
Abstract
Aims:
The objective of this study is to identify mechanisms for adipose stromal vascular fraction's (SVF) restorative effects on vasodilation in aging-induced coronary microvascular disease (CMD). We hypothesize that reactive oxygen species (ROS) diminish β1-adrenergic receptor (β1ADR)- and flow-mediated dilation (FMD) in coronary arterioles, reversible by SVF and adipose-derived stem cells (ADSCs).
Results:
SVF attenuates aging-induced chronic accumulation of ROS and pro-oxidant gene and protein expression with enhancement of antioxidant gene and protein expression and glutathione, but not nitric oxide. ADSCs attenuate hydrogen peroxide while restoring nitric oxide and glutathione. Mass spectrometry of SVF- and ADSC-conditioned media reveals abundant antioxidant proteins suggesting a paracrine mechanism. FMD and β1ADR-mediated dilation diminished with aging, restored with SVF and ADSCs. FMD was restored by a switch in the acute signaling mediator from hydrogen peroxide in aging to peroxynitrite with SVF and ADSCs. Vasorelaxation to β1ADR-agonism was mechanistically linked with hydrogen peroxide, nitric oxide, and glutathione. Exogenous ROS eliminates isoproterenol-mediated dilation in youth that is blocked by inhibition of pro-desensitization and internalization proteins while nitric oxide enhances isoproterenol-mediated dilation in aging.
Innovation:
We introduce a novel mechanism by which ROS impacts β1ADR trafficking: the ROS/RNS-β1ADR desensitization and internalization axis. Aging-induced ROS shunts β1ADR from the plasma membrane into endosomes. SVF reduces oxidative burden, restoring functional β1ADR.
Conclusions:
SVF (and ADSCs to a lesser extent) reduce oxidative stress, and restore flow- and β1ADR-mediated vasodilation in aging. SVF represents a promising therapeutic strategy for CMD by addressing root cause of pathology; that is, oxidative stress-mediated hyperconstriction. Antioxid. Redox Signal. 38, 261–281.
Introduction
Coronary microvascular disease (CMD) presents in postmenopausal, aging women with chronic angina due to microvascular hyperconstriction, as opposed to atherosclerotic blockage seen typically in men (Anderson et al, 2019). Clinically, CMD is defined as a coronary flow reserve (CFR) ≤2.5, vasoconstriction to acetylcholine, and <20% coronary dilation to nitroglycerin (Anderson et al, 2019). Coronary perfusion is compromised by ∼43% in advanced age with a negative correlation between advancing age and myocardial flow reserve, exacerbating CMD severity (Czernin et al, 1993; Hachamovitch et al, 1989).
Beta blockers may be utilized to preserve oxygen consumption by reducing the workload of the myocardium but may inhibit vascular dilative beta-adrenergic receptor (βADR) function (Colucci, 1989). Conversely, beta agonists would lead to desensitization over time and would increase inotropic myocardial workload (Colucci, 1989). Although nitrates may provide acute relief of symptoms, there is little evidence of reduced major adverse cardiac events or mortality; nitrates also cause rebound angina and oxidative stress (Gori and Parker, 2004; Takahashi et al, 2015).
Innovation
Current therapeutic options for coronary microvascular disease are of limited effectiveness; however, stromal vascular fraction (SVF) and adipose-derived stem cell (ADSC) therapy in a female rodent model reverses hyperconstriction by restoring effectiveness of flow-mediated and beta 1 adrenergic receptor (β1ADR)-mediated dilation. The mechanism of action of SVF and ADSC is to reduce chronic oxidative stress, which restores β1ADR function according to our novel conceptual reactive oxygen species/reactive nitrogen species-β1ADR desensitization and internalization axis (Fig. 1). SVF and ADSCs also facilitate an acute substrate switch in response to intraluminal flow from proatherogenic hydrogen peroxide-mediated, flow-mediated dilation (FMD) in aging to peroxynitrite-mediated FMD. This switch of FMD mediator is associated with FMD restored to levels seen in young control animals.

A novel strategy that holistically treats each facet of CMD pathophysiology, rather than merely treating the symptomatic consequences, is of significant clinical interest. Adipose-derived stromal vascular fraction (SVF) represents a heterogeneous cell population with regenerative potential, including mesenchymal and hematopoietic stem cells, endothelial cells, pericytes, fibroblasts, B and T cells, natural killer cells, dendritic cells, and macrophages (Alexander, 2016; Kelm et al, 2018; Morris et al, 2015; Rowe et al, 2019, 2022a).
We have previously shown that injected SVF incorporates into carotid arteries and aorta, and to a lesser extent the myocardium, lung, and brain (Kelm et al, 2018; Rowe et al, 2019). SVF reverses aging-mediated reduction in CFR and perfusion, improves maximal cardiac output 1-week postinjection, and reverses markers of diastolic dysfunction typical of aging (Kelm et al, 2018; Rowe et al, 2019).
One question our previous studies raise is which cellular population of SVF, in particular, is responsible for its regenerative effects (or alternatively whether synergy between cells within whole SVF is required)? Therefore, we will also compare the regenerative potential of the SVF cellular subpopulation of adipose-derived stem cells (ADSCs) with whole SVF in microvascular dysfunction. One possible mechanism for the regenerative effects of SVF or ADSCs could be due to antioxidant effects as has been shown in other settings via paracrine influences (Hogan et al, 2019; Hong et al, 2019; Lin et al, 2016; Liu et al, 2019; Teng et al, 2015).
A cornerstone aspect of vascular aging is increased oxidative stress (Donato et al, 2007). We recently reviewed how aging-associated oxidative stress impacts mitochondrial dynamics, flow-mediated dilation (FMD), and βADR function (Tracy et al, 2021). There is a known correlation between aging-associated oxidative stress and decline in vasodilation (Donato et al, 2007; Gurovich et al, 2014; Schutzer and Mader, 2003; Yurdakul et al, 2008). Therefore, targeting oxidative stress in age-related CMD may provide new therapeutic avenues to restore vascular function. In this study, we examine the interplays of oxidative stress in aging and two mechanisms for maintenance of microvascular patency: FMD and β-adrenergic-mediated dilation.
Increasing intraluminal flow causes shear stress that in youth activates endothelial nitric oxide synthase (eNOS) to produce nitric oxide, leading to vasorelaxation (Garland and Dora, 2017). During aging, this instead leads to production of hydrogen peroxide resulting in hyperpolarization-mediated vasorelaxation (Beyer et al, 2017). Overall, it is thought that the hyperpolarization-mediated pathways in aging are less efficient than nitric oxide-mediated during youth. Indeed, reductions in FMD are correlated with advancing age (Celermajer et al, 1994). Therefore, therapeutically restoring nitric oxide bioavailability in aging vessels may restore effective FMD signaling.
We previously showed decreased vasodilative β1ADR protein in aged coronary microvessels, but not β2-3ADR or constrictive alpha 1-2 adrenergic receptor (α1-2ADR) (Rowe et al, 2019, 2022b). Furthermore, β1ADR density is rescued to young control (YC) levels by the SVF therapy in aging female rats (Rowe et al, 2022b). Aging leads to catecholamine overload, with compensatory receptor desensitization [mediated by G-protein receptor kinase 2 (GRK2)] and internalization (mediated by beta-arrestin and dynamin) into endosomes for storage, eventual degradation, or recycling back to the membrane. We have recently shown an age-related increase in myocardial and coronary microvascular protein expression of GRK2 (Rowe et al, 2022b). SVF does not reduce expression of prodesensitization and internalization proteins, yet dilative β1ADR function is restored.
Regulatory post-translational modification may also be in play, as S-nitrosylation (SNO) sterically inhibits GRK2 to block desensitization (Vasudevan et al, 2011a). It is thought that thiol oxidation by reactive oxygen species (ROS) of the βADR facilitates desensitization and/or internalization (Guo et al, 2010; Lewis et al, 2005a, 2005b). ROS also activate phosphatidylinositol 3-kinase gamma (PI3kγ) signaling, causing inhibition of protein phosphatase 2A (PP2A)-mediated recycling of the βADR back to the plasma membrane (Vasudevan et al, 2011a). Therefore, it may be possible to influence βADR function by modulating oxidative stress and nitrosative potential.
Frame et al. (2018) found that nitrosative exposure can “uncover” β2ADR pools that increase isoproterenol potency (although with reduced efficacy) through GRK2 nitrosylation and inhibition, leaving functional β2ADR at the plasma membrane. The role of redox dysfunction in aging coronary microvascular β1ADR-mediated dilation and FMD with therapeutic potential of cell-based antioxidative strategies has yet to be elucidated.
We hypothesized that FMD and β1ADR-mediated dilation are reduced with aging, reversed by SVF and ADSC therapy through a paracrine mechanism to attenuate oxidative stress and restore protective nitrosative signaling. We predict FMD will be restored in aging with SVF via rejuvenated nitric oxide production (and reduced ROS that siphons nitric oxide). Reduced ROS and enhanced nitrosative signaling will also alleviate β1ADR desensitization and internalization according to our conceptually novel ROS/RNS β1ADR Desensitization & Internalization Axis (Fig. 1). The ability to therapeutically modulate vasodilative mechanisms through manipulating redox dynamics as shown in this study will provide exciting new avenues for addressing management of diseases such as CMD.
Results
Animal characteristics
Animal groups and methodology are illustrated in Figure 2. There was a significant increase in body weight and total ventricular weight between YC versus old control (OC), old treated with SVF (OSVF), and old treated with ADSCs (p < 0.001 for all) (Table 1). Left ventricular weight increased significantly between YC and OC, OSVF, and ADSC (p ≤ 0.001, <0.001, <0.001), and was reduced between OSVF therapy and OC and ADSC therapy (p = 0.047, 0.05). There was a significant increase in septum weight between ADSC and OSVF (p = 0.006), and nearly significant between YC and OSVF (p = 0.067).
