
Abstract
Significance:
Adaptor proteins control the spatiotemporal dynamics of cellular signaling. Dysregulation of adaptor protein function can cause aberrant cell signaling and promote cancer. The arrestin family of adaptor proteins are known to regulate signaling by the superfamily of G protein-coupled receptors (GPCRs). The GPCRs are highly druggable and implicated in cancer progression. However, the molecular mechanisms responsible for arrestin dysregulation and the impact on GPCR function in cancer have yet to be fully elucidated.
Recent Advances:
A new family of mammalian arrestins, termed the α-arrestins, was recently discovered. The α-arrestin, arrestin domain-containing protein 3 (ARRDC3), in particular, has been identified as a tumor suppressor and is reported to control cellular signaling of GPCRs in cancer.
Critical Issues:
Compared with the extensively studied mammalian β-arrestins, there is limited information regarding the regulatory mechanisms that control α-arrestin activation and function. Here, we discuss the molecular mechanisms that regulate ARRDC3, which include post-translational modifications such as phosphorylation and ubiquitination. We also provide evidence that ARRDC3 can interact with a wide array of proteins that control diverse biological functions.
Future Directions:
ARRDC3 interacts with numerous proteins and is likely to display diverse functions in cancer, metabolic disease, and other syndromes. Thus, understanding the regulatory mechanisms of ARRDC3 activity in various cellular contexts is critically important. Recent studies suggest that α-arrestins may be regulated through post-translational modification, which is known to impact adaptor protein function. However, additional studies are needed to determine how these regulatory mechanisms affect ARRDC3 tumor suppressor function. Antioxid. Redox Signal. 36, 1066–1079.
Introduction
To function normally, cells must properly respond to external stimuli. On stimulation, signals are transduced across the plasma membrane and activate signaling networks, leading to changes in cellular behavior that control normal physiology. The proper regulation of signaling networks is critical for the maintenance of organ and tissue homeostasis; dysregulation of these pathways, through either aberrant increase or decrease in signaling, is known to drive disease progression, including cancer.
One class of proteins that helps to maintain proper spatiotemporal control of cell signaling are adaptor or scaffold proteins. Adaptor proteins harbor no enzymatic activity, but instead function to coordinate signaling by bringing proteins in close proximity at distinct times and/or subcellular locations. Adaptor protein function, in turn, is regulated through various mechanisms that are driven largely by post-translational modifications, such as phosphorylation and ubiquitination, which serve to control protein-protein interactions, subcellular localization, and various other biological activities (49, 59).
One of the best studied adaptor proteins in mammalian cells are the ubiquitously expressed β-arrestins. β-arrestins are key regulators of G protein-coupled receptor (GPCR) function. After GPCR activation and coupling to heterotrimeric G protein signaling, β-arrestins are rapidly recruited to the receptor at the plasma membrane and facilitate uncoupling from G proteins, promoting receptor desensitization. In addition, β-arrestins control GPCR internalization, and they function as scaffolds to promote signaling independent of heterotrimeric G proteins (73).
As the largest family of surface receptors in mammalian cells, GPCRs control a vast array of physiological functions, and thus must be properly regulated. Dysregulation of GPCR signaling, much of which cannot be attributed to defects in β-arrestin function, is known to drive numerous diseases, including cancer (9, 50, 72). Although GPCRs are highly druggable, accounting for ∼33% of all Food and Drug Administration (FDA)-approved small-molecule drug targets, GPCRs have been underutilized as targets for therapeutic development in cancer, suggesting a need to better understand the molecular mechanisms that control GPCR function in cancer (64, 78).
β-arrestins are composed of two arrestin-fold domains, each a seven-stranded β-sandwich. Arrestin folds have subsequently been identified in two other families of proteins: the vacuolar sorting protein 26 (Vps26) family, which comprises Vps26A and Vps26B, and the newly discovered α-arrestin family, which comprises arrestin domain-containing proteins (ARRDC) 1–5 and thioredoxin interacting protein (TXNIP) (2, 7). Due to the presence of arrestin-fold domains in the α-arrestin subfamily, it is speculated that, similar to β-arrestins, they may function in regulation of GPCRs. Here, we provide an overview of a new family of mammalian arrestins, termed the α-arrestins, that have been proposed to play a key role in cancer by serving as multifunctional adaptor proteins to control receptor trafficking and signaling in a manner distinct from β-arrestins. We additionally provide a proteomic analysis of arrestin domain-containing protein 3 (ARRDC3) interacting proteins that suggests a broad role for α-arrestin function in various signaling and trafficking pathways, that may be related to breast cancer.
α-arrestins are important mediators of redox regulation and cellular metabolism
TXNIP was initially characterized as an important regulator of oxidative stress and metabolism, before it was known to be a member of the mammalian α-arrestin family. The most well-known feature of TXNIP is that it binds to thioredoxin, thus inhibiting thioredoxin antioxidant activity and promoting oxidative stress (42). Although all α-arrestin family members share high sequence homology, no other α-arrestin besides TXNIP has been demonstrated to interact with thioredoxin or to control redox regulation (51, 57). The presence of two key TXNIP cysteine (C) residues, C63 and C247, is required for interaction with thioredoxin, but they are not conserved in other α-arrestins and likely confer TXNIP-specific functions (55, 57). Thus, TXNIP is unique among the α-arrestins in its ability to regulate redox reactions.
However, TXNIP has a number of thioredoxin-independent functions, including regulation of glucose transport. Previous studies reported that depletion of TXNIP resulting from a spontaneous nonsense mutation in HcB-19 mice resulted in decreased blood glucose (13). Furthermore, elevated TXNIP expression is associated with type 2 diabetes (53), suggesting that TXNIP regulates glucose metabolism. Subsequent studies demonstrated that TXNIP depletion in adipocytes promoted glucose uptake, whereas TXNIP overexpression decreased glucose uptake (55). TXNIP effects on glucose were attributed to regulation of glucose transporters (GLUT) 1 and 4. TXNIP directly interacts with GLUT1 and 4 and facilitates endocytosis; depletion of TXNIP leads to increased GLUT1 and 4 at the plasma membrane and thus increased glucose uptake (46, 85, 87). These studies also showed that TXNIP regulation of GLUTs is independent of TXNIP ability to bind thioredoxin, as thioredoxin binding-deficient TXNIP mutants still regulate glucose uptake (55).
