BrdU+ after only 24 h (Fig. 4A) (26). It is worth noting that a similar CD4−CD8− TCR-αβ+ (non-NKT) subpopulation also exists in wild-type mice, although at a much smaller proportion, given that Fas usually promotes the elimination of these cells (26). Hence, the CD4−CD8− T cells that accumulate in lpr mice are not merely a peculiar subset unique to these mice. Rather, the lpr mouse contains large numbers of the T cells that Fas normally eliminates, allowing their study to determine why it is so important to eliminate this population of T cells in a timely manner.
Several additional observations by other groups support the concept that increased numbers of homeostatically expanded T cells, either by reduced T cell elimination as in lpr mice or by augmented proliferation, can lead to autoimmunity. A common element in many of these models is attempts to compensate for initial lymphopenia. Thus, neonatal thymectomy of several mouse strains results in autoimmune inflammation in specific organs accompanied by T cell infiltrates and autoantibodies (83, 89). Mice lacking the T cell inhibitory molecule, CTLA-4, develop a severe lymphoproliferative disorder and die from autoimmunity within 1 month of birth (98). Autoimmunity of various organs and lupus-like features have been observed in genetically lymphopenic mice, such as nu/nu, Rag-deficient, and SCID mice, following the adoptive transfer of small numbers of T cells, particularly those depleted of regulatory CD4+ FOXP3+ T cells (2, 36, 83, 89, 104). Interestingly, the transfer of large numbers of T cells can prevent autoimmunity, implying that it is the rapid homeostatic proliferation of small numbers of transferred T cells that upregulates an inflammatory diathesis. Similar observations have been made in humans following lymphopenia induced by chemotherapy (8), retroviral infections (23, 66), lethal irradiation, and bone marrow reconstitution (4, 8). Lymphopenia is also a feature of several murine models of lupus as well as human lupus (94, 95).
Increased Mitochondrial Mass and Oxygen Consumption by lpr CD4−CD8− T Cells
The strikingly high rate of proliferation by CD4−CD8− T cells was paralleled by equally striking changes in metabolism. Both the spontaneous oxygen consumption rate and glycolysis were considerably elevated in CD4−CD8− T cells compared with the CD4+ and CD8+ subsets from the same mice (25). Further analysis showed that the CD4−CD8− T cells also contained a much larger content of mitochondria than their CD8+ precursors (25). Closer inspection by electron microscopy revealed normal-sized sausage-shaped mitochondria in the CD8+ subset, but greatly enlarged balloon-shaped mitochondria in the CD4−CD8− T cells (25). The mechanism for this is under investigation. Preliminary studies reveal that the main fission protein of mitochondria, Drp1, is excluded from mitochondria in CD4−CD8− T cells, potentially contributing to their enlarged size. A very similar enlargement of mitochondria has also been reported in T cells from SLE patients, which was accompanied by increased ROS (29, 30). This was particularly prominent in the subset of CD4−CD8− T cells from lupus patients with antiphospholipid syndrome (47). The ROS of these cells was reversed by the antioxidant, N-acetylcysteine (NAC), in SLE T cells in vitro (22) and is now being used in a clinical trial in SLE patients. A similar decrease in mitochondrial mass and ROS production has been reported for the JAK1/3 inhibitor, tofacitinib, in primary synovial fibroblasts from rheumatoid arthritis patients (59), raising the possible consideration of JAK inhibitors also in lupus.
We previously reported that ROS alone was sufficient to drive oligomerization of MAVS protein, the downstream regulator of the RIG-I/MDA5 RNA-sensing pathway, in T cells from healthy individuals. It was thus perhaps not surprising to find that T cells from patients with active SLE manifested spontaneous MAVS oligomers (10, 88). SLE patients bearing a loss of function of MAVS (C79F), who manifest reduced expression of IFN-I and milder disease (74), also had reduced oligomerization of MAVS (10, 88). Based on these findings, we examined the presence of MAVS oligomers in lpr T cells. Similar to human SLE T cells, lpr T cells also manifested spontaneous oligomerization of MAVS, which was more pronounced in CD4−CD8− T cells than in CD4+ and CD8+ subsets and was not present in T cells from wild-type mice, even though they expressed similar levels of monomeric MAVS protein (25). This was accompanied by upregulation of several interferon-stimulated genes in CD4−CD8− T cells (25). The remarkable metabolic and MAVS similarities between human SLE T cells and lpr CD4−CD8− T cells suggest not only a potential molecular mechanism of oxidative stress-induced MAVS oligomerization and upregulation of the IFN-I pathway in SLE but also provide a cellular model that drives this transition during successive rounds of T cell homeostatic proliferation. MAVS has also been revealed to be necessary for all symptoms of autoimmune disease in the FcγRIIB−/− model of SLE (93).
