
Abstract
Significance:
Altered plasma triglyceride metabolism and changes in dietary fatty acid types and levels are major contributors to the development of metabolic and cardiovascular diseases such as fatty liver disease, obesity, diabetes, and atherosclerosis. Lipid accumulation in visceral adipose tissue and ectopically in other organs, as well as lipid-induced redox imbalance, is connected to mitochondrial dysfunction in a range of oxidative stress-associated metabolic and degenerative disorders.
Recent Advances:
Successful mitochondrial adaptive responses in the context of hypertriglyceridemia and dietary bioactive polyunsaturated fatty acids contribute to increase body energy expenditure and reduce oxidative stress, thus allowing several cell types to cope with metabolic challenges and stresses. These responses include mitochondrial redox signaling, mild uncoupling, and changes in network dynamic behavior.
Critical Issues:
Mitochondrial bioenergetics and redox changes in a lipid overload context are relatively well characterized. However, the turning point between adaptive and maladaptive mitochondrial responses remains a critical issue to be elucidated. In addition, the relationship between changes in fusion/fission machinery and mitochondrial function is less well understood.
Future Directions:
The effective mitochondrial responses described here support the research for new drug design and diet or nutraceutical formulations targeting mitochondrial mild uncoupling and effective quality control as putative strategies for cardiometabolic diseases. Antioxid. Redox Signal. 36, 953–968.
Overview
Hyperlipidemia is a primary or secondary feature of highly frequent metabolic diseases such as fatty liver disease, obesity, diabetes, and atherosclerosis. In this review, we indicate mitochondria as the center of cell adaptive (or maladaptive) responses in the context of hypertriglyceridemia and diets containing saturated or polyunsaturated fatty acids (PUFAs) that impact cardiometabolic disease development.
Mitochondria as Central Regulators of Cellular Homeostasis and Fate
Mitochondria are key bioenergetic organelles for cell life and death, and dysfunctional mitochondria are involved in a large range of diseases. They are responsible for macromolecule intermediary metabolism and adenosine triphosphate (ATP) synthesis, and play important roles in a variety of cell signaling processes such as calcium transport and buffering, redox homeostasis, cell proliferation, differentiation, and death (40, 113). These organelles are compartmentalized by a double-membrane system, in which the inner mitochondrial membrane (IMM) forms many folds (cristae) extending into the interior (matrix), and the outer mitochondrial membrane (OMM) delimits the intermembrane space.
At the cristae, the IMM contains many dehydrogenases such as glycerol-3-phosphate dehydrogenase, and the electron-transfer flavoprotein—ETF:Q oxidoreductase system that pass electrons from metabolites to the ubiquinone (QH2/Q pool), four redox protein complexes (I–IV) responsible for electron transport and oxygen consumption (respiratory chain), and a fifth complex that catalyzes ATP synthesis (FoF1 ATP synthase). The electron-transfer chain components may associate to form so-called supercomplexes. The higher level organization appears to be dynamic, presenting a distribution of free complexes and supramolecular assemblies of different compositions (19). The matrix compartment contains mitochondrial DNA, complex pyruvate dehydrogenase, enzymes of the citric acid cycle and fatty acid oxidation, among others.
While the OMM has a high degree of permeability promoted by many protein-based pores that allow the free diffusion of molecules <6000 Da, the IMM has a much lower permeability to ions and small molecules, a feature that is critical for bioenergetic functions, such as the electrochemical potential, including the H+ gradient that drives oxidative phosphorylation (OXPHOS). Substrates derived from glycolysis, fatty acid beta-oxidation, and amino acid metabolism are oxidized by mitochondria, feeding electrons into the QH2/Q pool directly (such as succinate, glycerol 3-phosphate, proline, and acyl-CoA), or indirectly through the NADH/NAD+ pool and complex I (mainly NAD-linked substrates from citric acid cycle and fatty acid beta-oxidation, pyruvate, 3-hydroxybutyrate, and 2-oxoglutarate) (18).
Electrons from NADH and FADH2 are transferred to complexes I and II, respectively, and then carried through ubiquinone to complex III and later through cytochrome c to complex IV, where they reduce molecular oxygen to water. The energy derived from the electron-transfer reactions is used to transport H+ across the IMM, creating an electrochemical H+ gradient (ΔμH+) in which the electrical component (membrane electrical potential [ΔΨm]) reaches 160–200 mV (negative on the matrix side). The energy derived from the H+ backflow to the matrix through ATP synthase is used to phosphorylate adenosine diphosphate (ADP) into ATP. This process was originally proposed by Peter Mitchell in 1961 as the “Chemiosmotic Theory” (71). Moreover, the electrochemical gradient drives passive ion movements across the IMM through selective channels, such as potassium, calcium, and anion channels; the translocation of adenine nucleotides; phosphate transport into the matrix; and protein import (99).
Pioneering studies in isolated mitochondria revealed that under certain conditions such as a high calcium concentration, mitochondria may undergo marked ultrastructure changes and swelling (105). These mitochondrial alterations are secondary to the formation of a proteinaceous mega-channel in the IMM, the permeability transition pore (PTP). The demonstration that inhibition of PTP opening prevents cell death confirmed the involvement of mitochondrial permeability transition (MPT) in the pathogenesis of many diseases. MPT inducers with biological relevance include inorganic phosphate, fatty acids, NADPH oxidants, and nitric oxide-derived species. The downstream direct MPT mechanism is IMM protein thiol oxidation. MPT induced by calcium is stimulated by exogenous oxidant-generating systems and by intramitochondrial oxidant production; in contrast, it is protected by a variety of antioxidants (105).
