
Abstract
Significance:
Alzheimer disease (AD) is an all-too-common condition in the aging population. However, aging does not automatically equal neurodegeneration and memory decline.
Recent Advances:
This review article involves metabolic changes in the AD brain that are related to oxidative stress. Selected pathways are identified as potential targets for intervention in AD.
Critical Issues:
One of the main factors of AD is the oxidative imbalance within the central nervous system, causing a disruption in metabolic processes. Reactive oxygen species (ROS) are a natural consequence of many cellular processes, especially those associated with mitochondria, such as the electron transport chain. Some ROS, when kept under control and maintained at reasonable levels, often play roles in cell signaling. The cellular damage of ROS arises when oxidative imbalance occurs, in which case ROS are not controlled, leading to a myriad of alterations in cellular metabolic processes. These altered pathways include, among others, dysfunctional glycolysis, calcium regulation, lipid metabolism, mitochondrial processes, and mammalian target of rapamycin pathway dysregulation.
Future Directions:
Understanding how ROS can lead to these alterations can, ideally, elucidate therapeutic options for retarding AD progression in the aging population. Antioxid. Redox Signal. 36, 1289–1305.
Introduction
The brain is optimized for learning and as humans live longer they accumulate more information. It is well known that neurons are involved in cognition, whereas glial cells help maintain homeostasis of the central nervous system (CNS) and are highly heterogeneous comprising oligodendrocytes, microglia, and astrocytes (110). Astrocytes are the most abundant and diverse glial cell, with roles in organizing and maintaining brain structure and function within all levels of CNS homeostasis such as molecular, subcellular, cellular, organ, and system (125). Astrocytes, which are intimately involved in glutamatergic and GABA-ergic neurotransmission (125), form a structurally interconnected network with long-distance signaling properties (82). Activation of astrocytes often leads to neuroprotection against the expression of pro-inflammatory molecules such as cytokines and chemokines (93). Neuron–glial interactions are important, as such interplay controls synaptic plasticity and consequent learning and memory formation. Astrocytes participate in the regulation of cerebral blood flow depending on neuronal activity and metabolism (68). Disruption in neuronal–astrocyte interactions and altered balance of signaling can lead to disturbances in homeostasis. Glial pathology occurs in the aging brain and is a major contributor to age-related neurogenesis (113).
As the brain ages, there is a decline in short-term memory; however, severe neuronal loss is more characteristic of neurodegenerative diseases. Aging is the greatest risk factor of neurodegenerative diseases, the most prominent being Alzheimer disease (AD) (110). The two types of AD are sporadic and familial, with sporadic AD comprising well more than 90% of the AD cases of patients who have no specific familial causal link. Genetic, environmental, and lifestyle factors may all be risk factors in the development of AD. Familial AD, however, makes up 2%–3% of cases and presents with an early onset of symptoms (age <65 years) with positive familial history (58).
AD is characterized by pathological hallmarks such as extracellular plaques that are rich in amyloid β-peptide (Aβ), intraneuronal neurofibrillary tangles (NFTs) composed primarily of hyperphosphorylated tau protein, brain atrophy, and increased neuroinflammation (68) (Fig. 1). Aβ is derived from the amyloid precursor protein (APP), a type I protein that resides in the plasma membrane. APP can be cleaved by γ-secretases and β-secretases, resulting in different products with different cellular functions. Through interactions, β-secretase, APP dimerizes and endocytoses into the cell where it is cleaved, producing sAPPβ and β-carboxyl terminal fragment (βCTF) (116). The component βCTF can then undergo cleavage via γ-secretase, resulting in Aβ (116) (Fig. 4). Presenilins act as components of membrane-bound aspartyl protease assemblies and sustain γ-secretase cleavage of APP, increasing Aβ production. Presenilins, not only expressed in the CNS mainly in neurons but also detectable in glial cells, form the catalytic core of γ-secretase (58). As noted, NFT are composed principally of highly phosphorylated tau and of medium- and high-molecular-weight neurofilaments (104). Tau is a neuronal protein that is encoded by the microtubule-associated protein tau (MAPT) gene. Disproportional phosphorylation of tau occurs in AD, with the longest isoform of human tau containing 85 potential phosphorylation sites (64). Neurofilaments and tau can form noncovalent aggregates in the absence of oxidation; however, they have a limited lifetime. The NFTs contain many covalent crosslinks that cannot easily be removed.

The brain is highly sensitive to homeostatic disturbances, and in AD these disturbances can occur as astrocytes localize around Aβ plaques, which potentially activate cells (93, 113). Neurogenesis is reportedly increased in the AD hippocampus, as seen by the expression of the immature neuronal markers doublecortin and TOAD (Turned On After Division)/Ulip/CRMP 4 (TUC-4) that are expressed in neurons in neuroproliferation. This may suggest a possible treatment option for those diagnosed with AD to give rise to new cells, replacing the lost neurons (75). Functional disconnect can also be observed between the hippocampus and other brain regions in AD (3). Normal, mild cognitive impairment (MCI) and AD subjects were analyzed via 18F-FDG PET/functional MRI and the MCI/AD patients showed reduced standardized uptake value ratios, indicative of decreased glucose metabolic utilization, along with reduced gray matter volume and reduced functional connectivity (131).
The earliest pathological changes within AD reportedly occur in the entorhinal cortex of the medial temporal lobe and the hippocampus, both of which are prone to NFT formation as well as Aβ plaques (96). Major loss of neurons can be seen in the cortical II layer of the entorhinal cortex in the AD brain. Synaptic loss associated with Aβ is also an early event in AD, and due to defects in synaptic plasticity, long-term potentiation and long-term depression of the synaptic strength are likely affected (53). Neurons in the entorhinal cortex that synthesize and innervate the hippocampus also die in AD (99). Entorhinal tau, not global tau, accumulation was correlated with hippocampal hyperactivation as shown among 15 MCI/AD patients given an item memory task during MRI scans (109). Our laboratory demonstrated that, compared with brains from people with no cognitive or pathological alterations obtained at very short postmortem intervals (PMI, less than 4 h), in the AD brain obtained at the same very short PMI, elevated oxidative stress occurred in brain regions rich in Aβ pathology, such as the hippocampus and frontal cortex, but not in Aβ-poor cerebellum (72).
