
Abstract
Significance:
Mitochondria play a critical role in the physiology of the heart by controlling cardiac metabolism, function, and remodeling. Accumulation of fragmented and damaged mitochondria is a hallmark of cardiac diseases.
Recent Advances:
Disruption of quality control systems that maintain mitochondrial number, size, and shape through fission/fusion balance and mitophagy results in dysfunctional mitochondria, defective mitochondrial segregation, impaired cardiac bioenergetics, and excessive oxidative stress.
Critical Issues:
Pharmacological tools that improve the cardiac pool of healthy mitochondria through inhibition of excessive mitochondrial fission, boosting mitochondrial fusion, or increasing the clearance of damaged mitochondria have emerged as promising approaches to improve the prognosis of heart diseases.
Future Directions:
There is a reasonable amount of preclinical evidence supporting the effectiveness of molecules targeting mitochondrial fission and fusion to treat cardiac diseases. The current and future challenges are turning these lead molecules into treatments. Clinical studies focusing on acute (i.e., myocardial infarction) and chronic (i.e., heart failure) cardiac diseases are needed to validate the effectiveness of such strategies in improving mitochondrial morphology, metabolism, and cardiac function. Antioxid. Redox Signal. 36, 844–863.
Introduction
Cardiac diseases are the major cause of morbidity and mortality worldwide, resulting in the death of 17.9 million people every year and costing ∼$300 billion in the United States (202). The pathophysiology of cardiac diseases is multifactorial and includes genetic (i.e., DNA mutations) and/or environmental factors (i.e., unhealthy lifestyle) (33, 185). More recently, mitochondrial dysfunction has been highlighted as a hallmark of both establishment and progression of cardiac diseases (32, 64, 105, 120).
Mitochondria represent ∼35% of the cardiomyocyte volume, categorized as subsarcolemmal, perinuclear, and intrafibrillar mitochondria. These highly dynamic organelles control many different aspects of the cell, including, but not limited to, ATP production, redox signaling, ion homeostasis, and gene expression (25, 56, 105). Moreover, mitochondrial metabolism regulates the majority of intracellular events in cardiomyocytes, ranging from redox signaling to posttranslational modification of proteins (28, 150).
Under stress conditions, mitochondria play a unique role in triggering and propagating damage throughout the cardiomyocytes (31, 105). As expected, disruption of mitochondrial metabolism and/or morphology negatively affects the viability and survival of cardiomyocytes under physiological and pathological conditions (32, 64). Indeed, genetic defects in mitochondrial DNA (mtDNA) cause inherited mitochondrial diseases and cardiomyopathies (83, 104, 195). Therefore, the tight control of mitochondrial number, shape, and metabolism becomes a promising strategy to tackle cardiac diseases.
This review focuses on the intrinsic mechanisms that control mitochondrial connectiveness and quality control as well as their effect on mitochondrial bioenergetics, oxidative stress, and mtDNA segregation in the heart. We also describe the contribution of such processes to the pathophysiology of cardiac diseases and highlight the main pharmacological approaches targeting mitochondrial dynamics to treat heart diseases.
Mitochondrial Bioenergetics
Mitochondria are double membrane organelles containing over 1300 proteins (69). The outer mitochondrial membrane is more permeable than the inner mitochondrial membrane, due to the existence of large-pore channels termed porins or voltage-dependent anion-selective channels, and molecules with molecular weight less than about 5000 Da are allowed free passage (19, 132). In contrast, the inner mitochondrial membrane is a tightly controlled diffusion barrier where molecules can only cross through specific membrane transport proteins, allowing for two discrete regions to form within the mitochondria (18, 166, 184). Furthermore, the inner mitochondrial membrane folds into the intramitochondrial space, termed mitochondrial cristae, which greatly increases its surface area and allows for the mitochondria to maximize ATP production.
Mitochondrial ATP production results from two closely coordinated metabolic processes, the tricarboxylic acid (TCA) cycle or Krebs cycle, and oxidative phosphorylation (OXPHOS) (23, 103). The TCA cycle involves a series of eight enzymatic steps located in the mitochondrial matrix that oxidizes acetyl coenzyme A from the catabolism of metabolites (e.g., fatty acids, glucose, and proteins) to generate the reduced electron carriers nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which transfer electrons to the electron transport chain (ETC) (Fig. 1) (23, 180).
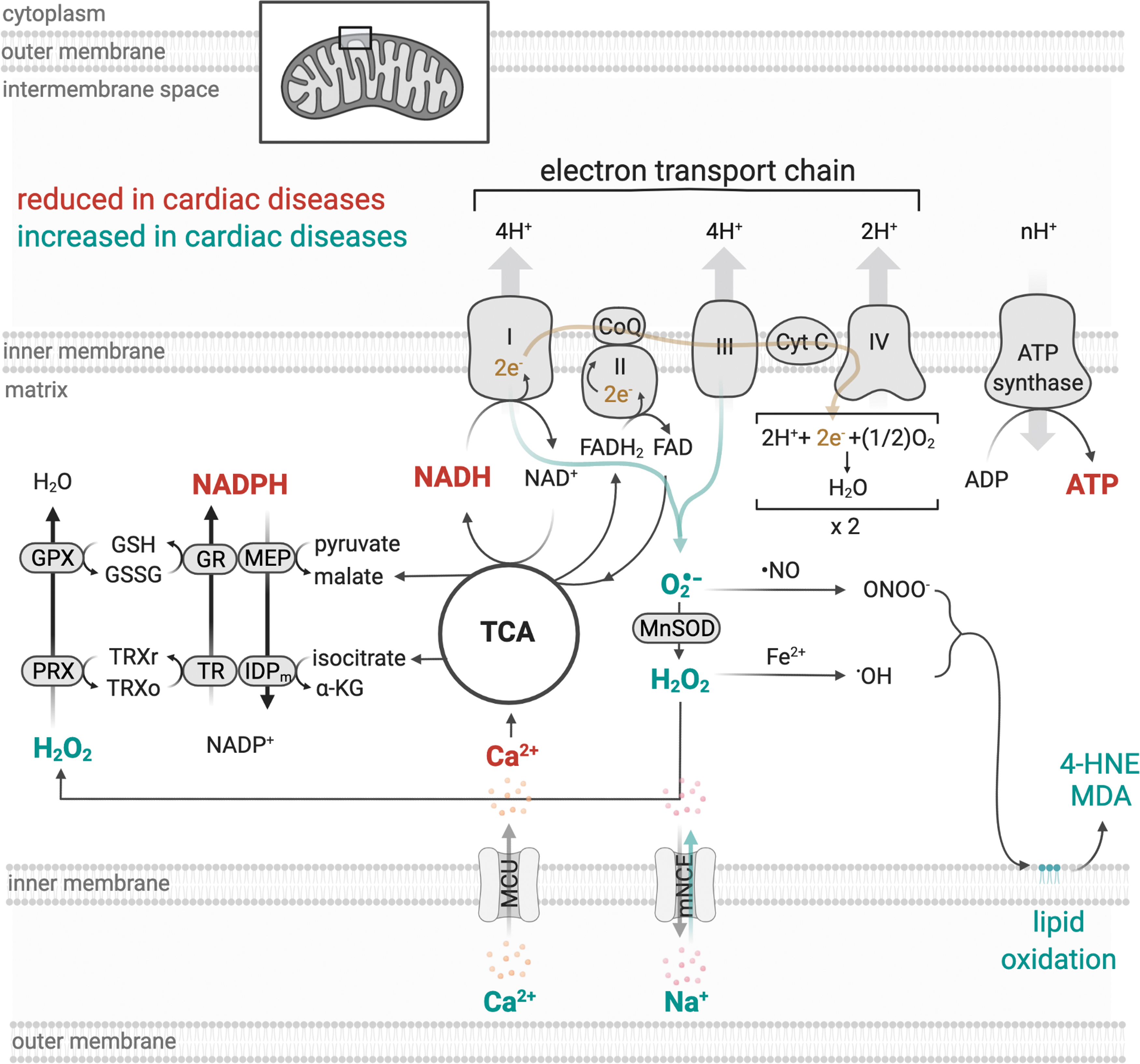
OXPHOS is a process that requires the orchestrated action of four ETC enzyme complexes, complex I–IV, embedded within the mitochondrial cristae of the inner mitochondrial membrane (Fig. 1) (71). Each of these complexes comprised several protein subunits that are associated with a variety of prosthetic clusters with redox potential (103). Complexes I, III, and IV form a “supercomplex,” physically interacting with one another, while complex II represents a point of interaction between TCA cycle and ETC, which occurs through succinate dehydrogenase (3).