Interestingly, dysregulation of GLUT expression has been linked to cancer. In particular, overexpression of GLUT1 and GLUT3 is negatively correlated with survival in multiple cancer types, including breast, colorectal, glioblastoma, ovarian, and others (17, 24). Many tumor types are known to increase their glucose uptake and rely on aerobic glycolysis to meet their metabolic demands in a process known as the Warburg effect (83). The overexpression of GLUT receptors in cancer likely facilitates the increased demand for glucose (3). TXNIP functions as a tumor suppressor, by regulating GLUT expression. TXNIP expression is downregulated in invasive breast cancer, and TXNIP depletion in vitro and in vivo led to an increase in GLUT1 mRNA expression (54). TXNIP tumor suppressor function appears to be, at least in part, thioredoxin-independent and therefore may be arrestin-domain related, but whether this is a conserved function among α-arrestins remains to be determined.
In addition to TXNIP, other α-arrestins have been implicated in glucose metabolism. ARRDC4, but not ARRDC2 or 3, was shown to inhibit glucose uptake in HEK293 cells through an unknown mechanism (55). Interestingly, ARRDC3 (also known as thioredoxin-binding-protein-2-like inducible membrane protein [TLIMP]) was shown to regulate peroxisome proliferator-activated receptor (PPAR) activity, which is known to regulate glucose metabolism. In NIH3T3 cells, overexpression of ARRDC3 suppressed ligand-induced PPARγ activation (51), whereas adipocyte-specific depletion of ARRDC3 in vivo led to an increase in the expression of PPAR target genes (15). However, a subsequent study reported that depletion of ARRDC3 in vivo did not alter glucose homeostasis (58), suggesting that ARRDC3 does not directly regulate glucose uptake, such as TXNIP, but rather may have a different role in regulating cellular metabolism. Since α-arrestins share structural homology with TXNIP and β-arrestins (2), they may similarly function as key regulators of cellular physiology and disease, including cancer. However, the role of α-arrestins in cancer remains poorly understood.
The α-arrestin ARRDC3 functions as tumor suppressor in invasive breast cancer
To explore α-arrestin function in cancer, Gene Expression Profile Interactive Analysis (80) (GEPIA,
α-Arrestin Expression in Human Cancers
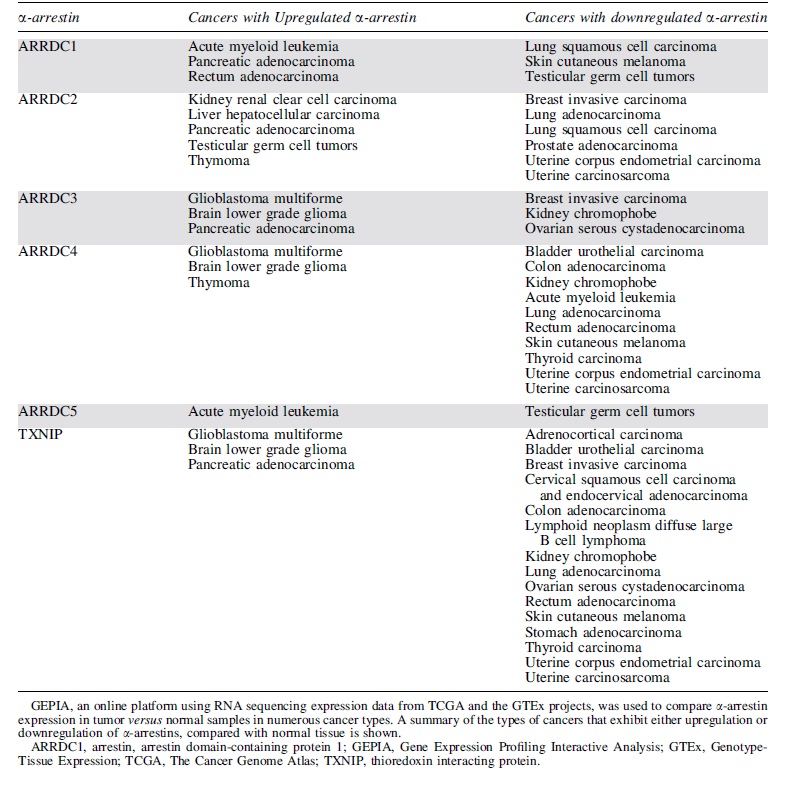
Of the mammalian α-arrestins, ARRDC3 has been most closely linked to cancer. Previous studies reported that in triple-negative breast cancer (TNBC) patients, diminished ARRDC3 expression correlated with increased breast cancer metastasis, tumor recurrence, and poor prognosis (1). The loss or suppression of ARRDC3 expression in TNBC patients and cell lines has been linked to gene deletion or epigenetic silencing (1, 36, 77), suggesting a role for ARRDC3 in tumor suppression.
Depletion of ARRDC3 in murine non-transformed breast epithelial cells increased the expression of markers of epithelial-to-mesenchymal transition (EMT), the process by which epithelial cells acquire migratory and invasive properties (91). Conversely, re-expression of ARRDC3 in MDA-MB-231 invasive breast cancer cells reversed EMT (75). In addition, re-expression of ARRDC3 sensitized cells to a chemotherapeutic agent in MDA-MB-231 invasive breast cancer cells (75), similar to the role of TXNIP in non-small-cell lung cancer, where in cisplatin-resistant A549 non-small-cell lung cancer cells, depletion of TXNIP attenuated the sensitivity of these cells to gemcitabine (14). However, the molecular mechanisms by which ARRDC3 exerts its tumor suppressor function in TNBC are poorly understood. Here, we review studies that examine ARRDC3 function as a tumor suppressor by acting as a multifunctional adaptor protein that regulates receptor trafficking and signaling. We further provide a discussion of how ARRDC3 function might be coordinated, which has not been thoroughly examined.