The aging brain itself is highly susceptible to disease due to a number of factors. Neurons are subject to damaged protein accumulation, increased oxidative stress, perturbed energy homeostasis, and mutations in nucleic acids (69). Neurons with relatively long axons are most damaged in AD as they have high energy requirements, rely on axonal transport for functional support, and have a large cell surface area. Astrocytes have significantly higher levels; they are various antioxidants, including glutathione, glutathione peroxidase (GPx), catalase, and thioredoxin reductase (10). Astrocytes shuttle glutathione precursors to neurons, and this process is instrumental in the neuroprotective effects of astrocytes toward neurons (10). Although astrocytes and neurons can synthesize the glutathione (GSH) tripeptide on their own, neurons are highly dependent on astroctyes for their supply of precursor amino acids for GSH synthesis (10).
Normal synapse function requires well-coordinated mechanisms that, in turn, require a sufficient supply and utilization of energy substrates (96). Energy is typically found in the form of ATP, which requires a steady supply of glucose to the brain. The fate of glucose, once inside the cells, depends on the cell itself: Neurons use oxidative phosphorylation mostly, whereas astrocytes are glycolytic. However, aging increases glycogen metabolic enzymes, causing a shift from astrocytes to neurons, elevating the capacity of neurons to oxidize glucose (47). This suggests that neurons become independent of astrocytic lactate and metabolic crosstalk becomes disturbed. Lactate from astrocytic glycogen supports memory function in hippocampi (47).
Studies show disturbed neuronal cytoskeletal network not only in axons but also in cell bodies, along with elevated vesicle accumulation within cell bodies in AD (140). Axonal blockage interferes with axonal transport, evinced by the reduction in numbers and the total length of microtubules. Microtubules are consumers of a large amount of ATP, as this energy source is required for transport and polymerization. Microtubule assemblies are depleted in AD, in large part due to the removal of hyperphosphorylated tau from the microtubules, thereby decreasing the stability of mictotubule assemblies. This, in turn, leads to a buildup of active mitochondria within the cell body and spent mitochondria at the presynaptic terminus, leading to both decreased ATP at the synapse needed to maintain cell potential and elicit neurotransmission, and elevated ROS accumulation (76, 140).
Oxidative Imbalance
Much of the interferences within AD can be attributed to the elevated oxidative stress within the brain. The brain is highly sensitive to oxidative imbalance as it is rich in easily peroxidizable fatty acids, has a high requirement for paramagnetic O2, and transitions metal ions (20, 21, 129). These three factors play an interwoven role in the regulation of oxidative balance within the brain. Oxidative stress is due to an imbalance of reactive oxygen species (ROS) and the ability of the antioxidant system to effectively remove them (34, 69, 71). The ROS generation is inevitable in oxidative ATP production and is the primary cause of macromolecular damage (12). However, ROS are essential for cell signaling (117). The ROS play a role in a range of signaling processes from protein phosphorylation to transport systems. Depending on their location and subcellular compartmental concentration, ROS can have either negative or positive effects (41, 69).
The ROS are oxygen-containing usually reactive molecules. Unstable, short-lived, and highly reactive ROS are either free radicals or molecules from which free radicals are easily generated, ranging from low reactivity to high reactivity (88). The most common ROS is superoxide radical anion, which is produced mainly in complex I of the electron transport chain (ETC) within the inner mitochondrial membrane. Superoxide radical anions can move within 30 nm of formation; however, when protonated, superoxide becomes neutral HO2, which is able to cross the inner mitochondrial membrane and accrue subsequently in the cytoplasm (91). A net effect of these oxygen radicals and ROS is damage to and death of vulnerable neurons in AD (104).
Although ROS usually have negative effects on the cell, they are also necessary for some cell processes (69, 94). The ROS act as signaling molecules, regulating and maintaining normal physiological function (69, 94). Redox-derived changes affect transcription, phosphorylation, and other signaling events. The ROS have also been shown to promote cell proliferation via redox reactions (69, 94).
Antioxidant proteins and small-molecule antioxidants perform neutralization of various ROS such as superoxide (21).One such antioxidant protein is the superoxide dismutase (SOD) family, which converts superoxide to hydrogen peroxide (H2O2) (88). H2O2 is also an ROS and due to its zero-dipole moment can easily diffuse across the inner mitochondrial membrane moving at least 1 μm from the site of production. Although H2O2 is less reactive than superoxide, it can interact with reduced iron (Fe2+), from iron–sulfur proteins or heme proteins, through Fenton chemistry forming one of the most highly reactive ROS, hydroxyl free radicals (OH·) (67, 88). Much of this work largely stems from the early work of Gutteridge and his colleagues. Hydroxyl radicals are the most reactive and can cause large amounts of lipid peroxidation within a short range from their formation (12, 22). Due to their high reactivity, OH· have a broad range of possible interactants. However, hydroxyl radicals have a higher site specificity due to being formed at the metal location (67). Disruption in metal metabolism has been seen in AD. Reportedly, there are higher copper ion concentrations in the CSF of AD patients as well as elevated levels of iron ions in the amygdala, hippocampus, and olfactory pathway (139). Iron regulatory protein (IRP)-2 is increased in AD and regulates the expression of the iron storage protein, ferritin, to maintain iron homeostasis. In AD brain, alterations in IRP interactions with the iron-responsive element of DNA cause an increase in iron ions (139).
As noted, ROS are generated in many areas within the cell; however, a large proportion of ROS are produced in the mitochondria. There are 11 sites within the mitochondria where ROS can be produced, with two sites being the most relevant. Within the respiratory chain in the inner membrane of the mitochondria, complex I and complex III produced superoxide (117). Electrons from NADH and FADH interact directly with O2 or other electron acceptors. Complex I is a multi-subunit complex comprising more than 46 proteins and contains at least one flavin mononucleotide and eight iron–sulfur groups (9). Complex I comprises a hydrophobic arm and a matrix-protruding hydrophilic arm. The hydrophobic electron carrier, Coenzyme Q10-binding is located at the tight junctions between the arms (12).
Two sites within complex I are proposed to be the major sites for ROS production. In isolated complex I, fully reduced flavin, site IF, produces ROS; however, under physiological conditions, ROS production is rather low (12). The quinone-binding site, site IG, has a higher capacity for superoxide production than the site IF (12). Complex III participates in the Q-cycle (9) and contains electron carriers (cytochromes b566, b562, c1, the Rieske Fe-S clusters, and quinones). The quinones are located at two centers, centers i and o; center o has a higher superoxide production. The superoxide from complex I is generated into the mitochondrial matrix, and from complex III it is split into the matrix as well as the inner membrane space (12).