During OXPHOS, NADH and FADH2 are oxidized, and their electrons are transferred to complex I (NADH-ubiquinone oxidoreductase) or complex II (succinate-ubiquinone oxidoreductase), respectively, and are transported to complex III (ubiquinol-cytochrome c oxidoreductase) via coenzyme Q. Complex III transfers the electrons from coenzyme Q to the mobile electron carrier, cytochrome c, which delivers electrons to complex IV (cytochrome c oxidase), the terminal complex of the ETC. Once in complex IV, the electrons are transferred from reduced cytochrome c to molecular oxygen, the final electron acceptor, to generate water (103). Recently, Spinelli et al. found that fumarate acts as a terminal electron acceptor when oxygen is not available (hypoxia) (181).
Concomitant with the transfer of electrons between complexes I, III, and IV, protons are pumped from the mitochondrial matrix across the inner mitochondrial membrane, to the intermembrane space using energy from electrons to create an electrochemical proton gradient (Fig. 1). This gradient determines the polarization of the inner mitochondrial membrane (mitochondrial membrane potential), which can be dissipated by the flow of these protons through the ATP synthase or complex V (FoF1-ATP synthase) (2, 134). The proton flow is coupled to the ATP synthesis and leads to the condensation of ADP and inorganic phosphate (Pi) into ATP, which, in turn, is the molecular currency of energy transfer in a cell (2, 81).
The heart has a high demand for ATP synthesis and elevated oxygen uptake rate, therefore relying mainly on mitochondrial OXPHOS to maintain its biochemical and mechanical properties (32, 34). The production of ATP by mitochondria and the use of this energy by cardiomyocytes are highly dynamic. Without oxygen, the mitochondrial-derived ATP supply is only sufficient to power the ATPase required for contraction (e.g., myosin) and relaxation (e.g., sarcoplasmic reticulum calcium ATPase, SERCA) for a few seconds (124, 135, 160). In healthy hearts, fatty acid catabolism, via β-oxidation, is the main substrate utilized for ATP synthesis (up to 70%), which can vary depending on physiological conditions [for review see Bertero and Maack (21)].
In pathological conditions, such as ischemia/reperfusion injury (196) and heart failure (191), glucose becomes the predominant substrate for ATP production. During energy depletion state, the activation of AMP-activated protein kinase (AMPK) stimulates translocation of glucose transporters (mainly GLUT-4) from intracellular vesicles to the sarcolemmal membrane, therefore increasing the rate of glucose uptake (167). In addition, the shift in cardiomyocyte ATP substrate dependence involves the suppression of peroxisome proliferator-activated receptor (PPAR)-α (PPARα) signaling (16) and the activation of both hypoxia-inducible factor 1α (HIF1α) and PPAR, therefore reducing heart exposure to fatty acids (70, 114). This increases glucose uptake and may contribute to cardiac dysfunction and cell death (114).
Moreover, impaired cardiac mitochondrial bioenergetics is associated with defective fatty acid oxidation (120), reduction of mitochondrial supercomplexes (164), and elevated oxidative stress (32, 170), which further leads to cardiomyocyte degeneration and establishment/progression of cardiac diseases in both humans and preclinical models.
In cardiac myocytes, mitochondria represent a major source of reactive oxygen species (ROS). Under physiological conditions, ∼0.12%–2% of the available oxygen is converted into ROS (24, 122, 182), which plays a key role in the cardiac redox signaling and affects several intracellular processes such as protein turnover (26, 29), calcium handling (107, 198), and contractility properties (148). However, under conditions of stress, electrons can prematurely react with oxygen in the ETC and generate excessive ROS, which can interact with different macromolecules (i.e., DNA, lipids, and proteins) (Fig. 1) (24, 78, 207).
Under this scenario, mitochondrial dysfunction-induced oxidative stress can disrupt other intracellular systems and propagate oxidative damage throughout cells, organs, and systems, therefore contributing to the establishment and propagation of cardiac diseases [for review see Bozi et al. (25), Burgoyne et al. (30), and Tsutsui et al. (190)]. To avoid the deleterious effect of excessive oxidative stress, mitochondria contain a variety of enzymatic and nonenzymatic components of the antioxidant defense systems capable of scavenging reactive molecules to nontoxic species, such as the glutathione and thioredoxin systems (Fig. 1) (9) [for details see review Figueira et al. (66)]. Pharmacological inhibition (183) or genetic disruption (85) of thioredoxin system compromises mitochondrial redox balance and cardiac physiology.
Mitochondrial Connectiveness
The mitochondrial network is constantly undergoing fusion/fission cycles, which together control the transport, size, morphology, and turnover of mitochondria (Fig. 2) (37). Collectively, these processes are referred to as mitochondrial dynamics, which directly affect cell viability, proliferation, and ATP production, especially in tissues with high metabolic demand, such as the heart (34, 42, 64). Moreover, mitochondria continuously exchange metabolites, solutes, proteins, DNA, and membranes through mitochondrial fission and fusion processes (169). Therefore, disruption of the mitochondrial fission/fusion balance commonly results in morphological and functional alterations of the mitochondria, which are tightly associated with several chronic disorders, including cardiac diseases (Fig. 3) (31, 177).
