
Abstract
Significance:
Reactive sulfur and nitrogen species such as hydrogen sulfide (H2S) and nitric oxide (NO•) are ubiquitous cellular signaling molecules that play central roles in physiology and pathophysiology. A deeper understanding of these signaling pathways will offer new opportunities for therapeutic treatments and disease management.
Recent Advances:
Chemiluminescence methods have been fundamental in detecting and measuring biological reactive sulfur and nitrogen species, and new approaches are emerging for imaging these analytes in living intact specimens. Ozone-based and luminol-based chemiluminescence methods have been optimized for quantitative analysis of hydrogen sulfide and nitric oxide in biological samples and tissue homogenates, and caged luciferin and 1,2-dioxetanes are emerging as a versatile approach for monitoring and imaging reactive sulfur and nitrogen species in living cells and animal models.
Critical Issues:
This review article will cover the major chemiluminescence approaches for detecting, measuring, and imaging reactive sulfur and nitrogen species in biological systems, including a brief history of the development of the most established approaches and highlights of the opportunities provided by emerging approaches.
Future Directions:
Emerging chemiluminescence approaches offer new opportunities for monitoring and imaging reactive sulfur and nitrogen species in living cells, animals, and human clinical samples. Widespread adoption and translation of these approaches, however, requires an emphasis on rigorous quantitative methods, reproducibility, and effective technology transfer. Antioxid. Redox Signal. 36, 337–353.
Introduction
Reactive sulfur and nitrogen species are small cellularly produced molecules that mediate signaling in healthy physiology (2, 39, 56). However, they can contribute to pathophysiological conditions when levels are too low or too high and thus precise measurement of these species is required to attain a complete understanding of their biological roles. Nitric oxide (NO•) was thrust into prominence in the 1980s when it was discovered that it could activate guanylyl cyclase and serve as an endothelium-derived relaxation factor to relax smooth muscle cells—discoveries that would eventually be rewarded with a Nobel prize in 1998 (63).
Nitric oxide is enzymatically produced by three isoforms of the nitric oxide synthase (NOS) enzyme—neuronal NOS (nNOS, NOS-1), inducible NOS (iNOS, NOS-2), and endothelial NOS (eNOS, NOS-3), each of which uses
Canonical nitric oxide signaling begins with nitric oxide binding to guanylyl cyclase, increasing the rate of conversion of guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP), which, in turn, can modulate signaling via cGMP-dependent protein kinases (PKGs), cGMP-gated cation channels, phosphodiesterases (PDEs), and other targets (40). In addition, nitric oxide is a reactive molecule that gives rise to a host of reactive nitrogen species (56) that can also participate in cellular signaling (2), including (but not limited to) S-nitroso proteins and small molecules (34), peroxynitrite (ONOO−) and decomposition products (38), nitroxyl (HNO) (41, 64), and nitrite/nitrate (31). Taken together, these progenies of nitric oxide are collectively referred to as reactive nitrogen species.
Hydrogen sulfide (H2S) is another small, reactive, and diffusible molecule that is enzymatically produced in mammalian systems by cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfur transferase (3-MST) (65). Endogenous levels of hydrogen sulfide are controlled, in part, by catabolic enzymes sulfide:quinone reductase (SQR) and persulfide dioxygenase, ETHE1. Hydrogen sulfide can also be produced by the microbiome via reduction of dietary sulfates to play important roles in mammalian biology (19). Hydrogen sulfide can mediate cellular signaling in a number of physiological scenarios by means of persulfide and polysulfide (H2Sn) formation, binding to metal centers, and interaction with other reactive sulfur, oxygen (O2), and nitrogen species (39).
Similar to nitric oxide, hydrogen sulfide has emerged as a ubiquitous mediator of physiology in a number of tissues and organs (39). Hydrogen sulfide is present in aortic tissue at levels that are 20 − 100 times higher than other tissues (77), and the actions of hydrogen sulfide have been well studied in the cardiovascular system (19, 101, 142). It has also been studied in the brain and gut, where some of the first evidence for hydrogen sulfide's signaling roles was observed (1, 71).
A related class of reactive sulfur species central to hydrogen sulfide signaling comprises polysulfide/sulfane sulfurs that contain sulfur in the S0 oxidation state with a sulfur–sulfur bond (86). Polysulfides can be formed by oxidation of hydrogen sulfide (24), but enzymatic synthesis by the same enzymes that form hydrogen sulfide, CBS, CSE (62), and 3-MST (97) are more likely to be physiologically relevant avenues of polysulfide formation via generation of cysteine persulfide (Cys-SSH).
Quite interestingly, cysteinyl-tRNA synthetases (CARSs) were found to be able to mediate persulfidation to form Cys-SSH, in addition to incorporating Cys-SSH into proteins, accounting for a large percentage of protein persulfidation (3). Other potential avenues for persulfide generation include heme proteins such as superoxide dismutase (SOD) (99) and myeloperoxidase (MPO) (44). The catabolism of polysulfides is less understood, but it has been shown that thioredoxin and glutathione systems can mediate this conversion (33). A number of proteins have been shown to be targets of persulfidation, including NF-E2 p45-related factor (Nrf2), Kelch-like ECH-associated protein (Keap1), heme oxygenase-1 (HO-1), phosphatase and tensin homolog (PTEN), and protein kinase G-1α (PKG1α) (86).
Indeed, there is an emerging realization that polysulfide signaling is a central component of reactive sulfur signaling in cardiovascular disease and other biological systems (73). It should be emphasized that polysulfide chemistry is quite complex (75), and that reactive sulfur and nitrogen species interact both chemically (39) and biologically (19). When combined with reactive oxygen and carbon species, they form a complex web of interacting species that has been referred to as the reactive species interactome (28).
Given the importance of reactive sulfur and nitrogen species in physiology and pathophysiology, their measurement and detection in biological systems is of the utmost importance to increase fundamental understanding and develop therapeutic approaches. Due to their reactive nature, one class of methods that is particularly well suited for measuring reactive sulfur and nitrogen species is chemiluminescence.
Chemiluminescence is the light generated from a thermal reaction that leads to the production of a molecule in an emissive excited state (128). Most (although not all) chemiluminescent reactions involve the cleavage of a weak oxygen–oxygen bond in a structure with a large ring strain, leading to a highly exothermic reaction. The key advantage of chemiluminescence is that the background signal is quite low due to the elimination of an external light source; this ultimately leads to a large increase in sensitivity and offers opportunities for deep tissue imaging (109).
