
Abstract
Significance:
Salt sensitivity of blood pressure (SSBP) is an independent risk factor for mortality and morbidity due to cardiovascular disease, and disproportionately affects blacks and women. Several mechanisms have been proposed, including exaggerated activation of sodium transporters in the kidney leading to salt retention and water.
Recent Advances:
Recent studies have found that in addition to the renal epithelium, myeloid immune cells can sense sodium via the epithelial Na+ channel (ENaC), which leads to activation of the nicotinamide adenine dinucleotide phosphate oxidase enzyme complex, increased fatty acid oxidation, and production of isolevuglandins (IsoLGs). IsoLGs are immunogenic and contribute to salt-induced hypertension. In addition, aldosterone-mediated activation of ENaC has been attributed to the increased SSBP in women. The goal of this review is to highlight mechanisms contributing to SSBP in blacks and women, including, but not limited to increased activation of ENaC, fatty acid oxidation, and inflammation.
Critical Issues:
A critical barrier to progress in management of SSBP is that its diagnosis is not feasible in the clinic and is limited to expensive and laborious research protocols, which makes it difficult to investigate. Yet without understanding the underlying mechanisms, this important risk factor remains without treatment.
Future Directions:
Further studies are needed to understand the mechanisms that contribute to differential blood pressure responses to dietary salt and find feasible diagnostic tools. This is extremely important and may go a long way in mitigating the racial and sex disparities in cardiovascular outcomes. Antioxid. Redox Signal. 35, 1477–1493.
Introduction
Rodents, other mammals, and most humans adapt to salt loading or salt depletion without significant changes in blood pressure (BP), and hence are called salt resistant (SR). In contrast, some members of the population exhibit increased or decreased BP with salt loading or are salt depletion, respectively, and are called salt sensitive (SS).
Salt sensitivity of blood pressure (SSBP) is a pathophysiological trait or abnormal phenotype, since SS normotensive or SS hypertensive humans have a documented increased cardiovascular morbidity and mortality, compared with their SR counterparts. Hence, in a study of 62 SS and 94 SR hypertensive subjects, classified by the presence or absence of a >10% increase in mean arterial pressure in response to a dietary salt load, SS subjects had more than double the rates of nonfatal and fatal cardiovascular events compared with SR over 7.3 years of follow-up, and SSBP was an independent risk factor for these outcomes in multivariate regression (84). In another study with much longer follow-up (27 years), 430 normotensive and 278 hypertensive subjects were initially classified into SS or SR with an inpatient protocol of rapid intravenous salt loading followed by furosemide-induced salt depletion, and SSBP was associated with an increased mortality risk ratio of 1.73. Survival curves showed the worst prognosis for the hypertensive SS subjects and the best for the normotensive SR, as expected, whereas surprisingly the survival curves for hypertensive SR and normotensive SS were intermediate and not different from each other, confirming that SSBP was a risk factor as powerful as, and independent of BP (136).
There is no specific therapy for SSBP because its mechanisms are not fully understood. Two major hypotheses have been proposed. One, derived from Guyton's physiological model of the circulation states that owing to the kidney's infinite gain for sodium excretion, an abnormality in natriuresis is indispensable for sustained salt-dependent hypertension. The other is based on observations in humans and animals, in which the normal vasodilatory response to a sodium load is lacking. This occurs immediately after a salt load that increases plasma volume and cardiac output equally in SS and SR, and therefore cannot be attributed to long-term autoregulation of organ blood flow.
Studies in rodents inbred for the phenotype, with resulting well-dichotomized SS and SR substrains, have reported abnormalities in many vasoregulatory and renal systems and in the genes for their main products (30), but a primary defect has not been established because many of these systems interact. In humans, in whom the trait is not dichotomous but is normally distributed instead, studies have relied on arbitrary classifications into SS and SR, employing different cutoffs in the magnitude of BP change in response to acute or chronic manipulation of salt balance.
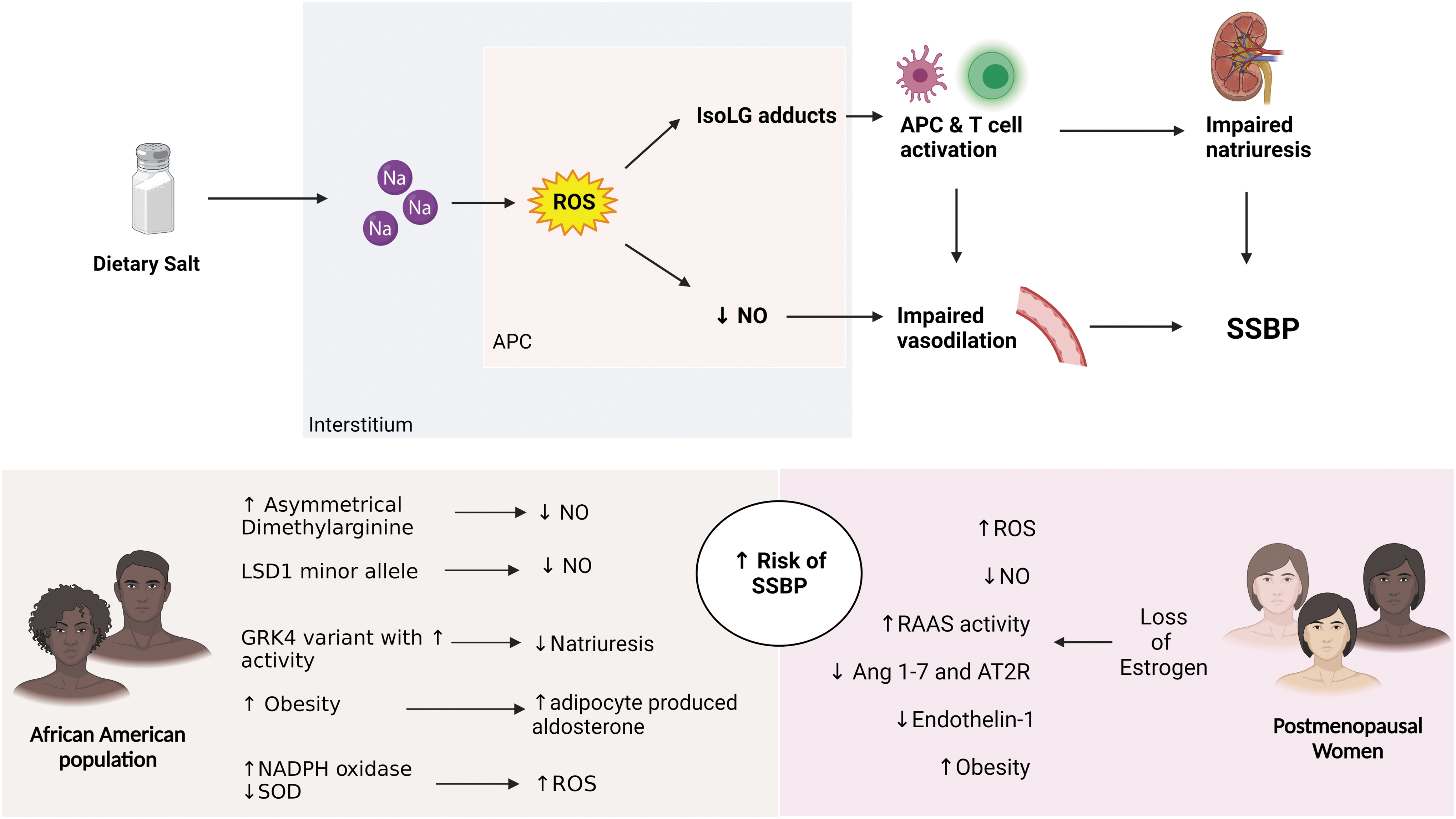
Despite these arbitrary classifications, a series of phenotypic characteristics have been described in SS humans that resemble those in pure strains of SS rodents (e.g., the Dahl-S rat). For example, their responses of vasoactive substances (e.g., renin, angiotensin, aldosterone, catecholamines, renal dopamine, endothelin, eicosanoids, natriuretic peptides, nitric oxide [NO]) and of sympathetic renal nerve activity to salt loading or depletion are blunted, and their target organ damage shows predominance of stroke, left ventricular dysfunction, and renal disease over ischemic heart disease. Also, an effect of environmental factors on the trait is strongly supported by clinical characteristics, such as increased prevalence in subjects with low birth weight (24), insulin resistance (65), obesity, reduced nocturnal BP dipping (42), chronic renal disease, and during aging. Finally, two demographic characteristics associated with increased prevalence of SSBP are self-defined African American race and female sex, particularly after menopause.