Mitochondria form a highly dynamic network within most cells, where they constantly undergo fission and fusion in response to extra- and intracellular conditions and stresses. These mitochondrial dynamic events appear to be central regulators of cellular activity and modify organelle functions, including susceptibility to MPT, respiratory properties of the electron transport chain, and reactive oxygen species production (86). Physiological processes associated with increased energy demand and decreased energy supply, such as acute stress and caloric restriction, are characterized by mitochondrial network elongation and the tight coupling of respiration and ATP synthesis. On the contrary, processes associated with decreased energy demand and increased supply (high levels of nutrients and obesity) are associated with mitochondrial fragmentation and decreased coupling (63). The main players in fusion–fission dynamics are mitochondrial fission 1 protein (FIS1), mitochondrial fission-related protein dynamin-1-related protein (DRP1), mitofusin 1 and 2 (MFN), mitochondrial fusion-related protein optic atrophy 1 (OPA1), among others.
Fusion and fission of the mitochondrial network are part of the organelle quality control system, which also includes mitochondrial biogenesis and mitophagy. This system regulates mitochondrial morphology, quantity, quality, and turnover to sustain cell homeostasis. Mitochondrial biogenesis is stimulated by environmental conditions of high-energy demand and in disease risk conditions, such as during inflammatory and degenerative processes, as a homeostatic mechanism response (25). Mitophagy is a cellular process that selectively removes aged and damaged mitochondria via the specific sequestration and engulfment of mitochondria for subsequent lysosomal degradation (68). Defective mitophagy is implicated in diverse oxidative stress-associated metabolic, proliferative, and degenerative disorders (30, 111).
In addition to undergoing mitophagy, dysfunctional mitochondria may be expelled from cells and cleared up by local immune cells. For instance, cardiomyocytes eject dysfunctional mitochondria in dedicated particles through a process driven by the autophagy machinery during cardiac stress. Macrophages lodged within the myocardium actively take up these vesicles, preventing activation of the inflammasome and ventricular dysfunction (76).
Mitochondria also interact closely with other cell organelles. An intracellular structure called mitochondria-associated endoplasmic reticulum membranes (MAM) is conserved in eukaryotes and participates in many intracellular processes, including lipid synthesis and transfer, calcium flux between organelles and mitochondrial bioenergetic functions (29, 67). Alterations in MAM dynamics have been associated with several metabolic diseases such as fatty liver disease and obesity (10), diabetes (103), rheumatoid arthritis and coronary artery disease (114).
In adipocytes, particularly in conditions of lipid droplet expansion, mitochondria are found associated with lipid droplets supporting ATP-dependent triacylglycerol synthesis. These “peridroplet mitochondria” have distinct protein composition and cristae structure, increased electron transport and ATP synthesis capacities, low β-oxidation capacity, and are segregated from the rest of the mitochondrial population due to a reduced fusion–fission dynamics (13). These recent data reveal a novel regulatory role of mitochondria in lipid metabolism of adipocytes.
Recent studies reveal additional signaling functions of mitochondria (69). For instance, release of citric acid cycle metabolites may change epigenetic landscape of the cells through chromatin modifications, DNA methylation, and post-translational modifications of proteins. These events include acetyl-CoA as a necessary cofactor in the histone acetylation, α-ketoglutarate as an essential cosubstrate for hydroxylation reactions, and succinate as an antagonist of this reaction, while fumarate mediates inhibition of histone and DNA demethylases.
In summary, our classical view of mitochondria as sites of energy production has markedly expanded in recent decades. Mitochondria are now appreciated as signaling hubs for the regulation of cell homeostasis and cell fate. Moreover, mitochondrial dysfunction causes stress responses, connecting the organelle to a wide range of mitochondrial diseases and aging.
Mitochondrial Uncoupling, Oxidant Generation, and Metabolism
OXPHOS is not completely coupled to respiration because protons can leak back across the IMM and return to the matrix, bypassing ATP synthase, and thus uncoupling substrate oxidation and ATP synthesis. This futile cycling of H+ correlates mainly with the fatty acyl composition of phospholipids (generally an unregulated process) or may be catalyzed by proteins (an inducible, regulated process) at the IMM (Fig. 1A). This energy-dissipating process apparently occurs in all eukaryotic cells and accounts for a high proportion of the cellular metabolic rate (up to 25% of the basal metabolic rate in rats) (90).

Another physiological significance of mild mitochondrial uncoupling is to protect cells and organelles from excessive oxidant production. Mitochondria continuously generate superoxide radical anions due to the monoelectronic reduction of molecular oxygen at intermediary stages of the electron transport chain (17), among other sites related to substrate oxidation, such as mitochondrial glycerol 3-phosphate dehydrogenase (18). Superoxide is then converted to hydrogen peroxide by mitochondrial superoxide dismutase (SOD), and subsequently to water by glutathione peroxidase and/or catalase. Other highly reactive short-lived species such as hydroxyl radicals may be formed in the presence of reduced transition metals. Although superoxide and hydrogen peroxide play an important role in cell redox signaling (40, 98), when produced in an excessive or uncontrolled manner they may cause oxidative damage to a wide variety of macromolecules and may lead to MPT, inflammation, and cell death (105).