ARRDC3 regulates receptor trafficking at the endosome
ARRDC3 functions as an adaptor protein to regulate trafficking of receptors, including GPCRs. In contrast to the role of β-arrestin at the plasma membrane, ARRDC3 functions primarily at the endosome to regulate GPCR trafficking. Previous studies using small interfering (si)RNAs linked ARRDC3 to the regulation of β2-adrenergic receptor (β2AR) trafficking in HEK293 cells (48, 65). However, it was later demonstrated that the ARRDC3 siRNAs used in these studies had off-target effects on β-arrestins, and therefore many of the effects of presumed ARRDC3 knockdown on β2AR endocytic trafficking were shown to be attributed to β-arrestins (26). Nonetheless, subsequent studies demonstrated that β2AR is trafficked to ARRDC3-containing endosomes (26, 82), where ARRDC3 regulates β2AR recycling to the plasma membrane (82). In HEK293 cells, ARRDC3 impedes β2AR entry into recycling tubules by preventing interaction between β2AR and sorting nexin 27 (SNX27), which controls β2AR recycling (82) (Fig. 1A). Thus, ARRDC3 is not the major regulator of β2AR trafficking, but rather functions in an ancillary role.
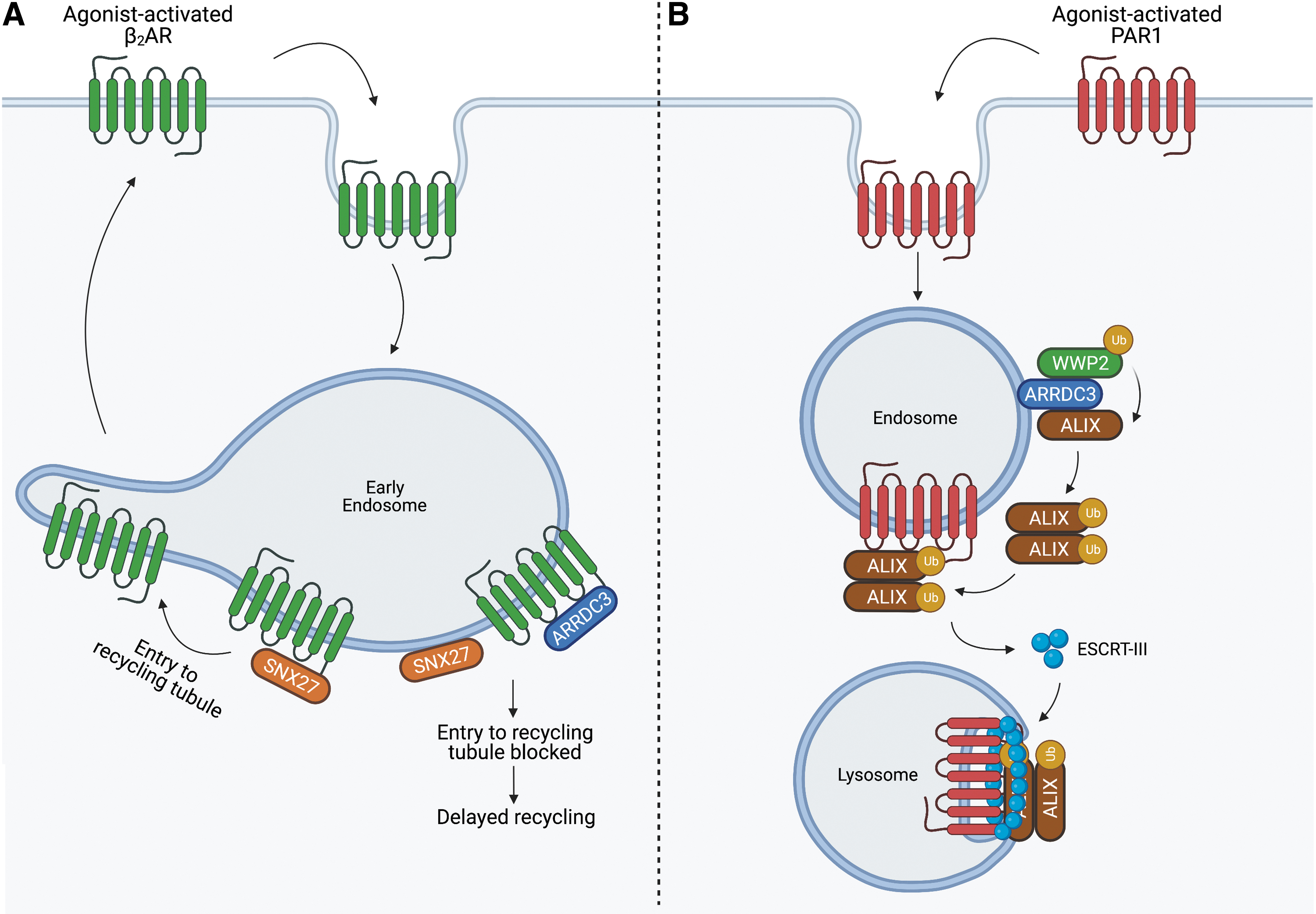
However, ARRDC3 has a critical role in regulating protease-activated receptor-1 (PAR1) endo-lysosomal trafficking via a non-canonical pathway. PAR1, as well as other GPCRs that contain an intracellular loop 2 (ICL2) YPXnL motif, interacts with ALG-interacting protein X (ALIX), an endosomal complex required for transport (ESCRT)-interacting protein (19, 21). ARRDC3 facilitates the ubiquitination of ALIX by recruiting the neuronal precursor cell-expressed developmentally downregulated 4 (NEDD4)-family E3 ligase WW domain-containing protein-2 (WWP2). Ubiquitination of ALIX is required for ALIX dimerization, which promotes interaction between ALIX and PAR1, facilitating entry into the ESCRT pathway (20) (Fig. 1B). This mechanism is particularly important in metastatic breast cancer, where, in multiple TNBC cells lines, loss of ARRDC3 expression results in aberrant PAR1 expression due to dysregulated lysosomal trafficking (4). In MDA-MB-231 invasive breast cancer cells, re-expression of ARRDC3 restores PAR1 endo-lysosomal trafficking, suppresses PAR1 persistent signaling, and blocks metastasis in vivo (4, 6).