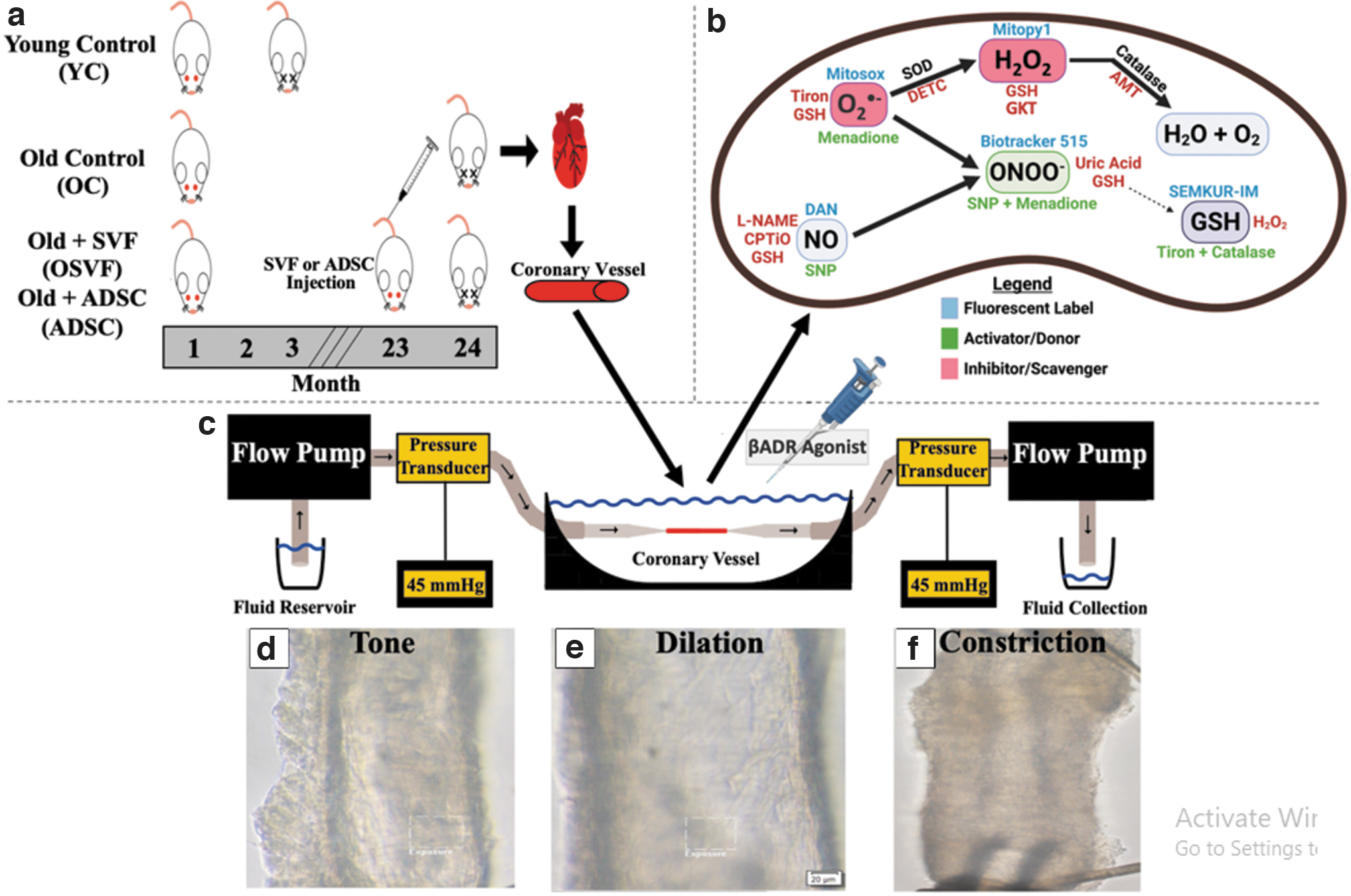
Rat Body, Cardiac and Vascular Characteristics
Significance set as p < 0.05 (*YC vs. OC, # YC vs. OSVF, $YC vs. ADSC, ^OC vs. OSVF, †OSVF vs. ADSC) via one-way ANOVA with the Holm–Sidak post hoc analysis. Echocardiographic measurements were procured on ADSC rats, and compared with historic data of echocardiographic analysis of YC, OC, and OSVF rats as previously published (Rowe et al, 2019).
ADSC, adipose-derived stem cell; OC, old control; OSVF, old + stromal vascular fraction; YC, young control.
There was no change in right ventricular weight, systolic or diastolic blood pressure, vessel wall thickness, maximal diameter, or percentage resting tone. Compared with historic data (Rowe et al, 2019), there was no recovery in echocardiographic parameters of ADSCs compared with OC. OSVF reversed the significant aging-mediated attenuation of CFR (YC vs. OC p = 0.047; OSVF vs. OC p = 0.005), isovolumic relaxation time (IVRT) (YC vs. OC p < 0.001; OSVF vs. OC p = 0.025), and E/A (YC vs. OC p < 0.001; OSVF vs. OC p = 0.001). CFR was significantly different between ADSC and OSVF (p = 0.009), but not YC (p = 0.069). Similarly, IVRT (p = 0.008), E/A (p < 0.001), and E/e’ (p = 0.002) were significantly different between ADSC and YC with E/A (p = 0.007) also being significantly different between ADSC and OSVF.
SVF reduces aging-associated oxidative stress
An illustrative guide for fluorescent dyes, inhibitors/scavengers, and activators of key ROS/RNS is shown in Figure 2b. There were significant increases in coronary microvascular hydrogen peroxide and superoxide in coronary arterioles from aged (p < 0.001, 0.048) vs. YC at baseline (Fig. 3a, b). Hydrogen peroxide and superoxide mean fluorescence intensity (MFI) were all decreased in OSVF compared with OC (p < 0.001, p = 0.002). ADSCs only reduced hydrogen peroxide (p = 0.004) with superoxide MFI being significantly greater in ADSC versus OSVF (p = 0.018).

There were decreases in nitric oxide (p = 0.006) and glutathione (p < 0.001) MFI between youth and aging. ADSC enhanced nitric oxide compared with all groups (p < 0.001) and glutathione versus OC (p < 0.001). SVF therapy was unable to significantly increase nitric oxide MFI, but nearly significantly increased glutathione MFI compared with OC (p = 0.057), although not fully restored to YC levels (p = 0.046 vs. YC). There was a nonsignificant increase in peroxynitrite with aging versus YC and OSVF; however, ADSC dramatically enhanced peroxynitrite MFI versus all groups (p < 0.001 for all).
Plasma thiobarbituric acid reactive substances (TBARS) assay revealed a nonsignificant increase in malondialdehyde in aging (21.64 ± 1.30, n = 5) and ADSC (22.40 ± 0.52, n = 8) versus YC (20 ± 2.07, n = 5) and OSVF (20.09 ± 0.14, n = 5). Validity of the fluorescent dyes was confirmed with appropriate positive and negative controls (Supplementary Fig. S1a–f). Mytopy1, Biotracker 515, Mitosox, 2,3-diaminonaphthalene (DAN), and SEMKUR-IM MFI were appropriately increased by exogenous hydrogen peroxide, cobalt chloride (CoCl2), SNP, menadione, and tiron + catalase, respectively.
Mytopy1, Biotracker 515, Mitosox, DAN, and SEMKUR-IM MFI were appropriately decreased by PEG-catalase, tiron, 2-4-carboxyphenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTiO), uric acid, GKT136901, and hydrogen peroxide, respectively. Repeatability was confirmed via linear regression and Bland–Altman analysis with blinded inter- and intraobserver reanalysis of ∼15% of the data (Supplementary Fig. S2).
To elucidate the redox environments between youth, aging, and OSVF, we utilized RNA sequencing to study transcriptional expression and fluorescence staining to extrapolate protein expression of various antioxidant and pro-oxidant enzymes (Fig. 4a–c). Among pro-oxidant protein gene expression, xanthine dehydrogenase was increased by 21% in OC versus YC and 28% in SVF versus YC (p = 0.042 and p = 0.006). Peroxisome proliferator-activated receptor gamma (coactivator-related), known to increase oxidative stress, was increased by 22% in OC versus YC (p = 0.042). SHC adaptor protein 1 expression was 18.9% greater in OC versus YC (p = 0.018). Monoamine oxidase A was increased by 27% in YC versus OC (p = 0.01) and 38.4% versus OSVF (p = 0.019).

Among antioxidant protein gene expression, there was significant increase in catalase by 19% (p = 0.049), (extracellular) superoxide dismutase 3 (SOD3) by 22% (p = 0.007), thioredoxin reductase 1 by 23% (p = 0.014), and glutathione-S-transferase mu 1 by 24% (p = 0.019) and pi 1 by 31% (p = 0.025) in OC versus YC. Antioxidant protein gene expression increases in YC versus OC included 26% more oxidation resistance protein 1 (p = 0.035), 40% more COX20 assembly factor (p = 0.002), and 15% and 21% more heat shock proteins 90 and 110 (p = 0.037 and p = 0.034). For OSVF versus YC antioxidant enzymes, catalase was increased by 19% (approaching significance, p = 0.057), SOD3 by 20% (p = 0.012), glutathione peroxidase 3 and 4 by 24% and 18% (p = 0.016 and p = 0.024), sirtuin 5 by 20% (p = 0.038), thioredoxin reductase 1 by 21% (p = 0.034), although with 39% less peroxiredoxin 4 (p = 0.015) and 40% decreased heat shock protein 110 (p = 0.003).