The rate of mitochondrial superoxide production is dependent on the mitochondrial membrane potential. As the mitochondrial membrane potential is lowered, electron transport rates and O2 consumption (respiration) are accelerated, decreasing local oxygen availability and decreasing the lifetime of intermediates capable of donating electrons toward superoxide radical formation (96). It was demonstrated in respiring mitochondria that H2O2 generation also decreases proportionally to the decrease in mitochondrial membrane potential promoted by chemical uncouplers (58). Thus, physiological processes that promote mild mitochondrial uncoupling, such as the activities of some ion channels and proteins at the IMM, in addition to activating metabolism, have been proposed as mechanisms of protection against oxidative damage in many diseases, including metabolic, cardiovascular, and neurodegenerative diseases (106, 107), and during the aging process (20).
Uncoupling proteins
Uncoupling proteins (UCPs) are inner membrane-carrier proteins able to dissipate the proton gradient, and were first identified in brown adipose tissue (BAT) by David Nicholls and collaborators in the 1970s (75). Later, other UCP homologous proteins were identified and included in a superfamily of mitochondrial anion-carrier proteins containing five members widely distributed in mammalian tissues (50, 59) and expressed in plants (104, 108). UCP1, UCP2 and UCP3 show high amino acid sequence identity, while UCP4 and UCP5 are less homologous (16, 112). Changes in mitochondrial energy metabolism promoted by high cellular free fatty acid (FFA) levels are often related to the activation of UCPs.
In BAT, UCP1 is a thermogenic protein mediating nonshivering thermogenesis (75) in response to cold, feeding status, and body energy reserves (21). The sympathetic nervous system releases norepinephrine and initiates lipolysis of triglycerides (TG) in BAT, primarily via β-adrenergic receptors and intracellularly via cAMP and protein kinase A. Released fatty acids play a dual role; they both directly activate UCP1 and provide a substrate for the maintenance of accelerated respiration rates to restore the mitochondrial membrane potential. Since the density of mitochondria and the expression of UCP1 in BAT are very high, the uncoupling process is intense, and the dissipation of the proton gradient generates a sufficient amount of heat to warm the blood flowing through BAT. Thermogenic activity of BAT seems to be also redox sensitive; that is, dependent on a substantial increase in the levels of mitochondrial oxidants, cysteine thiol redox oxidation, and sulfenylation of UCP1 Cys253 (26).
The mechanisms of action of UCP are still a matter of debate. The models are based on the requirement of long-chain fatty acids for UCP activity (Fig. 1B). One proposition, named the buffering model, introduced by Klingenberg, is that UCP1 conducts protons to the matrix side through a hydrophilic pathway lined with fatty acid carboxylic groups that buffer the protons as they move across the membrane (110). Another model, introduced by Garlid et al., which was also applied to UCP2 and UCP3, proposes that UCP does not conduct protons but translocate fatty acids to the intermembrane space, where they become protonated (high [H+]) and rapidly diffuse across the membrane back to the matrix (“flip-flop”), where deprotonation occurs due to low [H+], allowing fatty acids to behave as regulated cycling protonophores (42). Finally, a fatty acid-shuttling model has been proposed (38), where UCP1 functions as a fatty acid anion/H+ symporter, but as the fatty acid anions cannot dissociate from the protein due to hydrophobic interactions, UCP1 effectively operates as an H+ carrier activated by fatty acids.
In summary, UCP activity uncouples mitochondria, decreasing the electrochemical transmembrane potential, increasing respiratory rates, and reducing the OXPHOS efficiency (ADP/O ratio) (50, 51). This OXPHOS inefficiency consequently increases substrate consumption and energy expenditure, thus regulating metabolic rates and body mass. In fact, mice overexpressing UCP3 in skeletal muscle are hyperphagic and lean (28), and mice expressing UCP1 in white adipose tissue display a lean phenotype (57), while UCP1-deficient mice exhibit increased susceptibility to diet-induced obesity (56). Muscle UCP3 has also been proposed to protect mitochondria against fatty acid accumulation and may help maintain the muscular fat oxidative capacity (46, 94).
Adenine nucleotide translocase
Adenine nucleotide translocase (ANT) exchanges ADP and ATP across the IMM to provide ADP for OXPHOS and deliver ATP into the cytosol (54). ANT also promotes mitochondrial H+ leak by a mechanism similar to the UCP1, with fatty acids as cofactors and purine nucleotides as negative regulators. Bertholet et al. measured these two ANT transport modes, and showed that ADP/ATP exchange negatively regulates H+ leak without completely inhibiting it, but adjusting H+ leak in accordance with cellular ATP demand. Thus, ANT appears to serve as a master regulator of mitochondrial energy output, maintaining a delicate balance between ATP production and uncoupled energy conversion (e.g., thermogenesis) in mitochondria (14).
Mitochondrial ATP-sensitive potassium channels
Another mechanism of mitochondrial uncoupling results from the K+ cycle across the IMM due to the activity of mitochondrial ATP-sensitive potassium channels (mitoKATP). The properties of mitoKATP were first described by Inoue et al. (48) and Paucek et al. (82), and its molecular identity was recently demonstrated (80). mitoKATP is a protein complex composed of the pore-forming (MITOK) and ATP-binding (MITOSUR) subunits, and mediates K+ uptake into the matrix, driven by the negative mitochondrial membrane potential (Δψm). This protein complex is inhibited by physiological ATP levels. K+ uptake is accompanied by phosphate and water movement, resulting in increased mitochondrial volume, which in turn activates the K+/H+ antiporter (43). The net result is the entrance of one H+ for every K+ exchanged, resulting in a slight decrease in the membrane potential, which is re-established by an increase in the mitochondrial respiration rate, thus promoting mild uncoupling. mitoKATP is activated by oxygen species, and its activation modulates oxidant generation, as demonstrated in cardiac and cerebral tissues (36, 39). The loss of mitoKATP (knockout) increases oxidant production and cell death triggered by oxidative stress (80).
