
Abstract
Significance:
Ionizing radiation can damage cells either directly or through oxidative damage caused by ionization. Although radiation exposure from natural sources is very limited, ionizing radiation in nuclear disaster zones and long spaceflights causes inconspicuous, yet measurable physiological effects in men and animals, whose significance remains poorly known. Understanding the physiological impacts of ionizing radiation has a wide importance due to the increased use of medical imaging and radiotherapy.
Recent Advances:
Radiation exposure has been traditionally investigated from the perspective of DNA damage and its consequences. However, recent studies from Chernobyl as well as spaceflights have provided interesting insights into oxidative stress-induced metabolic alterations and disturbances in the circadian regulation.
Critical Issues:
In this review, we discuss the physiological consequences of radiation exposure in the light of oxidative stress signaling. Radiation exposure likely triggers many converging or interconnecting signaling pathways, some of which mimic mitochondrial dysfunction and might explain the observed metabolic changes.
Future Directions:
Better understanding of the different radiation-induced signaling pathways might help to devise strategies for mitigation of the long-term effects of radiation exposure. The utility of fibroblast growth factor 21 (FGF21) as a radiation exposure biomarker and the use of radiation hormesis as a method to protect astronauts on a prolonged spaceflight, such as a mission to Mars, should be investigated. Antioxid. Redox Signal. 37, 336–348.
Introduction
The phenomena of radiation and radioactivity, referring to high-energy electromagnetic spectra as well as high-velocity particles (10), were discovered in the late 19th century and opened a whole new era of physics. In the early days, radiation was marveled at because of its fascinating features and scientific novelty. For example, soon after Wilhelm Conrad Röntgen had discovered cathode tube radiation and taken the first famous X-ray image of his wife's hand, X-rays became a popular amusement in the early 20th century funfairs (86). Radioactive substances, such as radium, were hailed for their imagined health benefits. However, the adverse effects of radiation became quickly apparent.
Radiation injuries were noted already by the early workers Pierre Curie, Thomas Edison, and Nikola Tesla. Famously, Marie Curie, a Nobel laureate in chemistry and physics for her work on radium, died of leukemia likely caused by prolonged exposure to radiation (38). The dangers of radioactivity came to wider appreciation when radionuclides were weaponized in the mid- to late 1940s. Besides demonstrating the applicability of radionuclides for mass destruction, the nuclear era introduced concepts such as radioactive contamination and fallout to the wider audience. Being invisible, dangerous, and difficult to clean or neutralize, radiation has also a guaranteed spook factor for the public (52).
The biological effects of radioactivity are almost exclusively caused by ionizing radiation, which has the ability to kick off electrons from atoms (18), causing a cascade of reduction/oxidation reactions that damage and destroy biological macromolecules. Ionizing radiation can exist in the form of high-energy particles or X- and γ-rays (Fig. 1), the latter being capable of penetrating deep into the tissue and causing systemic effects. Furthermore, the absorption of β-particles (as well as larger particles with high-enough velocity) by matter will emit part of the energy as X-rays in a phenomenon known as Bremsstrahlung (18).

The main immediate effect of ionizing radiation in cells is the production of reactive oxygen species (ROS) from water. This oxidative stress is the major cause of physiological as well as pathological responses to radiation exposure. There are also major natural sources of ROS, such as the electron transport chain (ETC) in mitochondria (75), which have important roles for adaptive responses in living organisms (81). Cells have therefore a number of physiological pathways that respond to increased oxidative stress and cause, for example, metabolic alterations to maintain redox homeostasis.
In this review, we focus on the effects of radiation-induced ROS in cells, especially in the light of recent data from studies conducted in space and in areas contaminated by radionuclides. It is expected that chronic exposure to ionizing radiation will phenocopy the effects of mitochondrial dysfunction, as it should trigger similar pathways via excess ROS production. Studies from human mitochondrial disorders and their animal models might in fact help to understand some of the pathological and adaptive responses seen in radiation stress. More detailed knowledge of the physiological effects of ionizing radiation-induced ROS signaling and metabolic remodeling could help to device strategies mitigating the harmful effects of radiation exposure during spaceflights and medical imaging.
Sources of Ionizing Radiation
In contrast to UV radiation, ionizing radiation is not common in nature, apart for the low γ-ray and radionuclide emissions from thunderstorms (32, 110). The magnetic field of the Earth shields us from high-energy cosmic rays, and the naturally occurring sources of radioactivity, such as surfaced radioactive ores, are very rare and local. However, in areas of high natural radiation, exposure can be 55–200 times higher than the usual background radiation dose of 10 μSv per day (5). This is also roughly comparable with the radiation exposure in most parts of the Chernobyl Exclusion Zone (CEZ, some areas up to 6 mSv/h) (9, 59), which cordons the area worst affected by the nuclear disaster fallout in 1986.