Comparing antioxidant enzyme gene expression between OSVF versus OC, glutathione synthetase was increased by 28% (p = 0.018), thioredoxin-like protein 1 increased by 17.2% (p = 0.045), heat shock proteins 40 (member C7) and 70 increased by 21.4% and 22% (p = 0.01 and p = 0.07), selenophosphate synthase 1 increased by 24.5% (p = 0.023), and cytochrome oxidase assembly factor 3 increased by 21% (p = 0.032) in OSVF. A comprehensive listing of antioxidant and pro-oxidant transcriptional differences is illustrated in Figure 4a and Supplementary Table S1.
We selected proteins of potential relevance to evaluate protein expressional differences via immunofluorescence of isolated coronary microvessels (Fig. 4b, c). Antioxidant proteins catalase, glutathione synthetase, and heat shock protein 70 yielded no differences in MFI, although they were slightly elevated in the ADSC group. SOD2 MFI was nonsignificantly reduced with aging but was significantly increased with SVF therapy (p = 0.035) with no differences between groups for SOD1 or SOD3.
Pro-oxidant enzyme NOX4 was increased with aging (p = 0.019) with reversal to YC levels by SVF (p = 0.03) and ADSC (p = 0.009) with no differences in NOX2. General protein SNO was reduced with aging (p = 0.051) but was not restored with SVF (p = 0.006 vs. YC) with near significance between OSVF and ADSC (p = 0.053). Nitrotyrosine MFI was enhanced with aging (p = 0.025), reversed by OSVF (p = 0.004). ADSC was unable to attenuate nitrotyrosine levels (p = 0.001 vs. YC, p < 0.001 vs. OSVF).
Mass spectrometry of cultured SVF- and ADSC-conditioned media
Mass spectrometry analysis of SVF- and ADSC-conditioned media revealed 219 individual proteins, 30 of which were relevant for redox balance (20 with antioxidant effects, 6 with pro-oxidant effects, and 4 with mixed effects) (Fig. 5). Classic antioxidant enzymes such as glutathione peroxidase 3, glutathione-S-transferase (P and alpha 3), SOD3, thioredoxin, and peroxiredoxin 4 and 6 were present. There was significantly or nearly significantly greater secretion in SVF versus ADSC of decorin (p < 0.001), apolipoprotein E (p < 0.001), angiotensinogen (p < 0.001), serotransferrin (p = 0.026), metallothionein-2 (p = 0.014), carbonic anhydrase (p = 0.005), prosaposin (p = 0.001), and protein disulfide-isomerase (p = 0.093), ADSCs secreted more metalloproteinase inhibitor 1 (p = 0.041), latent-transforming growth factor beta-binding protein 1 (p = 0.044), and nucleoside diphosphate kinase B (p = 0.1).

Alterations of FMD signaling throughout the life span and with SVF
Introduction of intraluminal flow into isolated coronary microvessels in pressure myography experiments elicited different signaling responses to induce FMD between YC, OC, OSVF, and ADSC. In youth, flow elicited a significant increase in nitric oxide MFI at 10 and 25 μL/min (YC vs. OC: p = 0.01 and p = 0.025; YC vs. OSVF: p = 0.078, 0.026; YC vs. ADSC: p = 0.025, 0.012) (Fig. 6a). There was minimal change in nitric oxide MFI with flow in OC, OSVF, and ADSC.

In all groups, flow induced an increase in hydrogen peroxide, but the increase was greatest in the OC group with significance at 25 μL/min (OC vs. YC: p = 0.022; OC vs. OSVF: p = 0.046; OC vs. ADSC: p < 0.001) (Fig. 6b). There was also significantly less production of hydrogen peroxide in ADSC versus YC and OSVF (p = 0.048, 0.012).
Inhibition of catalase with 3-amino-1,2,4-triazole (AMT) without flow led to greater hydrogen peroxide MFI increases in OSVF versus YC, OC, and ADSC (p = 0.006, < 0.01, 0.012) (Fig. 6b). With AMT + flow at 10 and 25 μL/min, OSVF hydrogen peroxide MFI was significantly greater than OC (p < 0.001, < 0.001), YC (p = 0.033, 0.004), and ADSC (p = 0.018, 0.004). At AMT +25 μL/min, OC was nearly significantly reduced compared with YC (p = 0.084), and ADSC was nearly significantly increased (p = 0.1). These results with AMT suggest significantly greater catalase function in OSVF versus YC and OC.
Superoxide MFI decreased with intraluminal flow in all groups except ADSC, which exhibited a minor increase in MFI (p = 0.035, 0.027 vs. YC; p = 0.011, 0.009 vs. OC at 10 and 25 μL/min) (Fig. 6c). Inhibition of superoxide dismutase with DETC led to significantly greater superoxide accumulation in YC and OSVF groups compared with OC at baseline (p = 0.023, 0.006).
With flow + DETC, OC exhibited less superoxide production at flow rates of 10 (p = 0.009 vs. YC; p < 0.001 vs. OSVF) and 25 (p = 0.013 vs. YC; p = 0.04 vs. OSVF) μL/min, suggesting greater SOD expression and/or function in youth than aging, and that SOD function was restored with SVF therapy. In ADSC, DETC +10 μL/min was nearly significant from OSVF (p = 0.061). Peroxynitrite MFI was increased with intraluminal flow at 10 and 25 μL/min in YC, OSVF, and ADSC, significant for OSVF at 25 μL/min compared with minimal peroxynitrite production in OC (p = 0.009) (Fig. 6d).
Glutathione is an essential antioxidant that reflects the redox balance of the cell, quenching ROS such as hydrogen peroxide, superoxide, and peroxynitrite. The SEMKUR-IM dye indirectly reports the ratio between reduced and oxidized glutathione, with increased fluorescence changes indicating reduced glutathione, and decreased changes indicating oxidized glutathione.
At intraluminal flow rate of 10 μL/min, glutathione MFI was significantly decreased in OC versus YC (p < 0.001), OSVF (p < 0.001), and ADSC (p = 0.001) (Fig. 6e). At intraluminal flow rate of 25 μL/min, there was a significant difference between YC (p < 0.001) and OSVF (p < 0.001) MFI versus OC as well as between ADSC versus YC (p = 0.02) and OSVF (p = 0.032). These results are consistent with the results that flow in OC increased hydrogen peroxide (and superoxide transiently before it was converted to hydrogen peroxide by SOD).
These data indicate that the FMD signaling mediator is nitric oxide during youth, hydrogen peroxide in aging, and switching to peroxynitrite with SVF and ADSC therapy. To complement and strengthen the validity of these findings, we used pressure myography with flow while recording percentage vasorelaxation in each group with or without inhibitors/scavengers/enzymes of these redox signaling mediators. Overall, the effectiveness of FMD was significantly attenuated with aging at flow rates of 10, 15, and 25 μL/min (p = 0.005, 0.003, 0.002) but rescued to YC levels in the SVF (p = 0.043, 0.04, 0.022) and ADSC (p < 0.001, 0.023, 0.01) therapy groups (Fig. 7a).

In youth, FMD was significantly attenuated by preincubation with N-Nitro-L-arginine methylester (L-NAME), but not catalase or uric acid, at all flow rates (p = 0.005, < 0.001) (Fig. 7b). This signifies that nitric oxide production is responsible for FMD during youth, as L-NAME inhibits eNOS-mediated nitric oxide production. During aging, FMD was not further attenuated by L-NAME, uric acid, or catalase (Fig. 7c). OSVF's improvement in FMD was significantly abrogated by uric acid at all flow rates (p = 0.04, 0.04, 0.04, 0.001, and 0.001) and L-NAME at flow rates 15–25 μL/min (p = 0.027, 0.014, and 0.012) (Fig. 7d). Considering Figure 6a, we show that OSVF produces negligible nitric oxide with flow; this signifies that with SVF therapy, the FMD signaling mediator switches to peroxynitrite, at least partially depending on nitric oxide production.
In ADSC therapy, similar to OSVF, flow was inhibited by L-NAME at 10–25 μL/min (p = 0.008, 0.05, 0.036, < 0.001) and uric acid at 10–25 μL/min (p = 0.008, 0.108, 0.086, 0.02), suggesting a nitric oxide-dependent peroxynitrite-mediated FMD mechanism. In Supplementary Figure S3a–d, the effects of inhibiting NOX2- and NOX4-mediated hydrogen peroxide production with GKT136901 (also a peroxynitrite scavenger) on flow are also provided. GKT136901 had no effect on YC FMD with nonsignificant reduction in OSVF FMD. GKT136901 increased OC FMD at 10–15 μL/min (p = 0.077, 0.043) and abrogated ADSC FMD at 10–25 μL/min (p = 0.004, 0.01, 0.011, 0.003).
Baseline oxidative and nitrosative stress are mechanistically linked with β1ADR-mediated dilative function
Consistent with our laboratory's previous reporting, β1-adrenergic-mediated vasorelaxation was reduced with aging and restored to YC levels in aging + SVF therapy. Specifically, there were significant or near significant differences between YC versus OC dobutamine dose responses at 10−9–10−5 [M] (p = 0.043, < 0.01), OSVF versus OC at 10−9–10−7 and 10−5 [M] (p = 0.083, < 0.01) and ADSC versus OC at 10−8–10−6 M (p < 0.001, 0.007, 0.039) (Fig. 8a). Similarly, there were significant differences between YC versus OC norepinephrine concentration responses at 10−8–10−4 [M] (p = 0.046, < 0.001), OSVF versus OC at 10−8–10−4 [M] (p < 0.01), and ADSC versus OC at 10−7–10−4 M (p < 0.001, 0.002, 0.016, 0.027) (Fig. 8b).

Finally, there was significantly diminished dilation to isoproterenol in OC versus YC at 10−8–10−4 M (p < 0.001), OC versus OSVF at 10−11–10−4 M (p < 0.01), and OC versus ADSC at 10−8–10−5 M (p < 0.01) (Fig. 8c). In OSVF, the dilative response to isoproterenol evoked a stronger dilative response at 10−8 M versus YC (p = 0.004). The dilative response to isoproterenol by ADSC was significantly lower than YC at 10−5–10−4 M (p = 0.03) and OSVF at 10−8–10−4 M (p < 0.05). Since aging is associated with both increased oxidative stress and reduced β-adrenergic function, we next endeavored to determine if there was a true correlation between them.