The main mitochondrial uncoupling mechanisms are summarized in Figure 1.
Mitochondrial Responses to Hypertriglyceridemia and Bioactive Fatty Acids
TG-rich lipoproteins, hypertriglyceridemia, and cardiovascular diseases
TGs are the most abundant neutral lipid class present in our diet and body. These molecules are easily and efficiently absorbed from the diet, synthesized endogenously (liver and adipose), transported in the bloodstream, and stored preferentially in adipose tissue depots. TG-rich lipoproteins (LPs) are specialized systems to transport TG and distribute FFA among body tissues. The small intestine and liver assemble and secrete TG-rich LP, chylomicrons, and VLDL (very low-density lipoproteins), respectively. Apolipoprotein (apo) B is the structural protein component in TG-rich LP. They also contain other apolipoproteins, such as apo A-IV, apo A-V, apo C-II, apo C-III, and apo E, which play regulatory roles in plasma lipolysis and clearance of these LPs.
Lipoprotein lipase (LPL) is the critical enzyme mediating the hydrolysis of TG-rich LPs, releasing FFA and remnant LP. LPL is synthesized by most tissue parenchymal cells and is transported to the capillary endothelium, facing the vessel lumen and catalyzing the lipolytic processing of TG-rich LP. Abundant LPL expression is observed in adipose tissue, skeletal muscle, and heart, which use the released FFA either for energy storage after re-esterification (adipose tissue) or as an energy source for muscle contractions. LPL activity is stimulated by insulin, apo C-II, and apo A-V, and inhibited by apo C-III and angiopoietin-like proteins 3 and 4. TG-rich LP remnants are taken up by the liver through low-density lipoprotein (LDL) and LDL receptor-related protein (LRP) receptors using apo E as a ligand.
VLDL remnants are further hydrolyzed by LPL and hepatic lipase, generating LDL, a cholesterol-rich and TG-poor LP. TG-rich remnant uptake by the liver is a source of lipids for subsequent VLDL assembly and secretion. Other lipid sources for VLDL are FFA released from adipose tissue under the action of hormone-sensitive lipase (HSL) and de novo hepatic lipogenesis, which is often driven by overnutrition or the consumption of simple sugars (93, 95). Defective proteins involved in the overproduction, impaired lipolysis or clearance of plasma TG-rich LP contribute to elevated plasma TG levels. Plasma TG-rich LP metabolism is summarized in Figure 2.
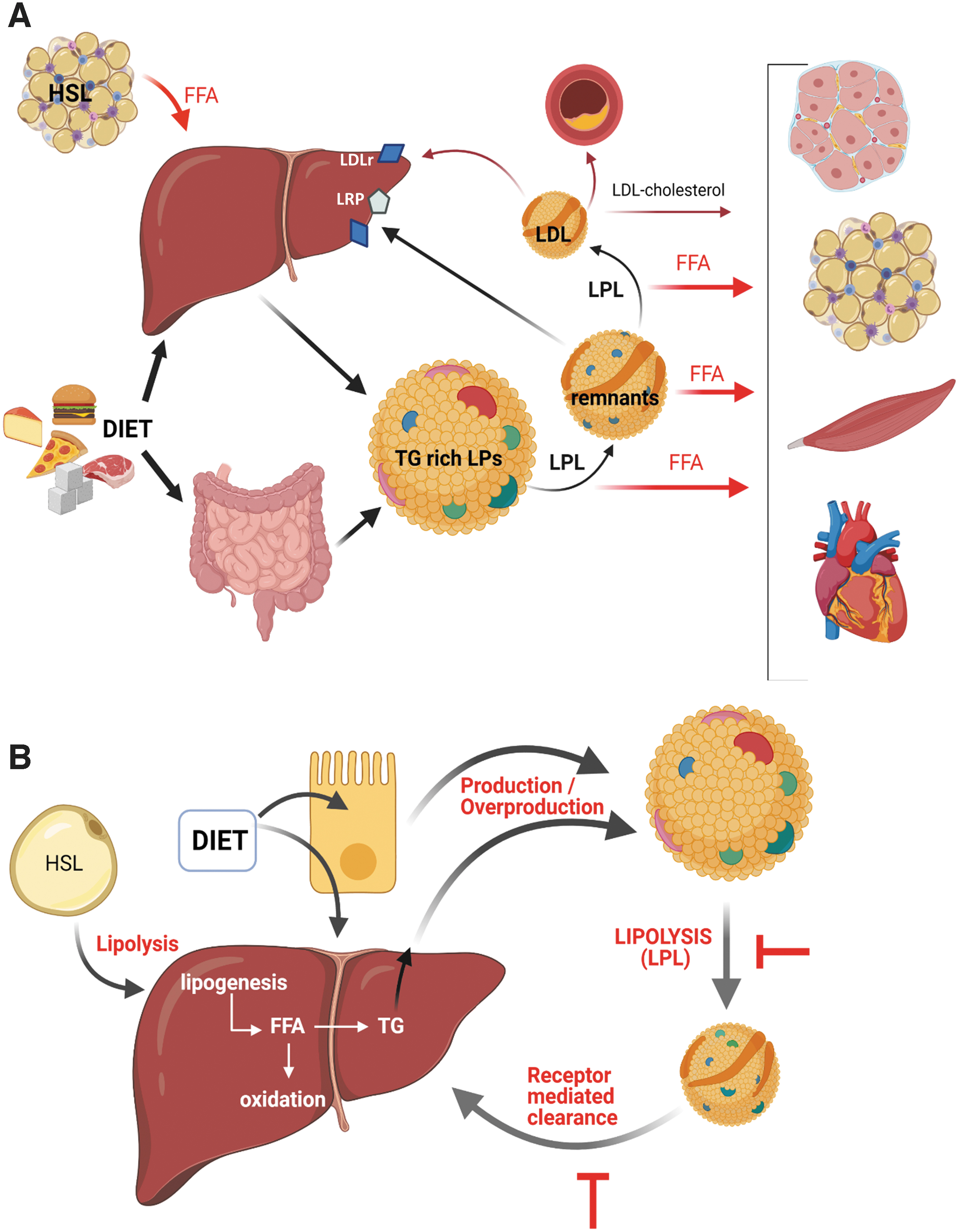
Hypertriglyceridemia is a highly prevalent disorder worldwide (95). The causes may be primary (genetic) or secondary to external factors (diet, alcohol, drugs, etc.) or pathological conditions, including insulin resistance (IR)-associated diseases (obesity and diabetes), nephrotic syndrome, hypothyroidism, among others. Moderately elevated TG concentrations are commonly due to diets enriched in simple carbohydrates and/or saturated fats, excess alcohol consumption, obesity, and sedentary behavior (93). High alcohol consumption is associated with increased adipocyte lipolysis and the flux of FFA to the liver, resulting in increased VLDL production (55).