The main difference between the two is that almost all naturally occurring radioactive sites are composed of the decay chain of primordial uranium-238, where the subsequent products radium-226, radon-222, and polonium-218 all decay via α-emission, having poor penetration, whereas in the CEZ, organisms are exposed to more penetrant forms of ionizing radiation, such as γ-emissions from the cesium-137 decay chain (9, 47). In contrast to the natural sources of radiation, many of the fission products associated with nuclear fuel, such as strontium-90, iodine-131, and cesium-137, are biologically active and can accumulate in living organisms (8). While α-radiation can cause a health hazard, such as radon-222 in poorly ventilated mines or buildings, its effects are more local compared with γ- and X-rays (10).
For most people, at least in Western countries, the main exposure to ionizing radiation comes from medical imaging and medical use of radionuclides (86). In fact, growth and technological development has expanded this field substantially, especially due to the higher utilization of computed tomography and nuclear medicine. It is estimated that during 2000–2007, ∼3.6 billion annual medical procedures with ionizing radiation were performed worldwide (69). The average annual per-capita effective dose from medical uses of ionizing radiation was then about 0.6 mSv, with a total of 3.0 mSv received from all sources and has probably increased since (106).
The final significant source for ionizing radiation is cosmic radiation. Although life-forms on Earth are generally well protected from this type of radiation, exposure to cosmic radiation increases with increasing distance from the Earth's surface. Charged particles, such as protons and α-particles, from space collide with atoms of the Earth's atmosphere and generate radiation in the form of photons and other subatomic particles. The atmosphere is, for example, an intense source of γ-radiation (28). The level of the galactic radiation can increase by a factor of 100 when charged particles are released from the Sun by solar flares. The significance of this exposure has increased during times of modern air travel, with the average air crew being exposed to 3–6 mSv annually (3), with the average exposure from all other sources being 3.2 mSv.
The further we go from the protective magnetic field of the Earth, the more dominant the highly energetic and penetrating ions and nuclei of the galactic radiation become (44). Together with solar X-rays, these particles constitute the main health hazard in interplanetary space and are a special concern in prolonged spaceflight, as they deposit more energy in the tissue that they penetrate, compared with other types of radiation (Fig. 1). For example, while the International Space Station (ISS) is still partially protected by the magnetic field of the Earth, so that the astronauts there are exposed to 100–200 mSv of radiation per year, the planned 3-year mission to Mars would dose the astronauts with up to 350 mSv in a single year (1).
Impact of Ionizing Radiation Exposure on Living Tissue
The effects of radiation on living beings have been traditionally classified into deterministic and stochastic effects (47). Deterministic effects manifest as acute damage on living cells and tissues resulting in cell death, ranging from mild skin burns (as witnessed by the early workers) to laceration and systemic radiation poisoning, while stochastic effects almost exclusively result from the consequences of genome damage. The genotoxic properties of radiation were noted early on (52), as they were possible to diagnose from mutagenesis experiments using plants (17) and animals (63, 74).
As these effects can potentially span over generations, genetic consequences have been the most studied and feared. However, apart for certain specialized types of cancer, such as thyroid cancer and leukemia, the additive risk from radiation exposure is surprisingly small, even in atomic bomb attack (47, 62) and nuclear disaster survivors (9, 12, 43, 70), and there seems to be no evidence for cross-generation effects (73). In fact, by all measures, the carcinogenicity of radiation is weak, and any excess cancer risk is low for even the highest survivable doses (106). Similar observations have been made from animals and people in the CEZ, which show no evidence of increased mutation rates (50, 109) or notable increases in DNA damage signaling (51), despite showing other phenotypic signs of radiation exposure (51, 55).
It is noteworthy that there are discrepancies between different epidemiological studies from occupational and environmental radiation exposures. It might be that the relatively low-energy radiation, such as X-rays used in medical imaging, may increase the risk for certain cancers more than high-energy γ-rays (45), which are the predominant type of exposure in both CEZ and atomic bomb survivors. However, as much of the research focus has been on acute and genomic effects of radiation exposure, it is likely that it's more subtle, but physiologically meaningful, impact on cells remains underexplored (73). For example, a correlation between radiation exposure and cardiovascular and kidney disorders has been found, but the causal relationships remain unclear (47, 55).
Radiation damages biological molecules directly, but also indirectly through the generation of ROS. While the direct damage on cell structures and nucleic acids can be important in acute high-intensity exposure, the majority of cell damage is caused by ROS. Most of the tissue of living organisms consists of water molecules (H2O), which are ionized into a range of free radicals, including highly reactive hydroxyl radicals (HO·)(4) (Fig. 2).

The type of radiation influences its linear energy transfer (LET) rate, which has great significance on both cell damage and ROS generation. Ionizing particles with high LET rate transfer more energy to the tissue than low LET electromagnetic photons. For example, 1 μs exposure to the γ-radiation emitted by 137Cs generates 60 molecules of ROS per nanogram of tissue, while one 3.2 MeV α-particle traversal through the tissue will generate ∼2000 ROS molecules (4). ROS in turn can react with a number of important biomolecules, including lipids, proteins, and DNA, causing oxidative damage as well as modifying the normal biochemical reactions in cells.