Using separate vessels from the same animals (of each group), we performed linear regression analysis with percentage relaxation to norepinephrine 10−4 [M] and ROS, nitric oxide, or glutathione MFI. There was a negative correlation between percentage relaxation to norepinephrine and MFI of hydrogen peroxide [r(35) = −0.59, p < 0.001], but not for superoxide [r(22) = 0.07, p = 0.75] or peroxynitrite [r(27) = −0.25, p = 0.19] (Figs. 9a–c). There was a nearly significant positive correlation between percentage relaxation to norepinephrine and nitric oxide MFI [r(24) = 0.38, p = 0.063] and a significant positive correlation with glutathione MFI [r(21) = 0.563, p = 0.002] (Fig. 9d, e).
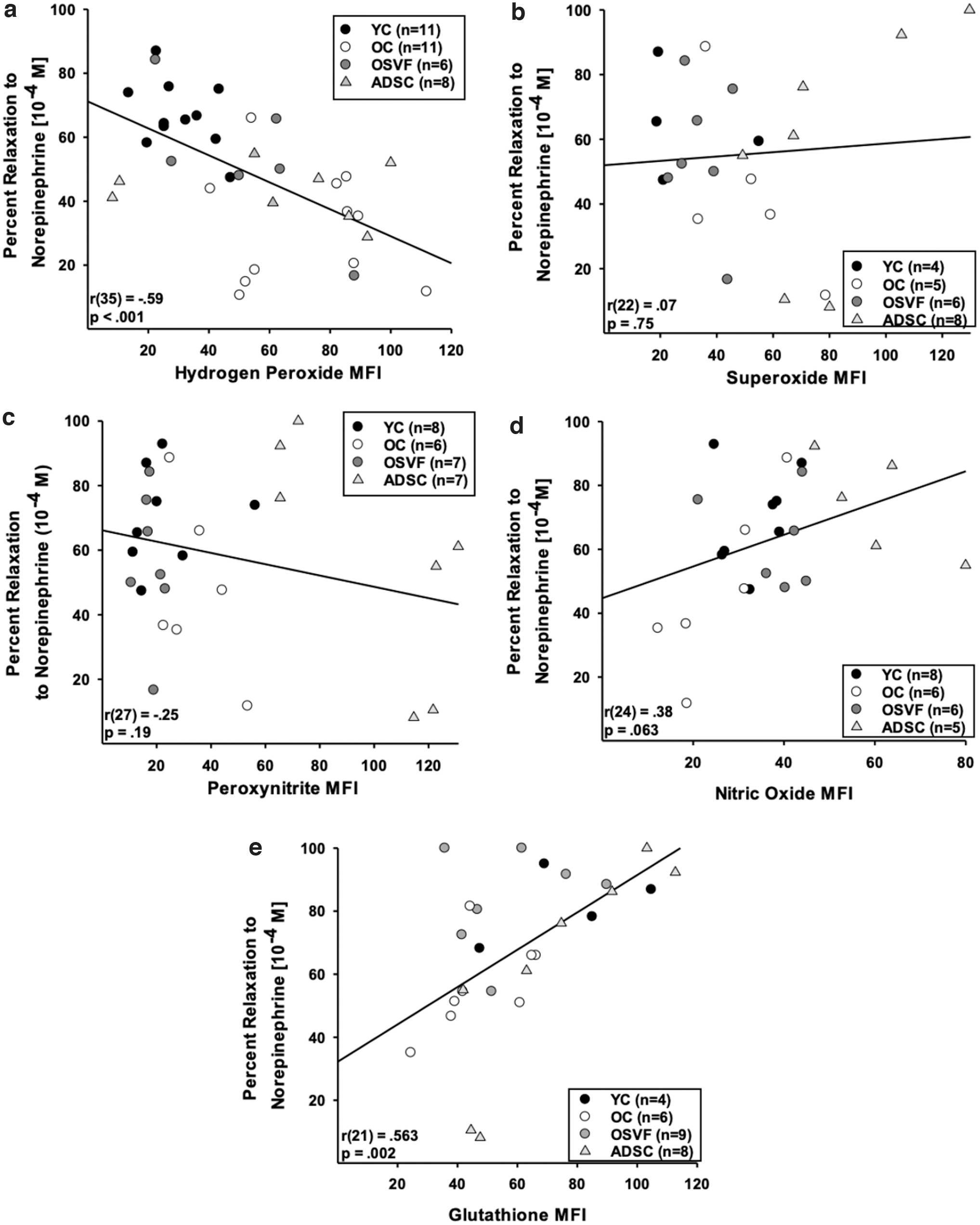
Since elevated hydrogen peroxide MFI was significantly associated with attenuated vasorelaxation to norepinephrine (and vice versa), we investigated the effect of NOX2 and NOX4 inhibition with GKT136901 preincubation on norepinephrine response in each group (Supplementary Fig. S3e–h). YC and ADSC norepinephrine response was not altered by GKT136901 preincubation. However, OC vessels preincubated with GKT136901 exhibited significantly rejuvenated dilative response to norepinephrine at 10−8–10−4 M (p = 0.041, < 0.01), while OSVF vessels exhibited attenuated dilative response at 10−5–10−4 M (p = 0.032, 0.011).
Exogenous ROS eliminates while nitric oxide rescues β1ADR-mediated vasodilation due to the ROS/RNS β1ADR desensitization and internalization axis
Isoproterenol concentration response was significantly abrogated in YC from doses 10−8–10−4 [M] (p = 0.003, < 0.001) and OSVF at 10−8–10−4 [M] (p < 0.001) when preincubated with exogenous hydrogen peroxide and superoxide donation via menadione (Fig. 10a, c). Postexperimentation, exposure to calcium-free solution + SNP still facilitated dilation, indicating that ROS-attenuated dilation is β-adrenergic specific (YC = 78.39% ± 9.76%; OSVF = 88.88% ± 6.37% dilation). In YC, the effect from ROS was nullified when coincubating with dynasore at 10−8–10−4 [M] (p = 0.005, < 0.001), but not restored to YC levels at 10−6–10−5 [M] (p = 0.037, 0.003). Paroxetine also nullified the ROS attenuation of dilation at doses 10−8–10−4 [M] (p = 0.02, < 0.001), although not completely restored to baseline levels for doses 10−7–10−4 [M] (p = 0.047, 0.011, 0.01, and 0.004).
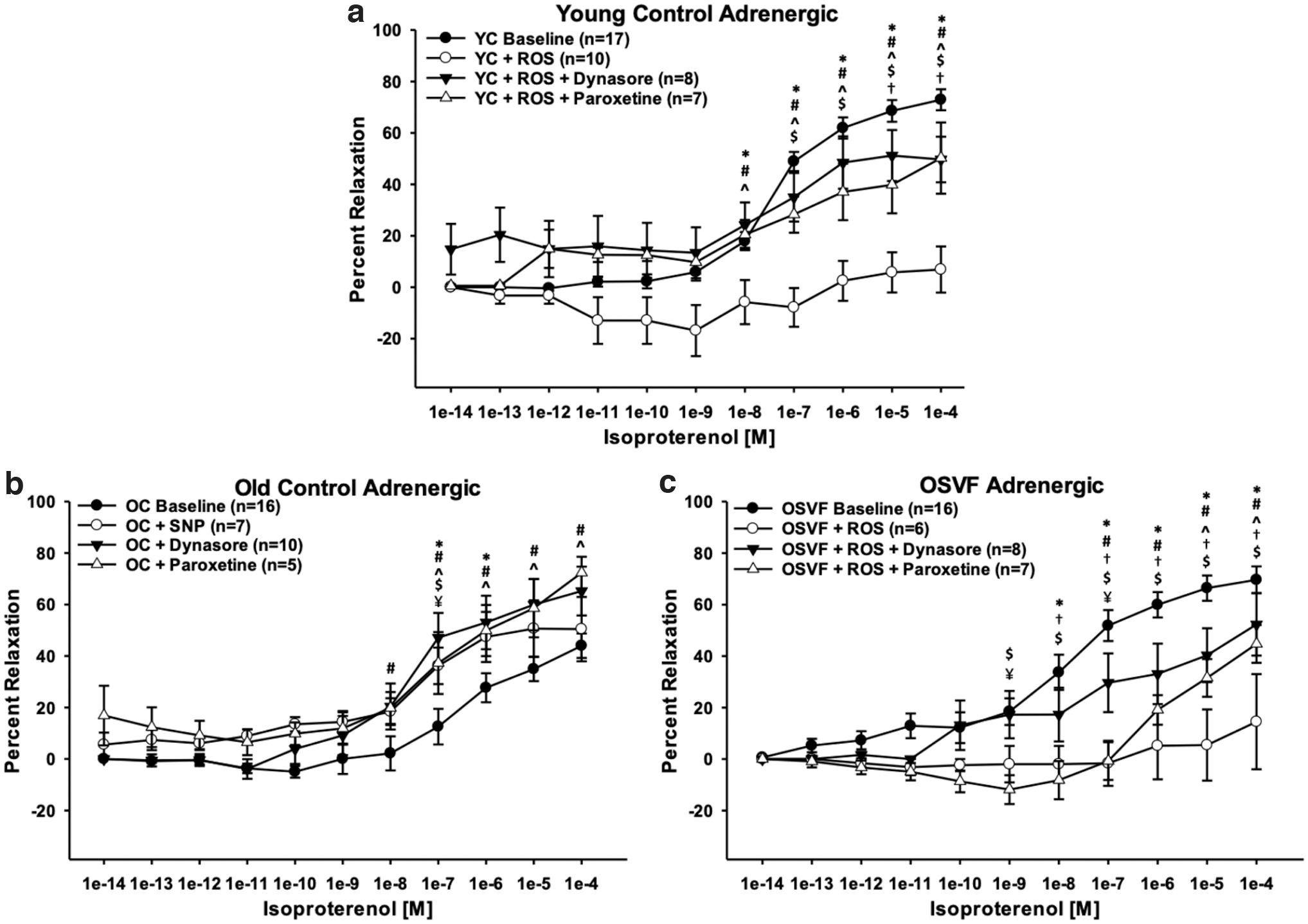
In OSVF + ROS, dynasore and paroxetine preincubation also rescued function from doses 10−7–10−4 [M] (dynasore: p < 0.01) and 10−5–10−4 [M] (paroxetine: p = 0.042 and p = 0.015). However, the recovery was partial, as dynasore and paroxetine were also significantly different from baseline at 10−8–10−4 [M] (p < 0.05) and 10−9–10−4 [M] (p = 0.004, < 0.001), respectively. Dynasore and paroxetine also significantly differed at 10−9 and 10−7 [M] (p = 0.004, 0.003). These reversing effects to attenuate ROS-blunted adrenergic dilation were therefore the result of inhibiting GRK2-mediated desensitization and dynamin-mediated internalization.