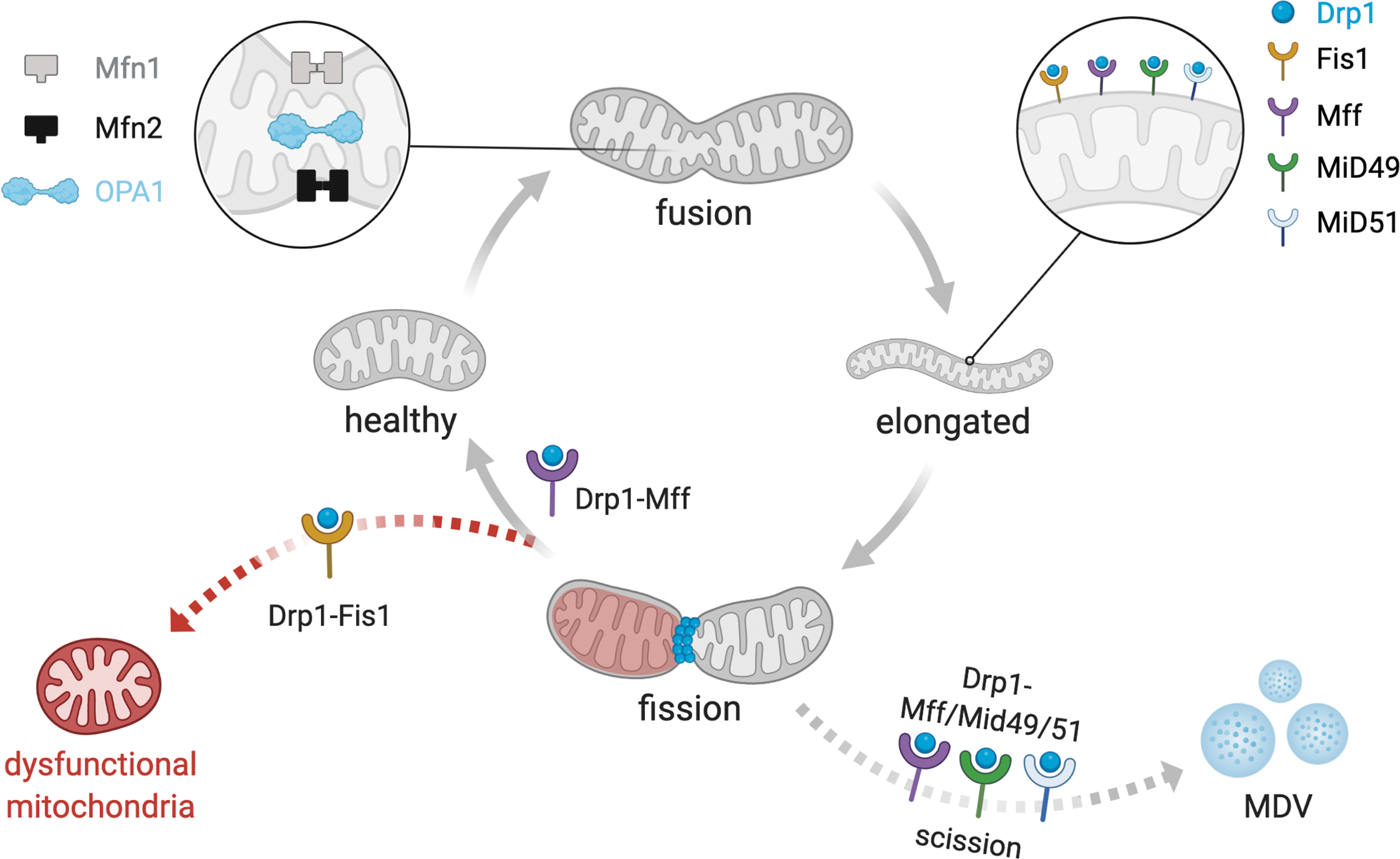
Mitochondrial fusion
Mitochondrial fusion is fine-tuned by three-membrane guanosine triphosphatases (GTPases), the mitofusins (mitofusin 1 [Mfn1] and mitofusin 2 [Mfn2]), located at the outer mitochondrial membrane, and the optic atrophy factor 1 (Opa1), located at the inner mitochondrial membrane (Fig. 2) (179).
Mitofusins contain a GTPase domain involved in the hydrolysis of GTP, which is required for mitofusin oligomerization and fusion of outer mitochondrial membranes to the adjacent mitochondria (112). Interestingly, mitofusins require membrane curvature for binding, which is promoted by the hydrolysis of phospholipase D-dependent cardiolipin (47, 51). In addition, inner membrane fusion requires a similar process, triggered by Opa1 and coordinated by several other proteins, including prohibitins (131, 179). The interaction between cardiolipin and Opa1 is critical for inner mitochondrial membrane fusion, forming interconnected and elongated mitochondria (14).
Mitofusins are responsible for regulating many cellular processes, including mitochondrial number and morphology (12, 40), biogenesis (152), calcium homeostasis (27), protein turnover (35), and apoptosis (118). Mfn2 regulates cell proliferation (52), oxidative metabolism (20), and autophagy (32, 46), and acts as a mitochondrial antiviral signaling protein (204). Mfn2 is also located at the endoplasmic reticulum and mitochondria contact sites, aiding in the maintenance of cytosolic and mitochondrial calcium homeostasis (27, 139).
The absence of MFN2 in the mitochondria decreases oxygen consumption (40), membrane potential (12, 157), and TCA cycle activity (157), which increase with MFN2 overexpression in embryonic and skeletal muscle cells (12, 157). Finally, Mfn1 and Mfn2 are required for the stability of mtDNA, as mitochondrial fusion prevents the accumulation of mtDNA mutations (40, 138). Of interest, mutations in MFN2 or OPA1 (but not MFN1) cause neurodegenerative diseases (62, 156).
In the context of cardiac physiology, mitofusins are essential for the maintenance of cardiac function and morphology under physiological and pathological conditions. The ablation of both mitofusins leads to mitochondrial fragmentation in cultured myocytes (42) and whole heart (178), resulting in embryonic lethality in mouse. Conditional disruption of MFN1 results in cardiac mitochondrial fragmentation, with no effects on mitochondrial bioenergetics and cardiac function under basal conditions (153). In contrast, MFN2 deletion results in the accumulation of enlarged and pleomorphic cardiac mitochondria, which are associated with a modest mitochondrial bioenergetic dysfunction and pathological cardiac hypertrophy (151).
The differences in cardiac phenotype between MFN1 and MFN2 knockout mice might be explained by their unique biochemical properties and role in mitochondrial dynamics. For example, MFN1-harboring mitochondria (but not MFN2-harboring mitochondria) are efficiently tethered in a GTP-dependent manner (93). Moreover, Mfn1 has higher rate of hydrolysis and a lower affinity for GTP relative to Mfn2 (93). Other individual characteristics of Mfn1 and Mfn2, including posttranslational regulation, protein/protein interaction, and location, seem to explain their distinct role in mitochondria fusion, trafficking, turnover, and contacts with other organelles.
Despite their biochemical and functional singularities, Mfn1 and Mfn2 present a high homology (∼80% sequence identity), therefore suggesting these mitofusins might be functionally redundant under adverse conditions, which might explain the lack of severe cardiac phenotypes of MFN1 or MFN2 deficiency mice. In fact, cardiac-specific ablation of MFN1/MFN2 results in massive accumulation of fragmented and dysfunctional mitochondria, which severely disrupts cardiomyocyte sarcomere architecture and results in left ventricle dysfunction in mice (77, 177). These results suggest a compensatory mechanism, not necessarily functionally redundant, between Mfn1 and Mfn2 in the maintenance of cardiac physiology under baseline conditions. However, the molecular mechanism behind such response remains to be elucidated.
Counterintuitively, individual or combined MFN1 and MFN2 deletion protects against ROS-induced cardiomyocyte death (77, 151, 153). Moreover, MFN2 or MFN1/MFN2 ablation protects mice against in vivo cardiac ischemia/reperfusion injury (77, 151). The role of Mfn1 during in vivo cardiac ischemia/reperfusion injury still needs to be determined. The cardiac compensatory effects triggered by ablation of mitofusins are associated with reduced interaction between the mitochondria and sarcoplasmic reticulum (151), mitochondrial calcium overload (77, 151, 153), oxidative stress (77, 151, 153), and delay in the opening of the mitochondrial permeability transition pore (mPTP) (77, 151, 153).
However, the molecular mechanisms behind such responses are still unknown. The contribution of the mitochondrial permeability transition to the cardioprotective effects triggered by deletion of mitofusins seems unlikely considering the lack of efficacy of cyclosporine, a pharmacological inhibitor of cyclophilin D (a major component of mPTP), in reducing myocardial infarction in patients with ST-segment elevation myocardial infarction (49).