Besides the oxidative damage and chemical alteration of metabolic substrates, radiation-induced ROS also trigger several different signaling cascades, which monitor and maintain the redox homeostasis of the cell. Compared with the acute effects as well as genotoxic properties of ionizing radiation, its effects on ROS signaling have been understudied. Some studies from Chernobyl have reported profound effects on wildlife already at dose rates as low as a few μSv/h, although this seems to be organism-dependent, and there is an overall controversy over the causality of population-level effects in the CEZ (9).
Interestingly, ROS stress has been implicated in some of the pathological changes observed in animals in the CEZ, including pigmentation defects and cataract (11, 12, 72). However, the ability of ionizing radiation to induce physiological reactions, such as elevation of antioxidant defenses, has been questioned (97). In fact, the source and context of ROS exposure play a major role in the downstream effects (35, 37, 49, 68, 83, 91, 98). A chronic increase in ROS could mimic developmental signals that induce cell- or organ-level physiological changes, whose interference with elevated antioxidant defenses alone might be maladaptive (81, 88, 98).
It is therefore likely that the developmentally important ROS signaling responses and their physiological outcomes play a crucial role both in adaptation and pathological changes occurring during chronic exposure to ionizing radiation. Also, radiation exposure might not necessarily manifest as a pathological phenotype, but rather as a transformed steady state in cellular physiology.
Universal Triggers for Radiation Stress Response
As pointed out earlier, organisms on Earth are normally not exposed to physiologically meaningful levels of ionizing radiation. Therefore, they are [with few exceptions (26)] not adapted to ionizing radiation. Instead, their response to ROS from ionizing radiation is likely resembling the response to more typical intracellular or environmental sources of ROS. It is noteworthy that while direct damage of DNA might occur from the ionizing radiation, the overwhelming stimulus triggering a DNA damage response for the cell is likely to come from the ROS damage. Furthermore, the whole redox state of the cell can be altered by rather low levels of radiation, which are unlikely to cause direct damage on cellular components.
Under normal conditions, the main source for cellular ROS is the ETC of the mitochondria (75, 81), although inflammation reactions can also constitute a major source for oxidative stress in tissues (88, 98, 99). Redox signaling and oxidative stress reactions are further complicated by the fact that different ROS molecules have different characteristics, affecting their half-life, diffusion rate, and reactivity with biological molecules. It is generally agreed that superoxide is the most important ROS molecule produced by the ETC (13).
Superoxide has a relatively long half-life and is diffusible, enabling it to affect also other cellular compartments besides the mitochondria. Superoxide is also able to convert to hydrogen peroxide, and both can produce highly reactive hydroxyl radicals via the Haber–Weiss and Fenton reactions (24). Notably, the same ROS molecules are also produced by ionizing radiation (Fig. 2). However, unlike in the case of ionizing radiation, organisms are adapted to the intrinsic ROS sources by a plethora of molecular triggers and signaling cascades. Intrinsic ROS also constitute an important source of physiological information.
For example, mitochondrially produced ROS increase upon suboptimal ETC function, caused by the sudden increase in energy requirement (81, 83), transient hypoxia (37), or dysfunctional respiratory complexes (57, 92). For cells, an increase in intracellular ROS is therefore usually a signature of mitochondrial dysfunction or increased energy demand, to which they respond accordingly. Cellular responses to ROS are complicated by the fact that the type, location, severity, as well as the duration of the exposure, all play a role in the redox signaling (35, 91). Interestingly, despite decades of work on the effects of ionizing radiation on cells, fairly little is known about the signaling outcomes of radiation-induced ROS.
Many studies have focused on the biomarkers of oxidative stress, such as accumulation of lipid peroxidation products or the pathology of late effects of ROS from ionizing radiation (113). Due to the importance of the medical use of ionizing radiation, studies have also largely focused on the effects after a large acute dose. Similarly, studies on chronic radiation exposure, while acknowledging the role of ROS, have focused on biological endpoints (1, 9, 44, 51, 106) such as chromosome aberrations, impairment of cognitive skills, and increased eye lens opacity. Although radiation-induced ROS signaling is poorly known, some inference can be drawn from the wealth of literature on other types of redox stress and the comparison of endpoint outcomes in these cases.
ROS and Cellular Stress Responses
As pointed out earlier, while ROS can trigger several molecular signaling pathways in cells, the duration and the context of the insult play a major role in the outcome. For example, the well-known tumor suppressor protein p53 (p53 or TP53) is a master regulator of many types of cellular stress responses, including oxidative stress (64). Under mild oxidative stress, p53 contributes to the redox homeostasis by upregulating antioxidant defenses, whereas under persistent and/or severe stress, p53 is hyperactivated, resulting in apoptosis or cellular senescence.
As whole-body apoptosis is not a viable option during systemic oxidative stress, the cell fate decisions need to have inputs also from other prosurvival pathways, where also the tissue context plays an important role. For example, heterozygous superoxide dismutase 2 (Sod2) knockout mice develop cardiomyopathy when aging without showing many other symptoms (107). As SOD2 is the mitochondrial superoxide dismutase, specifically involved in the dismutation of ETC generated superoxide to hydrogen peroxide, the heart phenotype underscores the importance of tissue differences in the tipping points of redox stress.
