
Abstract
Significance:
Cancer-associated tissue-specific lactic acidosis stimulates and mediates tumor invasion and metastasis and is druggable. Rarely, malignancy causes systemic lactic acidosis, the role of which is poorly understood.
Recent Advances:
The understanding of the role of lactate has shifted dramatically since its discovery. Long recognized as only a waste product, lactate has become known as an alternative metabolism substrate and a secreted nutrient that is exchanged between the tumor and the microenvironment. Tissue-specific lactic acidosis is targeted to improve the host body's anticancer defense and serves as a tool that allows the targeting of anticancer compounds. Systemic lactic acidosis is associated with poor survival. In patients with solid cancer, systemic lactic acidosis is associated with an extremely poor prognosis, as revealed by the analysis of 57 published cases in this study. Although it is considered a pathology worth treating, targeting systemic lactic acidosis in patients with solid cancer is usually inefficient.
Critical Issues:
Research gaps include simple questions, such as the unknown nuclear pH of the cancer cells and its effects on chemotherapy outcomes, pH sensitivity of glycosylation in cancer cells, in vivo mechanisms of response to acidosis in the absence of lactate, and overinterpretation of in vitro results that were obtained by using cells that were not preadapted to acidic environments.
Future Directions:
Numerous metabolism-targeting anticancer compounds induce lactatemia, lactic acidosis, or other types of acidosis. Their potential to induce acidic environments is largely overlooked, although the acidosis might contribute to a substantial portion of the observed clinical effects. Antioxid. Redox Signal. 37, 1130–1152.
Introduction
Nearly two centuries ago, in 1843, the German physician and chemist Johann Joseph Scherer (Fig. 1a) demonstrated the postmortal
However, the causative link between leukemia and the presence of lactic acid in blood appeared controversial (125). Indeed, in 1878, Georg Solomon from Berlin provided evidence of the presence of lactic acid in the blood of patients with other pathologic conditions, including pernicious anemia, congestive heart failure, chronic obstructive pulmonary disease, pleuritis, pericarditis, pneumonia, and a variety of solid tumors (188).
A century ago, in 1925, S. W. Clausen from St. Louis (MO) identified lactic acid accumulation as a cause of acid-base disorder (38). More recently, in 1978, the Canadian-American physiologist Peter A. Stewart from Providence (RI) postulated the physicochemical approach to acid-base physiology, which suggests that lactate has an acidifying effect similar to that of chloride. Lactate, therefore, causes a decrease in the strong ion difference, which can be calculated as [Na+] + [K+] + [Ca2+] + [Mg2+] - [Cl−] - [lactate] (156). Thus, according to the electroneutrality law, lactate causes a proportionate increase in [H+] and therefore acidosis (186). For every 1 mM increase in lactate, the standard base excess decreases by one unit (104). Currently, lactic acidosis is recognized as the most common form of metabolic acidosis.
In addition to metabolic acidosis, several other types of acidosis exist, including diabetic acidosis (due to the accumulation of ketones), hyperchloremic acidosis (due to loss of sodium bicarbonate), and renal tubular acidosis (due to limited secretion of acids into the urine). Another unrelated type of acidosis is respiratory acidosis, which occurs when CO2 is not eliminated from the body and carbonic acid accumulates in the blood. A decrease in the intracellular pH of brain cells is characteristic of epilepsy and MELAS (mitochondrial myopathy, encephalopathy with lactic acidosis, and stroke episodes) (4). Acidosis is often associated with pain and increased nociception, as the primary nociceptors are acid-stimulated ion channels (211).
To diagnose systemic
Type A typically manifests in patients with ischemic bowel syndrome, sepsis, cardiogenic shock, and hypovolemia. Type B lactic acidosis manifests despite aerobic conditions and is found in well-ventilated patients. Type B typically manifests in patients with hematologic malignancies, human immunodeficiency virus infection, diabetes mellitus, liver disease, thiamine deficiency, and hereditary mitochondrial enzymatic deficiencies and as a side effect of treatment with various compounds, including metformin (69). Type D-lactic acidosis is caused by excessive production of D-lactic acid by proliferating intestinal bacteria and is commonly noted in patients with short bowel syndrome or other forms of gastrointestinal malabsorption (138).
Systemic Lactic Acidosis in Patients with Cancer
Cancer-associated systemic lactic acidosis mostly manifests in patients with aggressive lymphomas and leukemias (e.g., 66, 198). Only sporadic cases have been reported from a broad range of other cancer types (52).