SSBP in Blacks
There is no controversy over the fact that hypertension is more prevalent (85), with disproportionate target organ damage (80) and more resistance to treatment (45) in African Americans than in non-Hispanic whites, observations that are also present in some African countries (21, 89). Whether such differences reflect environmental factors such as excess salt and fructose and reduced potassium intake, social issues such as access to health care or adherence to medication or biological and genetic factors is not known. Equally unknown are the reasons by which SSBP is much more prevalent (∼75%) in African American than in white (∼50%) hypertensives, and whether this difference contributes somehow to prognosis.
Possible inheritance of SSBP in African Americans was suggested in a study of 140 healthy adolescents, in whom the prevalence of SS assessed with a modest dietary salt load was 22% (141). There were no distinguishing features between SS and SR subjects in clinical characteristics, family history, BP, or heart rate. In contrast, the salt load increased body weight in SS but not in SR adolescents, which is consistent with observations made in hypertensive adults and attributed to a differential capacity to store Na+ in interstitial tissues.
In black SS hypertensive women, pressor responses to salt were significantly greater than in their white SS hypertensive counterparts. The distinguishing feature was increased erythrocyte concentrations of Na+ and Ca2+ with increased Na+/K+ and Ca2+/Mg2+ ratios in the former. Although this suggested differential activity of cation transporters in African Americans, this study did not find differences in the activity of the sodium pump (142).
In hemodynamic studies of normotensive SS (n = 19) and SR (n = 18) African Americans, the inability of the former to normally vasodilate in response to a dietary salt load was associated with salt-induced increase in the plasma levels of asymmetrical dimethylarginine, an effect not observed in SR. This compound interferes with the synthesis of NO from
In terms of genetic studies, G-protein receptor kinase 4 (GRK4) variants with increased activity are more common in people of African descent, and are linked to impaired stress-induced Na excretion in normotensive black men and to BP responses to salt restriction in African patients with essential hypertension, probably by diminished natriuresis owing to uncoupling of the dopamine receptor from its G protein (99). In the African American Study of Kidney Disease (AASK) and in other studies, men with GRK4 variants had adverse cardiovascular outcomes and reduced responses to beta-blockers.
A single-nucleotide polymorphism in lysine-specific demethylase-1 (LSD1), an enzyme that demethylates histones and is therefore an epigenetic regulator, was linked to SSBP in African American carriers of the minor allele in the HyperPath cohort. This association was not present in Caucasians and interacted with the increased prevalence of SSBP observed with aging. Hypertensive subjects had low-renin and aldosterone levels, and blunted renal blood flow responses to salt loading. Mice with heterozygous knockout of LSD1 also had low-renin-, low aldosterone-, and low epithelial Na+ channel (ENaC) activity-hypertension. Their hypertension was associated with increased vascular reactivity, decreased endothelium-dependent relaxation, diminished expression of endothelial nitric oxide synthase (eNOS) mRNA and protein, decreased responses to exogenous NO and reduced guanylate cyclase protein (63, 140).
Results of these physiological and genetic studies suggest that there may be pathophysiological mechanisms or gene abnormalities specific to the SSBP of African Americans. However, no unifying hypothesis to explain their excess prevalence of SSBP or the mechanisms underlying the pressor effect of salt on their BP has been formulated to date.
SSBP in Women
The prevalence of hypertension in women is less than that in men until about the age of menopause, after which this trend is reversed (38). Although prevalence of hypertension increases with aging in both sexes, such reversal suggested early on that there may be specific effects attributable to menopause. Whether excessive SSBP in women contributed to this observation was initially controversial because BP responses to salt were assessed by casual BP measurements, which overestimate the pressor effect assessed by continuous or repeated BP measurements with ambulatory monitors in women but not in men (48). However, many large studies have now confirmed that SSBP is more prevalent in women.
In the GenSalt study, 39% of Chinese adults were SS, but BP responses to low- and high-salt dietary interventions were larger in 896 women than in 1010 men (18). In 1592 subjects of the HyperPath cohort, women had 30% higher SSBP than men regardless of age or hypertension status (117). In the DASH-Sodium trial, reduction of Na+ intake produced a greater depressor response in women than in men (133). A Brazilian study of 12,813 subjects, 35–64 years old, demonstrated that the slope of the pressor effect of salt was steeper in women than in men, after adjustment for confounders and independent from having hypertension or using antihypertensive drugs (82). In a Japanese study of 3392 drug-naive normotensive subjects, a 10-mmol/day increment in Na+ excretion was associated with a +0.16 mm Hg increase in systolic BP in men versus +0.37 mm Hg in women in multivariate analyses adjusted for age, body mass index (BMI), creatinine, homeostatic model assessment index of insulin resistance, and K+ excretion (86).