These observations raised the question to what extent oxidative stress contributed in vivo to MAVS oligomerization, augmented IFN-I levels, and disease in MRL-lpr mice. Using a mitochondrially targeted antioxidant, MitoQ (60, 61), young MRL-lpr mice were administered MitoQ in their drinking water for 11 weeks. Although there were no observed effects on lymph node cell numbers or composition, nor on autoantibody titers, there was reduction in MAVS oligomers and serum IFN-I, as well as loss of deposition of IgG and complement in the kidneys (25). This is in general agreement with another study using a different antioxidant treatment of MRL-lpr mice (6). This reveals a possible new therapeutic strategy for human SLE using antioxidant therapy.
Inflammatory Gene Expression Program Parallels T Cell Homeostatic Proliferation
T cells in lupus murine models and in human lupus frequently manifest an activated phenotype and a state of replicative senescence (12, 82). This is reflected in the upregulation of certain phenotypic markers of activation/memory (CD44, CD122, and CD132), but not of conventional, antigen-induced early activation markers (CD69, CD71, CD25, or downregulation of CD62L) (14, 15, 28, 33, 67, 69). It was of some surprise that the majority of CD4+ and CD8+ kidney-infiltrating T cells in three murine lupus models were not effector cells, but rather expressed multiple inhibitory receptors and were defective in cytokine production and proliferative capacity (97). This hypofunctional profile was also linked to mitochondrial dysfunction. To examine the seeming paradox of senescence T cells coupled with autoimmune tissue injury, gene expression profiling during T cell homeostatic proliferation was performed on CD8+ T cell subsets bearing low, intermediate, or high levels of CD44, as well as the CD4−CD8− CD44high T cells. This was conducted using both B6-lpr and B6 wild-type mice to determine whether the findings were unique to lpr T cells. The B6-lpr strain was chosen over lupus-prone MRL-lpr mice as B6-lpr mice develop negligible lupus disease and hence the gene expression changes would be due solely to homeostatic proliferation and not confounding autoimmune disease. The findings revealed numerous gene expression changes with progressive homeostatic proliferation that were remarkably similar between B6 wild-type and B6-lpr mice (Fig. 4B). These included gene upregulation of several inflammatory molecules, such as Fas ligand, granzyme B, and interferon-γ, as well as immune inhibitory molecules, including PD-1, Lag3, and IL-10 (26). Although IL-10 is traditionally viewed as an immune inhibitory cytokine, it also boosts B cell proliferation and immunoglobulin class switching (79, 92). IL-10 also has immunostimulatory effects on CD8+ T cells and NK cells (11, 64). IL-10 is overproduced by the B cells and monocytes of SLE patients (35, 39, 51), has increased serum levels in SLE patients (39, 96), and is associated with increased disease activity (40). Furthermore, administration of IL-10 increased disease activity in murine lupus (42), and IL-10 blockade limited renal damage in animal models of lupus nephritis (75). This constellation of both inflammatory and inhibitory molecules suggested an explanation for two clinical immunology paradoxes. The first is the occasional observation that individuals with various immunodeficiency syndromes suddenly develop an autoimmune disorder. The classic example is the sudden appearance of psoriasis and psoriatic arthritis in individuals with HIV (3, 65). In this scenario, it is likely that the greatly accelerated T cell homeostatic proliferation that attempts to compensate for HIV-induced lymphopenia could result in upregulation of the same inflammatory genes, provoking psoriasis. Second, and conversely, SLE patients, who have an activated immune system, are nonetheless prone to infections, independent of medications, and often respond poorly to vaccinations (56, 80). In this instance, the possible increased T cell expression of PD-1, Lag3, and IL-10 could inhibit new T cell responses to infections or vaccines. Remarkably, nearly identical gene expression profiling was very recently reported for CD4−CD8− T cells in patients with autoimmune lymphoproliferative syndrome, also due to a Fas mutation (50). These collective findings add weight to the importance of Fas in regulating the accumulation of T cells undergoing excessive rounds of homeostatic proliferation.