Although the lowered levels of the SOD2 enzyme activity in the heterozygous knockout animals are enough to maintain the redox homeostasis in most tissues, the heart is affected due to the naturally high levels of ROS caused by the active oxidative metabolism in cardiomyocytes. Although the hearts of these mice show massive upregulation of p53 (82), the cardiomyocytes are lost only gradually, apparently due to age-related tipping points caused by the deteriorating cardiomyocyte function.
Other ROS stress-responsive transcription factors include hypoxia inducible factor 1α (HIF1α), nuclear factor erythroid 2-like 2 (NRF2 or NFE2L2), as well as a whole range of immunomodulatory factors, which themselves can be ROS sensors (HIF1α) or work downstream from the initial stress trigger (81). Many of these contribute to the restoration of redox homeostasis by upregulating antioxidant defenses, modulate cell fate decisions, or connect physiological ROS signals with developmental gene regulation (81).
The endoplasmic reticulum (ER) and mitochondrial stress response pathways are highly sensitive redox stress sensors, signaling either independently or by converging with the integrated stress response (ISR) pathway (66). The ER is specialized to protein folding and posttranslational maturation of membrane proteins as well as secreted proteins. The disulfide bond formation during protein folding is highly sensitive to the redox state of the ER lumen, and oxidative stress will result in accumulation of mis- or unfolded proteins (21).
The accumulation of unfolded proteins induces an unfolded protein response (UPR), an evolutionarily conserved stress response pathway functioning to restore the homeostasis for protein folding. The main sensors for the UPR in the ER are the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 α/β (IRE1α/β), and activating transcription factor (ATF) 6 (Fig. 3). These transmembrane proteins are inactive in their basal state, when their ER luminal domain is bound by the ATPase domain of the binding immunoglobulin protein (BiP or GRP78) (Fig. 4). Interaction of BiP with unfolded proteins in the ER lumen dissociates it from three sensor proteins. Reduced interaction with BiP causes PERK to oligomerize, resulting in its autophosphorylation (66).

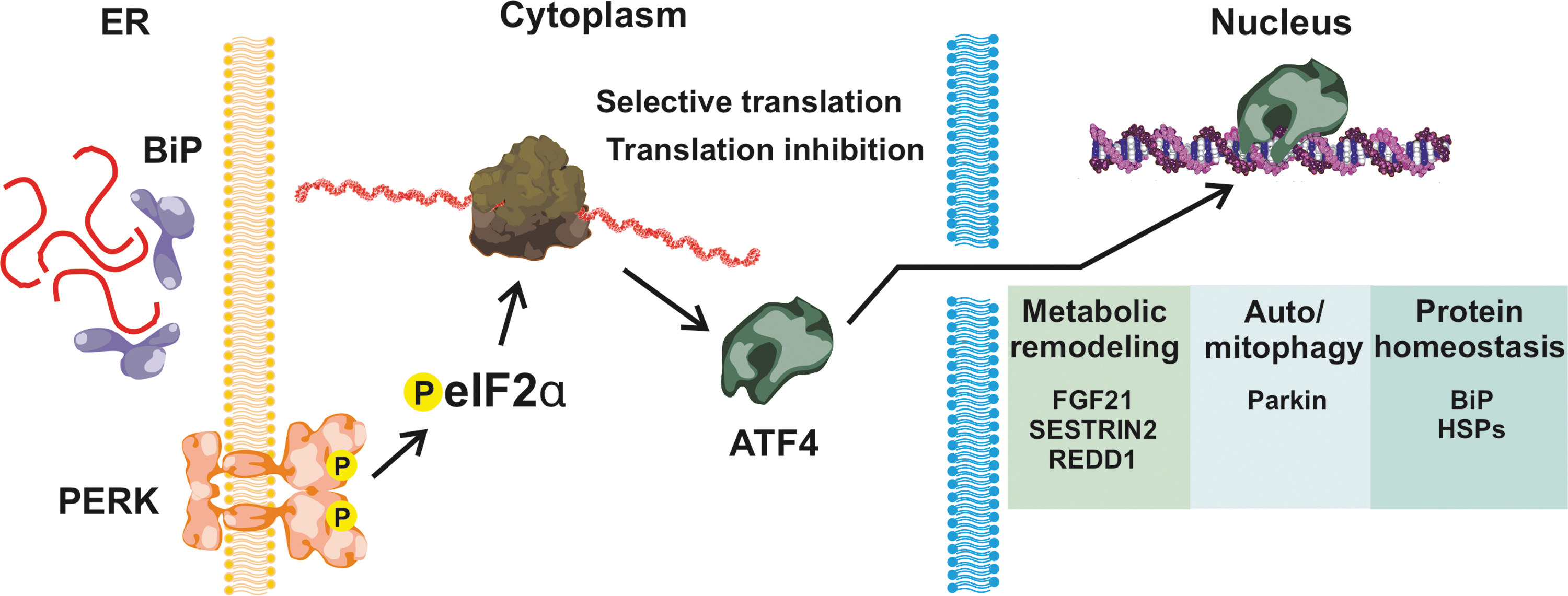
Activated PERK phosphorylates the eukaryotic initiation factor 2α (eIF2α), causing the cessation of translation for most transcripts, except those having a typical short open reading frame at their 5´-UTR, such as the basic leucine zipper protein family member ATF4. PERK activation also increases the elimination of nonfunctional or damaged proteins, stimulating autophagy by triggering NRF2 release from Kelch-like ECH-associated protein 1 (KEAP1) (111). This step is important for the selective autophagy controlled by the sequestosome 1(p62/SQSTM1)-KEAP1-NRF2 pathway in a mechanistic target of rapamycin complex 1 (mTORC1)-dependent manner (48).