Opa1 plays a critical role in regulating mitochondrial number, cristae organization, and bioenergetic efficiency (57, 94). Cleavage of Opa1 by Yme1l (ATP-dependent zinc metalloprotease) (7) or Oma1 (metalloendopeptidase) (59, 79) is sufficient to induce opposite phenotypes under baseline and stress conditions. Briefly, constitutive Opa1 cleavage by Yme1l results into long forms, which are critical to induce mitochondrial fusion and control mitochondrial crista dynamics (7, 87). On the contrary, stress-induced Opa1 cleavage by Oma1 results in short forms, which triggers mitochondrial fragmentation and cell death (7, 111).
Opa1 levels are reduced in failing human hearts, which is associated with accumulation of smaller mitochondria (43). The contribution of Opa1 to cardiac physiology was previously demonstrated in heterozygous OPA1 mutant mice (44). Aged OPA1 mutant mice display cardiac accumulation of fragmented and dysfunctional mitochondria along with excessive oxidative stress and left ventricle dysfunction when compared with wild-type littermates (homozygous OPA1 mutation is embryonic lethal) (44). Moreover, posttranslational modification of Opa1 through constitutive and inducible proteolytic cleavage by Yme1l and Oma1, respectively, plays a key role in cardiac physiology (199). Cardiac deletion of YMEL1L1 causes accumulation of short forms of Opa1 and fragmented mitochondria, which are followed by dilated cardiomyopathy and heart failure in mice (199).
Interestingly, the ablation of the stress inducible OMA1 is sufficient to neutralize the cardiac negative effects triggered by OPA1 deletion (199). Similarly, deletion of OMA1 reduces the number of fragmented mitochondria and mitigates the progression of heart failure induced by isoproterenol or hypertension in mice (4). These findings provide evidence that preventing mitochondrial fragmentation by deleting OMA1 protects against cell death and heart failure (199).
Mitofusins and Opa1 seem to play a key role in regulating apoptotic cell death. The proapoptotic proteins Bax and Bak colocalize with Mfn2 in the outer mitochondrial membrane, which seems to inhibit its pro-fusion effect and favors the mitochondrial release of cytochrome c and apoptosis-inducing factor (AIF) (141). Indeed, dominant active mutants of MFN2 repress Bax activation and cytochrome c release under proapoptotic conditions (141). Similarly, cells depleted of Opa1 are very sensitive to exogenous apoptosis induction (203). Mechanistically, Opa1 suppresses the proapoptotic action of fission 1 protein (Fis1) (203).
Considering the main role of mitofusins and Opa1 in controlling mitochondrial fusion and cardiomyocyte viability, it is expected that nonpharmacological and pharmacological approaches capable of rescuing the function of these proteins will have a positive impact during cardiac degeneration. In fact, reestablishing mitochondrial fusion through exercise (32, 33) or pharmacological intervention (64) is sufficient to restore mitochondrial bioenergetics and improve the prognosis of heart failure in preclinical studies. Indeed, an 8-week running treadmill exercise reestablishes cardiac mitochondrial morphology and autophagic flux, therefore improving cardiac oxidative capacity in rats with myocardial infarction-induced heart failure (32).
Mechanistically, the synergy between mitochondrial fusion and autophagy drives improved bioenergetic efficiency and cardiovascular fitness induced by exercise. Similarly, pharmacological sustained treatment with a selective antagonist of Mfn1-βIIPKC association (SAMβA) reestablishes Mfn1 GTPase activity, reduces mitochondrial fragmentation in heart failure rats, protects cultured neonatal and adult cardiac myocytes from stress-mediated cell death, and improves cardiac function (64).
Genetic interventions capable of correcting mitochondrial fusion might be another approach to treat inherited heart diseases. A recent article demonstrated that genetic correction of OPA1 variation by using CRISPR-Cas9 technology improves mitochondrial connectiveness, bioenergetics, levels of mtDNA, and resistance to stress in patient-derived OPA1 mutant-induced pluripotent stem cells (176). However, its therapeutic value to treat Opa1-related diseases, such as autosomal dominant optic atrophy and cardiac disorders, remains to be determined. Moreover, the potential use of genetics to correct the mitochondrial fission/fusion imbalance and the consequent mitochondrial dysfunction in cardiac diseases are still uncertain.
Mitochondrial fission
Mitochondrial fission is characterized by splitting one mitochondrion into two smaller mitochondria, which is critical for maintaining mitochondrial quality control and bioenergetics under baseline and stress conditions (Fig. 2) [for review see Chan (37)]. The mitochondrial fission process is orchestrated by the GTPase dynamin-related protein 1 (Drp1). Drp1 is a cytosolic protein that translocates to mitochondria upon activation and binds to specific proteins located at the outer mitochondrial membrane, including mitochondrial Fis1 (136, 205), mitochondrial fission factor (Mff) (145), mitochondrial dynamics protein of 49 kDa (MiD49), and mitochondrial dynamics protein of 51 kDa (MiD51) (123, 149).
Drp1 translocation to mitochondria is induced by many different factors in cardiomyocytes, including β-adrenergic stimulation (97), ischemia/reperfusion injury (53), and exercise (32). Upon Drp1 translocation and binding to aforementioned proteins at constriction sites, it polymerizes around the organelle and through the hydrolysis of GTP, changes its conformation to constrict both the outer and inner membranes, leading to mitochondrial fission and accumulation of smaller and spherical mitochondria (Fig. 2) (53, 92, 129).
Fragmented and dysfunctional mitochondria are not reincorporated into the mitochondrial network, inducing the autophagy-mediated removal of these organelles (Fig. 2). In this scenario, Drp1 plays a key role in cardiac autophagy-mediated mitochondrial removal, also termed mitophagy, and mitochondrial quality control (90, 178). These findings demonstrate the critical role of Drp1 in maintaining cardiac biochemistry homeostasis and contractility properties under physiological conditions.
Under pathological conditions, such as myocardial infarction, a pro-fission phenotype along with impaired mitophagy results in accumulation of fragmented and dysfunctional mitochondria, therefore facilitating the propagation of damage and cardiac degeneration over time (53). Cardiac ablation of DNM1L results in reduction of mitochondrial membrane potential, induction of mPTP (178), impaired mitochondrial respiration (96, 192), and disruption of mitophagy (178), therefore contributing to cardiomyocyte necrosis and dilated cardiomyopathy in mice (90, 177, 178). Mitochondrial translocation of Drp1 is also involved in cardiac ischemic damage, where pharmacological inhibition of Drp1 protects cardiomyocytes in culture and whole heart from ischemia/reperfusion injury (144).
Fis1 is another player involved in mitochondrial fragmentation under baseline and stress conditions (Fig. 2) (89). Interestingly, Fis1 is highly expressed in cardiac cells, therefore suggesting its important role in heart physiology (89).
During ischemia/reperfusion injury, both Fis1 and Drp1 are upregulated in neonatal and adult cardiomyocytes (84, 208). Under this scenario, upstream signaling events favor Drp1 activation and the consequent interaction with Fis1 at the outer mitochondrial membrane (205). This interaction results in an excessive accumulation of fragmented/dysfunctional mitochondria and redox imbalance (53). Fis1 also binds to mitochondrial fusion proteins (Mfn1, Mfn2, and Opa1) and inhibits their GTPase activity, thus blocking mitochondrial fusion in mammalian cells (206). These findings suggest that Fis1 triggers mitochondrial fragmentation also in a Drp1-independent manner. However, this novel role of Fis1 remains to be determined in the context of cardiac physiology.