Antioxidants are important to keep ROS in check within the cell, and an increased resistance to oxidative stress can improve longevity in mice (1). However, not all studies have found an increased antioxidant activity to correlate with extended life in rodents (1). In addition to SOD, catalase, GPx, and peroxyredoxins and other antioxidant proteins scavenge ROS (1). As implied earlier, SOD catalyzes the dismutation of superoxide to O2 and H2O2. The latter can be further converted to H2O via catalase and GPxs.
GPx is found within the cytosol and mitochondria. Glutathione peroxidase 1 (GPx1) is a main form of the GPx protein family and is ubiquitous in all cells. Its function is the neutralization of H2O2. However, GPx1 knockout mice show no evidence of increased oxidative damage to lipids and proteins compared with wildtype (111). GPx4 is resident in mitochondria. Knock-out of GPx4 is highly detrimental to cells (13, 70). The total antioxidant capacity is decreased in AD as well as MCI and may contribute to oxidative imbalance (25, 26, 81, 138).
Another frontline antioxidant is GSH, which resides in the cytoplasm in a dynamic pool of the reduced thiol and the disulfide oxidized form (GSSG) (1, 69). GSH in the presence of radicals is converted to GSSG, which can be converted back to GSH via glutathione reductase by using the reducing equivalents of nicotinamide adenine dinucleotide phosphate (NADPH). Neurons can also limit oxidative damage through the use of glucose metabolism via the pentose phosphate pathway (PPP) generating NADPH(H+), which helps maintain glutathione in the reduced state (77). However, there is evidence that PPP activity is lessened in apolipoprotein E4 (ApoE4) carriers, which would lead to decreased levels of NADH and, consequently, less reduced GSH and elevated oxidative stress (21, 23, 51, 74).
The ROS cause damage in AD and lead to advanced glycation end products, nitration, lipid peroxidation, and protein carbonyls (20, 21, 114). There is oxidative damage to every class of biomacromolecule, sugar, lipid, protein, nucleic acid, in AD (139). Usually, such damaged proteins can be removed by various degradative pathways, including proteasome, autophagy, and unfolded protein response pathways. However, in AD and MCI brains, each of these pathways is defective, likely due to oxidative damage (26). An increase in circulating ROS and protein carbonyls can be seen in F344 rats as early as middle age (15 months) (87). As previously stated, the brain is highly vulnerable to attack by ROS due to its high energy and oxygen consumption, high unsaturated lipid content, high level of transition metals, and relative scarcity of the antioxidant system (139).
There is widespread agreement across many laboratories that oxidative stress is a key contributor to the pathogenesis and progression of AD (15, 17, 20 –22, 90, 102, 114, 135). Despite this strong agreement, some reports suggest that oxidative stress is either not a major player in AD pathophysiology or secondary to other biological effects. For example, although simple eicosanoids were increased in the AD brain, the lists of oxidized products in AD brain and control brain determined by the authors reportedly were not different from each other (56). Of course, one should note that the PMI and cause of death may have influences on oxidative stress parameters (25). Another study demonstrated high oxidative stress indices in the AD brain, but not in people characterized as nondemented with Alzheimer's neuropathology. The authors ascribed these results to high levels of scavenging systems, including low levels of PGC1α miRNA-485 (54). The concept of cognitive reserve may be applicable with such people. Another recent paper suggested that micro-RNAs that regulated aspects of oxidative stress were the underlying reason for oxidative stress differences in prodromal AD (98).
Metabolic Alterations Occur During Alzheimer Progression
Glycolysis
The brain is only 2% of the body's total mass; however, 20%–25% of total body respiration occurs in the brain (139). Even a small interference in the brain's consumption of energy is correlated with signs of mental and neurological functions associated with disorders. Energy is consumed in the form of ATP, for which glucose metabolism and lipid metabolism are some of the major contributors. The brain consumes a large amount of glucose to maintain presynaptic and postsynaptic ion gradients. Phospholipid asymmetry of the bilayer requires a large amount of energy and has been shown to be lost in AD and MCI (8). Neurotransmission also requires constant phospholipid remodeling, which attributes to 26% net energy uptake of the brain's total energy (21, 34). The blood-brain barrier (BBB) allows for the transport of glucose, amino acids, and ketones from the blood to the brain by using specific transporters, while preventing bloodborne molecules and cells that could potentially harm neurons (32). On-resonance variable delay multiple pulse MRI was used to follow glucose uptake in AD mouse models. D-glucose uptake in the cortex was reduced and slower D-glucose uptake rate in CSF was shown, suggesting impaired glucose transporters (GLUTs) for the BBB and blood-CSF barriers (35).
Glucose is transported into the brain via GLUTs. GLUT1 (55 kDa) allows for glucose to cross the BBB into the extracellular space of the brain (132). This is followed by a second isoform of GLUT1 (45 kDa), which transports glucose in astrocytes. GLUT3 transports glucose into neurons (Fig. 5). Once inside astrocytes, glucose will continue to undergo anaerobic glycolysis where pyruvate is produced, continuing through net lactate extrusion (123). The first step of glycolysis is irreversible and ensures continuation of glucose-6-phosphate (G6P) into either the PPP or glycolysis. Neurons oxidize glucose via glycolysis, PPP, tricarboxylic acid (TCA), and the oxidative phosphorylation within the mitochondria. One glucose molecule produces CO2, H2O, and 30–36 molecules of ATP depending on proton leakage in the ETC and the shuttle system used to produce matrix-resident NADH (32).
Neurons prefer the use of PPP, which is an anabolic pathway converting G6P into five carbon sugars for nucleotides. This generates reduced NADPH, an important cofactor for fatty acid and myelin synthesis, neurotransmission, and redox homeostasis (32). Neurons lack the enzyme responsible for production of fructose-2,6-bisphosphate, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) as it is in continuous ubiquitin-dependent proteasomal degradation. Rather they express pyruvate kinase M1, which has a high affinity for phosphoenolpyruvate, ensuring higher levels of pyruvate. In conjunction with this, neurons also contain the low pyruvate affinity isoform of lactate dehydrogenase (LDH), limiting the conversion of pyruvate to lactate (32). Of course, under anaerobic conditions, such as a stroke, LDH is activated.