Response to isoproterenol in OC vessels was significantly rescued at doses 10−7–10−6 [M] by preincubation with a 1-min high-dose SNP preincubation, followed by 15 min of low-dose SNP and throughout the experiment (p = 0.004 and p = 0.023) (Fig. 10b). The response was also rescued by preincubation with dynasore at 10−8–10−4 [M] (p = 0.015, < 0.01) and paroxetine at 10−7–10−4 [M] (p < 0.05). Paroxetine elicited a significantly different response against dynasore and SNP at 10−7 [M] (p < 0.001). Colocalization analysis between β1ADR and plasma membrane or RAB5 (endosomes) did not reveal significant differences between groups at baseline (no beta agonist present) (Supplementary Fig. S4a–c).
β1ADR recycling is necessary in aging, but not youth or OSVF for maximal dilation to agonist
To determine the contribution of β1ADR recycling (from endosome back to plasma membrane), we inhibited recycling regulatory proteins PP2A (prorecycling) with okadaic acid, or PI3kγ (antirecycling) with BEZ235 in a separate cohort of coronary arterioles from each group. Okadaic acid did not significantly reduce YC dilation to isoproterenol but did significantly reduce dilation in OC at 10−5–10−4 [M] (p = 0.01, < 0.001) and OSVF at 10−7–10−6 [M] (p = 0.005, 0.045) (Supplementary Fig. S5a–c).
Inhibition with BEZ235 did not alter aged dilation but did significantly reduce dilation in YC at 10−7–10−4 [M] (p = 0.011, < 0.01) and OSVF at 10−6–10−4 [M] (p = 0.045, 0.008, and 0.017). BEZ235 partially negated the effects of exogenous ROS incubation in YC at 10−5–10−4 [M] (p < 0.001, 0.004), although differences were also significant against baseline at those doses (p < 0.001 for both) (Supplementary Fig. S5d). BEZ235 also negated the attenuation of dilation from exogenous ROS in OSVF at 10−6–10−5 [M] (p = 0.042, 0.017) (Supplementary Fig. S5e). There were no significant differences between groups for MFI of PP2A, protein phosphatase 2A inhibitor 2 (I2PP2A), or PI3kγ (Supplementary Fig. S6a, b).
Discussion
Aging induces a redox shift in coronary microvessels toward oxidative stress, accompanied by a decrease in antioxidants glutathione and nitric oxide. Oxidative stress occurs in aging despite an increase in transcriptional expression of catalase, superoxide dismutase 3, and thioredoxin reductase. This indicates attempted compensatory regulation that cannot compete with pro-oxidant processes, including enhanced transcription of xanthine dehydrogenase, monoamine oxidase A, and SHC1 (encodes p66SHC). These consequences with aging include abrogated FMD and β1ADR signaling. Intravenous SVF reverses these trends in old rats, except for nitric oxide. ADSCs attenuated hydrogen peroxide, but not superoxide or peroxynitrite while fully recovering glutathione and nitric oxide.
SVF upregulated transcription of additional antioxidant proteins such as glutathione synthetase, Sirt5, thioredoxin-like 1, glutaredoxin-like protein, selenophosphate synthetase 1, and several heat shock protein subunits compared with OC. Hydrogen peroxide negatively correlates whereas nitric oxide and glutathione positively correlate with β1ADR function; these associations are mechanistically linked. The major findings support that antioxidant effects of SVF and ADSC cell therapies restore vasodilation in aging by switching the acute mediator of FMD from hydrogen peroxide to peroxynitrite, as well as by attenuating desensitization and/or internalization of the β1ADR.
Immunofluorescence staining found greater NOX4 (but not NOX2) protein MFI with aging although no increase in gene expression was observed. This is consistent with the literature that NOX4-mediated ROS production is increased in vascular aging (Guzik and Touyz, 2017; Lee et al, 2020; Vendrov et al, 2015). Therefore, greater NOX4 could be a major driver of ROS production in aging coronary microvasculature. NOX4 MFI was reduced to YC levels by both SVF and ADSCs. Therefore, one mechanism for the antioxidant effects and vasodilatory recovery after SVF and ADSC therapy could be their reduction of hydrogen peroxide signaling from NOX4.
This is especially poignant since the only ROS that significantly correlated with β1ADR-mediated dilative function was hydrogen peroxide. Indeed, inhibition of NOX2/NOX4 with GKT136901 led to significant recovery of dilative response to norepinephrine (and FMD) in aging (while reducing hydrogen peroxide MFI). This mirrors what others have shown, indicating that NOX2 or NOX4 inhibition restores FMD in vascular pathology (Abdulmahdi et al, 2018; Katunaric et al, 2022).
FMD was significantly attenuated in aging, and significantly restored in both SVF and ADSC therapies. It has previously been reported that the mediator of FMD shifts throughout the life span from prostaglandin during youth, to nitric oxide during young adulthood, to hydrogen peroxide in coronary artery disease (Beyer et al, 2017). Our data confirm this trend, as youth produced significant nitric oxide MFI with flow and FMD was inhibited by L-NAME, whereas in aging hydrogen peroxide MFI was most significantly increased with flow. Catalase did not inhibit FMD in aged vessels because dilation was minimal to begin with.
With SVF (and to a lesser extent ADSC), there was increased peroxynitrite MFI in response to flow, and FMD was inhibited by uric acid scavenging of peroxynitrite as well as L-NAME in both OSVF and ADSC. This goes against our initial hypothesis, as we believed that nitric oxide would be restored to YC levels, and would mediate FMD in OSVF and ADSC vessels (Beyer et al, 2016). Although baseline nitric oxide was restored in the ADSC group, visually noticeable production of nitric oxide was not triggered by intraluminal flow. However, as nitric oxide is required to produce peroxynitrite (in combination with superoxide), and L-NAME inhibited OSVF and ADSC FMD, it stands to reason that peroxynitrite FMD response is dependent on enough nitric oxide production to combine with superoxide resulting in increased peroxynitrite.
Although peroxynitrite is a ROS, it has been speculated to have functional significance in geriatric population as a “new healthy vascular endothelium redox status” because peroxynitrite can donate nitric oxide, thereby acting as a reservoir and eliciting FMD, possibly without the proatherogenic effects of hydrogen peroxide (Muller-Delp et al, 2012).
To emphasize, the increase in OSVF peroxynitrite is acute, and limited to periods of FMD. During baseline (chronic status), aged vessels had slightly greater peroxynitrite MFI than YC or OSVF. These acute versus chronic redox signaling changes are likely of significance to cellular health and function. In contrast, ADSCs exhibited drastically enhanced peroxynitrite levels even at baseline, likely due to the combination of increased baseline nitric oxide and no attenuation of aging-induced superoxide.
Complementing these data was nitrotyrosine immunofluorescence, which revealed significantly greater nitrotyrosine in OC and ADSC versus YC and OSVF. This could be one contributing factor as to why ADSCs did not fully recover isoproterenol-mediated dilative response and overall cardiac function to the extent of SVF. In contrast, SNP-mediated improvement in β1ADR function has been attributed to nitrosylation and inhibition of GRK2 (Frame et al, 2018).
We analyzed general protein nitrosative status using a fluorescent S-nitrosocysteine antibody, believing that SVF may restore β1ADR function by reducing ROS, which would alleviate nitric oxide bioavailability to nitrosylate/inhibit GRK2. Instead, we found that SNO status is significantly reduced in aging and OSVF compared with youth (and greater in ADSC vs. OSVF). This complements our findings that SVF does not restore nitric oxide bioavailability and that ADSC did.
With intraluminal flow plus DETC inhibition of superoxide dismutase, superoxide accumulation was greater in YC and OSVF compared with OC (no differences with ADSC). This suggests that there is reduced SOD2 expression or function in aging, reversed by SVF (but not ADSC) therapy. RNA sequencing showed no difference in SOD1 or SOD2 transcription with increased SOD3 expression in OC and OSVF (vs. YC). Protein immunofluorescence showed no group differences in SOD1 or SOD3; however, there was a nonsignificant decline in SOD2 in OC versus YC/ADSC with significantly increased SOD2 availability in OSVF compared with OC.
Of note, superoxide itself was reduced in the SVF therapy but not in ADSC therapy. Other than expression changes in SOD2 described above, the function of SOD2 in aging can be inhibited by peroxynitrite-mediated tyrosine nitration (MacMillan-Crow et al, 1998). Chronic baseline peroxynitrite MFI was slightly increased in OC and drastically increased in ADSC, which could account for potential SOD2 dysfunction and superoxide accumulation in these groups. Interestingly, mass spectrometry showed appreciable secretion of SOD3 in SVF but not ADSC-cultured media.