A specific effect of menopause on SSBP was detected in 2212 women of the large, 25-country WHO-CARDIAC multicenter study. A positive relationship between Na+ excretion and BP, adjusted for age, BMI, and K+ excretion, was found in post- but not premenopausal women, indicating a tendency for SSBP to increase at the menopause (143). This was confirmed in a study of 40 normotensive women in whom elective hysterectomy–oophorectomy increased SSBP from 22.5% at baseline to 52.5% 4 months after surgery, without development of hypertension (114). Consistent with these observations, administration of replacement estrogen reduced SSBP in postmenopausal women (115).
Hypertensive disorders of pregnancy seem to accelerate the development of SSBP before menopause. This was documented in a study of 42 premenopausal women, in whom a history of pre-eclampsia increased high BP variability and the odds ratio for SSBP by 5.4 between 5 and 17 years after delivery (76). Similarly, premenopausal normotensive women studied ∼8 months after delivery exhibited SSBP and exaggerated BP and aldosterone responses to angiotensin II (Ang II) infusion if their pregnancy had been complicated by hypertension (112).
Many differences in vasoactive substances have been reported as possible causes for the different prevalence of hypertension and SSBP in men and women. In terms of the renin–angiotensin–aldosterone system (RAAS), premenopausal women have decreased plasma renin activity compared with men, possibly due to estrogen-induced NO generation (79). Also, women and female rats have enhancement of some depressor components of the RAAS (e.g., angiotensin 1–7 and angiotensin II type 2 receptor) (110), which may both be protective but sustain reversal after menopause (43). In contrast, SS rodents and premenopausal women who are SS may have activation of the adrenal RAAS, with excess aldosterone synthase expression and production of aldosterone, and increased expression of endothelial mineralocorticoid receptors (MCRs) leading to endothelial dysfunction, suggesting that they may benefit from MCR antagonists (33, 35). The SS of women in the HyperPath cohort was also linked to greater BP and aldosterone responses to Ang II infusion (117).
Findings in other vasoactive systems may explain protection of premenopausal women from hypertension and SSBP. Hence, estrogens stimulate endothelial NO synthase and NO generation, and whole-body production of NO in premenopausal women is 20% greater than that in men (36). Also, before menopause women have lower levels of oxidative stress owing to antioxidant effects of estrogens (106). Moreover, premenopausal women have 33% lower levels of plasma endothelin-1 (94), enhanced activity of the natriuretic endothelin B receptor, and concomitant decreased activity of the vasoconstrictor endothelin A receptor (56).
In contrast, the increase in copeptin (a marker of vasopressin release) in response to salt intake was negatively correlated with the magnitude of SS in women (not in men), suggesting that excess fluid intake and fluid retention may be associated with SSBP in women (125). Failure to normally increase the sodium pump inhibitor marinobufogenin in response to salt is observed in older women and may explain their increased prevalence of SSBP (4). Also, postmenopausal women have levels of oxidative stress similar to men, probably due to loss of estrogen beneficial effects (129) and an increase in circulating inflammatory markers, such as tumor necrosis factor α, interleukin (IL)-6, and plasminogen activator inhibitor 1 (92).
Many genetic studies have found sex-specific associations between gene polymorphisms and hypertension in women, including those in the angiotensinogen, angiotensin converting enzyme, aldosterone synthase, endothelial NO synthase, β1- and α2A-adrenergic receptor and estrogen receptor-1 genes. In contrast, very few studies have explored the genetic basis for SSBP in general and so, in women. One notable exception is the report that the minor allele of a variant at rs10144225 in the estrogen receptor-2 gene was associated with SSBP in premenopausal but not in postmenopausal women or men of the HyperPath and Mexican American Hypertension–Insulin Resistance Study cohorts (75), which is consistent with the multiple effects of estrogens described above.
New Insights on the Pathogenesis of SSBP and Their Implication for Blacks and Women
Tissue sodium regulation and immune system activation
Regulation of Na+ balance in the kidney is well studied. The traditional view that high-salt intake was followed by increased water intake, plasma volume, and cardiac output, with consequent flow autoregulatory responses leading to vasoconstriction and hypertension, has been challenged by recent observations. Actually, a high-salt diet is associated with endogenous water conservation and diminished fluid intake (97). Also, obligatory water losses in disease states may trigger antidehydration mechanisms that result in BP elevation (61). Regarding salt metabolism, recent studies showed that Na+ is stored in the interstitium of several tissues, including the skin, skeletal muscle, lung, and liver. Some studies reported that such storage is nonosmolar, by binding of Na+ to glycosaminoglycans that are polymerized by high-salt diet, increasing their negative charge density (23, 60, 108). Other studies support the view that Na+ accumulation is not hypertonic but the result of cellular rarefaction or edema in the assessed tissues (103). Machnik et al. found that skin Na+ accumulation associates with interstitial macrophage infiltration (74). Whether hypertonic or not, interstitial Na+ storage leads to increased expression of tonicity-responsive enhancer binding protein (TonEBP) by macrophages, which binds to and activates the vascular endothelial growth factor (VEGF) C gene. VEGF-C subsequently promotes lymphatic capillary network formation and increases expression of eNOS in interstitial cells, which leads to increased Na clearance from the skin (74).
Both the adaptive and innate immune systems play a major role in the pathogenesis of hypertension. Various hypertensive stimuli, including excess salt, catecholamines, angiotensin II, and aldosterone, lead to renal and vascular T cell infiltration. This infiltration along with the associated cytokine release promotes Na+ retention, vasoconstriction, increase in BP, and subsequent end-organ damage (26). Activation of T cells occurs by interaction with antigen-presenting cells (APCs), which include B cells, macrophages, and dendritic cells (DCs). Antigens are presented through the major histocompatibility complexes, which interact with the T cell receptor of T cells. Our laboratory has shown that antigen-presenting DCs have a key role in development of angiotensin II and deoxycorticosterone acetate (DOCA)-salt hypertension (55), and that inhibition of DC activation eliminated the development of hypertension in these animal models.