In addition, ARRDC3 functions as a tumor suppressor by regulating integrin β4 (Itgβ4) endo-lysosomal trafficking. In TNBC, Itgβ4 overexpression serves as a marker of poor prognosis (12), and ARRDC3 and Itgβ4 expression was shown to be negatively correlated in a panel of breast cancer cell lines (22, 76). In MDA-MB-231 invasive breast cancer cells, re-expression of ARRDC3 reduced Itgβ4 protein expression by restoring Itgβ4 endo-lysosomal trafficking and suppressed migration, invasion, and growth of invasive breast cancer cells in vitro and in vivo (22, 76). Thus, ARRDC3 functions as a tumor suppressor, at least in part, by controlling endo-lysosomal trafficking, and therefore surface expression of membrane receptors, including Itgβ4 and PAR1. ARRDC3 is also likely to regulate other membrane receptors in TNBC, but further studies are required to identify receptor candidates.
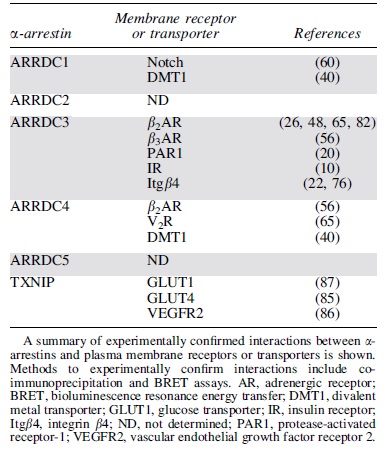
A summary of documented interactions between α-arrestins and plasma membrane receptors and transporters experimentally confirmed by co-immunoprecipitation and bioluminescence resonance energy transfer is shown in Table 2. ARRDC3 has been reported to interact with the most membrane receptors compared with other α-arrestins (Table 2). These studies indicate that α-arrestins can interact with GPCRs, receptor tyrosine kinases, integrins, and transporters, suggesting that α-arrestins have a broad role in regulating membrane protein function. In the budding yeast, Saccharomyces cerevisiae, α-arrestins are numerous—yeast expresses 14 different α-arrestins and does not express β-arrestins—and they regulate diverse transmembrane proteins, including GPCRs and transporters (29). However, the full complement of plasma membrane receptor partners of mammalian α-arrestins is not known.
α-Arrestin Interactions with Plasma Membrane Receptors or Transporters
ARRDC3 regulates receptor signaling via distinct functions
Beyond the role of ARRDC3 in endo-lysosomal trafficking of membrane receptors, ARRDC3 has been recently reported to regulate cellular signaling via a distinct function. In addition to ARRDC3 role in controlling β2AR recycling, ARRDC3 appears to promote endosomal β2AR-G protein signaling. In HEK293 cells, ectopic expression of ARRDC3 enhanced β2AR-induced endosomal cyclic AMP production and recruitment of activated Gαs, suggesting that ARRDC3 promotes β2AR-induced G protein signaling at early endosomes (82) (Fig. 2A). However, the underlying mechanism for enhanced endosomal β2AR-G protein signaling induced by ARRDC3 remains unknown.
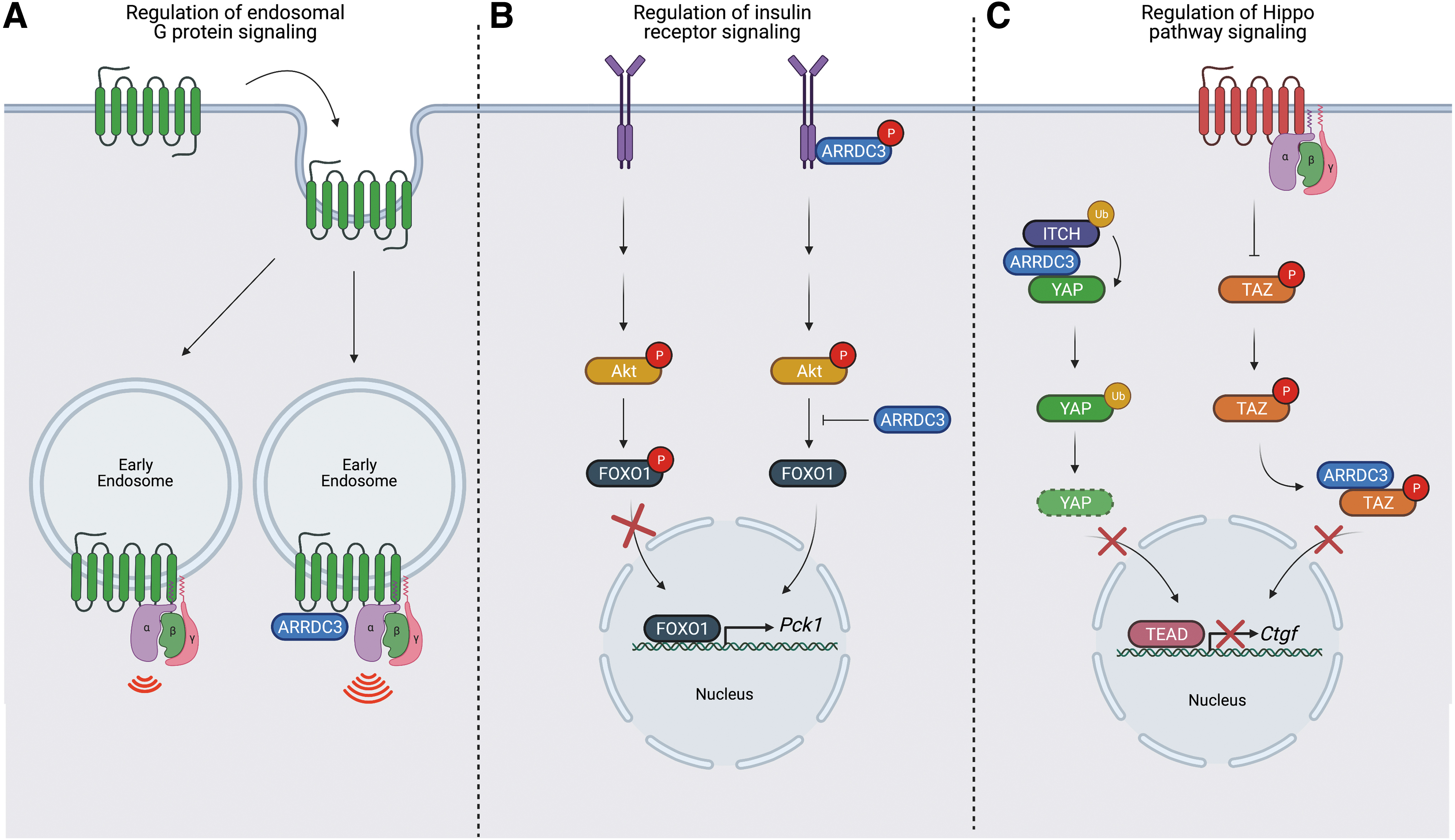
ARRDC3 also appears to control signaling of the insulin receptor (IR) from the plasma membrane. In a liver-specific ARRDC3 knockout mouse model, insulin-stimulated phosphorylation of the transcription factor forkhead box protein O1 (FOXO1) was enhanced and expression of FOXO1 target gene Pck1 was reduced compared with wildtype littermate control mice; however, phosphorylation of Akt, which acts upstream of FOXO1, was not affected (10) (Fig. 2B). Further, in HEK293 cells, ARRDC3 overexpression resulted in a reduction in insulin-induced IR phosphorylation, suggesting that ARRDC3 negatively regulates insulin-induced IR signaling at both the receptor and downstream (10) (Fig. 2B). ARRDC3 was also shown to co-associate with IR (10), but the mechanism by which ARRDC3 regulates IR signaling is not yet clear.