Astrocytes, on the other hand, mostly perform glycolysis with the generation of lactate contributing to their low levels of mitochondrial oxidative phosphorylation. Neuron-glial interactions actively control synaptic plasticity and neurotransmission (63). During chronic oxidative stress in AD, communication between astrocytes and neurons is impaired, disrupting memory consolidation (63). Implied from the earlier discussion, disrupted communication between astrocytes and neurons would lead to decreased GSH levels in neurons, that is, elevated oxidative stress. Astrocytes contain high levels of PFKFB3 maintaining high conversion rates to lactate and NAD+/NADH favoring aerobic glycolysis. Pyruvate is transformed into acetyl-CoA via the pyruvate dehydrogenase complex and follows into the TCA cycle. Reducing equivalents in the form of NADH and FADH are produced and shuttle electrons into the ETC located in the mitochondria (132).
With regard to glucose, there is a decrease in glucose uptake and an imbalance in O2 utilization causing loss of aerobic glycolysis. However, BBB integrity is compromised in AD evidenced by neurovascular dysfunction in the early stages of the disease (32). Mice overexpressing Aβ, but having GLUT1 deficiency, led to BBB breakdown, accelerated accumulation of Aβ, and decreased clearance of Aβ (129). As noted earlier, oxidative modification also affects glycolytic enzymes such as aldolase, triosephosphate isomerase, GAPDH, phosphoglycerate mutase 1, and α-enolase in AD (21), leading to reduced glucose metabolism in the AD brain. Within normal aging, levels of glycolytic products are decreased (132). GLUT1 and GLUT3 expression is reduced in AD, causing a loss in glucose transport, which may compromise cytoplasmic Ca2+ removal (4). Hexokinase, the first enzyme in glycolysis converting glucose to G6P, binds to the voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane. This interaction keeps the mitochondrial permeability transition pore (mPTP) closed and generates an outer membrane potential, causing neuroprotective effects. In the AD brain, hexokinase levels are decreased and VDAC levels are increased (132).
Insulin is needed for the regulation of glucose uptake. Secretion of this peptide hormone by pancreatic β cells is under neuronal control (112) (Fig. 2). However, rising levels of insulin can lead to extracellular regulatory kinase activation, which phosphorylates tau at Ser202, causing hyperphosphorylation (112). This causes destabilization of microtubules within the axons of neurons. Insulin also regulates the metabolism of APP into Aβ (112). Biomarkers of insulin resistance are well known in the AD and MCI brain (21, 121, 122).
Lactate, from astrocytic glycogen, supports memory function in the hippocampus. Aging increases concentrations of glycogen metabolism enzymes, causing a shift from astrocytes to neurons. This causes an elevated capacity of neurons to oxidize glucose but decreases fatty acid utilization (47). This suggests that neurons become independent of astrocytic lactate and metabolic crosstalk has become disturbed.
Mitochondrial metabolism
Mitochondria are double-lipid membrane organelles that are maternally inherited (1). They have drastically different morphologies that range from small spheres to short rods to even long tubules. Their length, shape, size, and number are all controlled by fusion and fission, which are in constant balance. Mitochondrial transport is required for distribution within the cell, a process that is highly regulated, especially in neurons (43). Mitochondria move along microtubules through the action of kinesin and dynein within the cell. Neurons are dependent on the proper control of mitochondria, as they have high energy demands to maintain synapsis and neurotransmission (43). Within neurons, mitochondria are concentrated in the presynaptic and postsynaptic spaces and axonal terminals. Mitochondria take part in highly interconnected and dynamic networks involving many of the organelles within the cell, such as the endoplasmic reticulum (ER), actin skeleton, and lysosomes. Many processes are in place to ensure mitochondrial homeostasis, including mitochondriogenesis, mitochondrial plasticity, autophagy (mitophagy), and the mitochondrial unfolded protein response (UPRmt) (105).
The internal structure of the mitochondria is highly dynamic with an inner membrane consisting of distinct regions, the inner boundary membrane, the cristae membrane, and the cristae junction (43). The inner boundary membrane resides where the inner membrane is within closest proximity to the outer mitochondrial membrane. These regions constitute separate functional domains.
The mitochondrial ETC spans within the inner membrane of the mitochondria and comprises five membrane-spanning enzyme complexes: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome bc1 complex (complex III), cytochrome c oxidase (complex IV), and ATP Synthase (sometimes called complex V) (96) (Fig. 3).

Mitochondria have an important function in aging. Their integrity declines with aging, and there is an age-dependent increase in damaged mitochondrial DNA (mtDNA). Studies demonstrate that old mitochondria are morphologically altered and produce more oxidants and less ATP (9). Mitochondria rely on fusion and fission for their optimal performance. Fusion enzymes include dynamin GTPase regulators optic atrophy 1 (OPA1), mitofusion 1 (MFN1), and mitofusion 2 (MFN2), whereas fission enzymes include the dynamin-like GTPase regulator dynamin-related protein 1 (DRP1) (52). Mitochondrial size directly relates to its function, distribution, quality, and interaction with other cell components and organelles. Mitochondrial dynamics are altered in AD evidenced by increased fragmentation, which is even more pronounced with increased Aβ and phosphorylated tau (52). Phosphorylated tau can promote mitochondrial dysfunction by interacting with DRP1 in the brain (64).
Mitochondrial quality control is a major contribution factor to aging (105). Oxidative stress can cause activation of mitophagy, resulting from the engulfment of dysfunctional mitochondrial into autophagosomes. Mitophagy impairment is seen in AD, with reductions of mitophagy program elements in the AD hippocampus (127). Damage to mitophagy can lead to buildup of damaged mitochondria, releasing the components into the cytosol or extracellular space triggering an immune response (105). Along with release of copious amounts of free radicals, mitochondrial dysfunction reportedly leads to inflammation within the brain (136).
A key site of ROS damage within the mitochondria is at the mtDNA. Bases of nucleic acids and the 2-deoxy ribose moieties are prone to free radical attack, possibly due to the lack of histones found with nuclear DNA (1). mtDNA is the source of specific subunits of the ETC, and without their expression the ETC would falter. Oxidative damage also affects individual ETC complexes and results in decreased ATP production as well as ROS elevation (17). Protein oxidation forms post-translational modifications such as hydroxylation, nitrosylation, carbonylation, disulfide bridges, and sulfhydrylation (1). Cytochrome c oxidase shows reduced activity in the AD brain (127).