Mechanistically, there are at least two functionally distinct mitochondrial fission molecular signatures that predict the biogenesis of new mitochondria (midzone division) and the clearance of fragmented and dysfunctional mitochondria (periphery division) (106). Mitochondrial fission in the midzone and periphery occurs through Drp1-Mff interaction and Drp1-Fis1 interaction, respectively (Fig. 2) (106). Moreover, midzone mitochondrial division is marked by endoplasmic reticulum (ER)/mitochondria contact sites (106) and relies on Golgi-derived vesicles (137). The function of these protein/protein interactions to mitochondrial fission, as well as its contribution to physiological and pathological conditions, has only recently been elucidated (100, 110).
The Drp1-Mff interaction plays a critical role during physiological mitochondrial fission and function (110). Disruption of Drp1-Mff interaction, using a rationally designed peptide, is sufficient to reduce ATP levels and impair mitochondrial morphology, leading to behavioral deficits and progression of Huntington's disease in mice (110). On the contrary, Drp1-Fis1 interaction seems to be important for mitochondrial division under adverse conditions such as mitochondrial oxidative stress, impaired calcium homeostasis, and reduced membrane potential (106). This type of mitochondrial fission must be linked to mitochondrial clearance through autophagy. However, autophagy is generally impaired under degenerative conditions, which might result in accumulation of fragmented/dysfunctional mitochondria.
Under these conditions, pharmacological inhibition of excessive Drp1-Fis1 interaction by a cell-permeable peptide termed p110 is sufficient to avoid accumulation of smaller/dysfunctional mitochondria (159) and protect against cardiovascular (53) and neurodegenerative diseases (67, 99, 100, 102). More recently, König et al. demonstrated that Mff, MiD49, and MiD51 are essential for Drp1-dependent mitochondrial scission and release of mitochondrial-derived vesicles (MDV) (Fig. 2) (109).
In summary, a coordinated response involving induction of key mitochondrial fission genes, upstream activation of Drp1 (137), and the bimodal mitochondrial division through Drp1-Mff interaction and Drp1-Fis1 interaction (106) dictates the mitochondrial fission rate in heart physiology (106) from embryonic development (41, 95), cardiomyocyte differentiation (99), to adult cardiomyocytes (53) under baseline and pathological conditions.
Mitophagy
Mitophagy is a process that selectively targets dysfunctional mitochondria for removal and is closely linked to mitochondrial fission and fusion (Fig. 4) [for review see Palikaras et al. (147)].

Briefly, under physiological conditions, PINK1 (PTEN-induced kinase 1) is targeted and imported to the inner mitochondrial membrane to be degraded by mitochondrial proteases (46, 128). However, under stress conditions, when the mitochondrial membrane potential is reduced, this process is disrupted and PINK1 accumulates on the outer mitochondrial membrane. PINK1 then phosphorylates and recruits Parkin (a cytosolic E3-ubiquitin ligase) to the mitochondrial surface (39, 108, 113). Parkin-mediated ubiquitination of outer mitochondrial membrane proteins results in the recruitment of autophagy adaptors (p62, OPTN, and NDP52) and initiates autophagosome formation through LC3 binding and the subsequent lysosomal degradation of autophagosome engulfed mitochondria (Fig. 4) (80, 162).
The PINK1-Parkin pathway affects mitochondrial fission and fusion at different levels. PINK1 indirectly triggers Drp1 translocation to the mitochondrial surface, therefore promoting fission of dysfunctional mitochondria, which enables their removal through autophagy (158). In coordination with Drp1 activation, PINK1-Parkin-mediated phosphorylation and proteasomal degradation of Mfn1 also contribute to mitochondrial fragmentation and removal of dysfunctional organelles (187). The activation of the PINK1-Parkin pathway results in Mfn2 phosphorylation and disruption of ER/mitochondria contact sites, which are critical for the maintenance of calcium handling, bioenergetic profile, and contractility properties of cardiomyocytes (46).
Mitochondrial fission/fusion balance and clearance are also regulated by mitophagy receptors located at the outer mitochondrial membrane, including NIX (NIP3-like protein X) (163), BNIP3 (BCL2 interacting protein 3) (119), and FUNDC1 (FUN14 domain-containing protein 1) (45). The activation of these receptors is critical to control mitochondrial number, size, and metabolism under both health and disease conditions (60). BNIP3 promotes fission and removal of damaged mitochondria in cardiomyocytes through PINK1 stabilization, inhibition of Opa1 assembly, and Drp1 translocation to the mitochondrial surface (119).
There are also other PINK1-Parkin-independent mechanisms of mitophagy [for review see Palikaras et al. (147)]. For example, mitochondrial protein FUNDC1 is located at the outer mitochondrial membrane and has been linked to mitophagy under hypoxia or mitochondrial membrane potential dissipation (45, 121). Phosphorylation of FUNDC1 and the consequent inhibition of FUNCD1-mediated mitophagy result in excessive accumulation of ROS, therefore impairing cardiac redox homeostasis and mitochondrial metabolism (208).
Overall, mitophagy plays a critical role in maintaining the pool of healthy mitochondria under physiological and adverse conditions [i.e., accumulation of mtDNA mutations (192)]. Recent studies provide evidence that dysfunctional mitophagy culminates in the accumulation of damaged mitochondria and degeneration of organs with high demand for ATP synthesis and elevated oxygen uptake rate, such as the heart (22, 32). For example, PINK1 protein levels are reduced in failing hearts (22), and mitophagy is impaired in both aged and failing hearts in mice (86).
There are several findings from preclinical studies supporting the key role of mitophagy in heart physiology. Parkin-deficient mice accumulate dysfunctional mitochondria and have elevated levels of oxidative stress, along with pathological cardiac remodeling and reduced left ventricle fractional shortening (74, 86). Furthermore, genetic disruption of PINK1-PARK2-mediated mitophagy results in lethal heart failure in mice (74), and PARK2-deficient Drosophila heart tubes display progressive cardiomyopathy (46). Lastly, Parkin knockout mice are more sensitive to myocardial infarction, have a higher mortality rate, and a bigger infarct size when compared with wild-type (115).
The PINK1-Parkin pathway is also involved in the cardioprotective effects of ischemic preconditioning. Genetic disruption of the PARK2 pathway abolishes ischemic preconditioning (the benefit of short ischemic events before prolonged ischemia) in mice (88). Pharmacological inhibition of autophagy and the consequent impairment of mitochondria clearance were found to prevent the positive effects of exercise in reestablishing the pool of healthy mitochondria, redox balance, and contractility properties of failing hearts in rats (31, 32).
Counterintuitively, mitophagy overactivation can negatively affect cardiac physiology. The overexpression of mitophagy receptors NIX and BNIP3 maximizes the cardiac damage induced by myocardial infarction (55). Moreover, genetic disruption of BNIP3 is sufficient to decrease apoptosis and improve cardiac remodeling and function in a model of postmyocardial infarction in mice (55).