Several studies describe a role for ARRDC3 in the regulation of Hippo pathway signaling. GPCRs are major regulators of Hippo pathway signaling in multiple organ and tissues, and dysregulation of Hippo signaling has been implicated in cancer progression (38, 44, 90). Leash, the Drosophila ARRDC3 homolog, was shown to inhibit Hippo pathway signaling by interacting with and facilitating degradation of Yorkie, the Drosophila Yes-associated protein (YAP) and WW domain-containing transcription regulator protein 1 (TAZ) homolog (33, 84). These studies suggest that ARRDC3 may control Hippo pathway signaling via protein degradation. Indeed, in renal cell carcinoma and colorectal cancer cells, ARRDC3 was shown to function as a tumor suppressor by regulating YAP stability (66, 88) through YAP ubiquitination mediated by the NEDD4-family E3 ligase Itch (88) (Fig. 2C).
However, in invasive breast cancer, we recently showed that ARRDC3 regulates GPCR-stimulated Hippo pathway signaling through a distinct mechanism. In MDA-MB-231 invasive breast cancer cells, ARRDC3 attenuates PAR1-stimulated Hippo signaling by inhibiting GPCR-induced TAZ dephosphorylation and nuclear translocation (6) (Fig. 2C). ARRDC3 controls TAZ transcriptional activity through cytoplasmic sequestration that requires interaction of ARRDC3 PPxY motifs and the TAZ WW domains (6, 88). We also showed that ARRDC3 suppresses Hippo signaling induced by other GPCRs including PAR2, LPA, and S1P receptors (6). These GPCRs are not known to require ARRDC3 for endo-lysosomal sorting. Moreover, ARRDC3 regulates Hippo signaling independent of ARRDC3-dependent PAR1 endo-lysosomal trafficking (6), consistent with an multi-functional properties of ARRDC3.
In addition to YAP/TAZ and NEDD4-family E3 ligases, ARRDC3 is likely to interact with other WW domain containing proteins, given the abundance of ∼100 WW domains identified in the human genome (79). Such ARRDC3-WW domain interactions likely require the ARRDC3 PPxY motifs and influence GPCR signaling, but this remains to be determined. Interestingly, the ARRDC3 PPxY motifs are dispensable for co-association with β-arrestins (65), suggesting that ARRDC3 can interact with other proteins utilizing other protein binding determinants. This further highlights the potential for ARRDC3 to function as a scaffold to facilitate the assembly of signaling complexes.
In a recent pilot study, we examined the ARRDC3 interactome in HEK293T cells using mass spectrometry. Among the 378 interacting proteins identified, several were previously reported ARRDC3-interacting proteins, including WWP1, Itch (also known as AIP4), and β-arrestin-2 (Fig. 3C, D). Protein analysis through evolutionary relationships (PANTHER,

Protein-modifying enzymes were the second most represented protein class and included kinases, phosphatases, ubiquitin E3 ligases, and proteases (Fig. 3A–C). These classes of enzymes are known for modulating β-arrestin activity (70) (Fig. 4) and may represent important regulators of ARRDC3 function. Among the kinases identified in the ARRDC3 interactome, c-Src was highly enriched (Fig. 3B). This is of particular interest, as c-Src has been shown to interact with β-arrestins through N-terminal domain PxxP motifs (39, 43). ARRDC3 may interact with c-Src similarly since ARRDC3 contains a PxxP motif at residues 119–122 within the N-terminal arrestin-fold, but this remains to be determined. GPCR-mediated activation of the NEDD4-family E3 ligase Nedd4–2 requires tyrosine phosphorylation by c-Src (25); thus, it is possible that ARRDC3 may function as a scaffold to facilitate the phosphorylation of NEDD4-family E3 ligases by c-Src.