Others have described varied effects of Na+ on the immune system. For example, its accumulation on the site of skin infection by Leishmania in mice enhanced macrophage function (49), and its activation of T helper 17 (Th17) immunity was important for protection against mucocutaneous infections in humans (32). In contrast, high-salt diets worsened experimental autoimmune encephalomyelitis in mice via activation and polarization of Th17 cells (57), and induced monocyte migration to tissues and macrophage polarization to an inflammatory phenotype in normal human volunteers (137). These studies were the impetus for our investigation of the possible immune-mediated mechanism of salt-induced hypertension described below.
Oxidative stress and inflammation
Reactive oxygen species (ROS) play indispensable functions in normal cell signaling and other biological functions. Oxidative stress is a phenomenon caused by an imbalance between the production and accumulation of free radicals in cells and tissues, and the ability of scavenging them by biological systems referred to as antioxidants. The optimal production of ROS by immune cells during inflammation is an important mechanism of host immune defense (78, 93). However, increased production of ROS or oxidative stress leads to oxygenation of polyunsaturated fatty acids, resulting in formation of highly reactive isolevuglandin (IsoLG). IsoLGs react with lysine residues on proteins to form adducts and crosslinks, which are immunogenic. IsoLG-modified proteins are processed in DCs and presented as neoantigens, which activate inflammatory cells in hypertension.
DCs from animal models of essential hypertension and hypertensive humans show increased ROS, which results in T cell activation. In 2014, we found that DCs present immunogenic IsoLG-modified proteins that promote T cell activation and the development of hypertension (55). In 2017, we reported a novel mechanism by which excess salt intake promotes activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and formation of isoLGs, resulting in production of IL-6 and IL-1β in DCs. Importantly, our in vitro studies showed that such activation was due to the concentration of Na+ in the solution employed, not to its osmolality, since equiosmolar mannitol solutions had no effect. Therefore, the controversy on whether interstitial Na+ accumulation is hypertonic or not does not alter the significance of our results (103).
Several studies have shown that levels of Na affect oxidative stress levels and BP (10, 44, 105). Beltowski et al. showed that elevated levels of ROS caused NO deficiency resulting in the abnormal Na+ handling and leptin-induced hypertension in Wistar rats (8). A study by Caroline et al. reported that reduced Na+ levels attenuated ROS levels, prevented increased heart weight index, and reduced albuminuria levels in rats fed with low Na+ diet and Ang II injection (98). Doxorubicin binds to the reductase domain of eNOS and generates superoxide. High-salt diet increased systolic BP significantly only in doxorubicin-treated rats. Doxorubicin-treated rats exhibited increased oxidative stress, suggesting eNOS dysfunction and impaired NO production in the kidney (41). Interestingly, carbon monoxide attenuated high-salt-induced hypertension by reducing proinflammatory markers (COX2, IL-1β, IL-6, NADPH oxidase 2 and 4 [NOX2 and NOX4]) in the hypothalamic paraventricular nucleus of male Dahl SS rats (146). Fructose-rich and high-salt diet increased oxidative stress, and inflammation-mediated hypertension in Fischer male rats (28).
Also, we discovered that in mice lacking serum glucocorticoid kinase-1 in DCs SS hypertension is prevented in an NADPH oxidase-dependent manner (127). DCs possess NADPH oxidase and are like specialized macrophages, and have been shown to modify cytokine production (51). Increased ROS have also been found to activate the inflammasome.
The role of the inflammasome
Low-grade inflammation generated by both the innate and adaptive immune systems contributes to the increase of mean BP that characterizes hypertension (15, 81). Interestingly, the immune system does not require the presence of a pathogen to induce this hypertension-associated inflammation (11). Instead, innate immune activation depends on pattern recognition receptors (PRRs) that not only detect pathogen-associated molecular patterns (PAMPs) but also damage-associated molecular patterns (DAMPs). PAMPs and DAMPs are detected by the best characterized inflammasome, NLRP3 (NOD-like receptor family pyrin domain containing 3), which belongs to the nucleotide-binding oligomerization domain leucine-rich repeat (NLR) PRR family and drives sterile inflammation in multiple pathologies (17, 67, 128). NLRP3 is also important as the only inflammasome studied in hypertensive patients.
The NLRP3 inflammasome signaling platform consists of the central NOD domain, the apoptosis-associated speck-like adaptor protein (ASC) containing a C-terminal caspase recruitment domain (CARD), and procaspase-1 (Fig. 1) (147). Working in concert, these three elements come together to form the active NLRP3 assembly, which in turn stimulates the catalytic cleavage of procaspase-1 into caspase-1 (37). The final step of inflammasome activation is the cleavage of the inactive pro-IL-1β and pro-IL-18 into the mature proinflammatory cytokines IL-1β and IL-18 (17, 37, 116). Activation of the NLRP3 inflammasome consists of two steps: priming and triggering (31). Priming occurs when Toll-like receptors interact with PAMPs or DAMPs. This signals downstream to nuclear factor-kappaB (NF-κB), which in turn upregulates transcription of both NLRP3 and pro-IL-1β (122, 147). This cascade of events results in the secretion of proinflammatory cytokines IL-1β and IL-18 to the extracellular space and blood stream. These cytokines are found in elevated concentration in the serum and monocytes of hypertensive humans (27, 69, 96), and are known inducers of renal and vascular dysfunction (25). Indeed, several findings suggest that NLRP3 inflammasome may have a crucial role in the development of hypertension and SSBP.

NADPH oxidase and ROS, important proposed factors in the development of SSBP, have been identified to trigger the activation of NLRP3 inflammasomes in monocytes and macrophages (1, 8, 22), which in turn would lead to increased IL-1β and IL-18 as seen in hypertensive patients (27, 69). Hypertensive patients are also known to have elevated levels of NF-κB in tissue and inflammatory cells (25), which may indicate higher activation of NLRP3. Furthermore, the blockage of NF-κB ameliorates hypertension and prevents hypertension-induced kidney damage in rodent models of hypertension (145). The eNOS and NO pathway, which is altered in SS and hypertensive patients, has also been suggested to play an important role in the regulation of NLRP3 inflammasome (118). NLRP3-mediated endothelial cell death mediates endothelial dysfunction, a great contributor to SS hypertension (123). Indeed, high-salt intake-induced endothelial dysfunction has been shown to be partly mediated by increased NALP3 inflammasome expression in mice (39).