Similarly to PERK activation, dissociation of BiP from IRE1α results in its oligomerization and activation via autophosphorylation (66). The cytosolic domain of the activated IRE1α is a ribonuclease, which degrades ER-localized mRNAs through a process known as regulated IRE1-dependent decay. IRE1 also specifically splices an intron of the X-box binding protein (XBP1) mRNA, increasing its translation. XBP1 is a transcription factor controlling several target genes, whose products modulate protein secretion, cell survival, apoptosis, and DNA repair.
The dissociation of ATF6 from BiP causes its translocation to the Golgi, followed by cleavage by site-1 (S1P) and site-2 (S2P) proteases (66). The proteolytic cleavage makes ATF6 an active transcription factor, translocating to the nucleus and inducing the transcription of a number of target genes, including XBP1. There is evidence that ionizing radiation might selectively activate the eIF2α/ATF4 branch of the UPR (54), but further studies are needed.
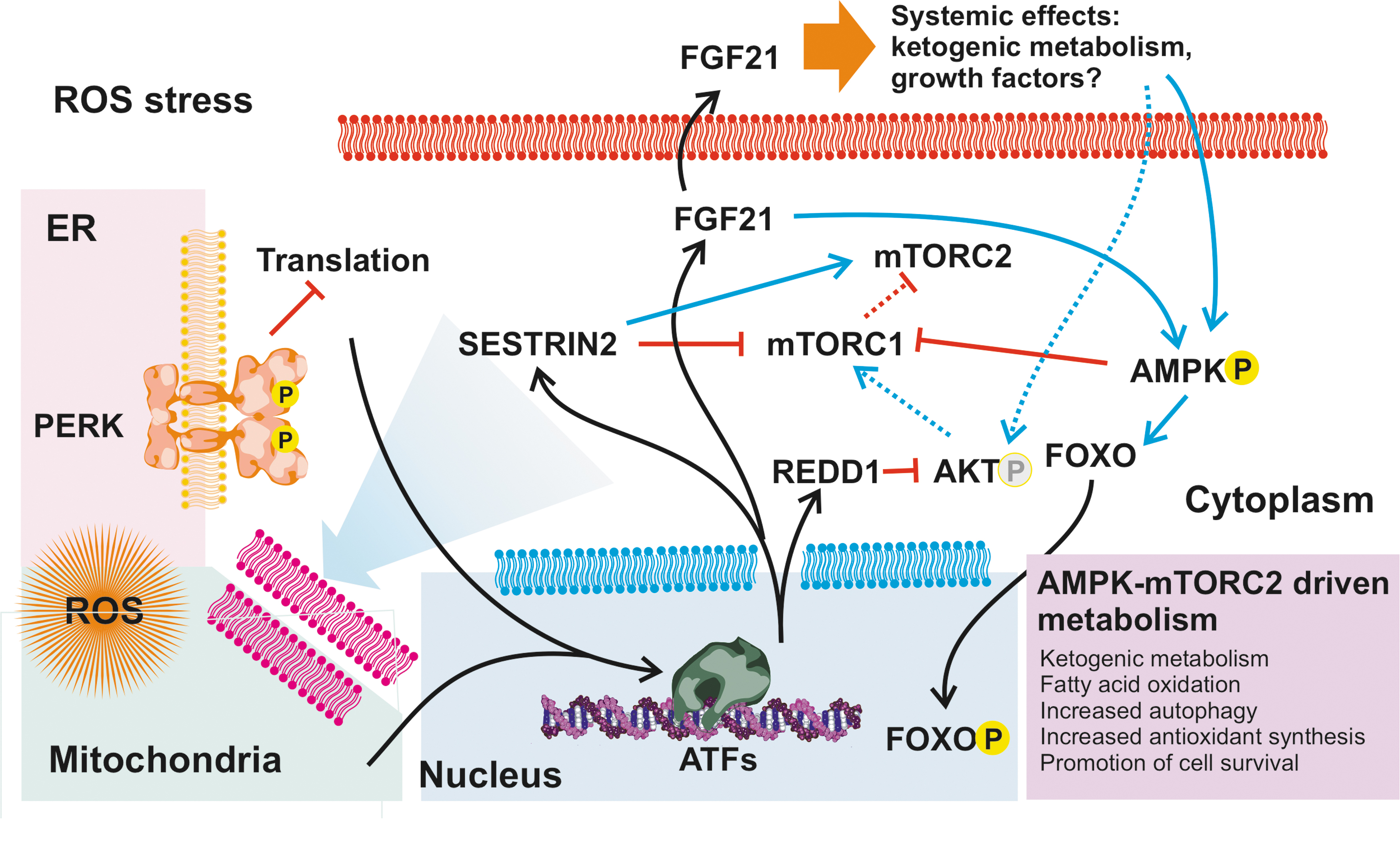
PERK activation plays also a role in both protein kinase B (AKT)-mediated (66) and mitochondrial prosurvival signaling (65) (Fig. 4). ATF4 targets include REDD1 and sestrin-2 (SESN2), both of which decrease AKT phosphorylation either directly (29) or via a switch from mTORC1 to mechanistic target of rapamycin complex 2 (mTORC2) signaling (19), promoting cell survival over proliferation and contributing to metabolic remodeling (61, 66) (Fig. 5), as discussed later. Activated PERK can localize to mitochondria-associated ER membranes, and evidence suggests that this influences mitochondria-mediated cell survival (108). In addition, ATF4 induces the expression of Parkin, a protein that mediates autophagy of mitochondria (mitophagy) and contributes to the maintenance of mitochondrial homeostasis (41).