This review article will focus on chemiluminescence methods for the detection of reactive sulfur and nitrogen species, which have been a critical tool in developing an understanding of hydrogen sulfide and nitric oxide in biological systems. We note that chemiluminescence methods for measuring reactive oxygen species have been recently reviewed (122). The structure of this review will be roughly chronological, starting with well-established and commercialized ozone (O3)-based and luminol chemiluminescence techniques and ending with emerging luciferin/luciferase,1,2-dioxetane, and nanotechnology strategies.
Ozone-Based Chemiluminescence
Ozone-based chemiluminescence detection of hydrogen sulfide
Gas chromatography with ozone-induced sulfur chemiluminescence has become a powerful method of choice for measuring biological hydrogen sulfide (131) and is the result of extensive fundamental and applied research (139 –141). The spectrum of the sulfur “afterglow” of sulfur dioxide (SO2) was first reported in 1934 (47), and the chemiluminescent reaction of sulfur monoxide (SO) with ozone to produce SO2 in the excited state was reported in 1966 (Fig. 1A, Equations 1 and 2) (54).

It was later shown that a range of sulfur-containing molecules, including SO, hydrogen sulfide, and other sulfur species, can undergo combustion and be reacted with ozone to form a species with a spectrum that is identical to that of SO2 (4, 74). This chemiluminescence emission spectrum has a maximum at ∼350 nm and extends from 280 to 450 nm (4, 54, 74, 131). These identical spectra lead to the conclusion that the reaction of ozone with sulfur species ultimately produces the same emitting species, excited state SO2. SO has been proposed as a common intermediate, and studies on the reaction of methyl mercaptan with hydrogen sulfide further support a mechanism with hydrosulfinyl radical (HSO•) and SO as important intermediates (Fig. 1A, Equations 3−5) (50).
Interestingly, experiments done at a low pressure to ensure single collisions showed that H2S can react directly with ozone to produce chemiluminescence (Fig. 1A, Equation 6) with the proposed mechanism as shown in Figure 1B proceeding through a strained 4-membered ring intermediate with two oxygen–oxygen bonds (49). Further, red-shifted chemiluminescence emission that extends to the near-infrared (NIR) spectrum has been observed for the reaction of hydrogen sulfide and ozone and attributed to emission from the excited radical species HSO• (115).
It was not long after these chemiluminescent reactions were understood that efforts turned toward harnessing them as an analytical technique for measuring sulfur compounds. It was first shown that a chemiluminescent aerosol spray consisting of O3/O2 mixtures could be used in conjunction with liquid chromatography for the detection of sulfur compounds (15). Later, researchers developed a Universal Sulfur Detection strategy by exposing sulfur-containing compounds, including hydrogen sulfide, to a hydrogen flame followed by a reaction with ozone to produce chemiluminescence via an SO intermediate (13). This detector was combined with gas chromatography separation and improved to form the basis of commercial gas chromatography/sulfur chemiluminescence systems (116, 117).
We also note that sulfur compounds can be reacted with fluorine gas to generate chemiluminescence and chromatographic systems with fluorine-based chemiluminescence detection have been developed (94). Safety issues with working with toxic fluorine gas, however, have likely made commercialization of these systems less viable.
On the development and commercialization of gas chromatography with sulfur chemiluminescence detection, methods and techniques were optimized for studying biological hydrogen sulfide. Early adoption of this technology for biological systems used it to study hydrogen sulfide production and metabolism in the digestive system (43, 78, 123).
This chemiluminescent technique was then used to revise reported estimates of micromolar tissue levels of free hydrogen sulfide to the now more widely accepted nanomolar levels (42). This study measured hydrogen sulfide in the headspace of tissue homogenates and in exhaled breath and was validated by recovery of 2.7 μM sulfide added to a buffer. In a convincing demonstration, an injection of 50 μM sulfide into brain homogenates (the putative estimate of biological sulfide levels at the time) completely swamped any signal from endogenous sulfide. The same group followed up on this discrepancy by showing that “acid-labile” sulfur pools were released at micromolar levels and explained the differing values that were measured (77). Interestingly, high levels of free hydrogen sulfide (∼1 μM) were observed in aortic tissues, suggesting important roles for hydrogen sulfide in cardiovascular function.
Gas chromatography with sulfur chemiluminescence was also used to identify contributions from CBS and CSE enzymes that were responsible for hydrogen sulfide production (66). This study examined mouse tissues, including liver, kidney, and brain, and showed both enhancement using S-adenosylmethionine, an allosteric regulator of CBS, and attenuation using propargyl glycine, an irreversible inhibitor of CSE. Further, this chemiluminescence method was used to provide quantitative measures of the kinetics of hydrogen sulfide production and metabolism in liver, brain, and kidney homogenates and provided steady-state estimates of tissue hydrogen sulfide in the nanomolar range (132). Polysulfides/sulfane sulfur can also be measured with this technique by first releasing hydrogen sulfide from sulfane sulfur by treatment with dithiothreitol (DTT) (72).
Ozone-based chemiluminescence is now a standard technique for measuring hydrogen sulfide in tissue homogenates and has become a method of choice in recent studies from several research groups (137, 138). We direct the reader to the work by Vitvitsky and Banerjee for procedures on using ozone chemiluminescence to measure biological hydrogen sulfide (131).
Ozone-based chemiluminescence detection of nitric oxide
Red to NIR chemiluminescence emission from the reaction of nitric oxide and ozone was first studied in 1959 (Fig. 1C, Equations 7 and 8), where an observed emission that spanned a wavelength range from 590 to 1085 nm was attributed to the excited state of nitrogen dioxide radical (NO2 •) based on spectral and thermodynamic analyses (52). Detailed kinetic (27) and mechanistic (26) studies confirmed a bimolecular reaction and second-order rate equation between nitric oxide and ozone to produce the emissive nitrogen dioxide species, with the overall emission intensity being proportional to the concentration of nitric oxide. In addition, a corrected spectrum with measurements up to 3200 nm showed that the peak emission was centered at 1200 nm and extended from 590 nm up to 2400 nm (26).
It is important to note that the wide spectral separation between the emission of SO2 and NO2 • enables selectivity between sulfur and nitrogen detection by using ozone-based chemiluminescence (139). An understanding of this chemiluminescence reaction led to its application as an analytical technique (8), initially for studying atmospheric, dissolved, and photolytically released nitric oxide in sea water (143). Soon after, its use for detecting biological nitric oxide proved to be transformative and was one of the methods used to confirm nitric oxide as the endothelium-derived relaxation factor (100).