Genetic hypertriglyceridemia is less frequent and more severe. Many monogenic and polygenic disorders causing severe hypertriglyceridemia have been identified; these include both loss-of-function and gain-of-function mutations in genes that drive TG-rich LP metabolism; for instance, deficiency of LPL or of its coactivator apo CII, increased expression of apo B, defective apo E, among others. Severe primary hypertriglyceridemia is also present in lipodystrophy due to defective genes involved in adipocyte development and differentiation, leading to hepatic steatosis and VLDL overproduction (95). Interestingly, a mutation of GPD1 gene, encoding glycerol-3-phosphate dehydrogenase 1 (cytoplasmic isoform), has been associated with moderate hypertriglyceridemia and fatty liver likely because of increased liver TG secretion (12). This case is particularly relevant considering the glycerol phosphate shuttle as a critical mitochondrial redox transfer mechanism in different tissues and conditions. Thus, GPD1 and 2 (mitochondrial isoform) system acts at the crossroads of glycolysis, OXPHOS, and lipid metabolism (72).
Recent epidemiological, genetic, and biological data strongly suggest that elevated TG-rich LP represents a causal risk factor for low-grade inflammation, atherosclerotic cardiovascular disease, and all-cause mortality (77).
Secondary hypertriglyceridemia, IR, and mitochondria
Hyperlipidemia secondary to chronic overnutrition, saturated fatty acid overfeeding, obesity, and diabetes are strongly correlated with the IR state. IR in turn contributes to elevated plasma TG (liver secretion) and FFA (lipolysis) concentrations and turnover, and thus to lipid accumulation in nonadipocyte tissues (ectopic lipid deposition), creating a vicious cycle between IR and ectopic lipid deposition. The latter is responsible for lipotoxicity causing dysfunction or failure of a variety of cell types (35).
Challenging mammalian cells with excess fuel alters the rates of the glycerolipids/FFA cycling, a constitutively active metabolic pathway that is relevant for many cell functions beyond the lipid storage (lipogenesis)/mobilization (lipolysis) and thermogenesis roles. This includes signaling to cell growth, gene expression, activation of energy homeostasis effectors, lipid excess detoxification, among others (87). High levels of certain metabolites such as FFA and other intermediates of this cycle are toxic to cells, and are rendered less toxic by re-esterification to TG and storage as lipid droplets or transport out of the cells. On the contrary, glycerolipid/FFA cycling is a high-energy requiring pathway and its enhanced operation causes acceleration of mitochondrial oxidation of FFA, thus functioning as a detoxification machinery. Imbalance in the operation of this cycle is associated with IR, diabetes, and fatty liver in several experimental models. This issue has been excellently reviewed by Prentki and Madiraju (87).
Saturation of the glycerolipid/FFA cycling increases the levels of intermediates from partial metabolism of TG such as diacylglycerol and ceramide and excess of saturated fatty acids in lipid-overloaded cells, which induce chronic inflammation, redox imbalance, and have harmful effects on multiple organs and systems (35). A robust body of evidence indicates that high-fat diets, obesity, and uncontrolled diabetes lead to FFA and other partially metabolized lipid induction of extra- and intramitochondrial oxidative stress, increased MPT, inflammation, and cell death (105). The main toxic mechanisms of FFA overload are summarized in Figure 3.

In recent years, studies have focused on mechanisms by which mitochondrial bioenergetics (41) and network dynamics (63) may be linked to diet-induced IR. For instance, high-carbohydrate and fat diet-fed Wistar rats show increased body weight, serum TG, and IR as well as decreased cardiac thioredoxin reductase activity and mitochondrial mitofusin-1 (MFN1), increased mitochondrial fission-related protein DRP1 (reducing MFN1:DRP1 ratio) and Bax expression (79).
Neufer's group proposed that mitochondrial bioenergetics are linked to IR via redox signaling. Nutrient overload induces mitochondrial H2O2 production as a primary signal connected downstream with insulin signaling. It is proposed that there is a continuous gradient in the cell redox environment (triggered by mitochondrial H2O2 production) associated with insulin sensitivity as a function of the energy balance. Thus, the chronic positive energy balance promotes gradual shifts to a more oxidized and less-insulin-sensitive state, ultimately leading to diet-induced IR (41).