The role of inflammasome activation in salt sensitivity in African Americans or postmenopausal women is yet unknown, while some findings should be noted. Monocytes from pregnant women during pre-eclampsia exhibit higher gene expression of NLRP3, caspase-1, and IL-1β compared with normotensive pregnant women (77). Importantly, elevated BP was found to positively correlate with the expression of IL-18 receptor accessory protein in hypertensive African Americans but not in Caucasians (2). While their frequencies in females or different ethnicities are not known yet, several polymorphisms in NLRP3 gene have been associated with increased susceptibility to hypertension (25) and may account for susceptibility to salt sensitivity in different populations.
Given this evidence, NLRP3 stands out as a potential target in antihypertensive therapy. In a recent study of uninephrectomized mice treated with DOCA and 0.9% salt, Krishnan et al. reported that hypertension was attenuated after treatment with MCC950, an assembly inhibitor of NLRP3. They also reported reduction in total leukocytes, interferon gamma (IFN-γ)-positive T cells, and the expression of proinflammatory genes in the kidney with treatment (62). In another study of DOCA/salt/uninephrectomy-induced hypertension model, anakinra, an anti-IL-1 receptor antagonist, was shown to significantly reduce BP and renal fibrosis independent of anti-inflammatory effects (70). In another recent study, ELABELA (ELA), a 32-residue hormone peptide was found to attenuate hypertension through inhibition of the NADPH oxidase/ROS/NLRP3 inflammasome axis in DOCA/salt-treated rats. This was of particular importance since DOCA/salt treatment greatly decreased ELA expression and increased NLRP3 expression in the renal medulla (19).
While the findings from animal studies are promising, no studies in humans have yet shown a beneficial effect of NLRP3 pathway blockage on BP. In the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS), 3 months treatment with canakinumab, an anti-IL-1β antibody, effectively decreased recurrent cardiovascular events in patients with elevated high-sensitivity C-reactive protein levels (100), while no effects on BP were shown in the follow-up period of 3.7 years (104). Further human studies are required to explore the role of the inflammasome in the development of hypertension.
The significance of ENaC in sodium regulation
In the kidney, ENaC fine tunes Na+ excretion by regulating reabsorption of this cation out of the amount delivered to the distal portions of the nephron. The importance of this renal channel in BP regulation is best exemplified by Liddle Syndrome, a severe but rare form of inheritable hypertension produced by gain-of-function mutations in ENaC. Because aldosterone is a major stimulator of ENaC, the channel participates in the hypertension of primary aldosteronism and in that of subjects with severe or resistant hypertension in which inappropriate aldosterone secretion has been described, particularly in association with obesity and sleep disorders (29). In addition, ENaC hyperactivity seems to be aldosterone independent in some severe hypertensive patients, particularly African Americans in the “stroke belt” of the southern United States. This is suggested by the fact that antihypertensive responses to amiloride were significant in these subjects, whereas those to spironolactone were of lesser magnitude in one series (107) or absent in another one (66). The mechanism for this ENaC-dependent, aldosterone-independent hypertension is unclear, but may relate to lack of normally negative regulation of the channel by epoxyeicosatrienoic acids, as suggested by some genetic studies (66), MR activation by the small GTPase Rac1 (5), changes in the regulation of ENaC by other factors (e.g., proteases), or by ENaC activation due to variants in the genes encoding its subunits (58, 87).
Although the channel was named for its discovery in epithelial cells, it is now known to be expressed in both epithelial and nonepithelial tissues. ENaC's expression in extrarenal organs influences BP. For example, endothelial ENaC is a shear sensor that leads to hypertension when the α-subunit is overexpressed in mouse endothelial cells (59). In contrast, systolic BP was unchanged when SCNN1A, the gene encoding the α-subunit, was selectively deleted in endothelial cells (121). ENaCs expression increases endothelial stiffness in vitro and in a mouse model of Liddle syndrome (50). The effect is mediated by ENaC-induced decreased NO via the PI3K/Akt signaling pathway, with resulting endothelial dysfunction and impaired vasorelaxation that can be reversed with amiloride (135). However, the effects of ENaC on NO production in endothelia are not consistent. For example, inhibition of ENaC in mesenteric arteries reduced NO levels induced by acetylcholine (124). Endothelial stiffness produced by activation of endothelial MCR requires ENaC (34). Normal regulation of endothelial ENaC in Sprague Dawley rats includes reduced expression and activity in response to high-salt diet with consequent compensatory vasorelaxation (71). This is disrupted in Dahl-S rats, in which the activity of the channel is increased despite high-salt intake, with increased endothelial stiffness and blunted salt-induced vasodilation, analogous to hemodynamic observations in men (64).
In the rat brain, the three ENaC subunits are expressed in many cardiovascular regulatory centers in variable combinations, suggesting that there is a heterogeneous population of these channels. They are involved in modulation of sympathoexcitation in response to salt (3) and in aldosterone-stimulated hypothalamic release of vasopressin (83). In a model of Liddle syndrome in mice, produced by knockout of Nedd4-2 with resulting increased expression of ENaC in the choroid plexus and brain nuclei, pressor responses to dietary salt and to Na+ injected into the CSF are enhanced, which can be blocked with benzamil (68). An indirect effect on brain BP regulation is exerted by ENaC channels located in the renal pelvis. When stimulated by urine hyperosmolality, these channels signal via renal nerve afferents, resulting in activation of forebrain and brainstem neurons such as those in the nucleus tractus solitarius and hypothalamic paraventricular and supraoptic nuclei (40).
Finally, ENaC expressed in tastebuds regulates salt taste in experimental hypertension (109), and ENaC expressed in intestinal cells mediates mineralocorticoid regulation of intestinal Na+ absorption. The latter participates in BP regulation, as demonstrated in mice with intestinal-epithelial-specific knockout of the MCR, which exhibit diminished colonic expression of ENaC, fecal Na+ losses compensated by diminished urinary Na+ excretion and lower BP (88).