Mitochondria have their own compact circular genome, containing genes for 13 subunits of the ETC and adenosine triphosphate (ATP) synthase, as well as tRNAs and rRNAs required by mitochondrial protein synthesis (80). Consequently, also mitochondria have their own UPR (UPRmt) (96). Imbalance between mitochondrial and nuclear-encoded oxidative phosphorylation (OXPHOS) subunits, ROS stress, and other perturbations in the mitochondrial proteostasis are known to activate UPRmt, although the mechanisms are not yet fully known. Mitochondrial UPR causes activation of basic leucine zipper domain (bZIP) transcription factor C/EBP homologous protein (CHOP) and another ATF (ATF5), which is translocated from the mitochondria to the cytosol and nucleus (93, 96).
ATF5 target genes are typically required for mitochondrial recovery from proteostatic stress, including chaperons, proteases, as well as factors promoting cell survival (93). ATF5 is highly interesting for chronic radiation stress, as besides its role in stress response, it is also an important factor for cell differentiation and organ development (93). As of note, it is closely interconnected with the eIF2α phosphorylation-mediated ISR (2), with both having overlapping target genes and a role in mitochondrial ROS-induced ER stress.
A common target for all three ATFs is the fibroblast growth factor 21 (FGF21). FGF21 is a peptide hormone and upregulated by various stress signals, with effects depending on the expressing tissue (34). FGF21 can work as an endocrine, but also paracrine, factor regulating metabolism, typically increasing oxidative metabolism, such as β-oxidation of fatty acids, at the expense of carbohydrates. A possible explanation for this feedback is that during tissue development (79) or changing environmental conditions (85, 89), ROS stress is typically caused by suboptimal mitochondrial OXPHOS, which can be restored by increasing mitochondrial biogenesis.
This might also explain why FGF21 is upregulated in a number of mitochondrial diseases (36, 105) and, at least in part, could contribute to the metabolic changes seen in both patients and mouse models (53). As of note, ROS-induced metabolic remodeling is sometimes attributed to other factors, such as HIF1α (112). While there clearly is convergence of and interconnections between the different ROS sensing pathways, metabolic remodeling with a marked increase in ATFs and FGF21 levels is expected to be UPR-elicited.
Parallels Between Responses to Oxidative Stress, Spaceflight, and Radiation Exposure
As pointed out earlier, only few systematic studies address the effects of chronic radiation exposure on oxidative stress responses. However, a recent multiomics study on the impacts of spaceflight revealed transcriptional and metabolic changes (25), which share a resemblance to the situation in mitochondrial myopathies (53, 105). Interestingly, the activation of ISR and UPRmt during spaceflight is tissue-specific (25), which most likely relates to the basal metabolic differences between the tissues and provides clues about the tissue-specific effects of radiation exposure.
Strikingly, spaceflight-induced stress resulted in a metabolic switch from carbohydrate oxidation to β-oxidation of long-chain fatty acids and an overall increase in tricarboxylic acid cycle (25), all indicators of increased oxidative metabolism rate. Consistent with elevated mitochondrial biogenesis, the study also found elevated levels of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a, PGC1α) in various tissues of mice exposed to prolonged spaceflight.
Although radiation exposure is not the only stress source during a spaceflight, the elevated levels of certain biomarkers, such as urinary 8-hydroxydeoxyguanosine (8-OHdG), indicate that oxidative stress is a major contributor to the physiological effects of spaceflight. Analogous to these spaceflight studies, also bank voles (Clethrionomys glareolus, a small rodent species) from the CEZ show a correlation between ionizing radiation exposure, oxidative stress markers, and increased lipid catabolism (51). If the oxidative stress caused by ionizing radiation is signaled by the same pathways as the stress caused by ROS released from dysfunctional mitochondria, radiation exposure should phenocopy the metabolic changes seen in mitochondrial disorders (53).