Discrete Loci of DNA Demethylation in CD4−CD8− T Cells Correspond to the Locations of Upregulated Genes and Endogenous Retroviruses
In considering a possible unifying mechanism to explain the particular pattern of genes upregulated during T cell homeostatic proliferation, epigenetic regulation by DNA demethylation was an obvious candidate. DNA demethylation been reported in human SLE T cells (49, 77, 105), and certain medications known to promote drug-induced SLE, such as procainamide and hydralazine, promote DNA demethylation (18). In addition, one of the DNA methyltransferases, Dnmt1, is redox regulated (49). Furthermore, increased expression of HRES/Rab4 in SLE T cells due to demethylation of the promoter and enhancer regions drives enhanced expression of mechanistic target of rapamycin (mTOR) upon T cell receptor stimulation, leading to augmented proliferation (32). Moreover, CD8+ T cells exhibit highly dynamic DNA methylation during normal immune responses (85, 102), and one of the genes that was upregulated during homeostatic proliferation, Pdcd1 (PD-1), is known to be methylation regulated (103). When we examined the two main regulatory loci of Pdcd1, we observed that they were considerably more demethylated in CD4−CD8− T cells than in CD8+ precursors.
These findings prompted a genome-wide study of demethylation sites in CD8+ and CD4−CD8− T cells. We found that although roughly 2.4% of the cytosines throughout the genome were more demethylated in CD4−CD8− T cells than CD8+ T cells, these CpG clustered at discrete sites whose locations overlapped extensively with the locations of several upregulated genes (Fig. 5) (87). In fact, 968 of the 1646 upregulated genes in CD4−CD8− T cells mapped to sites of DNA demethylation. The leading KEGG pathway for these 968 genes was TCR signaling. This was of interest given that CD4−CD8− T cells arise from CD8+ precursors during repeated rounds of homeostatic proliferation, which requires recurrent TCR stimulation by autologous MHC/peptide complexes (34, 68). Additionally, the upregulated genes were ranked according to the number of demethylated loci mapping to each gene. The gene with the most demethylation was Foxp1, a known regulator of quiescence in T cells (24, 76). This is consistent with observations that lpr CD4−CD8− T cells are small senescent cells that are unable to be activated in vitro (16, 17).