Aβ interacts with the mitochondria (140), which inhibits complex I and complex IV of the respiratory chain (5). Oxidative modifications to enzymes in the TCA cycle are reported (27). One of the most prevalent enzymes affected is the α-ketoglutarate dehydrogenase complex (KGDHC), the activity of which declines in AD (60). Chronic, long-term inhibition of KGDHC mimics AD-related changes to calcium as well as a decline in glucose metabolism (60). KGDHC is a rate-limiting enzyme complex in the TCA cycle and its dysfunction leads to an increase in ROS (139). KGDHC is sensitive to oxidants and is inhibited by H2O2, NO, peroxynitrite, chloramines, hypochlorites, and 4-hydroxynonenal (HNE). Addition of the antioxidant GSH after peroxynitrite treatment was shown to restore KGDHC activity (62). Modifications to KGDHC compromise metabolism associated with neuronal activity in AD, and cognitive decline derives from TCA abnormalities (62).
Decreased expression and oxidative modification of ATP synthase subunits are also reported in AD (18, 21). ATPase activity is inhibited by APP and Aβ, as they have sequence homology with the native ATPase inhibitor factor IF1 (48). Decreased ATP production is detrimental for neurons and diminishes the ability to maintain ion gradients, causing an influx of Ca2+ (21).
Calcium regulation and signaling
Calcium plays a pivotal role in mitochondrial remodeling (42). Ca2+ homeostasis displays other roles in control of neuronal activity such as, among others, neurotransmitter release, synaptic plasticity, memory storage, and neuronal death (30). The concentration of Ca2+ is finely regulated by cell surface receptors, channels, pumps, Ca2+ buffers, and Ca2+ sensors (33). Ca2+ signaling is controlled by influx through voltage-gated channels, N-methyl-D-aspartate (NMDA) receptors, and tRP channels. NMDA receptors have an influence on synaptic plasticity, cognition, and connectivity of neural networks (83). Early long-term potentiation is caused by membrane depolarization and glutamate binding to postsynaptic receptors that activate NMDA. Ca2+ then enters the postsynaptic neuron, continuing to stimulate the reactions required for early long-term potentiation (66).The major source of intracellular Ca2+ is released from internal stores located in the ER and mitochondria. In ER, two key releasing channels, the ryanodine receptor and the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R), are present. Phospholipase C activation mobilizes IP3, which interacts with the IP3R, causing the release of Ca2+ (58). Ca2+ balance within the cell is maintained through the buffering capacity of mitochondria (30). Mitochondria quickly and efficiently removed Ca2+ from hotspots within the cell. To maintain the resting concentration of Ca2+, this ion must be transported out of the cell or back into the intracellular stores (58).
Efflux of Ca2+ out of neurons is controlled by two transporters. The first is the high-affinity, low-capacity plasma membrane Ca2+-ATPase (PMCA), which hydrolyzes ATP to pump Ca2+ against its concentration gradient. The second is the low-affinity high-capacity Na+/Ca2+ exchanger (NCX). The NCX is abundant in neurons and uses the electrochemical gradient of Na+ to remove Ca2+ (49). In neurons, free cytosolic Ca2+ levels are kept at ∼100 nM whereas extracellular levels reach millimolar ranges (49). Once inside the cell, Ca2+ activates calmodulin (CaM) by causing a change in conformation on binding. CaM activates calcineurin (CaN), Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII has a role in synaptic strengthening (118).
Calcium dyshomeostasis is present in the AD brain, as evidenced by hyperactive neurons close to amyloid plaques (28, 89). Elevated mitochondrial Ca2+ concentration was found in transgenic mice after plaque deposition (28). Aβ impairs mitochondrial Ca2+ homeostasis and in vitro leads to Ca2+ uptake in mitochondria, causing overload (29). Progressive Aβ overproduction and accumulation cause dysregulation of ionic homeostasis (33). The mitochondrial Ca2+ uniporter is required for Aβ-driven mitochondrial calcium uptake. Intracellular free Ca2+ concentration rises after neurotransmitter release after depolarization, which causes the activation of voltage-dependent Ca2+ channels (30). However, Aβ promotes Ca2+ entry into the cytosol and mitochondrial uptake. Aβ accumulation causes the influx of Ca2+ from extracellular space, leading to dyshomeostasis (55). Aβ may activate NMDA receptors, or open permeable pores or nonspecific ion channels as part of this activation. Chronic mild activation of NMDA ultimately leads to excitotoxicity and neurodegeration (24). Studies have identified several receptors that can mediate Aβ oligomer-mediated synaptotoxicity, such as the NMDA receptors (65). Not only does Aβ allow for influx of Ca2+ into the cell but it alters efflux of Ca2+ through the inhibition of PMCA (49). Ca2+ influx is observed when as few as 2000 oligomers of Aβ were delivered to the cell surface via nanopipette. When dosing of Aβ was halted, intracellular calcium was returned to basal levels or lower (46).
When mitochondria buffer Ca2+ appropriately, mitochondrial metabolism and energy production are stimulated, whereas excessive uptake causes the opening of the mPTP and can lead to apoptosis of the cell (11). Cytosolic levels of Ca2+ are typically around 10 nM and can reach 10 μM; however, Ca2+ must be shuttled all over the cell for fine tuning (88). Disruption of this scenario occurs in the AD brain (29). Increased intracellular levels of Ca2+ in AD enhance CaN activity, which activates different phosphatases. Ca2+ also activates glycogen synthase kinase 3β (GSK3β), which plays a role in tau phosphorylation (118). Downregulation of the expression of mitochondrial influx Ca2+ transporter genes and upregulation of efflux pathways are reported in postmortem AD brains (31). This suggests an attempt to counteract the effect of Ca2+ overload. These changes in AD can be related to oxidative damage to enzymes associated with glucose metabolism and decreased production of ATP, with consequent loss of neuronal cell potential, opening of voltage-gated Ca2+ channels, and deleterious overload of Ca2+ (21).
High incidence of seizures occurs in mouse models before Aβ plaques are observed. This is confirmed by seizures noted in the earliest patients (119). Seizures can happen at any point in the course of AD (126). Epilepsy and AD share commonalities in that the depletion of calcium binding protein Calbindin-D28K has been observed in both (119). Ca2+ is regulated spatiotemporally through the buffering capacities of calbindin and parvalbumin (7). Seizures quicken cognitive impairment, with the predominant subtype being nonmotor complex partial seizures (119).