This method was further optimized for direct detection of nitric oxide in biological and clinically relevant scenarios, including nitric oxide release from nitrovasodilator drugs (17, 93). Although ozone-based chemiluminescence is used for the direct detection of nitric oxide, the short lifetime of nitric oxide in the presence of oxygen, metals, and other biomolecules is an obstacle because sampling or homogenizing tissues often takes longer than the lifetime of nitric oxide under these conditions. For this reason, emphasis has been placed on tracking more stable end products of nitric oxide such as nitrite, S-nitroso compounds, and N-nitroso compounds that can be re-converted to nitric oxide by using reductive (11, 58, 88) and photolytic methods (92, 119).
Early methods for environmental nitrite analysis used vanadium(III) (16) (Fig. 1C, Equations 9 − 11) or iodide (29, 45) (Fig. 1C, Equations 13 − 17) to reduce nitrite and nitrate to nitric oxide before detection with ozone-based chemiluminescence. Vanadium chloride can be used to reduce both nitrate (Fig. 1C, Equations 9 and 10) and nitrite (Fig. 1C, Equation 11) (16, 20, 29). It soon became appreciated that vanadium (36) and iodide (110) could also release nitric oxide from S-nitroso groups (Fig. 1C, Equations 12 and 16).
Photolytic methods were developed to homolytically cleave the S − N bond of S-nitroso thiols (Fig. 1C, Equation 22 and 23) (119). These methods were used to show that vanadium reactions could misidentify S-nitroso compounds as nitrate, and a copper-based assay for S-nitroso compounds was developed to solve this problem (Fig. 1C, Equations 18 and 19) (36). Although some vanadium-mediated release of nitric oxide from S-nitroso compounds is, indeed, observed, a more complete measurement of S-nitroso compounds was accomplished by first releasing nitrite from the S − N bond using mercury (35), via the Saville reaction (111), followed by ozone-based chemiluminescence detection.
For accurate S-nitroso compound measurements, interferences from nitrite can be mitigated by first reacting nitrites in the sample with sulfanilimide in a diazotization reaction, and interferences from nitrosation of free thiols can be mitigated by pretreatment with N-ethylmaleimide to cap any free thiols and prevent reactions that recapture released nitric oxide (88, 91). Carbon monoxide can be added to prevent recapture of nitric oxide by iron–heme complexes (11, 32). Iodide and hydroxyquinone were also shown to generate nitric oxide for ozone-based chemiluminescence, with hydroxyquinone being more selective for S-nitroso compounds (110).
Similar ozone-based chemiluminescence methods have been developed to measure nitrosylhemoglobin [Hb(II)NO] (96) and dintrosyl iron complexes (95). Photolytic methods to cleave the S − N bond are complicated by known photochemistry of nitrite (Fig. 1C, Equations 20 and 21) (89), whereas reductive methods are complicated by nitric oxide release from diverse nitrogen-containing species, including nitrate, nitrite, S-nitroso compounds, and N-nitroso compounds.
Although the precise interpretations and methods for quantifying nitric oxide, nitrite, and S-nitroso compounds has been the subject of healthy debate, direct comparisons of different methods suggest that all can provide useful information when interpreted correctly and the use of more than one method improves confidence in results (11). The interested reader is directed to the work of Basu et al. for procedures (11).
Luminol and L-012 Chemiluminescence Detection of Reactive Sulfur and Nitrogen Species
Luminol and derivatives such as 8-amino-5-chloro-2,3-dihydro-7-phenyl-pyrido[3,4-d]pyridazine-1,4-dione (L-012) (98) have been used for the sensitive detection of peroxynitrite (102), nitrogen dioxide (90), nitric oxide (70), and other oxidative species. Luminol can directly react with NO2 • without the need for a catalyst, and this reaction was used to develop an instrument to measure NO2 • and peroxy radicals in the atmosphere (Fig. 2A) (21, 24, 68, 90, 135). Luminol can also be used for peroxynitrite detection in a process that is enhanced by the addition of carbonate and inhibited by SOD (Fig. 2B) (103).
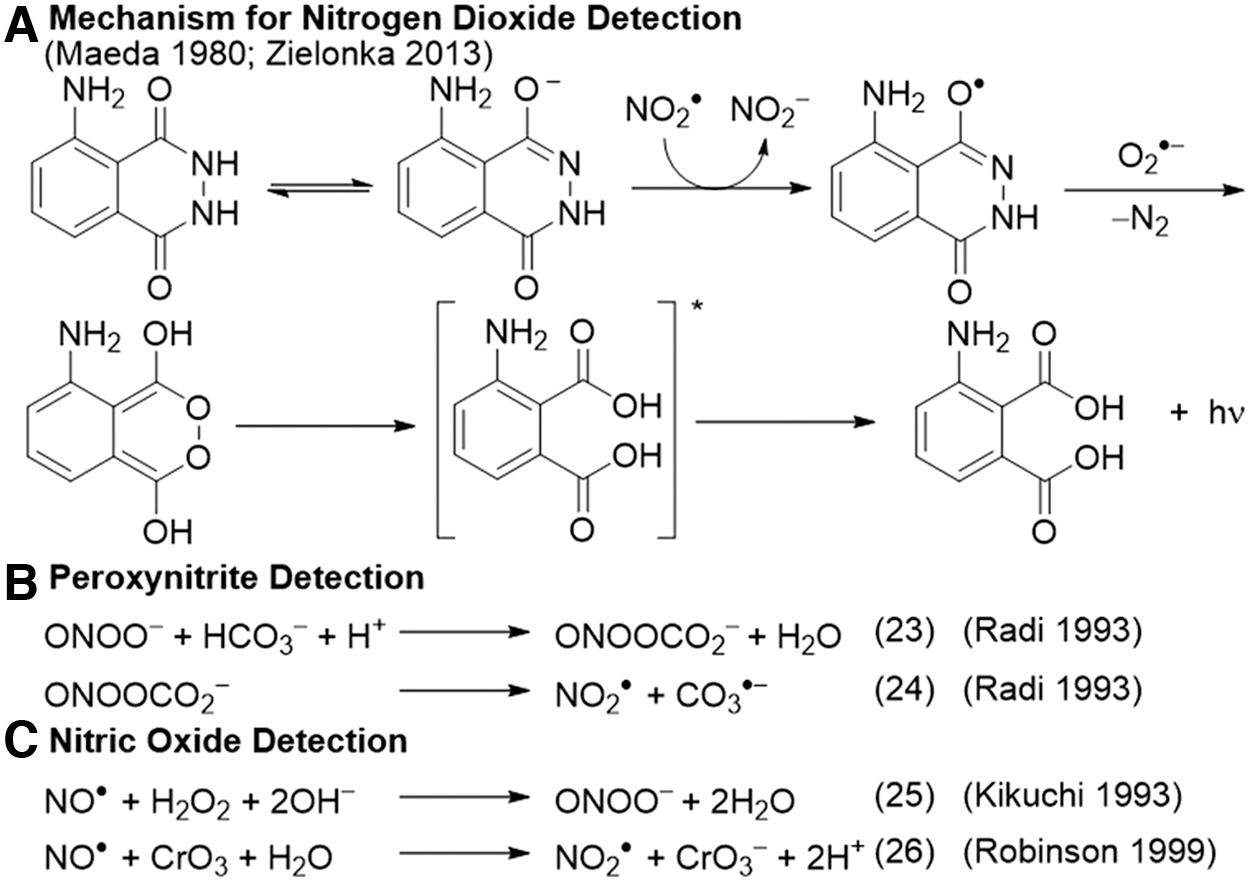
The originally proposed mechanism involves the adduct of peroxynitrite and carbon dioxide mediating a one-electron oxidation of luminol, with the formation of superoxide that reacts with the luminol radical to yield the light-emitting endoperoxide. Studies of the luminol derivative L-012 revealed that it likely reacts with radical species derived from peroxynitrite such as carbonate radical or nitrogen dioxide radical, as evidenced by an increase in signal with the addition of carbonate (30). Further, it was shown that superoxide can actually be formed in the course of the peroxynitrite oxidation of L-012, making it possible to form the endoperoxide in the absence of any additional superoxide (149). The complexity of these reactions demands careful evaluation of mechanistic considerations when using luminol and derivatives for analytical assays.