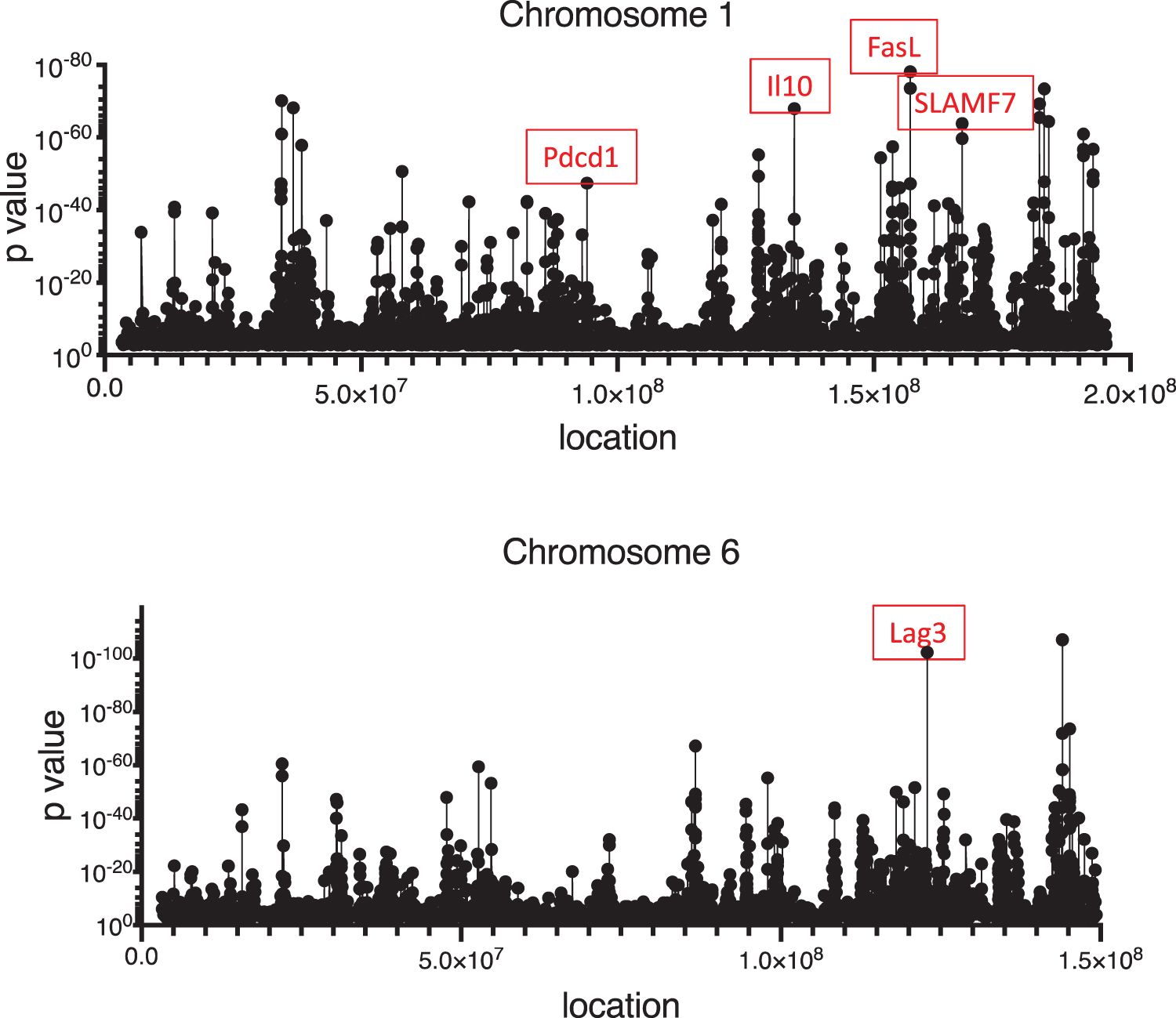
A closer analysis to identify potential common sequences surrounding the demethylated loci revealed a high frequency of binding sites for the transcription factors, AP-1, T-BET, and EGR2 (87). Demethylated CpG were enriched for loci with dynamic chromatin accessibility in CD8+ T cells responding to the lymphocytic choriomeningitis virus (84), indicating that demethylation may be more than a molecular passenger effect of homeostatic proliferation, but a heritable epigenetic record of previously activated CD8+ T cell states. These findings also have striking similarities to recent epigenetic and transcriptional analyses of B cells in human SLE (86). A subpopulation of IgD−CD27−CXCR5−CD11c+ (DN2) B cells is expanded in SLE and has been linked to disease (44). DN2 B cells also share some similarity with autoantibody-associated B cells (ABCs) described in mice (37, 81). Interestingly, the AP-1, T-BET, and EGR transcription factor binding motifs were also enriched in the accessible chromatin of DN2 B cells as well as other expanded B cell subsets from SLE patients during disease flares (86). Furthermore, a recent study of peripheral blood mononuclear cells (PBMCs) from 17 monozygotic and dizygotic twin pairs discordant for SLE revealed extensive demethylation at 807 CpG sites corresponding to 49 genes in the affected twin compared with their healthy twin (43). This was not observed in twins discordant for rheumatoid arthritis or diabetes mellitus. This study also found that SLE patients had reduced messenger RNA (mRNA) levels of the DNA methyltransferase, Dnmt3b, which we also observed to be decreased in lpr CD4−CD8−TCR-αβ+ T cells. An additional similarity was that several of the immune genes that were both demethylated and upregulated in the SLE twin study were also demethylated and upregulated in lpr CD4−CD8−TCR-αβ+ T cells, including Il10, Grb10, Gfi1, Padi4, Cd9, and Aim2 (26, 43).