Lipid involvement
Nervous tissue has rich lipid content, with the brain comprising a large amount and diversity of lipid classes such as phospholipids, sphingolipids, cholesterol, and its metabolites (78). These lipids and their components are essential for the structure and function of the brain (14). Essential fatty acids, those that must be ingested, such as linoleic and alpha-linoleic acids, are involved in energy production, cell growth and division, and nerve function. A high concentration of these essential fatty acids can be found within the brain (134). Lipids and other amphipathic compounds are transported via lipoproteins. These contain a hydrophobic lipid core made up of cholesterol, esters, and triglycerides (36).
Cholesterol is one of the most prominent lipids within the brain, as it is a main lipid component of neuronal and glial membranes. Cholesterol is the key constituent in myelin, plasma membrane compartments, signaling, myelination, and formation and maintenance of synapses.
The high lipid content in the brain, coupled with acyl chains of phospholipids being unsaturated, are highly susceptible to lipid peroxidation-mediated damage induced by lipid bilayer-resident free radicals (15, 78). The lipid composition of the two monolayers of the bilayer is different, containing phospholipids and proteins (79). Lipid rafts are dynamic structures within all cell membranes. The β-cleavage site of APP is cut by the β site APP cleaving enzyme 1 (BACE1), which is the major β secretase (36). Vitamin D deficiency has also been shown to be prevalent in AD (50). Vitamin D is a fat-soluble vitamin. APP/PS1 mice were fed a vitamin D-deficient diet, promoted glial activation, as well as increased Aβ production and deposits due to the elevated expression of APP and BACE1 (50). Middle-aged rats, over a period that led to old age while on a vitamin D-deficient diet, led to elevated 3-nitrotyrosine modification of proteins consistent with decreased cognitive performance (80). It is well known that autophagic and endosomal–lysosomal functions are diminished in the AD brain (38).
APP and BACE1 are associated with lipid rafts in membrane domains of cholesterol, sphingolipids, and gangliosides. Homeostasis of lipid composition is important for APP processing (36). Lipid rafts are the location where Aβ interacts with ApoE4 (79). Apolipoprotein E (ApoE) is a genetic risk factor for AD and has three main isoforms. The expression of ApoE4 has the greatest risk factor of the three alleles for developing AD with a 10-fold higher risk than the other two isoforms (39, 74, 128). Glucose uptake is impaired in astrocytes that express ApoE4 (128). ApoE4 disrupts regular neuronal function, including fatty acid oxidation, which decreases ATP production considerably (106). Fatty acids are sequestered in lipid droplets and internalized by astrocytes; however, ApoE4 reduces this sequestering as well as transport efficiency. With decreased fatty acid oxidation, lipid accumulation increases in astrocytes and hippocampus (106). Although ApoE4 has been shown to contribute to greatly increasing the risk of developing AD, another isoform ApoE2 has been shown to be highly protective against developing AD and may have possible therapeutic effects (6). E4FAD mice containing human ApoE4 were treated with rapamycin, which normalized bodyweight and restored BBB activity for Aβ transformation, neurotransmission levels, and neuronal integrity as well as reduced Aβ retention (85). (see discussion of Mammalian [aka Mechanistic] target of rapamycin [mTOR] signaling below).