In line with this proposal, Fazakerley et al. found that the mevalonate-coenzyme Q (CoQ) biosynthesis pathway is defective in in vitro and in vivo models of IR. They showed that loss of mitochondrial CoQ impairs insulin action via complex II-derived H2O2. Supplementation of CoQ restores mitochondrial CoQ and insulin sensitivity (37). On the contrary, correct insulin signaling in specific organs is necessary for mitochondrial functioning, independent of unbalanced diets. Double knockout of insulin and IGF-1 receptors in BAT causes severe brown fat atrophy, loss of mitochondrial mass, mitochondrial crista disruption and loss of proteins involved in mitophagy, mitochondrial quality control, and dynamics (109).
Mitochondrial responses to hypertriglyceridemia in the absence of IR
We have previously investigated mitochondrial function in genetic hypertriglyceridemia. We used a genetically modified mouse model overexpressing the apolipoprotein CIII enconding gene (49), a particularly useful model to study the effects of elevated plasma levels of TG and FFA per se, independent of other metabolic factors such as IR, inflammation, and obesity caused by chronic hypercaloric diet feeding regimens. These hypertriglyceridemic (HTG) mice present a three- to fivefold increase in TG levels and a two- to threefold increase in FFA plasma levels. Surprisingly, under standard laboratory conditions and a chow (low fat) diet, the mice show normal growth, body weight and composition, reproduction and unaltered glucose homeostasis and insulin secretion (9). However, as expected, under a high saturated fat diet that induces IR and inflammation, HTG mice showed more atherosclerosis (45), obesity (92), and fatty liver disease (81) than their littermate controls.
The intriguing normal phenotype for glucose homeostasis and body fat mass of HTG mice led us to investigate possible mitochondrial adaptations in this context. Compared with those of the controls, the content of total glycerolipids in the liver of these HTG mice was twofold greater, and the mitochondrial respiration rates supported by lipid substrates were higher. These data indicated lipid accumulation together with accelerated beta-oxidation in the liver of these HTG mice (4). Liver mitochondria from these HTG mice showed elevated oxygen consumption during resting nonphosphorylating respiration, which was associated with higher activity of the mitoKATP (2, 5). Although intracellular availability of FFA is frequently associated with the induction of UCP activity, liver mitochondria from HTG mice presented no changes in the OXPHOS efficiency (ADP/O ratio), UCP2 content and activity, and adenine nucleotide carrier activity, the latter of which is another possible uncoupling mediator (2). The elevated resting respiration was re-established to the control levels when measured in the absence of K+ and was sensitive to mitoKATP antagonists (ATP, 5-hydroxydecanoate and glyburide). Similarly, the mitochondrial swelling mediated by these channels was also re-established by mitoKATP antagonists (5).
Liver mitoproteome analysis showed a general upregulation of inner membrane proteins in HTG mice (34), which supports the observed increased respiration rates and possibly mitochondrial biogenesis. In addition to the liver, increased mitoKATP activity was observed in spleen mononuclear cells (6) and in the brain but not in the skeletal muscle of HTG mice (7). In vivo studies of HTG mice clarified the mechanisms that induce mitoKATP activation in the liver. This was shown to be dependent on the high intracellular availability of FFA since hypolipidemic ciprofibrate treatment restored the resting respiratory rate (2). mitoKATP activation is also dependent on the cell oxidized state since treatment with the antioxidant N-acetylcysteine (NAC) reversed the higher mitoKATP activity and oxygen consumption in the liver as well as the redox imbalance (elevated levels of malondialdehydes, carbonylated proteins, and oxidized to reduced glutathione [GSSG/GSH] ratio) found in HTG livers. This oxidized profile was associated with elevated extramitochondrial oxidant production promoted by higher activities of xanthine and NADPH oxidases (4), enzymes that can be activated by intracellular FFA accumulation (61) or their oxidized products (27).
In agreement, liver proteome analysis showed upregulation of several cytosolic reactive oxygen species detoxifying enzymes, suggesting that the cytoplasm of HTG mice is subjected to oxidative stress (34). Nevertheless, the mitochondrial compartment of HTG livers was found to be protected from oxidative stress, as shown by lower H2O2 release in a manner sensitive to mitoKATP inhibition, indicating that these channels also work as redox sensors in the liver (4). This cross-talk between extra- and intramitochondrial compartments keeps the mitochondria in a more reduced state, which can avoid amplification of overall cell oxidative stress in lipid-overloaded states.
HTG mice also exhibited increased whole-body metabolic rates, as indicated by their higher CO2 production rates and significant elevation in body (rectal) temperature than those of the controls (5). However, the mice were not leaner than the controls because they showed increased food ingestion that compensated for the higher metabolic rates (5). An increased relative abundance of the dietary fatty acid, margaric acid, in the liver lipid profile confirmed the higher food consumption of HTG mice (3). The high body metabolic rates of HTG mice were sensitive to in vivo antioxidant (NAC) treatment, pointing to a new biological role of mitoKATP in the regulation of body energy homeostasis (5). This metabolic role was reinforced by findings that mitoKATP activity can be modulated by the diet, as observed in mitochondria isolated from nonalcoholic fatty livers of animals on a high-fat diet (22).
These data support the concept that mild mitochondrial uncoupling is an endogenous homeostatic mechanism that could be targeted to counteract or alleviate oxidative stress and lipid-related metabolic stresses. In fact, long-term, low-dose treatment of Swiss mice with 2,4-dinitrophenol (DNP), a protonophore with high affinity for the mitochondrial membrane, improved plasma glucose and TG levels, and reduced the body weight in these wild-type mice (20). Goldgof et al. (44) showed that DNP treatment attenuated obesity in mice acclimated to thermoneutrality. Niclosamide ethanolamine, an anthelmintic drug, uncouples mitochondria and protects against high-fat diet-induced hepatic steatosis, IR, and glycemic control in db/db diabetic mice (101).