Luminol has been used to detect nitric oxide in a luminol-H2O2 (hydrogen peroxide) system, through the reaction of NO• and H2O2 to form peroxynitrite (Fig. 2C) (70). The authors provided ultraviolet/visible evidence of peroxynitrite production, showed that the assay is independent of superoxide inhibition by SOD, and neither NO2 • nor hydroxyl radical (HO•) was detected. The assay was used in a flow organ perfusion system to measure NO• in the kidneys. A later study was unable to detect NO• by using the same luminol-H2O2 system but it did observe that the oxidation of NO• to NO2 • with chromium trioxide (CrO3) before exposure to luminol-H2O2 greatly enhanced the signal. This chemistry was used to develop an instrument for measuring NO• in the exhaled breath (Fig. 2C) (106).
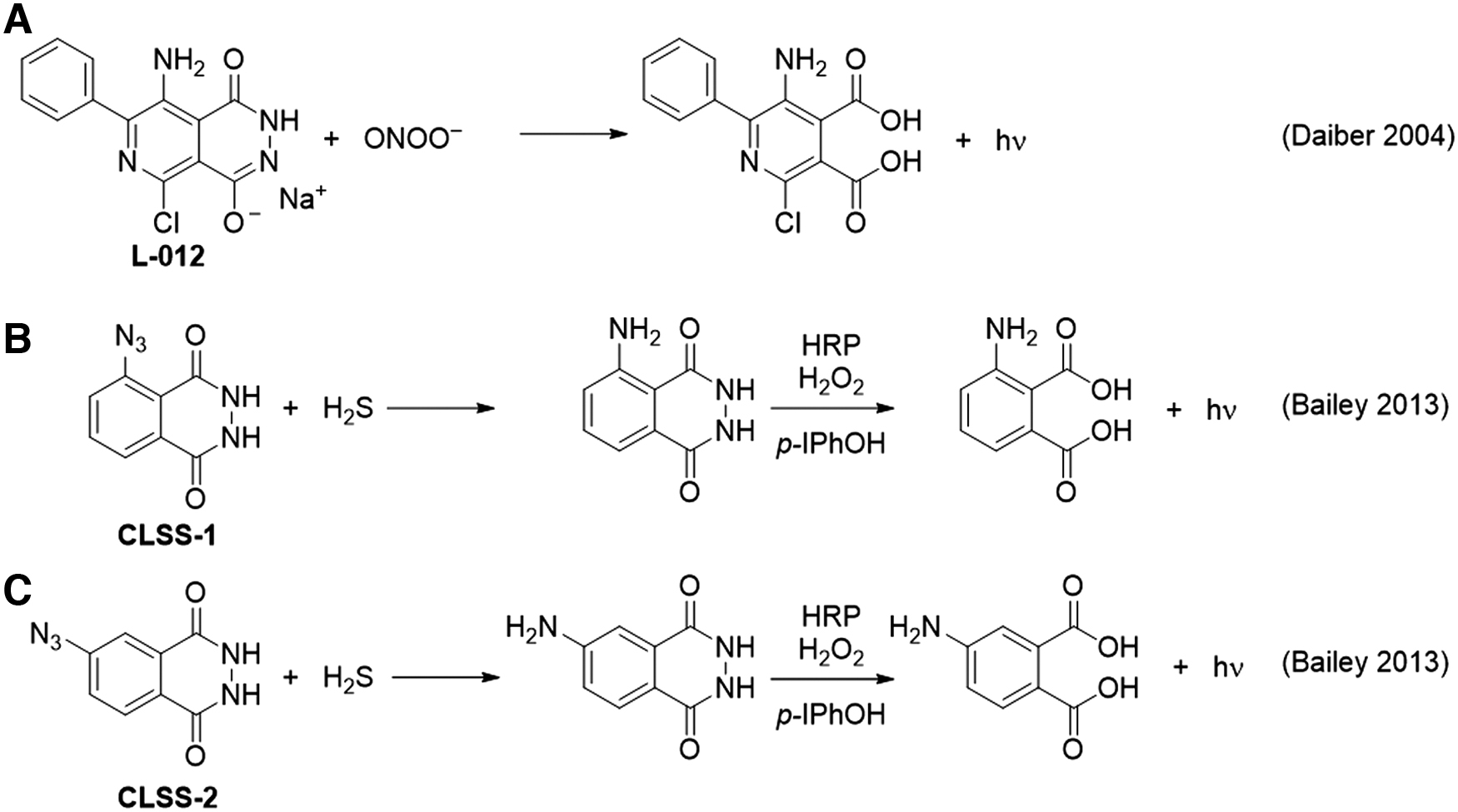
The luminol derivative L-012 was shown to be capable of detecting peroxynitrite and gave a signal ∼100-fold higher than that of luminol (Fig. 3A) (30). The system showed a response from synthetic ONOO−, the peroxynitrite donor 3-morpholinosydnonimine (SIN-1), and a continuous enzymatic superoxide production system [hypoxanthine/xanthine oxidase and diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA NONOate)]. The response was validated and compared with other chemiluminescence assays and high-performance liquid chromatography (HPLC) analysis of dihydroethidium. Controls using the NO• scavenger 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxy 3-oxide (PTIO), the superoxide scavenger SOD, and an iNOS inhibitor provided a selective assay and it was demonstrated that L-012 could detect peroxynitrite formed from superoxide generated in isolated mitochondria.