Circadian Rhythms and Oxidative Stress from Radiation
Another striking finding in the study on the biological effects of prolonged spaceflight was the upregulation of circadian rhythm pathways in all internal organs except the liver, although the experimental animals were maintained in a constant 12/12-h light/dark cycle (25). As many variables might affect the circadian genes in space, the authors did not explore the connection between the circadian rhythm and oxidative stress from radiation. The circadian rhythm or the so-called biological clock is a fundamental biological phenomenon in all known organisms, referring to innate oscillation of gene expression, metabolic flux rates, signaling molecules, and even cellular structures, with a rhythmic period of approximately one solar day (33).
Circadian rhythms are genetically determined by several genetic clocks, whose purpose is to adapt organisms to anticipated, daily occurring environmental changes by synchronizing physiological processes, cell cycle, and energy metabolism. This internal clock needs to be synchronized between different tissues and with the environmental time. While it has been long known that the redox state of the organism changes over the circadian time (42), its significance has not been fully appreciated until fairly recently (33, 102, 103). Although the precise mechanism remains unclear, ROS stress can reset the circadian rhythm in many organisms (40, 102, 103).
While ROS-elicited resetting of the expression rhythm of the circadian gene period 2 (Per2) has been suggested to promote prosurvival pathways controlled by heat shock factor 1 (HSF1), NRF2, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (102), these are also targets for other types of redox signals, and not much can be concluded about the biological significance of the resetting.
So far, it is unclear what the systematic upregulation of the circadian clock genes might mean in the context of chronic radiation exposure. Upregulation of circadian genes per se can be seen as countersynchronization, and while it can provide protection against radiation stress (27), for example, by boosting antioxidant defenses and repair, it could represent a maladaptive response to increased oxidative stress at the organism level. After all, the sensitivity of the circadian clock to the normal daily fluctuations in the redox state provides a natural synchronization input for the clock genes, and any disturbances in the internal timing result in problems with cell cycle control, apoptosis, and life span shortening (42, 95, 103).
Desynchronization of the circadian clock has been associated with the disadvantageous health effects of shift work and jet lag, which might result in a higher risk of cardiovascular diseases such as myocardial infarction and increase in coronary events (7). In the context of space travel, the dysregulation of the circadian rhythm has been suggested to explain some of the hormonal and sleep cycle-related abnormalities in astronauts (25). While the effects of chronic radiation exposure on the circadian genes still needs to be explored, for example, in the CEZ setting, several studies suggest that the circadian rhythm can be utilized to improve the effectiveness and reduce the side effects of radiotherapy (22, 27).
Turtles All the Way Down: Metabolic Adaptation or a Radiation Syndrome?
As pointed out earlier, cellular stress responses to increased ROS result in metabolic remodeling, an increase in mitochondrial biogenesis, and a ketogenic energy metabolism. The metabolic switch to more oxidative metabolism (25, 51), at least in some tissues, might mimic glucose deprivation, driving further metabolic reprogramming via AMP-activated protein kinase (AMPK) and promoting catabolism (glucose uptake, fatty acid metabolism, and increased autophagy) (114). The AMPK pathway is also specifically activated by FGF21 through fibroblast growth factor receptor 1 (FGFR1)/β-klotho signaling or by stimulating the secretion of the secretion of adiponectin and corticosteroids (90) (Fig. 5).
It is noteworthy that the AMPK signaling converges with the effects of ATFs and FGF21-mediated metabolic remodeling, including the interconnection with mTOR-controlled pathways (53, 114), and also with the prosurvival pathways under the circadian genes (56, 89, 95, 114), meaning that the causes and the consequences along the signaling cascades cannot be distinguished by looking at the endpoint gene expression or metabolism. Notably, the AMPK–AKT pathways are also important epigenetic modulators via sirtuin proteins (20, 39, 46), offering a possible explanation for the genome methylation and gene regulation changes observed in astronauts (1), as well as in animals and people exposed to radiation (67).
It is noteworthy that fibroblasts isolated from radiation-exposed voles from the CEZ show increased resistance to oxidative stress, even when being cultivated in conditions of normal background radiation (76). While it is possible that natural selection has adapted the voles to the higher background radiation levels of the CEZ during the last 30 years, this seems unlikely due to the geographical patterning of the fallout (varying and incremental fitness effects over a small geographic scale) and free gene flow between contaminated and uncontaminated areas. It is more plausible that the radiation stress has epigenetically primed the voles in the contaminated areas to absorb ROS stress better, and the investigation of this phenomenon might reveal interesting aspects about adaptational mechanisms as well as cross-generational effects of radiation exposure.
Studies from the CEZ (9) as well as space (1) have shown that animals appear remarkably resilient against low levels of ionizing radiation. While many of the physiological changes are reversible once the stress is removed (1), the seemingly subtle changes in gene regulation and metabolism might induce long-standing consequences. Many of the components of the oxidative stress response pathways have roles in cellular differentiation (23, 93) and tissue development (114). Especially brain development is highly sensitive to radiation exposure (45).