A controlled-release mitochondrial protonophore that produces mild liver-targeted mitochondrial uncoupling reversed diabetes and steatohepatitis in rats (84), decreased hypertriglyceridemia, and reversed nonalcoholic steatohepatitis (NASH) and diabetes in a mouse model of severe lipodystrophy and diabetes (1). Another mitochondrial uncoupler, BAM15, induced antiobesity effects and increased insulin sensitivity in mice fed a Western-type diet (8). In addition, sorafenib, a drug used to treat hepatocellular carcinoma that elicits serious side effects, when employed at a 1/10 ratio of the clinical dose, induces mild mitochondrial uncoupling and prevents the progression of NASH in mice and monkeys, without adverse effects (52). A recent study demonstrated that long-term, low-dose treatment with DNP is effective in reducing spontaneous and diet-induced atherosclerosis in the LDL receptor knockout mice (31).
The effects of mild mitochondrial uncoupling induced by mitoKATP and chemical uncouplers are summarized in Figure 4.

Although this growing evidence supports mild uncoupling as a therapeutic target, it is important to emphasize that controlling the degree of mitochondrial uncoupling is a critical issue since ATP deficiency and membrane depolarization may lead to cell damage and death. Another possible drawback is the possibility of undesirable uncoupling in off-target tissues (for instance, brain and heart). Thus, the challenge is designing tissue-specific uncoupling therapies to avoid off-target metabolic effects.
Bioactive fatty acids and mitochondrial bioenergetics and dynamics
PUFAs are dietary bioactive lipids able to attenuate IR and other factors involved in the pathogenesis of metabolic syndrome, obesity, and diabetes states. PUFAs, either as diet components or as pills, are also employed to reduce TG plasma levels and cardiovascular risks. The molecular mechanisms of action of PUFA in different tissues appear to involve modulation of mitochondrial function and efficiency, leading to attenuation of proinflammatory and pro-oxidant states and ectopic lipid accumulation present in these disturbances. One mechanism by which PUFA regulates energy metabolism is as ligands of peroxisome proliferator-activated receptors (PPARs). PPAR-delta regulates glucose metabolism and mitochondrial biogenesis by inducing FOXO1 and PPARγ coactivator-1α (PGC1-α) transcription factors (74).
Diverse dietary fat sources differentially affect mitochondrial bioenergetics and dynamics. Understanding how these bioactive lipids modulate mitochondrial function and contribute to body energy metabolism may provide an attractive therapeutic target for metabolic diseases (15, 24). For this purpose, diet supplementation with bioactive fatty acids has been considered a potential strategy for counteracting body weight gain by several mechanisms (73), including stimulation of body energy expenditure with conjugated linoleic acids (CLAs, 18:2 n-6) (78, 100), and the omega-3 PUFAs: docosahexaenoic acid (DHA, 22:6 n-3) and eicosapentaenoic acid (EPA, 20:5 n-3) (66).
CLA-supplemented mice showed elevated body metabolic rates and high levels of liver UCP2 messenger RNA (mRNA) (85, 102). Isolated liver mitochondria of CLA-treated mice showed indications of increased UCP activity, such as decreased ADP/O ratio in association with increased resting respiration in the presence of exogenous linoleic acid (UCP substrate) and sensitivity to the UCP inhibitor guanosine diphosphate (GDP) (11, 83, 91).
These mitochondria also presented increased oxidant generation detected at the extramatrix side (by Amplex Red, H2O2) and even more by the matrix-side oxidant probe, without leading to a redox imbalance in the liver. Enhanced oxidant generation occurred when mitochondria were energized with glutamate, malate, and pyruvate (citric acid cycle substrates) or palmitoylcarnitine (β-oxidation substrate) but not with succinate (complex II substrate) or β-hydroxybutyric acid (complex I substrate, NADH linked), demonstrating the contribution of oxireduction reactions involving NAD+/NADH in the citric acid cycle to oxidant generation (83). This mechanism can be involved in the uncoupling process since superoxide anions generated within the mitochondrial matrix can promote UCP activation (32, 33). In fact, in vivo antioxidant therapy with NAC abolished CLA-induced liver UCP expression and activity, and the elevation of body metabolism. In addition, the treatment of hepatocytes with catalase mitigated CLA-dependent UCP2 expression (11). Taken together, these results indicate the participation of an oxidative pathway in CLA-mediated UCP expression, activity, and body energy expenditure.
CLA diet supplementation also enhances the mitochondrial fission machinery, as detected by immunostaining of the FIS1 in the liver, suggesting a shift toward mitochondrial fragmentation (91). A possible interpretation is that numerous small organelles offer a high surface area for the accessibility of the metabolic substrate to carrier proteins, increasing mitochondrial intake and oxidation of substrates. Similar effects have been reported after treatment with high glucose or high fatty acids in cell and animal models (63, 64). In addition, CLA treatment significantly increased the expression of sirtuin 1, PGC1-α, and the key transcription factor needed to stimulate mitochondrial biogenesis, nuclear respiratory factor 1 (Nrf1), suggesting increased mitochondrial biogenesis in skeletal muscle (53). The main effects of CLA supplementation are depicted in Figure 5.