ENaC, oxidative stress, and immune cell activation
ENaC is expressed in DCs and may have a role in linking a high Na+ diet, inflammation, and hypertension (9). Rodents fed a high Na+ diet accumulate Na+ in the interstitium, with concentrations reaching 190 mM in the skin although plasma [Na+] is unchanged (74, 126, 139). DCs respond to increases in extracellular Na+ in an ENaC-dependent manner, resulting in Ca2+ influx and activation of a signaling pathway that includes protein kinase C and NADPH oxidase, superoxide production, and formation of immunogenic IsoLG-protein adducts (Fig. 2). This leads to T cell activation, release of inflammatory cytokines (IL-17 and IFN-γ), and salt-dependent increase in BP (9). Other studies have shown that a high-salt diet polarizes T cells and macrophages to an inflammatory phenotype (26). However, the mechanisms contributing to variability in the BP response to elevated Na+ are not clearly understood. As shown in Figure 3, we propose that the variability in monocyte Na+ sensing leads to variable IsoLG formation, T cell activation, and infiltration into vasculature and the kidney, leading to variability in SSBP. It is conceivable that circulating monocytes transmigrate into regions of elevated interstitial Na+, including the skin, muscle, and kidney, and are activated via IsoLG adduct formation leading to SSBP.

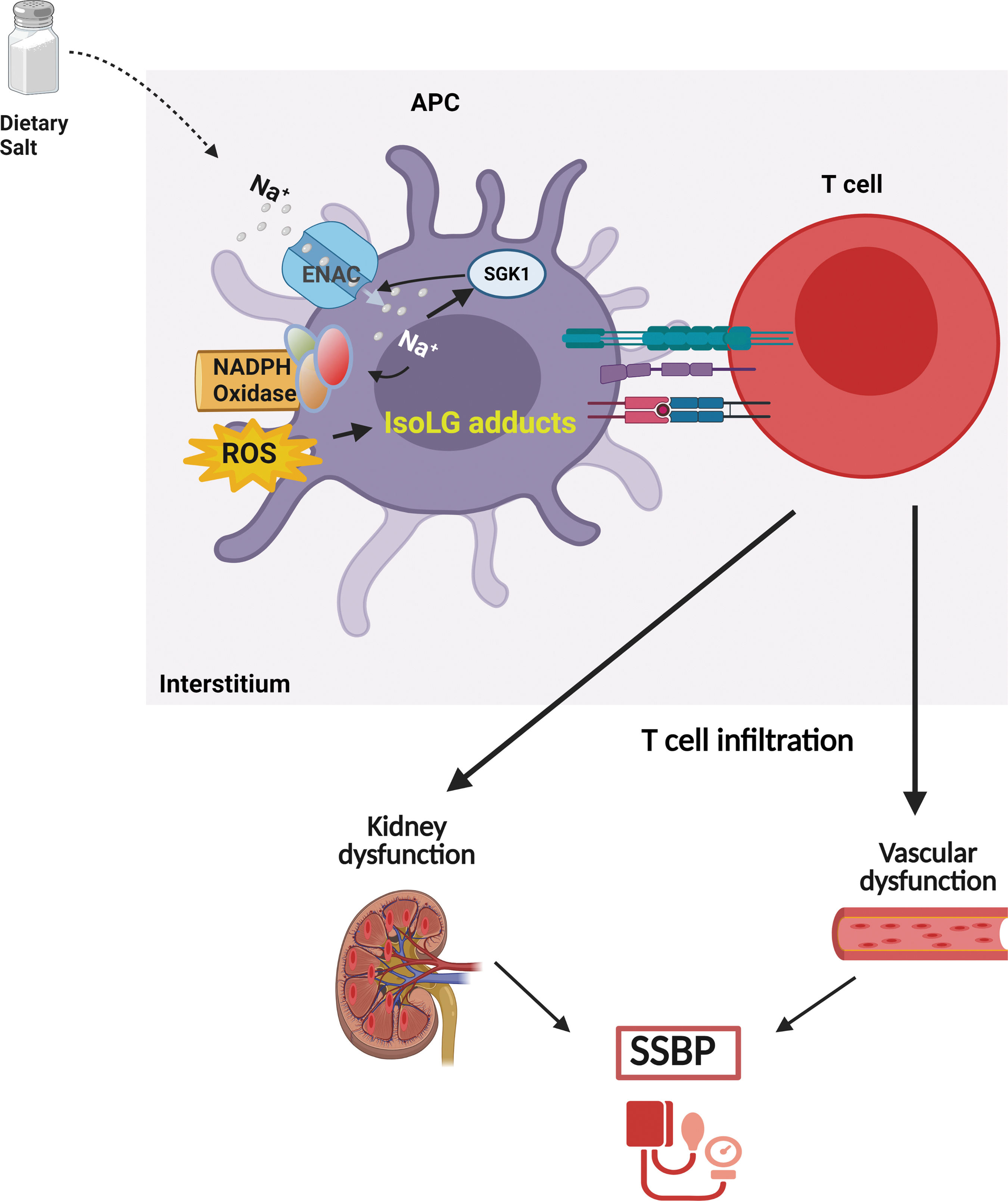
ENaC regulation, oxidative stress, and SSBP in blacks and women
Sodium and water handling are established key regulators of BP control. Although the mechanisms are not fully understood, dysregulation in Na+ transport may contribute to the pathogenesis of SSBP. ENaC expressed from the late distal collecting tubule through the connecting segment and cortical collecting duct is composed of α, β, and γ subunits. Several ENaC β-subunit nonsynonymous variants associated with a hypertensive phenotype have been identified in blacks, including βT549M and βR563Q (6, 52). Individuals with the βT549M exhibited a significant lowering of BP with the ENaC inhibitor amiloride (7).
Aldosterone plays a major role in regulating ENaC expression and activity. Aldosterone: renin ratio is an emerging potential prognostic tool for SS hypertension, and African Americans have elevated levels (111). Reports indicate a significant association of aldosterone: renin ratio in blood with high-salt intake, suggesting that insufficiently suppressed aldosterone may contribute to salt sensitivity in African Americans (46).
Sex discrepancies in aldosterone production may also play a role in SSBP in females. Female Balb/C mice on a high-salt diet also exhibit higher aldosterone: plasma renin activity ratio potentially mediated through aldosterone mechanisms involving impaired endothelium-dependent relaxation, effects that are not observed in males (35). Analysis of the hypertensive Pathotype cohort revealed that Ang II infusion resulted in significantly higher aldosterone release in women regardless of the salt intake. The greater aldosterone response corresponded to higher BP increase in women compared with men only in the hypertensive subgroup (117). Pharmacological blockade of aldosterone effects using spironolactone in hypertensive African American women controlled BP better than in African American women who were on different antihypertensive regimens, and African American men and white women who also received spironolactone (20). Spironolactone treatment in obese female mice prevented the development of diastolic dysfunction (12).