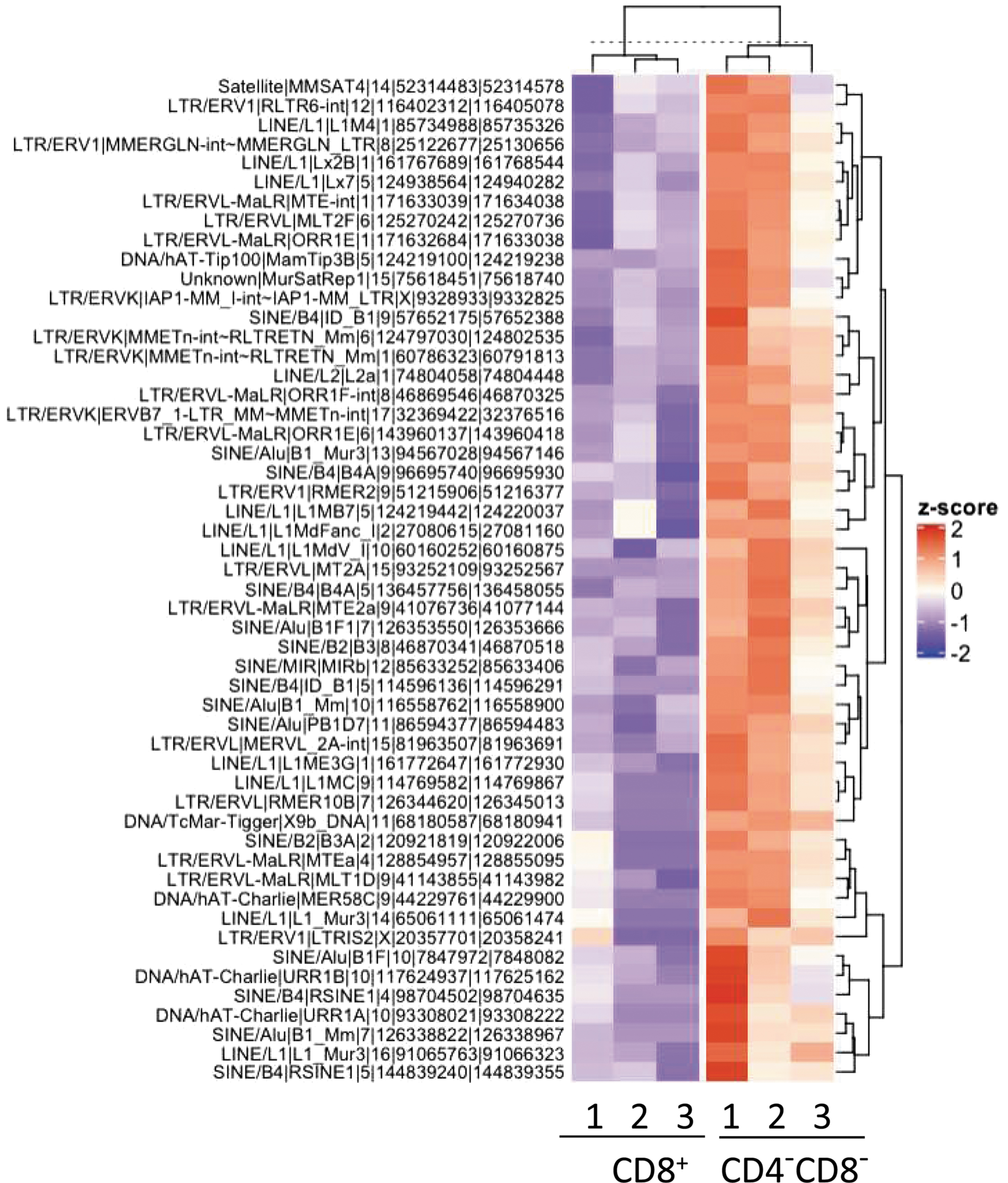
Of additional interest is that within the regions of DNA demethylation reside 52 potential endogenous retroviruses (ERVs) whose expression is also increased in the CD4−CD8−TCR-αβ+ T cell subset compared with CD8+ T cells (Fig. 6). Expression of ERVs has been associated with pathogenesis of both murine (46, 72, 100, 101) and human (5, 7, 52, 71) SLE, and the expression of some ERVs induces IFN-I (58). Conceivably, some of these ERVs may have retained the RNA structure of current viruses and are able to activate RNA sensors such as TLR7/8 and RIG-I. It is of some interest that in patients with a different autoimmune disorder—multiple sclerosis (MS)—seemingly reverse observations have been made in mitochondrial metabolism and methylation. PBMCs and B cells of MS patients revealed increased DNA methylation compared with healthy controls (54, 73), and T cells from primary progressive MS patients manifested lower mitochondrial mass and membrane potential (21). This contrast between MS and SLE patients parallels the use of IFN-β and demethylating agents in MS, both of which are known to induce SLE symptoms in some cases (55, 73).