Inhibiting catalase with AMT led to significantly greater hydrogen peroxide in OSVF compared with OC. This indicates a reduction in catalase expression or function with aging that is instead enhanced in SVF therapy. Interestingly, RNAseq showed increased catalase gene expression in both OC and OSVF when compared with YC, while protein immunofluorescence showed no difference between groups. Catalase function can be activated by phosphorylation at tyrosine 231 and 386, and can be ubiquitin degraded, which may also contribute to these results in lieu of our transcriptional or translational findings (Rhee et al, 2005).
β1ADR-mediated dilation with dobutamine, norepinephrine, and isoproterenol was reduced with aging; all restored by SVF and only the former two being fully restored with ADSC therapy. Using two vessels from the same animals, we determined that there is negative correlation between hydrogen peroxide and β1ADR dilative function, and a significant positive correlation between glutathione and nitric oxide with β1ADR function. Thus, oxidative stress reduces functional dilation with aging. We next sought to experimentally confirm a causal mechanistic link.
Incubating vessels with exogenous hydrogen peroxide and superoxide (mimicking aging conditions) completely attenuated YC and OSVF β1ADR dilation to isoproterenol. This effect was diminished if coincubating ROS with paroxetine (inhibiting GRK2, i.e., desensitization) or dynasore (inhibiting dynamin, that is, internalization). OC vessels preincubated with SNP (mimicking YC nitrosylation status), dynasore, and paroxetine significantly improved β1ADR dilation to isoproterenol. These results indicate a role for ROS and RNS (nitric oxide) in β1ADR desensitization and/or internalization.
βADR recycling from the endosome is known to be inhibited by ROS via its activation of PI3kγ (Vasudevan et al, 2011a, 2011b). We found that inhibition of PI3kγ with BEZ235 countered the poor dilatory effects produced by exogenous ROS in YC and OSVF vessels. This did not translate to BEZ235 improving OC vessel dilation. Interestingly, inhibiting β1ADR recycling with okadaic acid led to significant reduction in isoproterenol-mediated dilation in OC but not in youth. This signifies that recycling is not necessary in youth for successful maximal response to agonist, likely due to plentiful sensitized receptor density at the plasma membrane.
This is not the case in aging, indicating that a greater density of receptors has been internalized, and maximal dilative response to agonist depends in part on recycling. OSVF vessels exhibit partially attenuated dilation with okadaic acid, but at high doses of isoproterenol this is not significant. Overall, this indicates that OC vessels are most reliant on recycling, followed by OSVF, then YC. Protein levels of PP2A, I2PP2A, and PI3kγ were unchanged with aging or SVF therapy, indicating that the mechanisms of recycling themselves are unchanged.
In the colocalization experiment, there was no significant difference between groups of overlap between the β1ADR with either the plasma membrane or RAB5-containing endosomes. This indicates no difference in internalization between groups. However, it should be noted that this experiment was performed during baseline conditions (no beta agonist stimulation). Considering that dynasore-mediated inhibition of dynamin restores (or preserves) β1ADR-mediated dilation in aging or exogenous ROS exposure, it is then likely that differences (if any) in internalization are pronounced during times of agonism.
Rambacher and Moniri (2020) recently described the effects of oxidized cysteine thiols on human airway epithelial cell β2ADR function. Critically, overoxidation of the β2ADR to a redox-deficient state (which the authors model using hydrogen peroxide followed by dimedone) led to attenuated isoproterenol-induced cAMP formation, CREB response, reduced ERK1/2 phosphorylation and beta-arrestin-2 signaling, and reduced internalization of the β2ADR.
Alternatively, the authors provide evidence that there is a homeostatic level of receptor oxidation that is most conducive to optimal function. The authors conclude that excessive oxidative stress in the settings of asthma/COPD and therapeutic β2ADR agonism could be a cause of β2ADR overoxidation and therapeutic desensitization (but not internalization). Further study utilizing the techniques in the Rambacher et al's study in the context of aging coronary microvascular β1ADR would be beneficial to further clarify these complex and understudied redox-adrenergic dynamics.
Echocardiographic analysis of ADSCs was compared with historic data from our YC, OC, and OSVF groups (Rowe et al, 2019). CFR (a clinical indicator of CMD) was significantly decreased in aging, rescued by OSVF; however, ADSCs were unable to rescue to levels seen in YC. Similarly, IVRT, E/A, and E/e’ (parameters of diastolic dysfunction) that were pathologic in aging were rescued by OSVF but not ADSCs. Therefore, despite functional recovery at the level of the microvasculature this did not translate at the cardiac level for ADSC therapy. This could in part be due to secretome differences.
Mass spectrometry revealed six antioxidant proteins that were significantly greater in SVF-conditioned media than ADSC. There were only two antioxidant proteins that were significant or nearly significantly greater in ADSC versus SVF. Altogether, SVF attenuated all ROS, while ADSC only reversed hydrogen peroxide (but not superoxide or peroxynitrite). Therefore, there are currently unidentified constituents in SVF, or possibly synergistic effects between SVF cells, which allow it to outperform ADSC therapy. However, ADSCs likely make up one important constituent of SVF's full regenerative ability. Considering extra isolation steps and cost associated with isolating ADSCs from SVF, our study suggests that utilization of whole SVF in the context of CMD would be preferred.
The clinical translation of these findings would suggest a cell transplant from younger to older individuals in an allogenic manner. Autologous injection of SVF is limited by reduced yield and therapeutic effectiveness of aged SVF (Aird et al, 2015; Dos-Anjos Vilaboa et al, 2014). Certain strategies to rejuvenate aged SVF therapeutic potential, such as priming and preconditioning, could be further explored.
However, since SVF has low immunogenicity and has already been used in an allogenic manner we believe this is the most optimal therapeutic option (Diana et al, 2019; Gu et al, 2014; McIntosh et al, 2006; Zhou et al, 2019). Discussion comparing the benefits and deficits of allogenic versus autologous stem cell therapies for vascular pathologies has recently been conducted elsewhere, including the context of biological variables such as aging and sex (Tracy et al, 2022).
We hypothesized that FMD and β1ADR-mediated dilation are reduced with aging and reversed via SVF and ADSC therapy by attenuating oxidative stress and restoring protective nitrosative signaling. SVF and ADSCs (to a lesser extent) produce antioxidant proteins within their secretome and reduce oxidative stress in aged coronary microvasculature. FMD was abrogated in aging with reversal by SVF and ADSC therapy via enhanced peroxynitrite signaling. In the ROS/RNS β1ADR desensitization and internalization axis, young vessels experience limited ROS and plentiful nitric oxide bioavailability, resulting in sensitized β1ADR at the plasma membrane potentially via greater GRK2 nitrosylation, less β1ADR thiol oxidation, and less ROS-mediated blockade of receptor recycling.
In aging, this axis shifts such that there is reduced nitric oxide bioavailability, allowing functional GRK2 to influence desensitization and internalization, and there is greater accumulation of ROS to oxidize β1ADR-thiols and inhibit recycling. With SVF therapy, although nitric oxide bioavailability is not restored, ROS are reduced to alleviate β1ADR thiol oxidation-mediated desensitization and/or internalization, and restored receptor recycling. With ADSC therapy, nitric oxide is restored (potentially inhibiting GRK2), and hydrogen peroxide is attenuated to alleviate ROS-mediated desensitization and/or internalization, although superoxide and peroxynitrite levels are still comparable with aging.
In conclusion, aging-mediated coronary microvascular dysfunction and hyperconstriction can be ameliorated by targeting oxidative stress. SVF therefore has significant translational potential for patients with CMD, and is theoretically superior over current clinical standards (e.g., beta blockers) that treat symptoms but do not address the pathologic cause of microvascular dysfunction and hyperconstriction.
Materials and Methods
Animal model, groups, and endpoint procedures
Surgeries for all animals were carried out in accordance with approved protocols from the University of Louisville Institutional Animal Care and Use Committee and the NIH Guide for the Care and Use of Laboratory Animals (2011). Female Fisher 344 young control (YC, 3–6 months old; Harlan Laboratories, Indianapolis, IN) and aged (23–24 months old; National Institute on Aging, Bethesda, MA) rats were housed in pairs, and allowed free access to food and water.
Rats were accustomed to a regular 12-h light/dark cycle and acclimated to facility environment for ∼1 week before endpoint procedures. Aged rats were randomly divided into three groups, old control (OC, 24 months), or old treated with either SVF (OSVF) or ADSCs. SVF or ADSCs was injected at 23 months of age, with sacrifice at 24 months of age. Euthanasia was achieved by removal of the heart under deep anesthesia (lack of tactile toe reflex and lower respiratory rate) consisting of 5% inhaled isoflurane with 1.5–2.0 L/min O2 flow in an induction chamber.
Blood pressure measurements and echocardiography
In brief, the rat was placed in a supine position on a 37°C pad and anesthetized with 5% inhaled isoflurane with 1.5–2.0 L/min O2 flow. The right carotid artery was isolated and then secured using silk sutures. The cranial aspect of the carotid artery was ligated, and a microsurgical clip was placed on the proximal carotid artery for hemostasis. An arteriotomy was performed with microsurgical scissors, and a 1.9 F PV 6.00 mm conductance catheter (Transonic, London, ON, Canada) calibrated to current atmospheric pressure was introduced into the carotid artery. The catheter was then secured into the carotid artery with sutures and advanced retrograde. The catheter was allowed to equilibrize for 7 min before a 1-min recording of blood pressure.
Immediately after recording, the catheter was removed, and the animal was humanely euthanized via vital organ removal. Data recording and analyses were performed using LabChart Pro software (ADInstruments, Colorado Springs, CO). Echocardiographic measurements were procured on ADSC rats, and compared with historic data of echocardiographic analysis of YC, OC, and OSVF rats as previously published with detailed methodology described previously (Rowe et al, 2019).