Interestingly, L-012 has also been used for in vivo imaging of inflammation (69), where it showed significant increases in luminescent signal versus controls in mice injected with L-012 and various inflammatory stimulators including lipopolysaccharide (LPS), phorbol 12-myristate 13-acetate (PMA), and in an arthritis inflammation model. A strong signal was observed in the intestine, even under baseline conditions, and a weaker signal was seen in the lungs and spleen. Strong inhibition by the nitric oxide inhibitor N (ω)-nitro-
A luminol-based system has been used for the detection of hydrogen sulfide by masking the aniline nitrogen of luminol with an azide group that can be reduced by H2S (Fig. 3B, C) (9, 10). The masked luminol derivative is first incubated with a sample containing hydrogen sulfide, which reduces the azide to an amine to form the free luminol derivative. The sample is then mixed with horseradish peroxidase, H2O2, and para-iodophenol to oxidize the luminol derivative and generate chemiluminescence. The para-iodophenol is a chemiluminescence enhancer that increases the rate of formation of luminol radicals. It was observed that the isoluminol derivative with the azide at the meta position, named
Luminol systems offer high sensitivity for reactive species and can provide useful information when combined with the appropriate controls. This caged luminol strategy is an innovative approach that provides ample opportunity for further exploration.
Bioluminescent Caged Luciferin Probes for Reactive Sulfur and Nitrogen Species
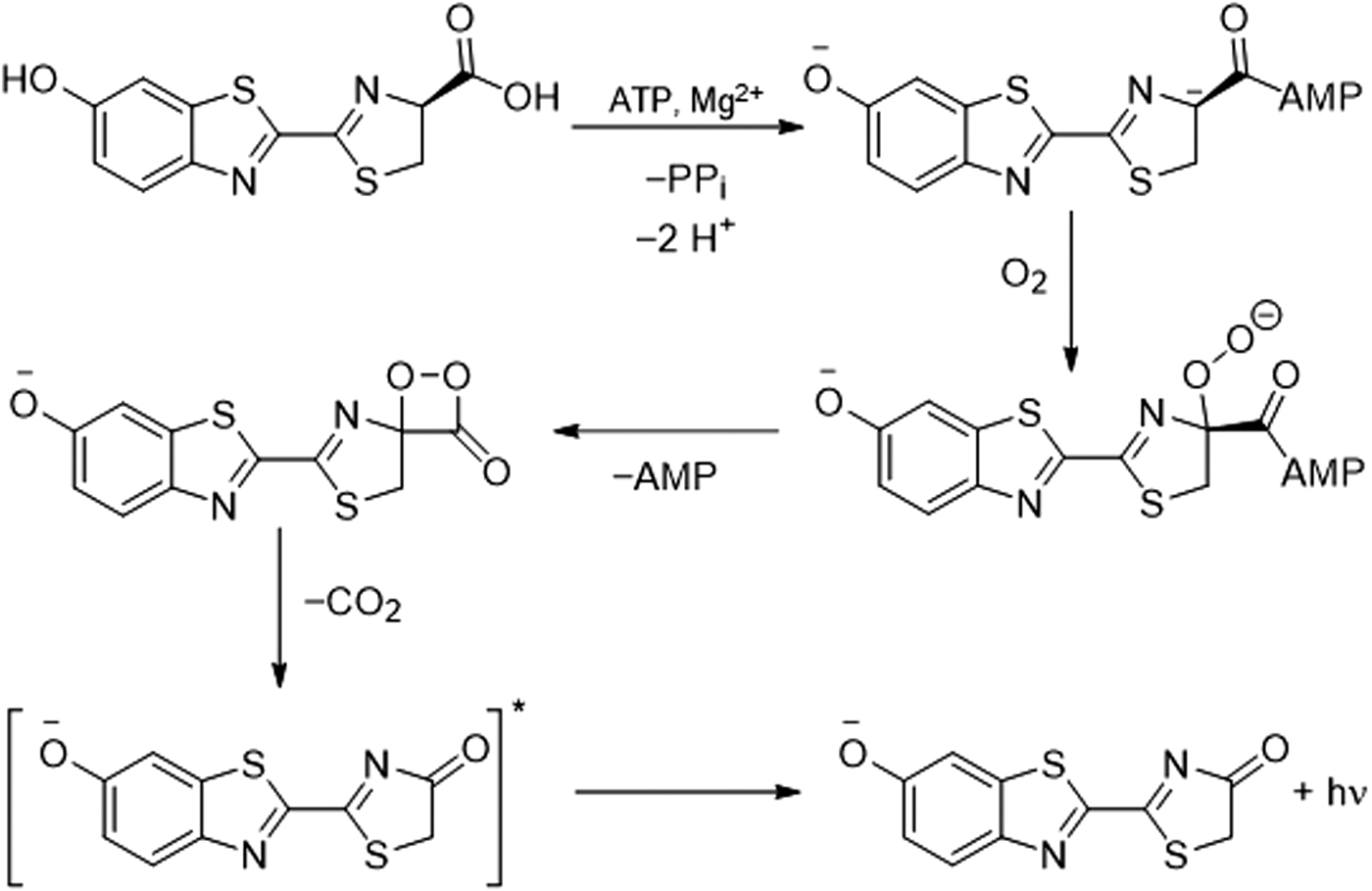
A “caged” luciferin is a luciferin derivative that is not a luminescent substrate for a bioluminescent enzyme but, after reaction with a targeted analyte or set of conditions, is “uncaged” and converted into a substrate that can generate bioluminescence. Luciferase catalyzes the chemiluminescent reaction of luciferin by using adenosine triphosphate, magnesium, and oxygen via an intermediate dioxetanone structure (Fig. 4). Caged luciferins offer a chemiluminescent signal that can be combined with genetically modified models for cellular studies and in vivo imaging (121). Given the versatility of this method, many examples of using caged and modified luciferins for reactive sulfur, oxygen, and nitrogen species have been reported (Figs. 5–7) (144).