Other important ARRDC3 interacting proteins include scaffold and adaptor proteins, membrane traffic proteins, protein binding and activity modulators and transporters (Fig. 3D–G). In general, the motifs and/or domains that facilitate ARRDC3 protein interaction as well as the impacts of ARRDC3 protein interaction on biological function are poorly understand, and future studies investigating the mechanisms of ARRDC3 protein interactions are important.
Regulation and coordination of α-arrestin function by post-translational modification
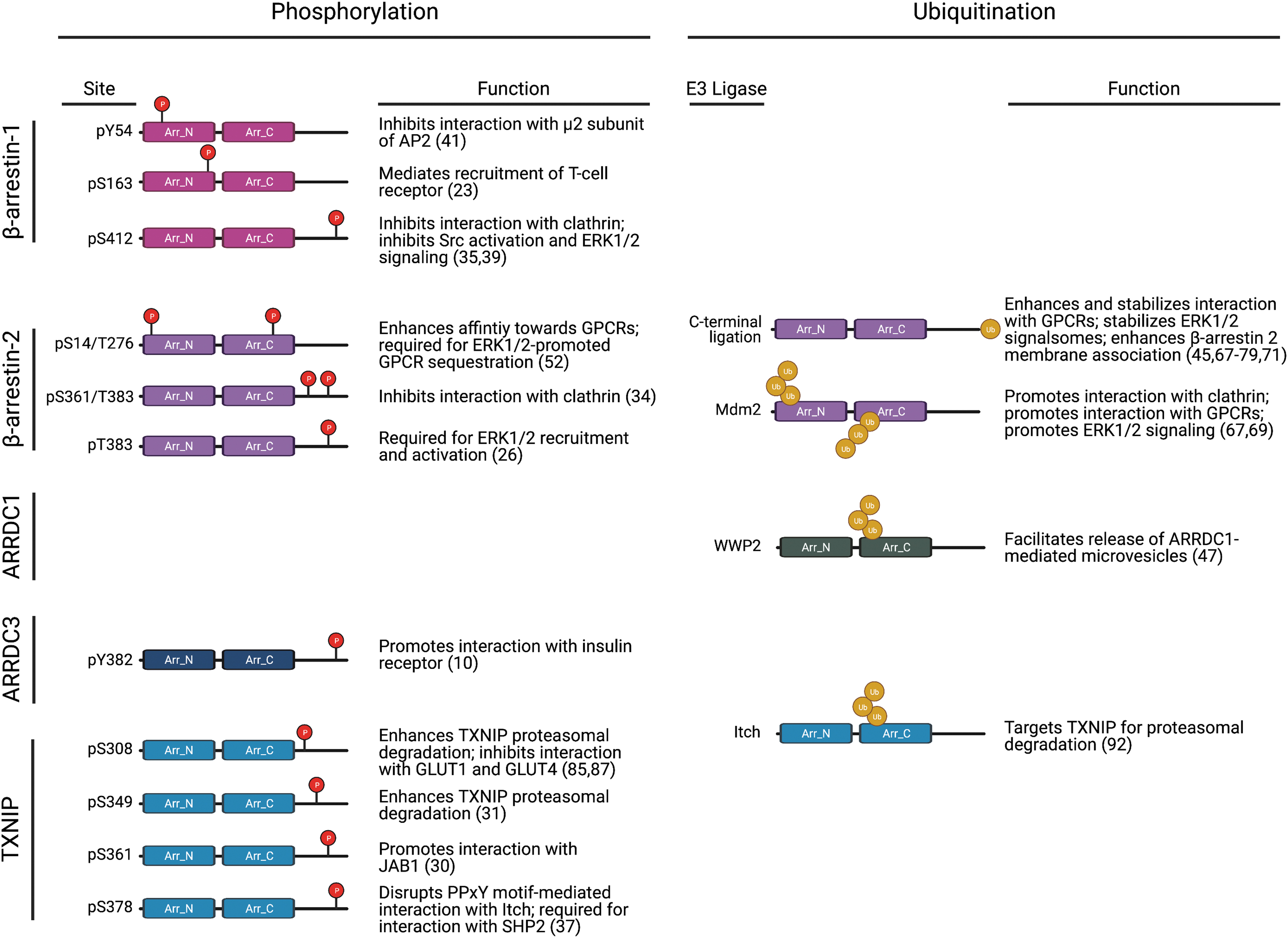
β-arrestin function has been studied since the 1980s and is well characterized. Post-translational modifications, including phosphorylation and ubiquitination, are known to influence β-arrestin conformation and function (Fig. 4) (70). Although α- and β-arrestins share minimal sequence homology, they are structurally similar, as demonstrated by predictive homology modeling strategies (7, 11) as well as by x-ray crystallography of TXNIP (28) and the ARRDC3 N-terminal arrestin domain (61). These studies revealed that α-arrestins contain two arrestin-fold domains as well as the finger loop, such as β-arrestins, but they lack the short α-helix that is present in the β-arrestin N-terminus, which is important for β-arrestin interaction with GPCRs (7) (Fig. 5). However, ARRDC3 structural analysis suggests a potential mechanism for interaction with GPCRs that involves the interaction of a basic patch on ARRDC3 with phosphorylated GPCR C-tail or an acidic patch (61).

In addition, in contrast with β-arrestins, α-arrestins harbor two PPxY motifs in their C-tail region (Fig. 5), which are important for the recruitment of WW-domain containing proteins, such as homologous to E6-AP C-terminus (HECT) domain-containing NEDD4-family of E3 ubiquitin ligases (48, 63). Although these structural differences between α- and β-arrestins may contribute to their unique functions in regulating GPCR signaling and trafficking, the structural similarities between α- and β-arrestins suggest that they may be subject to similar regulatory mechanisms. Thus far, both ubiquitination and phosphorylation have been linked to α-arrestin function, and other mechanisms, such as subcellular localization and control of protein expression, may also play a role in regulating α-arrestin activity.