SVF and ADSC isolation, cell culture and injection
Young (3–6 months) rats from a green fluorescent positive (GFP+) Fischer-344 colony (original breeders obtained from University of Missouri RRRC, strain #307, colony maintained in house) were utilized to obtain SVF cells as described before (Kelm et al, 2018; Leblanc et al, 2012; Rowe et al, 2019). In short, a 1:1 mixture of male epididymal and female ovarian fat pads was isolated. The fat was harvested, washed, and finely minced with subsequent digestion in 0.75 mg/mL Type 1 collagenase solution (Vitacyte, Indianapolis, IN, 011-1030) and 1 mg/mL DNAse (Sigma, Burlington, MA, 9003-96-9) in 0.1% BSA-PBS.
Two centrifugation cycles at 400 rpm for 4 min were performed to separate and then remove buoyant adipocytes. The SVF cell pellet was resuspended in 0.1% BSA-PBS, followed by a second centrifugation cycle for 5 min and removal of remaining supernatant. The cell pellet was resuspended in 0.1% BSA-PBS once more, followed by gravity filtration through 20 μm mesh. The final SVF solution was collected, and the cells were counted by hemocytometer, followed by a final centrifugation, removal of supernatant, and resuspension in normal saline to allow for 1 mL injections with 10 million SVF cells.
For mass spectrometry experiments, frozen P9-P10 young (3 months) Fisher ADSC (JangoCell ADSC-RFM-01, characterized by CD90, CD105, CD29, CD45, and CD34) and frozen P1 SVF (freshly isolated in house previously and stored in liquid nitrogen) were thawed 3 min in 37°C water bath, added to 10 mL prewarmed Mesenchymal Stem Cell Medium (ScienCell #7501) + 1% mesenchymal stem cell growth supplement (MSCGS, ScienCell #7552) + 1% Pen/Strep +5% FBS (MSCM Complete), centrifuged at 500 g × 5 min, resuspended in warm MSCM Complete, and plated one million cells per T75 flask (n = 3 ADSC, n = 3 SVF).
Cells received full medium change with MSCM Complete every 2–3 days until 90% confluence. Before mass spectrometry, a subset of cells were serum starved by removing the medium, rinsing each flask shortly with sterile DPBS, then treated with MSCM +1% MSCGS +1% P/S with no FBS. After 24 h of serum starvation, medium was harvested from each flask into separate sterile tubes, placed on ice, and mass spectrometry was performed. In ADSCs designated for therapeutic injection, the serum starvation step was skipped. The final ADSC solution was collected, and cells counted by hemocytometer, followed by a final centrifugation, removal of supernatant, and resuspension in normal saline to allow for 1 mL injections with 10 million ADSC cells. The SVF- or ADSC-saline solution was warmed to 37°C and injected in the tail vein of rats under isoflurane anesthesia.
Heart and subepicardial arteriole isolation
Hearts were isolated from each euthanized rat, and coronary arterioles were dissected and transferred into a vessel chamber pressure myography system as performed previously (Rowe et al, 2019, 2022b). In brief, each end of the vessel was attached to a glass pipette, and subjected to physiologic temperature (37°C) and pressure [45 mmHg (Chilian et al, 1986)] as shown in Figure 2a, b. Vessels were allowed to achieve spontaneous tone of >20% constriction from initial diameter (Kelm et al, 2018; Leblanc et al, 2012; Rowe et al, 2019). Whole heart, left and right ventricle, septum, total body weight, initial tone achieved, maximum dilation, and systolic and diastolic blood pressure (and MAP) were acquired, and are presented in Table 1.
Pressure myography with concentration or flow response
The following experiments were randomized to each vessel. Baseline concentration–response curves were generated for each group for dobutamine at 10−10–10−5 [M] (primary β1ADR agonist, partial β2ADR agonist; Sigma-Aldrich, Burlington, MA, D0676), isoproterenol at 10−14–10−4 [M] (nonspecific βADR; Sigma-Aldrich, I6504), and norepinephrine at 10−9–10−4 [M] (primary β1ADR, β2ADR, α1ADR, and α2ADR agonist; Sigma-Aldrich, A9512). Vessels were monitored for 2 min at each concentration, and diameter recorded manually using video calipers.
Percentage dilation was calculated as ([intraluminal diameter at specific concentration – diameter at baseline]/[maximal diameter – diameter at baseline]) × 100. Once experiments were concluded, vessels were washed with calcium-free physiologic salt solution twice for 15 min each time, followed by sodium nitroprusside (SNP, 10−4 [M]; Sigma-Aldrich, 13451, 2 min) to achieve maximum dilation. Tone was calculated as 1 – (initial diameter/maximum diameter) × 100.
Concentration–response curves for norepinephrine were generated in each group with 30-min preincubation with NOX2 and NOX4 inhibitor GKT136901 (1.5 μM; Millipore Sigma, Burlington, MA, 955272-06-7). Percentage dilation responses for norepinephrine were also plotted against MFI values for hydrogen peroxide, peroxynitrite, superoxide, nitric oxide, and glutathione (described further later).
Isoproterenol concentration–response curves were also generated for vessels from YC and OSVF vessels preincubated for 1 h with exogenous hydrogen peroxide (10−4 [M]; Sigma-Aldrich, 7722841) plus menadione (superoxide donor, 10−5 [M]; Sigma-Aldrich, 130-37-0) plus or minus dynasore (inhibits dynamin-mediated internalization; 1.6 μM; Abcam, Cambridge, United Kingdom, 120192) and paroxetine (GRK2 antagonist, 5 μM; VWR, Radnor, PA, 89160-896).
Additional isoproterenol concentration–response curves were generated for OC vessels with either incubation of dynasore, paroxetine, or SNP pretreatment. Vessels were incubated with SNP (10−4 [M]) for 1 min, followed by a 15-min wash without SNP, followed by isoproterenol concentration–response generation plus low-dose SNP (10−9 [M]).
In a separate cohort, vessels from each group were incubated with inhibitors of β1ADR recycling, including okadaic acid (PP2A inhibitor, 10−9 [M], APeX Bio A4540) and BEZ235 (PI3Kγ inhibitor, 10−6 [M]; MedChemExpress, Monmoth Junction, NJ, HY-50673), and concentration curves for isoproterenol were generated. In separate YC and OSVF vessels, concentration curves for isoproterenol were generated with exogenous hydrogen peroxide and menadione plus or minus BEZ235 coincubation.
Percentage dilation to varied flow rates (0, 5, 10, 15, 20, 25 μL/min, 2 min each) was assessed in separate vessels from each group. Following baseline FMD, flow rates were reassessed with addition of uric acid (peroxynitrite scavenger, 1 mM; Sigma-Aldrich, 69-93-2 for 20 min), catalase (hydrogen peroxide scavenger, 500 U/mL; Sigma-Aldrich, C4963, 1 h), and L-NAME (eNOS inhibitor, 10−5 [M]; Sigma-Aldrich, N5751 for 20 min), and GKT136901 separately.
Pressure myography with fluorescence imaging
Isolated coronary arterioles from each group were infused with fluorescent dyes for superoxide (Mitosox, 0.1 μM; Invitrogen, Waltham, MA, m36008, 30 min), peroxynitrite (Biotracker 515, 20 μM; Sigma-Aldrich, SCT035, 30 min), hydrogen peroxide (Mitopy1, 10−5 [M]; Tocris, Bristol, United Kingdom, 1041634-69-8, 1 h), nitric oxide (DAN, 10−4 [M]; Alfa Aesar, Haverhill, MA, 771-97-1, 10 min), and glutathione (SEMKUR-IM, 250 μM; ProteinMods, Waunakee, WI, 1 h). Experiments were carried out in physiologic salt solution without albumin.
Flow (10 and 25 μm/min) was applied to infused vessels plus or minus either diethyldithiocarbamic acid (DETC, 1 mM; Fisher, Hampton, NH 140-61-7, 1 h), 3-amino-1,2,4-triazole (AMT, 100 μM; Sigma-Aldrich, A8506, 1 h), or GKT136901 with images taken after 2 min at each flow rate. MFI for each image was calculated from the average of two regions of interest (ROIs) of consistent size (20 × 100 μm), applied over the left and right vessel walls using NIS Elements AR Analysis software (Nikon Instruments, Melville, NY).
Data supporting the specificity of the fluorescent probes utilized in this study are provided elsewhere (Probes for Mitochondria, 2010, Dickinson and Chang, 2008; Miles et al, 1995; Moller et al, 2019; Ueno et al, 2006). Positive and negative controls for each probe were conducted to ensure probes behaved in predictable manner respective to each ROS, nitric oxide, or glutathione. For Mitosox, cobalt chloride (generates superoxide through hypoxia-inducible factor 1, 0.5 mM; Sigma-Aldrich, 15862, 1 h) and menadione (10−5 [M]; 130-37-0) represented positive controls, whereas Tiron (superoxide scavenger, 10 mM; Acros Organics, Geel, Belgium, 174140250, 1 h) represented a negative control.
For Biotracker 515, menadione + SNP (10−4 [M]) for 1 h represented positive control, whereas Tiron (10 mM) + catalase (10 mM, 500 U/mL) for 1 h, GKT136901 for 30 min, as well as uric acid for 20 min represented negative controls. For Mytopy1, exogenous hydrogen peroxide (10−4 [M], 20 min) represented positive control, whereas catalase (500 U/mL, 1 h) and GKT136901 represented negative controls. For DAN, exogenous SNP (10−4 [M], 2 min) represented positive control, whereas CPTiO (1 mM; Sigma-Aldrich, C221, 20 min) represented a negative control. Controls are illustrated in Supplementary Figure S1. Simplified illustration of ROS and the fluorescent labels, enzyme activator/donors, and inhibitors/scavengers is summarized in Figure 2.