High-fat diets rich in fish oil (mainly ω3-PUFA), compared with high-fat diets rich in lard (mainly saturated fatty acids), have been shown to induce improvements in mitochondrial function and fusion processes associated with reductions in reactive oxygen species production in rat gastrocnemius skeletal muscle (65, 88) and liver (64) as well as in testicular tissue (70). In addition, rats fed a PUFA-enriched diet showed reduced energy efficiency of subsarcolemmal mitochondria, increased AMPK activation, and reduced endoplasmic reticulum and oxidative stress. Increased mitochondrial respiration was related to increased mitochondrial biogenesis in skeletal muscle, as shown by the increase in PGC1-α and PGC1-β expression (23).
Mice supplemented with an EPA/DHA-enriched oil exhibited increased body energy expenditure, but in contrast to CLA this was not associated with mitochondrial uncoupling in the liver, muscle (soleus), brain (hippocampus), or BAT (91). This diet did not modulate H2O2 generation. However, EPA/DHA supplementation induced mitochondrial biogenesis, as indicated by augmented citrate synthase activity (mitochondrial content marker) and elevated levels of PGC1-α mRNA expression (91). However, in contrast to CLA, dietary supplementation with EPA/DHA promoted enhancements of mitochondrial fusion protein detected by the immunostaining of mitofusin 2 (MFN2) in the liver, suggesting a condition required to ensure maximum oxidative metabolism (89).
When a murine diet is supplemented with both CLA and EPA/DHA, the body metabolic rates remain elevated, and some effects of each fatty acid are maintained, such as improved mitochondrial biogenesis and fusion (attributed to EPA/DHA) and high levels of UCP2 mRNA and fission (attributed to CLA) in the liver (91).
The positive mitochondrial effects of omega-3 PUFA appear to extend to several tissues under varying stresses. The neuroprotective effects of DHA on mitochondrial dynamics were demonstrated in in vivo and in vitro models of brain injury. DHA alleviated oxidative stress-induced apoptosis, as shown by decreased malondialdehyde levels and SOD stress, increased Bcl2 and Bcl-xl levels, and decreased Bax and cleaved caspase-3 levels in vivo. DHA ameliorated mitochondrial dysfunction by upregulating the OPA1 and downregulating the mitochondrial fission-related protein DRP1 both in vitro and in vivo (115).
Subcutaneous adipocytes from overweight females treated with EPA presented increased mitochondrial content, accompanied by increased mRNA expression of indicators of mitochondrial biogenesis including the following: Nrf1, mitochondrial transcription factor A, and cytochrome c oxidase IV. In these cells, EPA also promoted the activation of sirtuin 1, PGC1-α, and AMPK (60). In cardiomyoblasts, EPA was shown to counteract palmitic acid-induced apoptosis by re-establishing autophagic flux and improving mitochondrial function and dynamics, as shown by the augmented mitochondrial membrane potential and a reduction in the mitochondrial fission protein DRP1 (47).
Importantly, partial replacement of a high saturated fat diet with fish oil given to severe hyperlipidemic apoE knockout mice contributed to improving endothelial cell function. These findings were associated with lower levels of mitochondrial oxidative stress, cytochrome c release, and caspase-3 activity as well as higher expression levels of MFN 2 and OPA1 but lower expression levels of FIS1 in the aorta of fish oil-treated mice (97). Dietary EPA also protected rat hearts against septic damage through modifications that preserve mitochondrial integrity. This effect was shown by decreased oxidative stress of the mitochondrial matrix associated with increased UCP3 expression. EPA also displayed increased mitochondrial sirtuin-3 protein expression, which preserves OXPHOS (62). The main effects of omega-3 PUFA supplementation are depicted in Figure 6.

These results show that mitochondrial uncoupling, biogenesis, and changes in network dynamics are adaptive responses elicited by bioactive lipids that allow several cell types to cope with various metabolic challenges and stresses.
Final Considerations
Hypertriglyceridemia, cell lipid excess, and lipid-induced redox imbalance are connected to mitochondrial dysfunction in a range of metabolic and degenerative disorders. In this review, we discussed that mild mitochondrial adaptive uncoupling, as an endogenous mechanism elicited by hypertriglyceridemia or by mitochondrial uncouplers, is an example of a reliable strategy to lessen the harmful effects of the intracellular overflow of lipids and oxidative stress. We highlighted evidence that stimulation of this mechanism may be beneficial in a variety of pathological metabolic contexts such as fatty liver disease, obesity, diabetes, and atherosclerosis. Considering that intense uncoupling may impair the energy supply and cause mitochondrial and cell dysfunction, the turning point between adaptive and maladaptive mitochondrial uncoupling remains a critical issue. In addition, the tissue-specific target of mild mitochondrial uncoupling remains a worry to be technically overcome to avoid off-target effects. Dietary bioactive lipids have been shown to be effective modulators of several physiological functions and molecular mechanisms that contribute to preventing and ameliorating IR-associated states. Here, we described their effects on mitochondrial bioenergetics, uncoupling, and morphological dynamic behavior, leading to mitochondrial improvements in the context of high-fat diet and oxidative stress. Primarily, omega-3 PUFA and CLA mediate positive mitochondrial adaptations. Mechanistically, the relationship between changes in fusion/fission machinery and mitochondrial function is less well understood and less consistent in the literature. New methodological tools that allow turning on and off fusion and fission events may shed light on this issue.
Finally, the successful mitochondrial responses described here point to mitochondria as a putative therapeutic strategy for cardiometabolic diseases, and support research on new drug design and diet and nutraceutical formulations targeting mitochondrial uncoupling and effective quality control.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
The authors have their research supported by the Brazilian State Agency Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), grant numbers 2017/17728-8 and 2013/07607-8 (H.C.F.O.) and 2018/10089-2 (L.C.A.).