In summary, functional mitophagy, as well as its interdependence with mitochondria fission and fusion, plays a critical role in the maintenance of cardiac mitochondrial morphology, mtDNA content, bioenergetics, and cell death (Fig. 3) (31), therefore affecting cardiac metabolism, redox homeostasis, and contractility properties in health and disease.
mtDNA Heteroplasmy
The mitochondria of most eukaryotic organisms contain their own circular DNA, which has a high density of genetic information and is maternally inherited (133, 143). In humans, mtDNA is circular, double-stranded, ∼16.6 base pairs in length, and located in the mitochondrial matrix (133). mtDNA encodes 37 genes, of which 13 encode peptide subunits critical for complexes I, III, and IV and ATP synthase, while complex II is encoded only by a nuclear genome (8). In addition, 22 transfer RNAs and 2 ribosome-coding RNAs are encoded by mtDNA (143). Unlike the nuclear genome, mtDNA is not protected by histones—it is organized into nucleoids (an mtDNA-protein complex). Since mtDNA is located in the mitochondrial matrix, it is more vulnerable to ROS oxidation produced during OXPHOS, making the mitochondrial genome more susceptible to mutations (174).
mtDNA is characterized by a high copy number, and the coexistence of wild-type and mutated mtDNA in the cell is defined as heteroplasmy (Fig. 5) (174). The prevalence rate for mtDNA mutations is ∼1 in 5000 individuals (75). In healthy humans, low-level heteroplasmy is usually detected but well tolerated. Most mtDNA mutations are recessive and must accumulate to negatively affect mitochondrial bioenergetics (188). The maximum threshold to tolerate mutations differs among cell types, tissue, or individual (133, 155, 197). Heteroplasmy levels are dynamic and constantly changing, since mtDNA replication occurs throughout the cell cycle. Many point mutations and deletions of mtDNA may arise at embryonic development or over time as heteroplasmy increases (68, 185).

Leber hereditary optic neuropathy (LHON) was the first evidence that mutations of mtDNA maternally inherited could trigger the development of chronic diseases (83), such as cardiomyopathy (82) and diabetes (13). Mothers with high rates of heteroplasmy are more likely to have children with mtDNA mutations. However, the symptoms will only show up according to the percentage of heteroplasmy (197).
In somatic cells, pathogenic mtDNA mutations can arise from replication errors, oxidative stress-induced DNA damage, and/or a defect in the mtDNA repair machinery. Indeed, accumulation of mtDNA mutations (∼60%–90% mutant to wild-type DNA) has a negative impact on mitochondrial bioenergetics, especially in tissues highly dependent on mitochondrial ATP (68, 143, 185). Furthermore, mutations in nuclear genes responsible for mtDNA replication and transcription, such as mitochondrial transcription factor A (TFAM), DNA polymerase POLG, and RNA polymerase (POLRMT), can promote mtDNA mutation and be linked to several chronic diseases (6), including aging phenotypes and reduced life span (189). Therefore, the levels of heteroplasmy are the main determinant for the onset and severity of mitochondrial diseases.
Accumulation of mtDNA mutations in genes that encode subunits of the ETC complex likely results in mitochondrial dysfunction and redox imbalance. This phenotype has a major effect in organs highly dependent on mitochondrial ATP synthesis, including the central nervous system (168), skeletal muscle (197) and heart (195). Clinical studies demonstrate the relationship between cardiac diseases and mtDNA mutations, although the molecular mechanisms are still poorly understood (82, 91, 104, 195). Cardiomyopathy affects ∼20% of individuals with mitochondrial disease and has been related to high heteroplasmy levels in the cardiac tissue and early mortality in children (82, 91).
In a recent study, unique mutations were found to be enriched in stroke and myocardial infarction patients when compared with healthy individuals (195). Briefly, mtDNA mutations (m16145G>A and m.16311T>C) detected in stroke patients were positively associated with potential genetic risk factors. Contrasting, another set of mtDNA mutations (m.72T>C, m.73A>G, and m.16356T>C) found in myocardial infarction individuals was inversely associated with potential genetic risk factors (195). Different mitochondrial mutations have been found in leukocytes from the Russian (m.5178C>A and m.14459G>A) and Mexican (m.13513G>A and m.652insG) populations, and are associated with increased cardiovascular risk factors (104).
mtDNA mutations have also been associated with disrupted mitochondrial morphology, which may contribute to the development of heart failure (11). Taken together, these data suggest that accumulation of mtDNA mutations contributes to the establishment and progression of cardiac diseases. Therefore, evolutionary conserved systems capable of controlling mitochondrial heteroplasmy, such as mitochondrial fission, fusion, and clearance, are important to counteract or mitigate mtDNA mutation-induced heart diseases.
mtDNA Segregation
mtDNA can be segregated along the maternal germ line or in somatic cells and tissues (68, 185). The segregation is highly dynamic, and heteroplasmy levels constantly shift between daughter cells (Fig. 5) (185). During germ line segregation, a reduced number of mtDNA is transmitted from the mother to the offspring within each oocyte, a phenomenon known as a “genetic bottleneck,” which explains the different heteroplasmy levels and clinical disease severity between generations (48, 68). As discussed, mtDNA mutations may also increase during the lifetime through random replication in nondividing (postmitotic) cells, such as cardiomyocytes, a phenomenon that is related to aging (185). For these reasons, both mitochondrial fusion and fission, and mitophagy, are critical to ensure proper selection and/or transmission of mtDNA (1, 143, 192).
mtDNA copy number and turnover, as well as mtDNA segregation rate, are directly affected by changes in mitochondrial number and the continual exchange of mitochondrial content during mitochondrial fission, fusion, and clearance (142, 192). mtDNA is continuously replicated and exchanged between mitochondria during the lifetime through mitochondrial fission and fusion events. This process favors the distribution and propagation of mitochondrial genetic information throughout the cell during mitochondrial network reorganization (38, 42, 200). In the case of dividing cells, mitochondrial dynamics is crucial for the transmission of mtDNA content to daughter cells (Fig. 5).
The effectiveness of mitochondrial dynamics in regulating mtDNA distribution and segregation seems to be tissue- and cell-type-specific, and is affected by the mitochondrial bioenergetic state and redox balance (15, 142, 192). However, the molecular mechanisms regulating these processes remain elusive.
Mitochondrial fission/fusion and mtDNA segregation
Mitochondrial fusion plays a key role in regulating both stability and tolerance of mtDNA mutation in skeletal muscle (42) and mtDNA replication in the heart (161). Patients with mutation in MFN2 and OPA1, responsible for mitochondrial fusion, display increased mtDNA instability (5, 165), which is associated with Charcot-Marie-Tooth type 2A and dominant optic atrophy degenerative diseases, respectively (50, 209). Furthermore, mitofusin-deficient mice show a marked reduction of mtDNA levels and an increase in both mtDNA point mutations and deletions, which are accompanied by mitochondrial myopathy in skeletal muscle. These findings suggest that the maintenance of mtDNA copy number relies on mitochondrial fusion, which is essential to maintain mitochondrial function and muscle contractility properties (42).
Mitochondrial fission is also important in controlling mtDNA number (Fig. 5). Defective mitochondrial fission in DNM1L knockout HeLa cells reduces both mtDNA stability and ATP production, correlating with inhibition of cell proliferation and viability (154). Of interest, mitochondrial fission often occurs adjacent to nucleoids, which suggests a role for mitochondrial division during mtDNA replication and distribution. Indeed, deficient mitochondrial fission results in nucleoid clustering and the consequent dysfunctional mitochondrial bioenergetics (15, 116, 142). Finally, mtDNA segregation seems to be regulated by midzone mitochondrial division through Drp1-Mff interaction, but not Drp1-Fis1 interaction, which is likely associated with peripheral mitochondrial fission and clearance of mitochondrial debris (106).