Obesity is a strong risk factor for hypertension and disproportionally affects women more than men (16, 134), with central obesity affecting ∼60% of women in the United States compared with 38% of men (134). Obesity is even more prevalent in black women compared with white women, with >70% of black women affected (134). Importantly, this racial divergence can be observed as early as 12 years of age (14, 54). Adipocytes produce aldosterone, which in people with increased adiposity like African Americans and women may further contribute to increased susceptibility to salt sensitivity (13). Chronic leptin receptor antagonism reduces aldosterone levels BP, and improves endothelial function in female mice supporting a reported association between leptin- and aldosterone-mediated mechanisms of hypertension (47, 102).
Premenopausal women exhibit cardioprotection against diseases such as hypertension implicating the role of sex steroids. Testosterone increases renal α-ENaC expression in men and male rodents (95). In females, however, testosterone and estradiol decrease the expression of α-ENaC (53). This androgen-dependent β-ENaC expression suggests that postmenopausal state may be associated with increased ENaC activity in women, which needs further investigation.
Studies in rodents suggest greater activation of ENaC in females relative to males evidenced by higher expression of active forms of ENaC subunits (130). Although female rats have higher renal expression levels of the cleaved, or active forms of α- and γ-ENaC subunits, males may have a more active sodium transport through mechanisms independent of renal expression levels of the channel and sex hormones warranting more studies to uncover these mechanisms (119).
Blacks and women are also at a heightened risk of increased oxidative stress (15, 33), the major driver of IsoLG formation. In a study using human umbilical vein endothelial cells (HUVECs), Feairheller et al. reported that compared with Caucasian HUVECs, African American HUVECs exhibited increased expression of NADPH oxidase, IL-6, and lower superoxide dismutase activity (33). Furthermore, Gardner et al. showed that African American women with symptomatic peripheral artery disease exhibit exaggerated oxidative status and heightened proinflammatory profile of circulating biomarkers compared with men (37). In addition, aldosterone-mediated activation of ENaC has been deemed responsible for the increased SSBP in women (33).
New Therapeutic Strategies for SS Hypertension
Hypertension, including Ang II-induced but also DOCA-salt and norepinephrine-induced hypertension, is dependent on T cell activation (91). Treatment with ACE inhibitors and angiotensin receptor blockers, while benefiting Ang II-induced hypertension, might not affect other forms of hypertension. The recent paradigm shift in our understanding of SSBP and the interplay between salt, immune response, and hypertension (Fig. 4) can provide new therapeutic approaches for the treatment of SS hypertension specifically, which affects up to 50% of all hypertensive patients (127). Several steps in the proposed mechanism of SSBP can be targeted, including APC ENaC channels, ROS, IsoLG adduct formation, and inflammasome activation (Fig. 5).
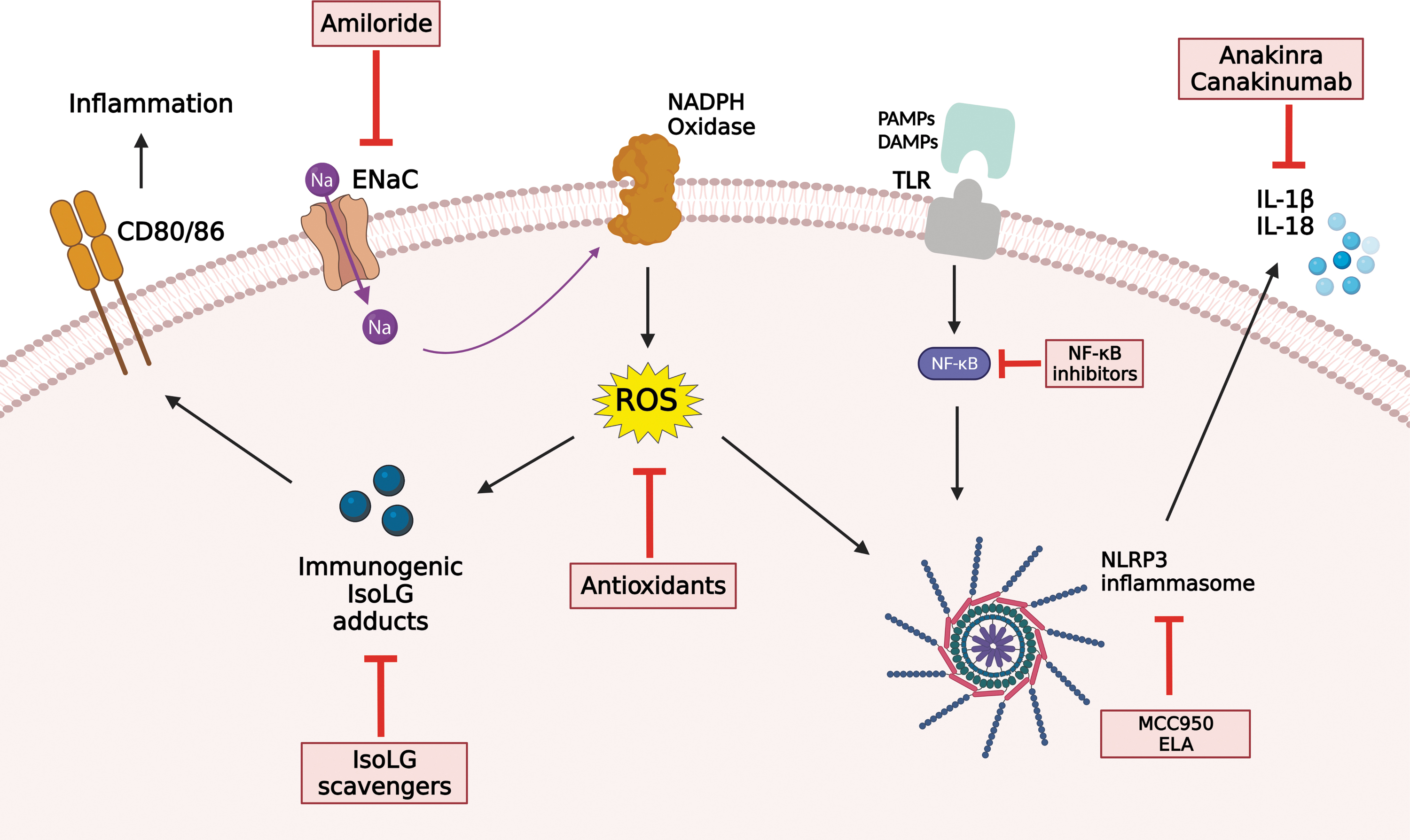
Oxidative stress contributes to inflammation and hypertension, but antioxidants have proven very ineffective in large clinical trials in the treatment of hypertension (26, 41, 72, 73, 98, 120, 131, 132, 144). This might be in part because their rate constants (∼104 M/s) are far below the rate constant of reactions of free radicals with endogenous antioxidants (109–1010 M/s). One thousand six hundred international units of vitamin E is required to suppress isoprostane levels (the precursor of IsoLG) in humans (101). These very high doses have adverse effects in several clinical trials and therefore should not be used (138). Thus, a more targeted approach including IsoLG scavenging or targeting ENaC may provide or serve a better therapeutic approach, and modify the increased cardiovascular risks found in SSBP. Using multiple murine models of hypertension, we found that pretreatment with IsoLG scavengers including 2-hydroxybenzylamine and pentylpyridoxamine prevents activation of APCs and development of experimental hypertension (55). Although amiloride, an ENaC inhibitor, is a well-recognized add-on treatment for hypertension owing to its diuretic function in the renal tubules (90), its utility specifically for SSBP as a monotherapy should be investigated. Thus, further studies are needed to determine if targeting ENaC or IsoLG can serve as a potential therapeutic strategy for salt sensitivity, specifically in blacks and women.