Plasma cholesterol levels reportedly increase, and cholesterol accumulates in amyloid plaques and nerve terminals in AD brains (59). Cellular cholesterol increase stimulates the accumulation of Aβ. Higher cholesterol increases AD risk. Studies have shown the presence of some oxysterols increase in the AD brain compared with the healthy brain (59). One such oxysterol, 27-OHC, increase into the brain can be favored by hypercholesterolemia, which induces oxidative stress and BBB permeability. The 27-OHC is also considered a marker for reduced brain glucose metabolism in AD, as it modulates the activation of IRAP and GLUT4 (59). Plasma levels of total and low-density lipoprotein correlate with the amount of Aβ in AD brains (130).


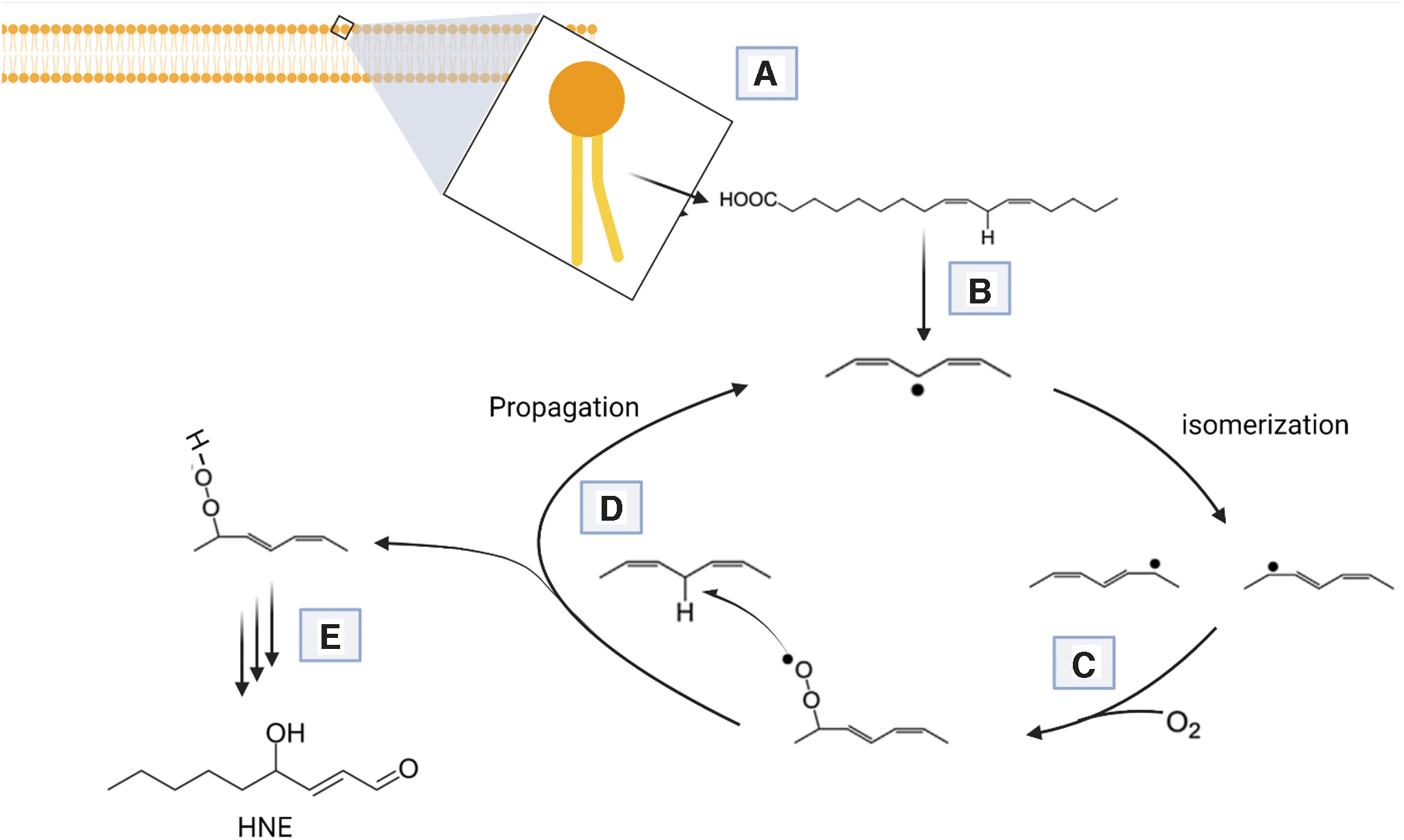
Increased ROS causes lipid peroxidation forming hydroperoxides and endoperoxides, which undergo fragmentation producing reactive intermediates such as HNE, acrolein, dialdehydes, and ketoaldehydes (78). HNE is neurotoxic and rich in AD and MCI brains (15, 19, 22, 25, 81, 84, 90, 92), and in oxidative stress conditions, HNE is found in CSF as well (95, 115). The lipid peroxidation process consists of three phases: initiation, propagation, and termination (21). Products of lipid peroxidation are classified depending on the modified lipid; isoprostanes/isofurans from arachidonic acid, neuroprostanes/neurofurans from docosahexaenoic acid, and di-homo-isoprostanes/di-homo-isofurans from adrenic acid (101). Lipid peroxidation products, including HNE (Fig. 6), are elevated in the AD brain. A study reported increased levels of specific HNE-histidine Michael adducts in the AD hippocampus (16). Other proteins that are modified by HNE include mitochondrial ETC proteins, ion and nutrient transporters, growth factor and neurotransmitter receptors, protein chaperones, and proteasomal proteins (107). mtDNA is also susceptible to oxidative stress due to the proximity to the respiratory chain as well as the absence of histones (107). Brain aging induces changes at all levels of biological organization, and longer optimal membrane lipid compositions are maintained by better cell survival and function (78).
mTOR axis
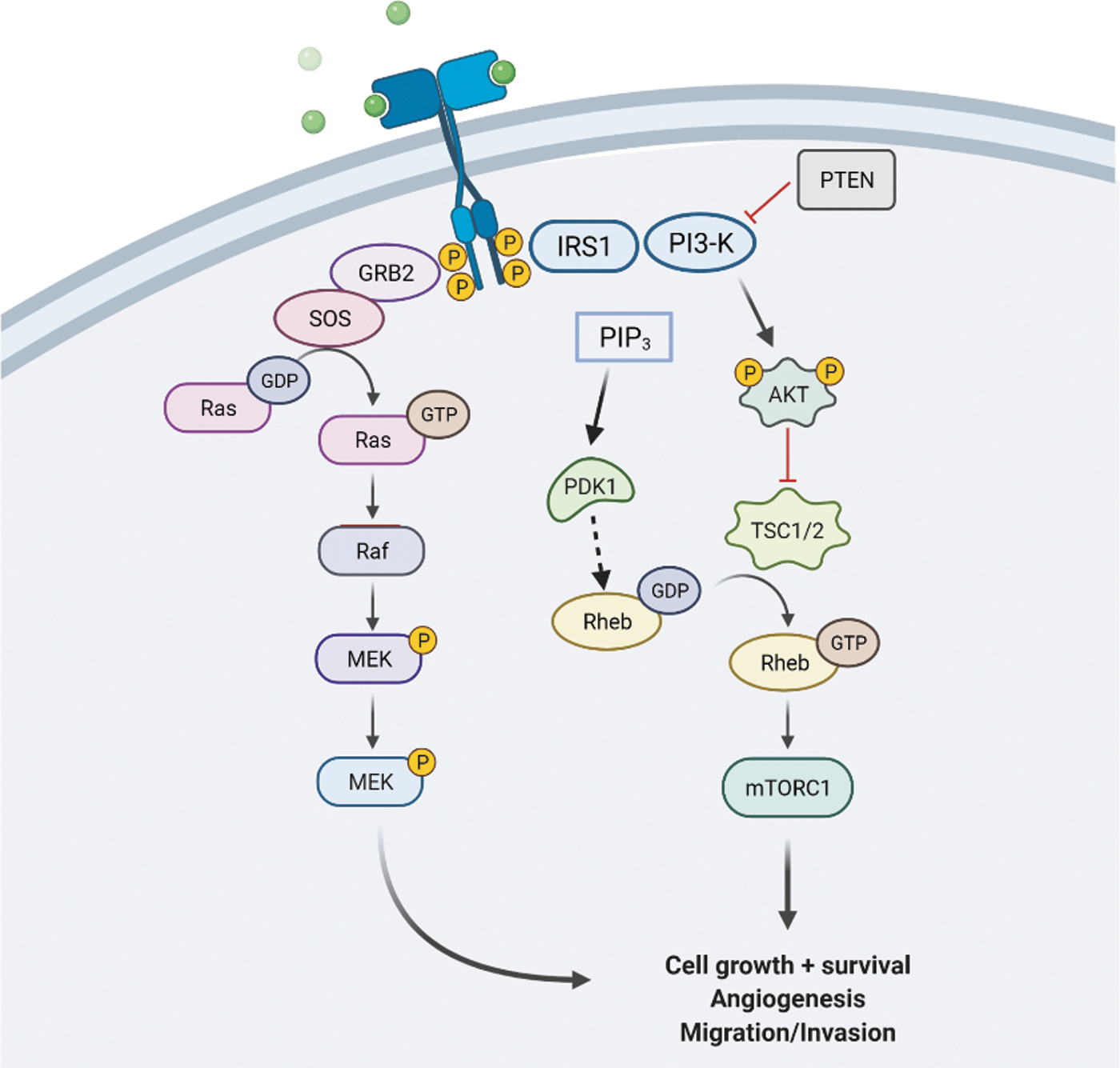
The mTOR is a 289 kDa serine/threonine protein kinase and is the catalytic subunit of two complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (86, 97, 103). These complexes regulate a number of cellular processes such as proliferation, transcription, and translation (97). mTORC1 phosphorylates substrates that increase the production of proteins, lipids, and nucleotides, and it limits autophagic breakdown (86). The first complex, mTORC1, is the more studied and contains many core components, among which are mTOR, mLST8, and regulatory protein associated with mTOR (RAPTOR). RAPTOR is required for subcellular localization (86). Upstream of mTOR is phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt), glycogen synthase kinase 3 (GSK3), AMP-activated protein kinase (AMPK), and insulin-like growth factor 1 (IGF-1) (97). Akt is a serine threonine kinase that is activated by PI3K in response to growth factors. Akt phosphorylates mTORC1 at the tuberous sclerosis complex (TSC) 2 (TSC2) and proline-rich AKT substrate 40 kDa (PRAS40) (97, 103). IGF-1 activates mTORC1 through the phosphorylation and inactivation of TSC. TSC is a negative regulator that inactivates the Ras homolog enriched in brain (Rheb) GTPase (133). mTOR is nutrient sensitive, sensing nutrient availability to promote cellular growth and protein synthesis (133). mTOR activation, however, inhibits autophagy that often is disrupted in age-related diseases (38).