Muscle-specific DNM1L knockout mice display severe mtDNA nucleoid clustering, which is associated with reduced mitochondrial bioenergetics, dilated cardiomyopathy, and neonatal lethality during heart development (96). In contrast, Jokinen et al. demonstrated that reduced mitochondrial fission in DNM1L mutant animals does not affect cardiac mtDNA segregation (98). This discrepancy may be due to the difference in the time-window that was examined in these studies; in the later study, measurements were conducted before the onset of dilated cardiomyopathy and heart failure. Therefore, whether mitochondrial fission and fusion play a role in cardiac mtDNA segregation under pathological conditions remains to be determined.
Mitophagy and mtDNA segregation
Mitophagy, another critical system involved in mitochondrial quality control, plays an important role in the maintenance and selection of mtDNA. In general, dysfunctional and depolarized mitochondria are identified, targeted, engulfed by autophagosome, and degraded by lysosomes, thereby contributing to selected removal of mtDNA (Fig. 4) (72, 186). It is also expected that a coordinated response between mitochondrial fission/fusion and mitophagy facilitates the clearance of damaged mitochondria, therefore mitigating the propagation of mtDNA mutations and mtDNA heteroplasmy (Fig. 5).
A seminal study by Twig et al. demonstrated that mitochondrial fission followed by selective fusion segregates dysfunctional mitochondria, allowing for elimination through mitophagy (192). In fact, induction of mitochondrial depolarization or overexpression of Parkin optimizes the clearance of deleterious mtDNA in heteroplasmic cells (186). As expected, defective mitophagy increases mtDNA heteroplasmy by accumulating mtDNA mutations, which is related to several genetic diseases, such as hereditary Parkinson's disease (42, 186).
The role of mitophagy in removing deleterious mtDNA has been underexplored in the context of cardiovascular diseases. Induction of mtDNA damage in a transgenic mouse model overexpressing the mutant uracil-DNA glycosylase (mutUNG1) causes a rapid congestive heart failure and premature death (117). These animals display accumulation of dysfunctional mitochondria, therefore suggesting a disruption of the mitochondrial fission/fusion balance and/or mitophagy (117). However, the contribution of mitochondrial dynamics to mutUNG1-induced cardiac phenotype was not addressed in the study.
In another study using a mouse model that accumulates mtDNA damage due to a proofreading-defective mtDNA polymerase γ (POLG), cardiac Parkin protein levels were reduced with aging (201). However, these transgenic mice do not present impaired mitochondrial bioenergetics or cardiac dysfunction compared with wild-type. Under these conditions, neither cardiac-specific overexpression nor global deletion of PARK2 affects the cardiac phenotype of POLG mice (201). These findings suggest that Parkin-mediated mitophagy is not essential for the maintenance of mitochondrial quality control in a preclinical model of cardiac mtDNA damage and aging.
Overall, the mechanisms underlying the clearance of deleterious mtDNA under health and disease conditions are still under debate. However, there is a reasonable amount of evidence suggesting that a coordinated response between mitochondrial fission/fusion and mitophagy plays an important role in controlling mtDNA heteroplasmy, which might have an impact on the onset and progression of cardiac disease.
Potential Therapeutics Targeting Mitochondria Fission and Fusion in Cardiac Diseases
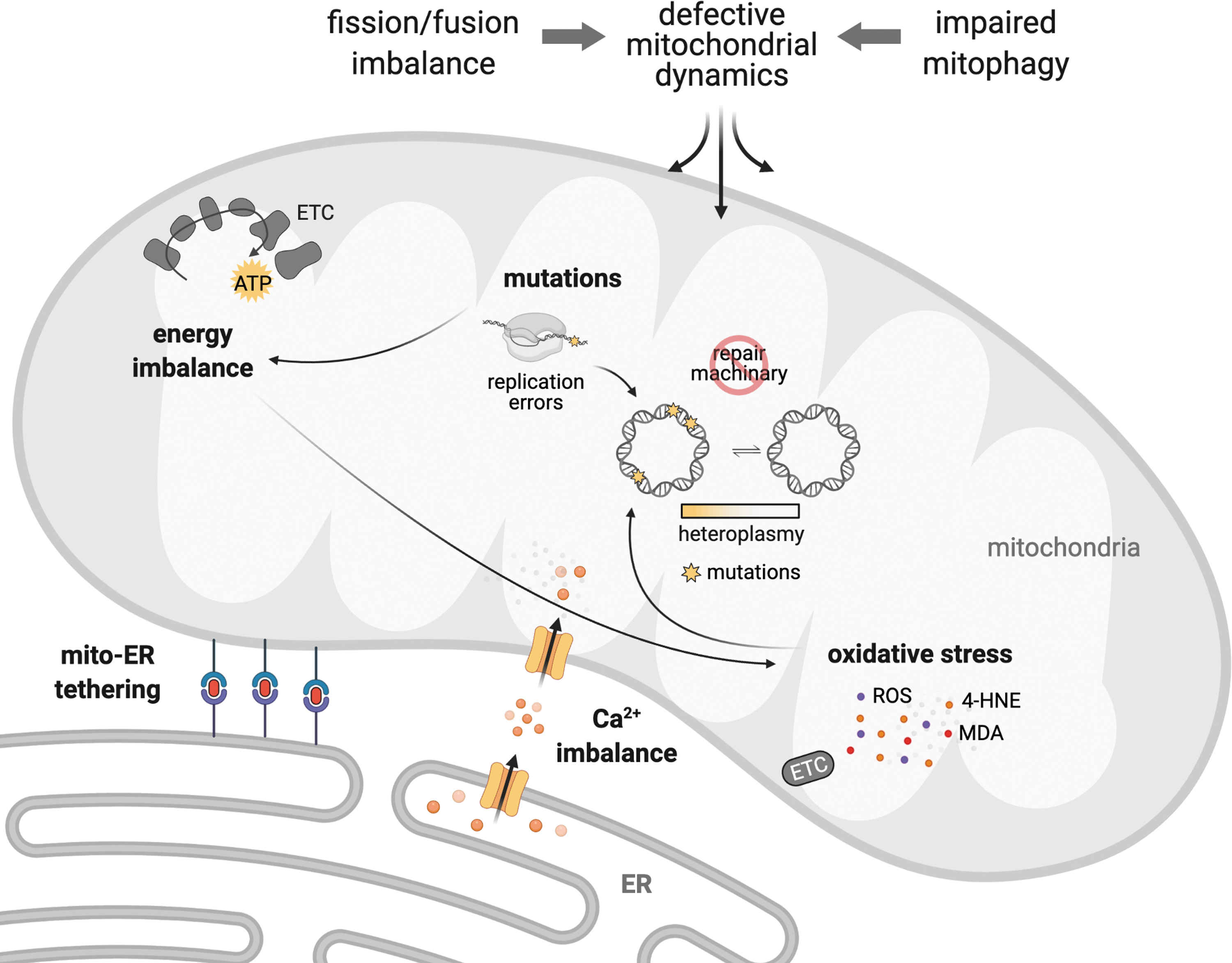
As previously discussed, disruption of mitochondrial quality control through impaired mitochondrial fission, fusion, or defective mitophagy negatively affects mitochondrial number, size, and shape. These changes ultimately result in excessive accumulation of damaged mitochondria, compromised mitochondrial segregation, impaired bioenergetics, and oxidative stress, which together contribute to the establishment and progression of cardiac diseases (Fig. 6). Therefore, it is expected that pharmacological strategies that reestablish mitochondrial connectiveness through inhibition of pathological mitochondrial fission, boosting mitochondrial fusion, or facilitating the clearance of dysfunctional mitochondria will abolish or mitigate cardiac mitochondrial dysfunction and therefore become a promising approach to improve the prognosis of cardiac diseases (193).