Although the β-arrestin C-tail contains a clathrin-binding DDIVFE motif that is critical for mediating GPCR internalization (32), α-arrestins harbor two PPxY motifs in their C-tail region (Fig. 5), which are important for the recruitment of WW-domain containing proteins, such as NEDD4-family E3 ubiquitin ligases. Indeed, ARRDC3 has been shown to interact with this class of HECT E3 ligases (62, 63), which facilitates the ubiquitination of other proteins, such as YAP (88) or ALIX (20), and also ubiquitination of ARRDC3 itself (65, 82). This appears to be true for other α-arrestins as well since the PPxY motifs of both ARRDC1 and TXNIP are required for self-ubiquitination (18, 48, 63, 92).
Multiple studies have reported that mutation of both ARRDC3 PPxY motifs, which also leads to loss of ARRDC3 ubiquitination, appears to stabilize ARRDC3 protein expression (4, 48, 65), but this has not been directly tested. In addition, both endogenous and exogenously expressed ARRDC3 seems to exist in cytoplasmic puncta that partially co-localize with endosomal and lysosomal markers (6, 20, 48, 82), however mutation of both ARRDC3 PPxY motifs causes relocalization of ARRDC3 to the plasma membrane in HEK293 cells (26, 48, 65, 82). The ARRDC3 PPxY motifs are also required for ARRDC3 function in regulating GPCR signaling to the Hippo pathway as well as for ARRDC3 tumor suppressor function in vivo (6). β-arrestin ubiquitination is also critical for regulating GPCR signaling and trafficking, and ubiquitination has been implicated in ARRDC1 and TXNIP function (Fig. 4). However, the exact mechanism by which the PPxY motifs and ubiquitination control ARRDC3 activity remains unknown.
It is well established that phosphorylation plays an important role in regulating β-arrestin function (Fig. 4), but the role of phosphorylation in α-arrestin function is largely unknown. Although PhosphoSite mining has identified multiple ARRDC3 phosphorylation sites (27), only a single study has suggested a role for phosphorylation in ARRDC3 function. Mutation of a single tyrosine (Y) residue to alanine (A) at position 382 (which was identified as a phosphorylation site by PhosphoSite mining) within the ARRDC3 C-tail was shown to reduce the association between ARRDC3 and IR in HEK293 cells, suggesting that ARRDC3 Y382 phosphorylation is required for interaction with IR (10). Phosphorylation sites on all α-arrestins have been identified by PhosphoSite mining (27), but of these, only TXNIP phosphorylation has been studied.
TXNIP is phosphorylated at multiple serine (S) and threonine (T) sites in the C-tail region and is a substrate for AMP-activated protein kinase (AMPK) (87), Akt (85), extracellular signal-regulated kinase (ERK) (31), and p38 mitogen-activated protein kinase (30). In these studies, phosphorylation enhanced TXNIP degradation; when TXNIP phosphorylation was blocked either by mutagenesis or by pharmacological inhibition of the upstream kinase (AMPK or ERK), TXNIP expression was stabilized and agonist-induced degradation was blocked (31, 87). In addition, TXNIP phosphorylation by AMPK or ERK leads to dissociation of TXNIP from GLUT 1 and 4, respectively (85, 87). These studies suggest that phosphorylation may negatively regulate α-arrestin function (Fig. 4), but further studies are necessary to determine whether this is the applicable to ARRDC3 and the implications in cancer progression.
Conclusions
α-arrestins are multifaceted adaptor proteins that control diverse biological functions. ARRDC3 is the most well-characterized example, where it is clear that ARRDC3 functions as a tumor suppressor by regulating receptor trafficking and signaling of important drivers of cancer progression, namely Itgβ4 and PAR1 (4, 6, 22, 76). The ARRDC3 interactome may consist of hundreds of proteins, including kinases and phosphatases, E3 ubiquitin ligases and deubiquitinases, adaptor and scaffold proteins, and protein binding and activity modulators suggesting that similar to β-arrestins, α-arrestins have broad biological functions (Fig. 3). In a similar proteomic analysis of the β-arrestin interactome, it was demonstrated that β-arrestins interact with many of the same classes of proteins (89); however, most identified proteins were unique to either β-arrestins or ARRDC3, further confirming the hypothesis that β-arrestins and α-arrestins have distinct and non-overlapping biological functions.
Although β- and α-arrestins are structurally similar, they have unique features and properties; the most pronounced is the presence of PPxY motifs in the α-arrestin C-tail that are absent in β-arrestins. The PPxY motifs appear to confer unique properties to α-arrestins: They are able to interact with WW-domain containing proteins, which include the NEDD4-family of E3 ligases (63). This suggests that ubiquitin—and the coordination of ubiquitin by α-arrestins—might be critical to their function. In addition, the PPxY motifs appear to be important for normal α-arrestin subcellular localization. In contrast to β-arrestins, for which the major site of function is the plasma membrane, α-arrestins appear to function primarily at the endosome. It is possible that these distinctions in subcellular localization may partially contribute to the differences in β- and α-arrestin function. β- and α-arrestins may function in a complementary manner to spatiotemporally coordinate receptor signaling and trafficking.
In contrast to β-arrestins, it remains unclear how α-arrestin function is regulated. Recent studies have demonstrated the potential for post-translational modifications to regulate α-arrestin function, but in general, the modes of α-arrestin activation are not known. Similar to β-arrestins, α-arrestins contain a finger loop domain, but it is unknown whether α-arrestins also engage activated GPCRs via this domain (61). Unlike β-arrestins, α-arrestins do not contain a polar core or an N-terminal α-helix (7, 28, 61), both of which are important for regulating activation of β-arrestins (73). Thus, α-arrestin function is likely governed by a separate mechanism that has yet to be determined. However, similar to β-arrestins, α-arrestins contain two arrestin folds that are predicted to be flexible and connected by a linker that may allow for conformational change similar to that observed for β-arrestins (7, 73), suggesting that α-arrestin activation could also be linked to conformational change. The mechanisms that control α-arrestin activation are not known but may include post-translational modifications, as growing evidence discussed here suggests.