Perspectives
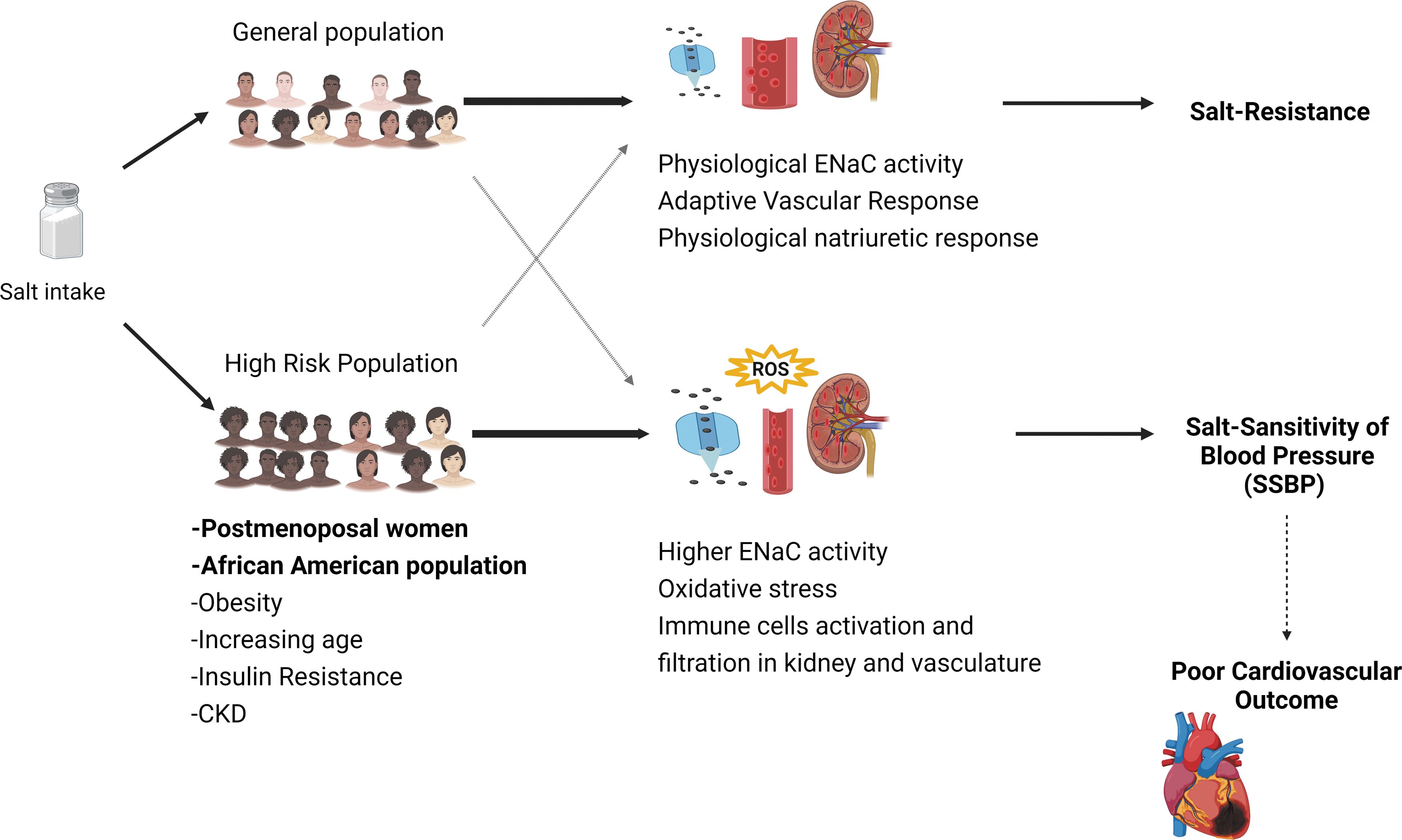
SSBP is a likely culprit of the health disparities existing in terms of cardiovascular disease morbidity and mortality in different populations. SSBP disproportionately affects blacks and women, but the unifying mechanisms are not fully understood (Fig. 6). While social determinants including poverty, which contributes to lack of medication adherence, low birth weight, lack of access to healthy food, transportation, education, access to health care, health insurance, and other barriers due to systemic racism, may play a role, there are inherent genetic factors that may contribute to SSBP in blacks and women. Excess dietary salt intake is a known environmental factor contributing to adverse cardiovascular outcomes, especially in blacks. The role of tissue sodium, oxidative stress, and immune activation in the predisposition for salt sensitivity in blacks and women should be investigated to provide effective therapeutic approaches specific to these high-risk groups. Emerging evidence suggests that targeting downstream pathways impacted by excess sodium such as IsoLG scavenging or ENaC blockade may provide therapeutic approaches to address health inequalities that are due to SSBP.
Footnotes
Authors' Contributions
M.S., F.E., L.A.E., J.I., A.P., M.S., N.M., T.R.K., C.L.L., and A.K. wrote the draft, revised and approved the article. T.R.K. and A.K. obtained funding for the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Institutes of Health grants K01HL130497 (Annet Kirabo), R03HL155041 to Annet Kirabo, R01HL144941 (Annet Kirabo), and R01HL147818 (Thomas R. Kleyman and Annet Kirabo).