Evidence of Increased T Cell Homeostatic Proliferation in Active Human SLE
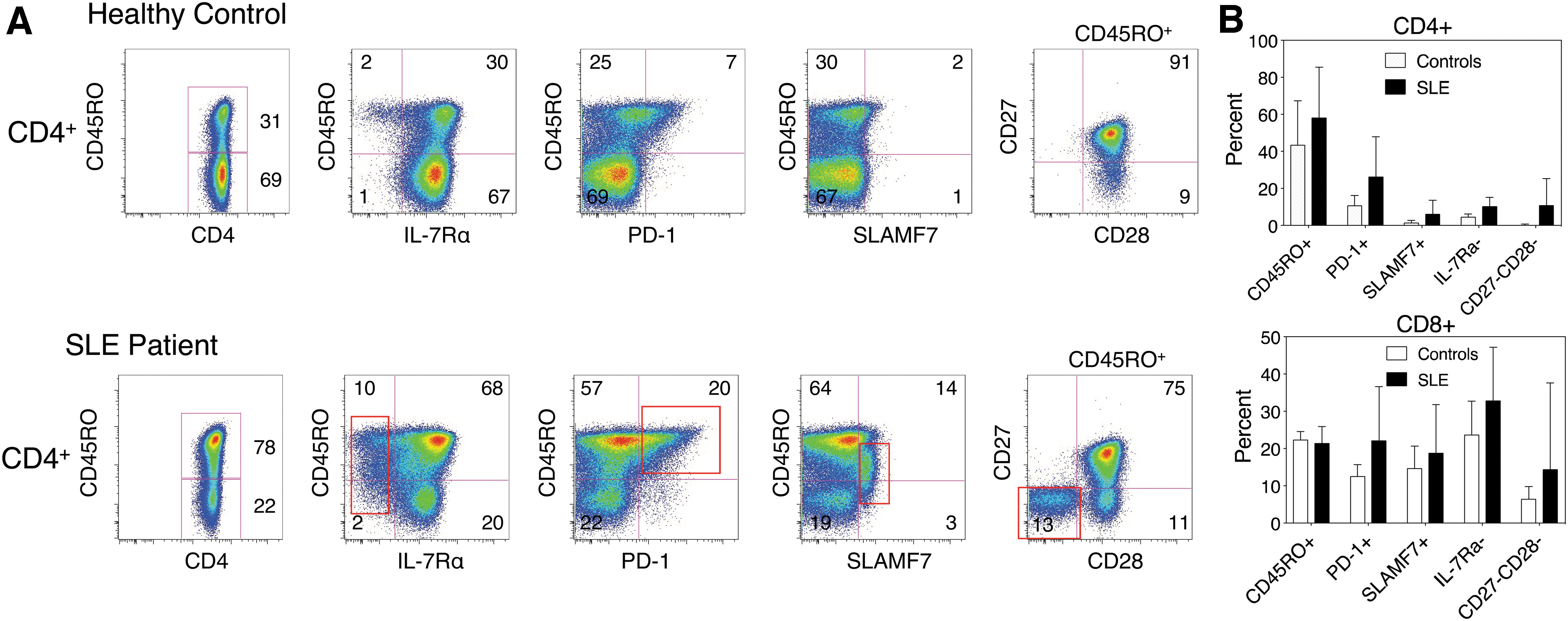
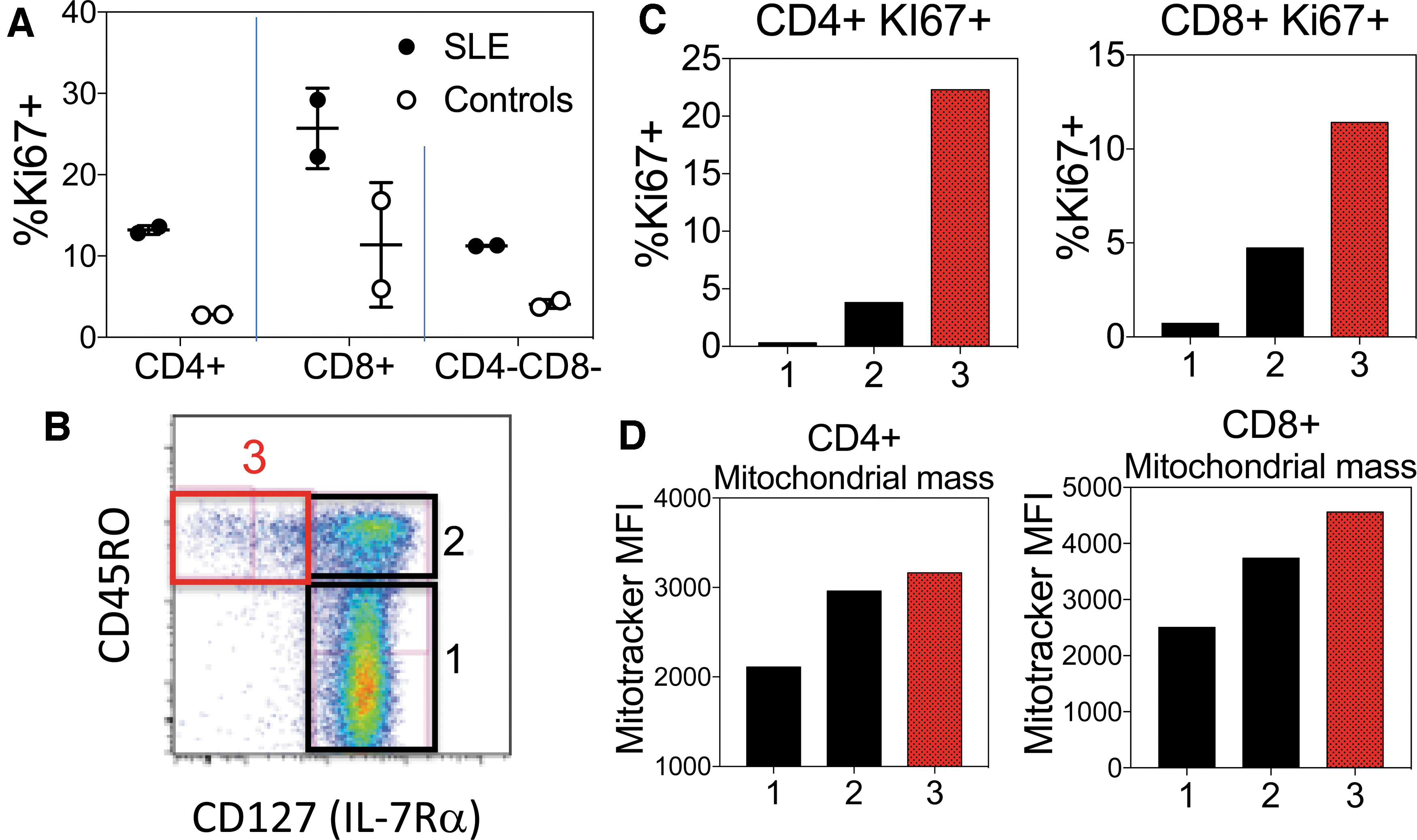
We have begun to take the various changes in surface phenotype of murine T cells during homeostatic proliferation and apply them to a study of T cells in human SLE. To examine this possibility, we developed a multicolor flow cytometry panel for several surface molecules whose genes were upregulated or downregulated in lpr CD4−CD8− T cells. Our initial studies on a limited number of active SLE patients showed that compared with age- and sex-matched healthy controls, SLE patients manifest an increased population of CD4+ and CD8+ T cells bearing the phenotype CD45RO+ IL-7Rα− PD-1+ SLAMF7+ CD27− CD28− (Fig. 7). Closer inspection of this subset revealed that it is enriched with Ki67+ cycling T cells, particularly in the IL-7Rα− subset (Fig. 8). This is of interest given that IL-7Rα is rapidly downregulated after activation by IL-7, and IL-7 is the principal cytokine that drives T cell homeostatic proliferation (38). A previous study also noted the expansion of CD8+ IL-7Rα− T cells in SLE patients, which correlated with their activity score (45), although the mechanism driving their expansion in SLE was not investigated. This subset also manifested higher levels of 2B4—a SLAM (signaling lymphocytic activation molecule) family member—perforin, granzyme B, and cytotoxicity than the CD8+ IL-7Rαhigh subset. These findings are very similar to our observations in murine CD8+ CD44high and CD4−CD8− T cells (26). Future studies will examine whether the human IL-7Rα− T cell subset also manifests DNA demethylation and upregulation of genes similar to those observed in lpr CD4−CD8− T cells.