The α-arrestins represent a new class of multifunctional adaptor proteins, and they potentially could serve as therapeutic targets in human diseases such as cancer. However, to advance the status of α-arrestins, further studies are essential to gain a better understanding of the full α-arrestin interactome and to define the molecular mechanisms that control α-arrestin activation and function.
Materials and Methods
Cell culture
HEK293T cells were grown in Dulbecco's modified Eagle's media supplemented with 10% fetal bovine serum. For DNA transfection in HEK293T, poly-ethyleneimine (PEI) was used at a ratio of 3 μL PEI (1 mg/mL) to 1 μg of plasmid and assays were performed at 48 h post-transfection.
Isolation of ARRDC3 complexes from HEK293T cells
HEK239T cells transiently expressing HA-ARRDC3 were lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40) supplemented with protease (cat. no. 11697498001; Roche) and phosphatase (cat. no. 04906837001; Roche) inhibitor cocktail. Cell lysate from HA-ARRDC3 expressing cells was immunoprecipitated for 5 h by using anti-HA magnetic beads (cat. no. 88837; Thermo Fisher Scientific). After washing, immunoprecipitates were eluted by 100 μL 0.1 M glycine, pH 2.0 at room temperature (RT) for 10 min and neutralized with 15 μL 1 M Tris, pH 8.5.
Protein/peptide extraction of isolated ARRDC3 complexes
Samples received were immediately desiccated in a Thermo SpeedVac and stored at -80°C until processing. For all samples, protein isolation was performed by using the standard Protifi S-trap micro protocol, with minor modifications. Briefly, samples were resuspended in 100 μL sodium dodecyl sulfate (SDS) resuspension and denaturing buffer (7% SDS, 6 M urea, 50 mM Tris, pH 8.1), protease inhibitor cocktail (cOmplete™ Protease Inhibitor Cocktail; cat. no. 4693124001), and phosphatase inhibitor (Roche PhosStop; cat. no. 4906837001). After resuspension, samples were reduced by adding 5 μL 500 mM dithiothreitol for 30 min at 47°C, followed by 15 μL 500 mM iodoacetamine for 30 min in dark conditions, and quenched by adding 5 μL 500 mM dithiothreitol at RT for 15 min. Samples were then acidified by adding 7.5 μL 12% phosphoric acid. Samples then had 600 μL S-trap binding buffer (90% MeOH, 10% triethylammonium bicarbonate [TEAB]) added. Samples were briefly vortexed and pipetted into the S-trap micro 200 μL at a time until all sample was spun through (3000 relative centrifugal force, RT, 1 min). After samples were loaded onto S-trap, each column was washed 5 × with 125 μL binding buffer. After this, each sample was digested by using 5 μg trypsin resuspended in 30 μL 50 mM TEAB at 47°C for 3 h. Digested peptides were eluted by using 50 μL 50 mM TEAB, followed by 50 μL 5% formic acid (FA), and finally 50 μL 50% acetonitrile. Samples were desiccated in the Speedvac and resuspended in 100 μL 5% FA. Peptides were then desalted by using 200 μL pipette tips with 4 c-18 disks. These samples were then desiccated and resuspended in 5 μL 5% acetonitrile and 5% FA.
Mass spectrometry
Samples were loaded into the high-performance liquid chromatography (HPLC) autosampler (Thermo Easy-nLC 1000, 4°C tray temp). Each sample was then injected into the mass spectrometer (MS) (Orbitrap Fusion Tribrid; Thermo Fusion) by using an in-house packed c18 column (30 cm long, SePax 1.8 μm, 10 angstrom, c18 bead slurry). Each sample was acquired on an 85 min gradient. Buffer A of the mobile phase contained 0.125% FA and 3% acetonitrile in HPLC-grade water, whereas buffer B contained 0.125% FA in acetonitrile. Gradient flowing 6% B was followed by a linear increase up to 25% B for 70 min, and it was then increased to 100% B over 10 min. The HPLC flow rate was 0.300 μL/min. The MS data were collected in the positive ion mode within the 375–1500 m/z range. An initial Orbitrap scan resolution of 120,000 was employed followed by collision-induced dissociation and analysis in the ion trap by using “Top Speed” dynamic identification with dynamic exclusion enabled (repeat count of 1, exclusion duration of 30 s). The automatic gain control for FT full MS was set to 4e5 and for ITMSn it was set to 1e4. Ion trap with collision-induced dissociation was used in the MS2 method.
Sample bioinformatics
The resulting mass spectra raw files were first searched by using Proteome Discoverer 2.1. using the built-in SEQUEST search algorithm. Samples were searched against Uniprot Swiss-Prot Homo sapiens (downloaded June 2020). Target-decoy searching at both the peptide and protein level was employed with a strict false discovery rate cutoff of 0.01 by using the Percolator algorithm employed by Proteome Discoverer. Enzyme specificity was set to full-tryptic with static peptide modifications set to carmbamidomethylation (+57.0214 Da). Dynamic modifications were set to oxidation (+15.995 Da) and n-terminal protein acetylation (+42.011 Da). Only high-confidence peptides and proteins (q-value <0.01) were used.
Footnotes
Authors' Contributions
H.W. and J.T. wrote the draft, reviewed and edited the article; C.C.G., W.A.P., and H.W. performed research; and C.C.G., W.A.P., D.J.G., and H.W., conducted data curation and formal analysis.
Authorship Confirmation Statement
This article has not been published nor is under consideration for publication elsewhere.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by NIH/NIGMS R35 GM127121 (J.T.), a Tobacco-Related Disease Research Program Predoctoral Fellowship T31DT1550 (H.W.) and NIH/NIGMS K12 GM068524 (C.C.G.).