Generally, radiation-exposed animals have smaller brains and declined cognitive skills (1, 45, 55, 71, 104). This is probably not directly due to the death of brain cells due to the radiation, but more likely due to perturbation of normal developmental signals during organ development. For example, ROS have an important role as signaling molecules in neuronal development and function, guiding neuronal polarity and pathfinding (77). Similarly, metabolic regulators affected by oxidative stress, such as AMPK (87), have important roles in brain development and functional maintenance. Notably, FGF21-mediated metabolic remodeling influences the one-carbon metabolism (30, 36), producing an excess of formate, an important developmental regulator in many tissue types (15).
In adults, some tissues such as the heart and kidney might be sensitive to radiation due to their naturally high oxidative environment (47, 70, 106). In these cases, the situation would be similar to the heterozygous Sod2 knockout mouse discussed earlier (82, 107), where metabolically highly active cells are unable to handle any extra ROS burden from radiation. The postmitotic nature of these cells contributes to the injury as the damage caused by cell loss is permanent.
Finally, metabolic remodeling alone can have systemic effects by developing into a starvation phenotype (101, 105), which might explain some of the phenotypic features in the animals living in the CEZ (12, 51, 55, 60, 104). Although the physiological changes caused by the radiation exposure can be life span expanding (53, 89) and not be considered pathological per se, allocating resources to the maintenance of redox homeostasis and overall cellular survival means that these resources are not available for other important purposes, such as reproduction.
Mitigating the Impacts of Radiation Stress
As stress from ionizing radiation is essentially oxidative stress, at least some of the adverse effects of radiation exposure can be mitigated by administration of antioxidants (16, 84), a fact potentially relevant for clinical use (14). Our poor understanding of the causes and consequences of radiation-related physiological and metabolic changes currently hinders the development of more targeted pharmacological solutions.
Therefore, it is important that future studies investigate the effects of space and environmental radiation on the different signaling pathways on protein levels and try to dissect the relationships between the different affected pathways. For example, both the UPR and AMPK–AKT signaling axes are highly relevant in the context of ROS and radiation exposure, as they not only contribute to the maintenance of redox homeostasis, cell survival, and developmental gene regulation, but also offer putative pharmacological targets to mitigate the effects of radiation exposure. For example, the mTOR inhibitor rapamycin (53) and the AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (78), both seem to improve some of the pathological alterations seen in mitochondrial dysfunction.
However, it should be emphasized that as the metabolic outcomes of the radiation stress represent cellular adaptations, metabolism-altering drugs can also worsen the stress outcome. For example, the mitochondrial biogenesis activator and pan-peroxisome proliferator-activated receptor (PPAR) agonist bezafibrate have been considered an interesting option to improve mitochondrial function and redox homeostasis in several disorders (31). While being potentially beneficial in the short term, long-term use of bezafibrate seems to phenocopy the effects of metabolic dysregulation seen in mitochondrial dysfunction (100).
An interesting possibility to buffer the negative effects of ionizing radiation would be to take advantage of the body's own ability to adapt. It has been long known that exposure to low levels of ionizing radiation might offer protection against subsequent insults (6, 94, 106). This phenomenon, also known as radiation hormesis, is evident in the improved ROS resistance of the CEZ voles (76) and could be biologically linked to epigenetic modifications, as pointed out earlier. Consequently, participants of a 3-year space mission to Mars might benefit from prior exposure to space radiation. It is obvious that the existence of hormetic health benefits would need to be experimentally verified in animal and human studies and weighed against other health risks of radiation before their practical application.
Conclusion
Oxidative stress is likely the main cause of the physiological alteration seen in men and animals exposed to ionizing radiation (Fig. 6). As oxidative stress triggers many types of converging and intersectional cellular responses, the causality of different endpoint results can be difficult to pinpoint. However, correlative evidence between FGF21 expression and metabolic remodeling suggests that ATF-mediated stress responses play an important role in cellular response to radiation. In general, these stress responses promote cell survival under radiation stress, but as a trade-off result in metabolic remodeling, which quite likely has detrimental physiological effects at the organism level.

Better understanding of the metabolic switches as well as other adaptive mechanisms in cells, such as radiation hormesis, could help to understand the long-term consequences of radiation exposure and enable the development of new radioprotective strategies.
Footnotes
Authorship Confirmation Statement
Conception and design: J.L.O.P. Drafting and revising critically for important intellectual content: J.L.O.P. and S.G. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: J.L.O.P. and S.G.
Acknowledgments
We thank Dr. Jenni Kesäniemi and Prof. Phillip Watts, University of Jyväskylä, for giving the inspiration to explore the topic. The Jane and Aatos Erkko foundation and the Finnish Cultural foundation are thanked for their generosity in supporting our mitochondrial hormesis project.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Some of the ideas presented in this article originate from the mitochondrial hormesis project supported by the Jane and Aatos Erkko foundation (project 1/2016 to J.L.O.P.) and the Finnish Cultural Foundation (project 1/2018, S.G.).