Conclusions
The collective remarkable metabolic and epigenetic similarities between the lpr CD4−CD8− T cells and T and B cells from SLE patients suggest that the numerous seemingly disparate T cell abnormalities described in SLE may be linked mechanistically through increased homeostatic proliferation. Aberrations in either T cell death, as in the lpr mouse, or increased proliferation, as in human SLE, could lead to a similar outcome with accumulation of T cells having undergone numerous rounds of homeostatic proliferation. Based on the model in Figure 1, there is a potential continual loop in which T cell homeostatic proliferation leads to upregulation of cytolytic machinery that can cause increased cell death. Release of RNA and DNA from dying cells (from immune cytolysis or environmental causes such as ultraviolet irradiation) can induce IFN-I via, respectively, the RIG-I and cGAS pathways. This can induce IL-15 and MHC upregulation in dendritic cells, which promotes T cell homeostatic proliferation, completing the circle. Hence, this proposed loop could be activated at any of these points and feed forward to sustain and amplify the circuit. The model not only unifies several previously disparate observations in SLE T cells but also suggests pathways that could lead to increased homeostatic T cell proliferation and therapeutic targets in SLE, which have received little attention to date. For example, numerous infectious or environmental insults that can trigger SLE flares can increase cell death and, through the release of damage-associated molecular patterns, can activate the innate immune system. The resulting cytokine production, including IFN-I, can upregulate MHC expression and IL-15 production by dendritic cells (31, 57), both of which can enhance T cell homeostatic proliferation. The resulting metabolic and redox changes in T cells would favor MAVS oligomerization and IFN-I production, as well as DNA demethylation and upregulation of inflammatory machinery. Therapeutically, mitochondrially targeted antioxidants, such as MitoQ, might prove effective in SLE, as they did in lpr mice. Reducing the rapid proliferation of T cells by targeting glycolysis with rapamycin is another potential target. Finally, inhibiting homeostatic proliferation upstream with anti-IL-7 or anti-IL-15 blocking provides another possible therapeutic intervention. Lymphopenia is associated with increased circulating IL-7, which drives the expansion of autoreactive T cell clones in patients with type 1 diabetes undergoing islet allo-transplantation (63). In addition, single-nucleotide polymorphisms of IL-7Rα are associated with an increased risk of developing type 1 diabetes and MS. Clinical trials are underway with some of these agents and they are likely to find a place in combination therapy of SLE.
Footnotes
Authors' Contributions
All authors contributed to the literature review and writing of this article.
Author Disclosure Statement
The authors declare that they have no conflicts of interest with publication of this article.
Funding Information
This work was supported by National Institutes of Health Grants (GM118228, and AI11997) to R.C.B.