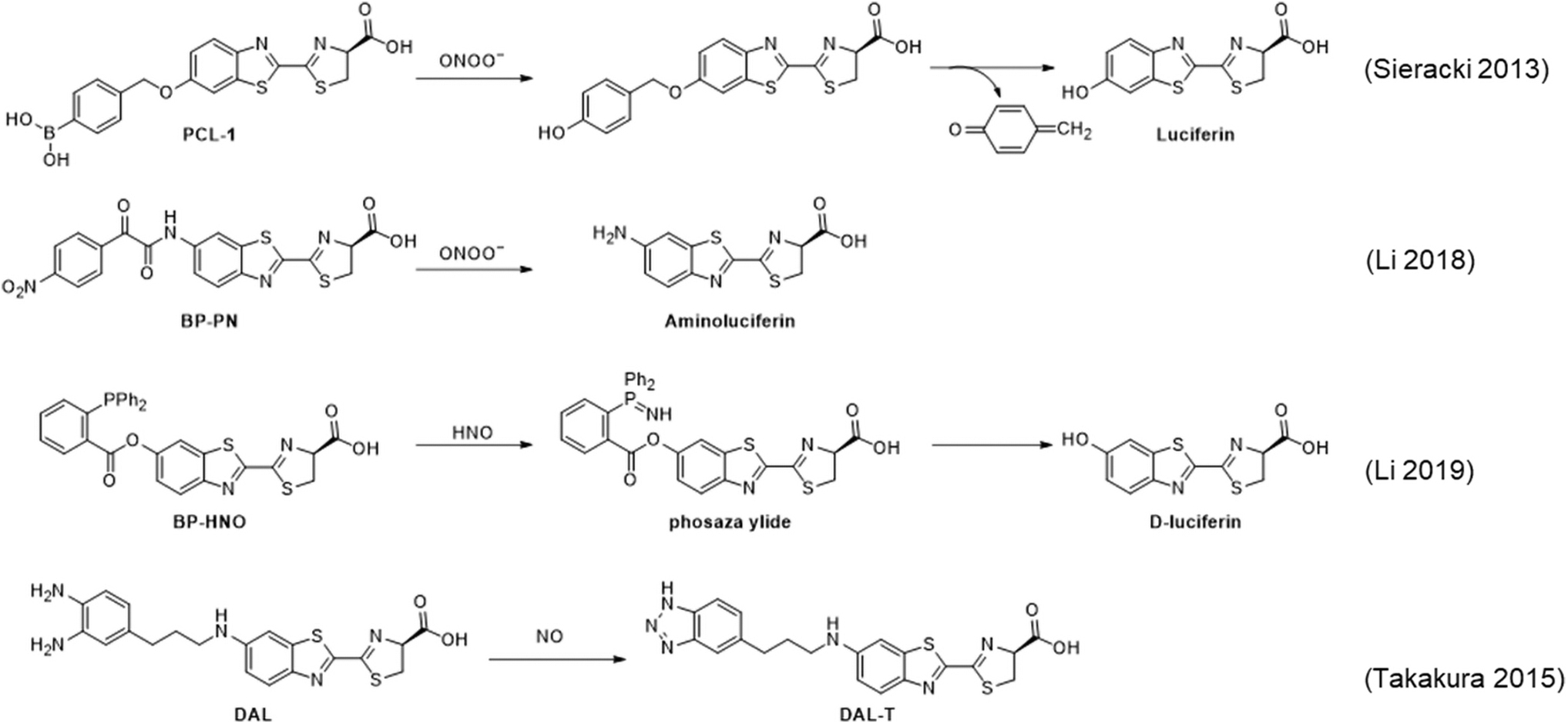

Originally demonstrated to be a bioluminescence probe for H2O2 (129, 130), the boronate-based probe
Detailed studies of the reaction products with H2O2, HOCl, and ONOO− show clean conversion to luciferin with H2O2, formation of luciferin and a chlorinated product with HOCl, and formation of luciferin and a nitrated product with ONOO− (150). This study further shows that combining bioluminescence imaging with HPLC analysis of product distributions coupled with proper controls can be used for more rigorous identification of which species are generated during an experiment.
The bioluminscent probe
Before reacting with nitric oxide, the diamino phenyl group quenches the chemiluminescence from the enzymatically produced excited state of the luciferin derivative. After reacting with nitric oxide to form a triazole, PeT quenching is reduced and an increase in luminescence emission can be observed. This mechanism was referred to as Bioluminescent Enzyme-Induced Electron Transfer (BioLeT), and is essentially identical to PeT, accept that the transition state is accessed through a bioluminescent enzymatic reaction. This bioluminescent nitric oxide probe
Bioluminescent probes for H2S have been developed by using an azide trigger to directly release aminoluciferin, named

A bioluminescent probe for H2Sn,
Bioluminescent probes for cysteine have been developed by caging luciferin with an acrylate (Compound
Chemiluminescent 1,2-Dioxetane Probes for Reactive Sulfur and Nitrogen Species
Triggered chemiluminescence from sterically hindered 1,2-dioxetanes was first accomplished by Schaap et al. and these structures were soon after commercialized for in vitro assays (112 –114). These molecules undergo a triggered chemiluminescence reaction that is believed to proceed through a chemically initiated electron exchange luminescence (CIEEL) mechanism (128), with a solvent cage mediated back electron transfer as the central excitation step (Fig. 8). Although some experimental evidence supports this mechanism, other mechanisms have been proposed and there is still debate (128).

It was only decades later when it was realized that these could actually be used for live cell experiments and in vivo imaging (23, 87). Soon after, key modifications of the molecular structure were developed to red-shift the emission and increase the chemiluminescence quantum yield in aqueous systems, leading to a surge of interest in these structures (53, 84). Many new biological imaging probes have now been developed (55), including chemiluminescent 1,2-dioxetane probes for reactive sulfur and nitrogen species (Figs. 9–11).