Immunofluorescence staining
Vessel immunofluorescence staining was performed as previously described (Rowe et al, 2019, 2022b). In brief, coronary arterioles measuring 750–1000 μm in length and <250 μm in diameter were isolated and fixed in 2% paraformaldehyde for 1 h. After fixation, vessels were washed 2 × 15 min in DCF-PBS followed by 20 min in 0.5% Triton-X/DCF-PBS, all at room temperature with mild rotation.
Next, vessels were placed in blocking solution containing 5% donkey serum, 0.5% BSA, and 0.5% Triton-X/DCF-PBS for 1 h, followed by incubation with primary antibodies, including copper-zinc superoxide dismutase 1 (SOD1, 1:100 dilution; Novus Biologicals, Littleton, CO, NB100-60944), manganese superoxide dismutase (SOD2, 1:100 dilution; Novus Biologicals, NB100-1992SS), extracellular copper-zinc superoxide dismutase (SOD3, 1:100 dilution,
The following day, the vessels were washed × 3 in DCF-PBS, followed by 1-h incubation in donkey antirabbit IgG Alexa Fluor 594 (1:300; Invitrogen, A21207), goat antimouse IgG Alexa Fluor 488 (1:300; Abcam, 150113), or donkey antigoat IgG Alexa Fluor 488 (1:300; Abcam, A11055) as appropriate in blocking solution. Nuclei were stained with DAPI (Life Technologies, Carlsbad, CA, R37606). Vessels were placed on slides with mounting media and coverslip, then imaged with Olympus FV1000 confocal microscope system (Olympus America, Center Valley, PA) with a 405 and 562 nm laser.
The pixel density set for image capture was 1024 × 1024 with 2 μm Z-step with 20 × magnification, generating a maximum intensity projection of ∼12 slices postimage capture. Immunofluorescence intensities for each antibody were quantified using the Nikon NIS Elements AR Analysis software with 20 × 100 μm area. ROI boxes were placed on the right and left vascular walls (endothelial + smooth muscle) excluding cardiomyocytes with MFI averaged.
For imaging of β1AR localization, coronary microvessels were stained with RAB5 endosomal marker (1:100, LSBio LS-B12415-300) and β1AR (1:100; Abcam, ab3442). For secondary staining, donkey antirabbit IgG Alexa Fluor 594 (1:300; Invitrogen, A21207) and donkey antigoat IgG Alexa Fluor 488 (1:300; Abcam, A11055) were used. Staining steps were the same as above, except for the use of 0.1% Triton-X instead of 0.5% Triton-X. After this initial staining with antibodies, microvessels were stained with red CellBright plasma dye (1:125; Biotium, 30023) for 20 min, followed by three washes in PBS and mounting with PBS. Colocalization of β1ADR with plasma membrane or RAB5 was quantified using the EzColocalization protocol (Stauffer et al, 2018).
TBARS assay
Blood was collected from rats of each group from the right ventricle immediately before euthanasia via vital organ removal. Plasma was separated via centrifugation at 2000 rpm for 10 min at 4°C, and then frozen at −80°C before use within a 1-month time frame. Plasma malonaldehyde was measured utilizing the TBARS assay kit (Cayman Chemical, Ann Arbor, MI, 10009055) according to the manufacturer's instructions and using the colorimetric method. The BioTek synergy 4 multimode microplate reader (Winooski, VT) was utilized for colorimetric analysis.
RNA sequencing
Additional coronary arterioles were allocated for RNA sequencing via flash freezing; samples were stored at −80°C until samples from all groups were collected as done previously (Rowe et al, 2022b). Six isolated vessels were pooled from two animals in the same group for n = 3 biological replicates/group. RNA was harvested with RNAqueous Micro kit (Invitrogen, AM1931) and quantified on a NanoDrop (ThermoFisher Scientific, Waltham, MA).
Samples were sent to the University of Louisville Genomics Core for quality control analysis using the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA) and quantified using a Qubit fluorometric assay (Thermo Fisher Scientific) as previously described (Mboge et al, 2019). Poly-A enrichments, barcoding, and Illumina Next-Gen sequencing for library preparation were performed as previously described (Lanceta et al, 2020; Mboge et al, 2019). Gene transcripts were analyzed by the University of Louisville Bioinformatics Core for differential expression analysis between group comparisons as previously described (Miralda et al, 2020).
Mass spectrometry
Cell culture supernatants were concentrated (2.5 mL to 0.1 mL) using Vivaspin-2 5000 MWCO HY filters (Sartorius, Goettingen, DE) by centrifugation at 3200 g and 4°C in a swinging bucket rotor. Samples were desalted by diafiltration using 2.4 mL 0.05 M triethylammonium carbonate (TEA-BD) pH 8.5 (Sigma Aldrich) three times. After the final diafiltration step, the retentate was recovered using 0.1 mL 10% sodium dodecyl sulfate in 0.05 M TEA-BC, pH 8.5 supplemented with 2 × HALTTM (Thermo) protease & phosphatase inhibitor cocktail. The recovery protein yield was estimated by DC assay (BioRad) using a BSA standard curve.
Protein samples (25 μg) were reduced, alkylated, and digested by the S-trap Micro Column digestion protocol provided by Protifi, Inc. (Farmingdale, NY) using MS-grade trypsin protease (Promega, Madison, WI). Before LCMS analysis, samples were dried by SpeedVac, resuspended in 20 μL chromatography buffer A (2% v/v acetonitrile/0.1% v/v formic acid). 1D-LC-MS/MS analysis was carried out using a Proxeon EASY-nLC 1000 UHPLC (ThermoFisher) and nanoelectrospray ionization into an Orbitrap Elite mass spectrometer (Thermo) using an Nth Order Double Play method with scan event one an FTMS MS1 scan (normal mass range; 120,000 resolution; full scan type) for the 300–2000 m/z with charge state screening and monoisotopic precursor selection were enabled.
Scan event two obtained HCD ITMS MS2 scans (normal mass range, rapid scan rate, centroid data type) on up to 20 peaks that had a minimum signal threshold of 5000 counts from scan event one. The lock mass option was enabled (0% lock mass abundance) using the 371.101236 m/z polysiloxane peak as an internal calibrant. Proteome Discoverer v2.4.0.305 (ThermoFisher) was used to direct the analysis of the RAW files with SequestHT and the 4/11/2022 versions of the UniprotKB reviewed reference proteome canonical Bos Taurus and Rattus norvegicus sequences (Proteome IDs UP000009136 and UP000002494, respectively).
Trypsin (KR|P) digestion with up to two missed cleavages was assumed with the dynamic modifications Oxidation (M), Acetyl (Protein N-term), Met-loss (Protein N-term) and Met-loss+Acetyl (Protein N-term), and the static modification Carbamidomethyl (C). Precursor and fragment mass tolerances were 10 ppm and 0.6 Da, respectively. In the consensus step, proteins were quantified from the summed abundances of all high confidence unique and razor peptide intensities. Proteins were excluded if in every sample they triggered recognition of protein sequence but were below the limit of detection (relative abundance of 6.51 × 10−5).
Statistical analysis
SigmaPlot 14 (Systat) was utilized for statistical analyses, with significance level set at p ≤ 0.05 for all analyses. Concentration– and flow–response curves as well as MFI evaluations were analyzed with two-way repeated-measures ANOVA with the Bonferroni or Holm–Sidak post hoc testing as appropriate.
Data are presented as means plus or minus standard error of the means as indicated. For pressure myographic fluorescence analysis, ∼15% of the data were reanalyzed by blinded inter- and intraobservers. Linear regression and Bland–Altman Plots were generated to assess for bias in interpretation of data (Supplementary Fig. S2). For confocal images, the images were coded and analyzed in a blind manner with the code being broken only after all MFI quantifications were acquired. Electronic laboratory notebooks were not used for this study.
Footnotes
Acknowledgments
We thank Mr. Ayomikun Samuel Oyeleye, Mr. Daniel Benson, Mr. Anirudh Devara, and Ms. Pallavi Katragadda for their technical assistance. We acknowledge Mr. Oyeleye and Ms. Katragadda for their role in interobserver analysis. We also thank Dr. Michael Merchant and Mr. Daniel Wilkey of the University of Louisville Proteomic Core and Clinical Laboratories for their assistance with mass spectrometry and experiments and analysis. Finally, we thank Dr. Eric Rouchka and Dr. Xiahong Li of the University of Louisville Bioinformatics Core for their assistance with RNA sequencing experiments and analysis.
Authors' Contribution
E.P.T. and A.J.L. conceived of the research, analyzed the data, and designed the research; E.P.T., G.R., R.N., M.D., and J.E.B. conducted experiments; E.P.T. wrote the article; E.P.T., M.D., G.R., J.E.B., and A.J.L. reviewed and edited the article. All authors read and approved the article.
Data Availability
Data supporting our findings will be made available upon reasonable request to the corresponding author (AJL). RNA sequencing data have been uploaded to the repository Gene Expression Omnibus with accession number GSE184674 and can be accessed at
These data include (A) the primary data files (.RAW) for the six 1D-LCMS runs comprising a pilot study containing three ADSC and three SVF samples; (B) sample key; (C) the sequence databases (4/11/2022 versions for the UniprotKB reviewed reference proteome canonical Bos Taurus (residual serum contamination) and Rattus norvegicus sequences (Proteome IDs UP000009136 and UP000002494, respectively); and (D) an excel file exported from Proteome Discoverer v2.4.0.305 (ThermoFisher) containing the SequestHT protein assignments filtered for 1% FDR (uploaded data maybe be accessed by reviewers using the following link: ftp://MSV000089689
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funding for this project was provided by NIH grants: R01 AG053585 (A.J.L.), R01 DE030103, P30 ES030283, Department of Defense: W81XWH-19-RTRP-IDA, and the Gheen's Foundation (A.J.L.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.