Different strategies targeting mitochondria biology, such as small interfering RNA (siRNA), CRISPR-Cas9 gene editing, small molecules, or short peptides, have been successfully tested in preclinical studies of cardiac diseases. The mitochondrial fission inhibitors mdivi-1 (58, 126), P110 (76), Drp1 siRNA (172), or calcineurin inhibitor (175) efficiently reverse the acute damage induced by ischemia/reperfusion injury in different tissues, including the brain, heart, and kidney. Treatment with mdivi-1, a Drp1 inhibitor, before ischemia attenuates mitochondrial damage and myocardial infarct size in mice subjected to transient coronary artery occlusion (126). Of interest, mdivi-1 poorly inhibits recombinant Drp1 GTPase activity in vitro (58, 125).
A recent study demonstrates that mdivi-1, at the same concentration that reduces mitochondrial fragmentation, reversibly inhibits mitochondrial complex I-dependent oxygen consumption and ROS production, even in the absence of DNM1L (125). Moreover, mdivi-1 induces mitotic spindle abnormalities and mitosis arrest by inhibiting tubulin polymerization in a Drp1-independent manner (61), which might affect fibroblast-mediated cardiac remodeling, function, and inflammation during pathological conditions (194). In summary, Drp1-independent effects of mdivi-1 remain a major issue limiting its use as a promising candidate to treat cardiac diseases.
Inhibition of Drp1-Fis1 interaction by the short peptide P110 (53, 102) reduces Drp1 enzymatic activity and efficiently blocks mitochondrial fragmentation in different cellular and animal models of cardiac diseases (Fig. 6) (53, 76, 101). The administration of P110 at the onset of reperfusion reduces cardiac accumulation of fragmented and dysfunctional mitochondria induced by ex vivo ischemia/reperfusion injury in rats (53). A single dose of P110 peptide at reperfusion is sufficient to inhibit mitochondrial fragmentation and increase mitochondrial size and bioenergetic profile after in vivo transient coronary artery occlusion in animals (53). These P110-mediated changes result in improved cardiac contractility properties when measured 3 weeks after myocardial infarction surgery (53).
Importantly, in contrast to mdivi-1, P110 does not inhibit basal activity of Drp1 in vivo, even after 5 months of treatment (54). P110 blocks pathological Drp1-Fis1 interaction and does not affect Drp1 binding to other adaptor proteins involved in physiological mitochondrial fission (i.e., Mff) (159). In summary, inhibition of DNM1L, specifically by targeting Drp1-Fis1 protein/protein interaction, seems to have a therapeutic potential in preventing ischemia-induced cardiac injury and the subsequent establishment of heart failure (Fig. 6). Similar protective results are observed in other degenerative diseases, including diabetes (173), Huntington's disease (54, 76), amyotrophic lateral sclerosis (102), Parkinson's disease (67, 159), and Alzheimer's disease (101).
The therapeutic use of peptides targeting mitochondrial fission and fusion has promising opportunities due to their high specificity and affinity to their targets as well as ease of modification and high biocompatibility, therefore having relatively few off-target effects.
The use of rationally designed peptides targeting selective protein/protein interaction has been successfully used in clinics to treat cancer [for review see Araste et al. (10)] and is well tolerated in patients with cardiac diseases (17). However, there are still some challenges for peptides as therapeutic entities, including a short half-life and lack of intracellular penetration compared with small molecules. Overall, both opportunities and novel strategies to circumvent the therapeutic challenges place rationally designed peptides targeting mitochondria, such as p110, as promising candidates to treat cardiac diseases.
Another strategy was recently developed to address mitochondrial fragmentation in failing hearts. In this case, the excessive accumulation of fragmented and dysfunctional mitochondria occurs throughout an excessive interaction between protein kinase C βII (PKCβII) and Mfn1 at the outer mitochondrial membrane, which results in PKCβII phosphorylation and the consequent inhibition of Mfn1 in a rodent model of myocardial infarction-induced heart failure (64, 65).
Under this scenario, the design and synthesis of a selective short peptide capable of attenuating PKCβII-Mfn1 interaction, termed SAMβA, are sufficient to counteract excessive mitochondria fragmentation in heart failure (Fig. 6) (64). Targeting intermolecular associations is a promising strategy to mitigate detrimental biological processes in a highly selective manner (63, 64, 146). The development of short peptides derived from protein interfaces that transiently affect intermolecular interaction, and therefore act as competitive inhibitors, becomes a useful tool to counteract the establishment and progression of cardiac diseases (25, 64, 130).
SAMβA protects primary culture of cardiac myocytes against angiotensin II-induced mitochondrial dysfunction and cell death (64). SAMβA treatment also improves heart failure outcome in a rat of model of myocardial infarction-induced heart failure (64). Sustained SAMβA delivery improves both morphological and functional properties of failing hearts compared with vehicle-treated heart failure animals. These changes in SAMβA-treated heart failure animals are paralleled by a series of biochemical modifications in failing hearts, including reduction in the number of fragmented mitochondria, improvement of mitochondrial OXPHOS efficacy, and redox homeostasis.
These findings unravel the contribution of PKCβII-Mfn1 association in disrupting the mitochondrial fission/fusion balance in heart failure, therefore highlighting PKCβII-Mfn1 interaction as a promising target to develop novel pharmacological interventions capable of improving mitochondrial health and heart failure outcome in patients. Note that cardiac overactivation of PKCβII and accumulation of dysfunctional mitochondria are also present in both heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF) patients (36).
Summary and Perspectives
The unique role of mitochondria as intracellular nodes and multieffector players involved in the maintenance of cardiac homeostasis in health and disease has been recently recognized. Under this scenario, extensive work has been performed to decipher the most critical processes involved in mitochondrial dysfunction and its contribution to cardiac diseases. As discussed above, the quality control mechanisms that regulate mitochondrial fission/fusion balance and removal play a critical role in the maintenance of mitochondrial health, therefore becoming potential novel therapeutic targets for cardiac disease. New targets affecting mitochondrial fission/fusion balance and clearance have been recently tested in many preclinical models, therefore paving a new road for the development of interventions capable of improving mitochondrial function to treat cardiac diseases.
Finally, many efforts have been made to identify and validate promising targets affecting mitochondrial quality control. However, the development and improvement of available strategies to increase tissue and cell permeability, as well as drug delivery inside mitochondria, are critical for the translation of these molecules targeting mitochondrial quality control to the clinic. Therefore, combined strategies to approach and treat defective mitochondrial fission/fusion balance and mitophagy seem to be a promising way to design effective interventions and develop new therapies for complex and multifactorial diseases such as cardiac diseases.
Footnotes
Authors' Contributions
D.L.S. and J.C.B.F. wrote the article. A.A.G., and L.L. designed the figures and contributed to the writing. J.C.B.F. designed the article and D.M-R. and J.C.B.F. revised it. All the coauthors have reviewed and approved the final version of the article.
Author Disclosure Statement
Patents on the design and application of mitochondrial fission peptide inhibitors have been filed. The authors claim that there is no conflict of interest related to this work.
Funding Information
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) numbers 2019/22204-2, 2018/18627-3, 2019/25049-9, 2013/07937-8, and 2015/22814-5, Conselho Nacional de Pesquisa e Desenvolvimento—Brasil (CNPq), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) Finance Code 001 (to J.C.B.F.), and by NIH R01HL52141 (to D.M-R.).