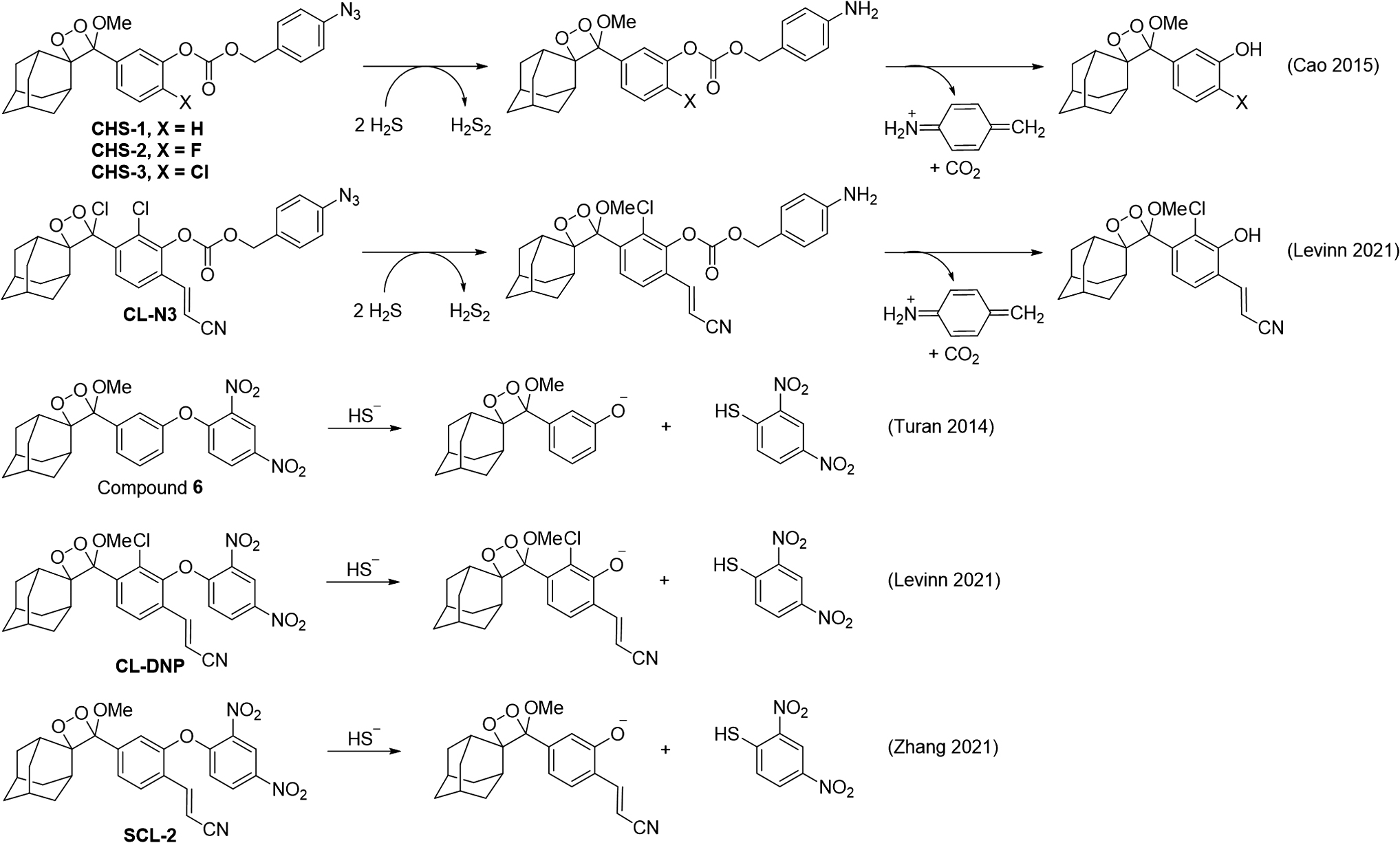
The peroxynitrite-mediated oxidative decarbonylation of an isatin (18) was used to develop an acrylonitrile 1,2-dioxetane chemiluminescence probe for peroxynitrite, called
A series of probes based on the oxidative decarbonylation of a formyl ester trigger and various 1,2-dioxetane scaffolds for the detection of peoxynitrite have been developed (60, 61). An NIR dicyanometh-ylene-4H-benzopyran 1,2-dioxetane scaffold was equipped with the formyl ester trigger and appended with a (2-hydroxypropyl)-β-cyclodextrin to make the renal clearable probe
Recently, this concept was expanded to develop a series of even more red-shifted NIR probes,
A 1,2-dioxetane probe for HNO,
The relevant rate constants for the reaction of the probe with HNO and the rate-limiting step of the CIEEL mechanism were measured, and the chemiluminescence emission from the phenol scaffold was carefully calibrated. An equation was derived to convert the raw chemiluminescence emission into a concentration of HNO and used to measure picomolar concentrations of HNO generated in the reaction between hydrogen sulfide and nitric oxide. The probe was further used to detect HNO release from donor compounds in living cells and live mouse models.
An early example of detecting H2S with a 1,2-dioxetane used a dinitrophenyl group that could release the dioxetane phenol via a nucleophilic aromatic substitution reaction (Compound
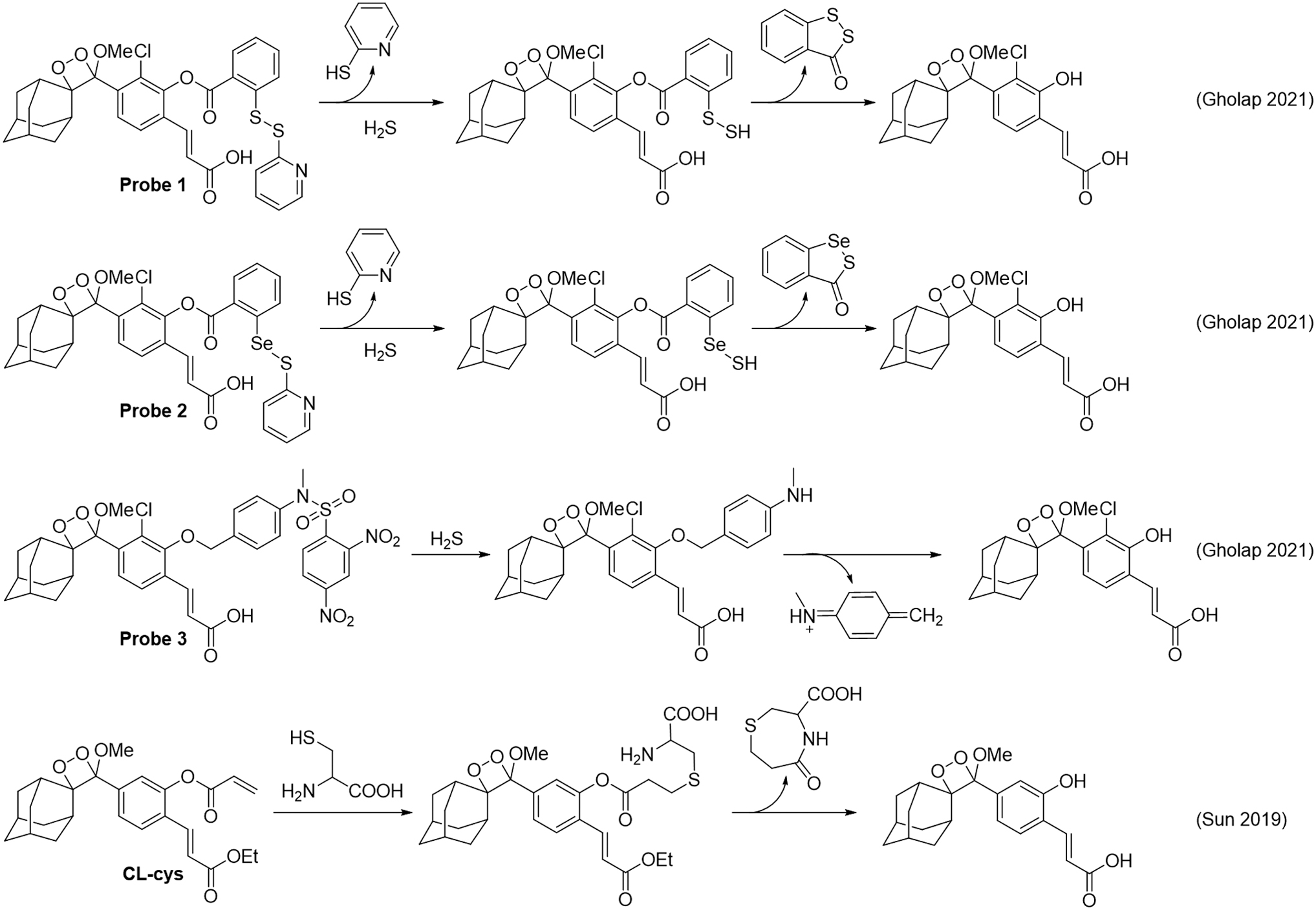
A series of chemiluminescent probes for hydrogen sulfide were reported to be using a disulfide, seleno-sulfide, or dinitrosulfonyl amide trigger (Fig. 11, Probes
Nanoparticle Chemiluminescence Approaches
There are now several examples of nanoparticle-based chemiluminescence systems that have been used for the detection and measurement of reactive sulfur and nitrogen species. Cadmium telluride (CdTe) quantum dots (147) and carbon dot nanoparticles (148) have been used for chemiluminescence peroxynitrite detection based on a proposed mechanism of generating a hydroxyl radical/superoxide radical-pair that leads to the generation of luminescence in the nanoparticles. Another carbon dot system was used for detecting nitrite by first oxidizing it to peroxynitrous acid, followed by reacting with the carbon dots to initiate chemiluminescence (83). Carbon dots have also been reported to have chemiluminescence emission when treated with acidic potassium permanganate in a process that is enhanced by sulfide (85).
Nanoparticles composed of an O-pentacene molecule display chemiluminescence on reaction with peroxynitrite via a proposed mechanism that invokes the formation of an O-pentacene peroxide species (134). These nanoparticles were used to image peroxynitrite in several in vivo models and represent an emerging chemiluminescence approach for the detection of reactive sulfur and nitrogen species.
Conclusions
Chemiluminescence is a powerful approach for the detection, measurement, and imaging of reactive sulfur and nitrogen species. Ozone-based chemiluminescence is a well-established and widely adopted method for monitoring hydrogen sulfide and nitric oxide in biological systems. High selectivity and sensitivity for hydrogen sulfide can be achieved when combining ozone-based chemiluminescence detection with gas chromatography. Selectivity for nitric oxide is generally achieved by using chemical treatments that release nitric oxide from specific biological stores, block reactivity from unwanted species, and cap other biological molecules to keep them from recapturing any released nitric oxide.