The AD and MCI brains show strong evidence of mTORC1 activation, with consequent decreased autophagy and onset of insulin resistance (103, 121, 122). On activation, mTORC1 kinase phosphorylates p70S6K, which then becomes a kinase, one of whose substrates in IRS-1, phosphorylating Ser-307 to initiate insulin resistance (45, 108). Animal models replicate human findings. In APP/PS1 mice, at 18 months old, the Aβ plaque load and mTOR activation were positively correlated, whereas autophagic activity was negatively correlated (124). Moreover, intranasal rapamycin treatment of a Down syndrome mouse inhibited mTORC1 activity, decreased markers of oxidative stress and Aβ levels as well as phosphotau levels, all of which correlated with improved cognition (44, 120). In a different study, APP/PS1 mice were treated with geniposide, an extract from gardenia fruit that modulates the activity of mTOR, for 8 weeks. Mice showed improved cognitive scores and reduced Aβ deposition. In yet another study, downregulation of mTOR enhanced autophagy and lysosomal clearance, resulting in beneficial effects (137). As noted earlier, damaged mitochondria are cleared via a type of autophagy called mitophagy, which is inhibited on mTOR activation (2).
Accumulation of oxidatively damaged proteins results from alterations in many processes, including mTOR-dependent autophagy. Early aberrant hyper-phosphorylation of mTOR with consequent reduction in autophagosome formation is associated with increased 3-NT and HNE Ts65Dn mice (102). This result suggests the involvement of altered autophagy with elevated protein oxidative damage (102).
AMPK, a heterotrimeric protein, helps to maintain cell homeostasis. Phosphorylation at threonine 172 exposes the active site and is inactivated by a high ATP/AMP ratio. Inactivation of AMPK leads to the inactivation of TSC and therefore the activation of Rheb. This chain of events causes the activation of mTOR (97). AMPK activity is decreased with age and could be a link to AD pathology (97).
Deregulation of insulin and IGF-1 signaling has been shown to lead to neurodegeneration and therefore AD (97). The insulin-PIK3-Akt signaling impairment is a hallmark trait of the AD brain (57, 103, 121). Insulin binding to the insulin receptor activates either the Ras-mitogen-activated protein kinase (MAPK) or PI3K/Akt pathways (57) Disturbances in insulin signaling through the PI3K/Akt pathway affect downstream factors such as mTORC1, activating the complex and inhibiting autophagy (57). Graphene oxide is a carbon-based nanomaterial that demonstrates neuroprotective effects by effectively increasing the clearance of abnormally aggregated proteins. The AD mice were treated with graphene oxide for 2 weeks. Memory and learning impairment were ameliorated as well as the PI3K/Akt/mTORC1 pathway was downregulated, inducing autophagy (37).
GSK3-β is a prime factor in cell cycle regulation, glycogen metabolism, transcription, and translation. In glycolysis, GSK3 dissociates hexokinase from VDAC, contributing to apoptosis by allowing the mPTP to be opened (132). GSK3-β also hypophosphorylates tau (97). Hyperactive GSK3 has been considered as a factor in AD pathology and it is also connected to the PI3K/Akt/mTOR signaling cascades (97).
Possible therapeutic interventions
There have been many different therapeutic approaches suggested to slow down the progression of AD. Nutrients and metabolites that raise blood NAD+ levels or blood NAD+/NADH ratios improve the energetic status of the brain (40). DNA repair-deficient 3xTgAD/POlβ +/− mouse models have a reduced cerebral NAD+/NAHD ratio, indicating impaired cerebral energy metabolism (73). When treated with nicotinamide, riboside phosphorylated tau pathology was decreased. DNA damage, neuroinflammation, and apoptosis of hippocampal neurons were also reduced. Interventions that increase neuronal NAD+ levels have therapeutic potential (73). The TCA cycle intermediates give rise to bioactive molecules such as acetylcholine, which is decreased in AD (40, 61). Patients with AD and 3xTgAD mouse models show low glucose metabolism that precedes memory loss and cognitive decline (100). A dietary supplement of D-β-hydroxybutyrate and R-1,3 butanediol, also known as ketone ester (KE), was incorporated into the rodent diet for 8 months. Higher concentrations of TCA and glycolytic intermediates compared with controls were observed, as well as lower levels of oxidized proteins and lipids (100). KE therapy has the potential to counter fundamental metabolic deficiencies common to patients and transgenic models.
Conclusion
The AD pathology is characterized by a myriad of factors, all linking back to a common characteristic: oxidative stress. Through the increased oxidative stress in the brain, Aβ oligomer, Aβ plaques, and hypophosphorylated tau accumulation and altered metabolism are found in the AD brain. Highly reactive ROS, when not kept in balance, cause damage at many sites via lipid peroxidation, mitochondrial dysfunction, and alteration of protein structure, ultimately affecting function. Because pathways within the cell are interconnected, oxidative stress can lead to increased damage in many brain functions and altered brain pathology with only a small amount of ROS. Better understanding of the relationships among oxidative damage and altered metabolism, we opine, will lead to new therapeutic strategies to slow or retard AD progression and thereby improve the quality of life for millions of people worldwide.
Footnotes
Authors' Contributions
Ms. Rummel and Professor Butterfield wrote the article. Ms. Rummel prepared the figures. Both authors agreed to the final version of the submitted article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by a grant from the National Institutes of Health, National Institute on Aging [AG060056].