Although ozone-based chemiluminescence is very well suited for sampled and homogenized tissues, it is not a viable technique for making measurements in living intact specimens due to the need to use highly reactive and toxic ozone to generate a signal. Luminol systems have also been well studied and are a useful technique for measuring reactive nitrogen species when combined with careful controls to determine which reactive species is leading to signals. Hydrogen sulfide has been measured by using an azide-caged luminol derivative and this relatively unexplored strategy may be amenable to the detection of other types of analytes. Further, in vivo detection of reactive nitrogen species has been accomplished by using luminol derivatives and careful control experiments to determine which species are generated during the biological process being studied.
Caging strategies have been successful in using luciferin to generate bioluminescent probes and spiroadamantane 1,2-dioxetanes to generate non-enzymatic chemiluminescent probes. A key advantage of these systems is that they can be used in living cells and animals with a selectivity that is imparted by the design of chemoselective reaction-based sensing triggers. This represents a versatile strategy that could be applied to a wide range of analytes. Bioluminescence requires genetically modified organisms, which can be an advantage or disadvantage depending on the experiment. Though quite promising, caged luciferin and dioxetanes are not as well established as ozone-based luminescence and the sensing triggers need to be carefully investigated and cross-validated.
The development of quantitative methods using ratiometric (5, 107) or kinetics-based (6, 108) approaches should further aid in the validation and general adoption of these probes. Another challenge is that most caged luciferin and dioxetane structures require complex multi-step organic synthesis, which sometimes limits their use to the group that developed the probe or close collaborators. It should be noted that 1,2-dioxetanes have been commercially available for many years, so commercialization of new molecular structures should not be an insurmountable problem.
An overview of the chemiluminescence literature brings some comparisons to light. Although ozone-based and luminol chemiluminescence have been used for measuring reactive sulfur and nitrogen species for several decades, the use of caged luciferin and 1,2-dioxetanes has only emerged in the past 10 years or so. The development of ozone-based chemiluminescence was marked by healthy scientific debate, particularly for the measurement of S-nitroso compounds, and eventually leads to the careful evaluation and construction of effective, reproducible methods. This has also been seen to some degree with luminol systems and select caged probes such as the bioluminescent probe
The field of caged probes, including caged luciferin and caged 1,2-dioxetanes, is largely driven by synthetic chemists. Because of this, there is often an emphasis placed on new molecular structures with brighter and red-shifted emission, sensitivity, solubility, and other molecular properties. Although these are certainly important, they often overshadow the critical need to develop rigorous quantitative methods, transparent studies of reproducibility, and cross-validation among different researchers (14). Nevertheless, given time, careful studies, and a continuation of healthy constructive debate, the prospects for using chemiluminescence for the analysis and imaging of reactive sulfur and nitrogen species in living systems are far reaching and sure to deeply impact our understanding of the roles they play in physiology and pathophysiology.
Footnotes
Acknowledgment
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.
Authors' Contributions
A.R.L. conceived the presented review. B.L., Y.L.K., and A.R.L. contributed to the writing of the article.
Author Disclosure Statement
A.R.L. declares a financial stake in BioLum Sciences, LLC. All other authors have no competing financial interests.
Funding Information
The authors acknowledge funding from the National Institute of General Medical Sciences of the National Institutes of Health under Award number R15GM114792-02 and the National Science Foundation under Award number CHE 1653474.