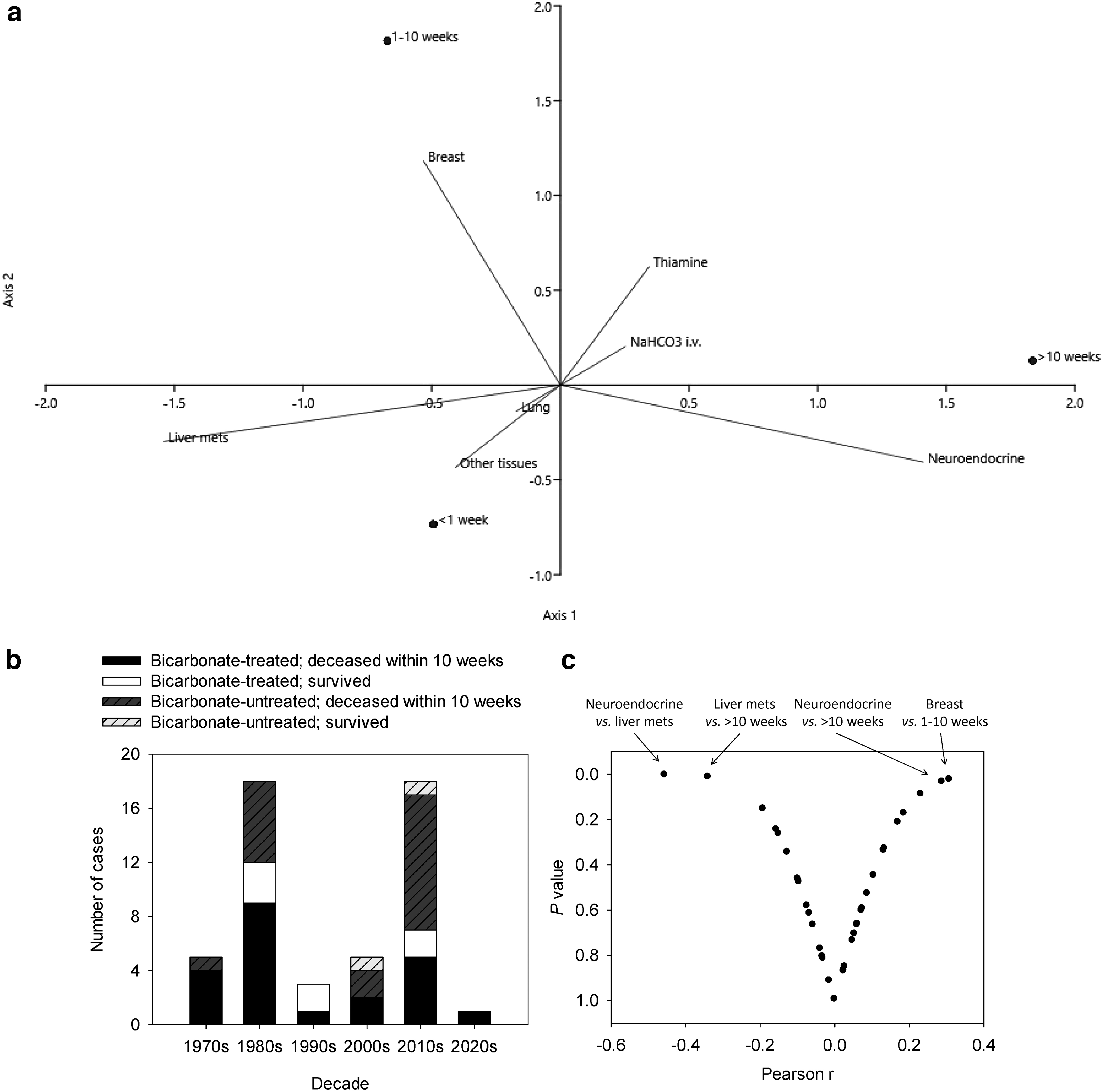
Cancer-associated lactic acidosis is occasionally resolved with the regression of the tumor, including its metastases, as reported by Rice & Schwartz (177) or, more recently, by Imperiale et al. (115). However, successfully resolved lactic acidosis cases are mostly associated with neuroendocrine tumors, particularly with pheochromocytochroma.
The β agonism of catecholamines increases blood glucose levels (by increasing glucagon and adrenocorticotropic hormone secretion as cortisol decreases tissue uptake of glucose) and increases lipolysis (100). Catecholamines also induce vasoconstriction, which results in peripheral tissue ischemia and thus may induce anaerobic glycolysis in peripheral tissues. Importantly, catecholamines are more effective in glycogenolysis and glycolysis stimulation than in gluconeogenesis stimulation. The difference in increases in glycogenolysis and gluconeogenesis is significant (approximately three orders of magnitude). This difference is responsible for the excessive production of lactate in many patients with pheochromocytochromas, particularly in epinephrine-secreting pheochromocytomas.
The pathogenesis of the
The mechanisms of wound healing resemble those exploited by proliferating tumors. This finding was reported in 1924 by Montrose T. Burrows (23), although it became famous only after Harold F. Dvorak coined the phrase
When these patients were stratified into those in whom arterial lactate decreased by at least 60% during the first 6 h and those in whom arterial lactate decreased by less than 30%, the first group had a mortality rate of 7.5%, whereas the latter group had a mortality rate of 30% (157). Therefore, systemic lactic acidosis is associated with poor prognosis in patients with both cancer and trauma.
Tissue-Specific Lactic Acidosis in Solid Cancer
Cancer-associated tissue-specific lactic acidosis has many parallels with wound-induced tissue-specific lactic acidosis. Wounds are characterized by transient ischemia-driven tissue acidification, which resolves as soon as the wound heals and the vascular supply is reinstated (8). The physiological effects of wound acidification are unclear; some studies suggest increased production of inflammatory cytokines by the surrounding endothelial or stromal cells (58).
These factors induce angiogenesis. Then, acidosis is resolved, and inflammation disappears. However, in the tumor environment, acidification never resolves. Thus, regarding tissue-specific acidosis, the tumors clearly resemble wounds that do not heal. Because acidification never resolves, T cells are unable to be activated unless the extracellular pH is artificially increased (166). Therefore, tissue-specific lactic acidosis in cancer is a potential target of future therapies exploiting ways to improve the host body's own anticancer defense.
Aerobic glycolysis, which is characteristic of proliferating tumor cells, leads to the overproduction of lactate and acidification of the
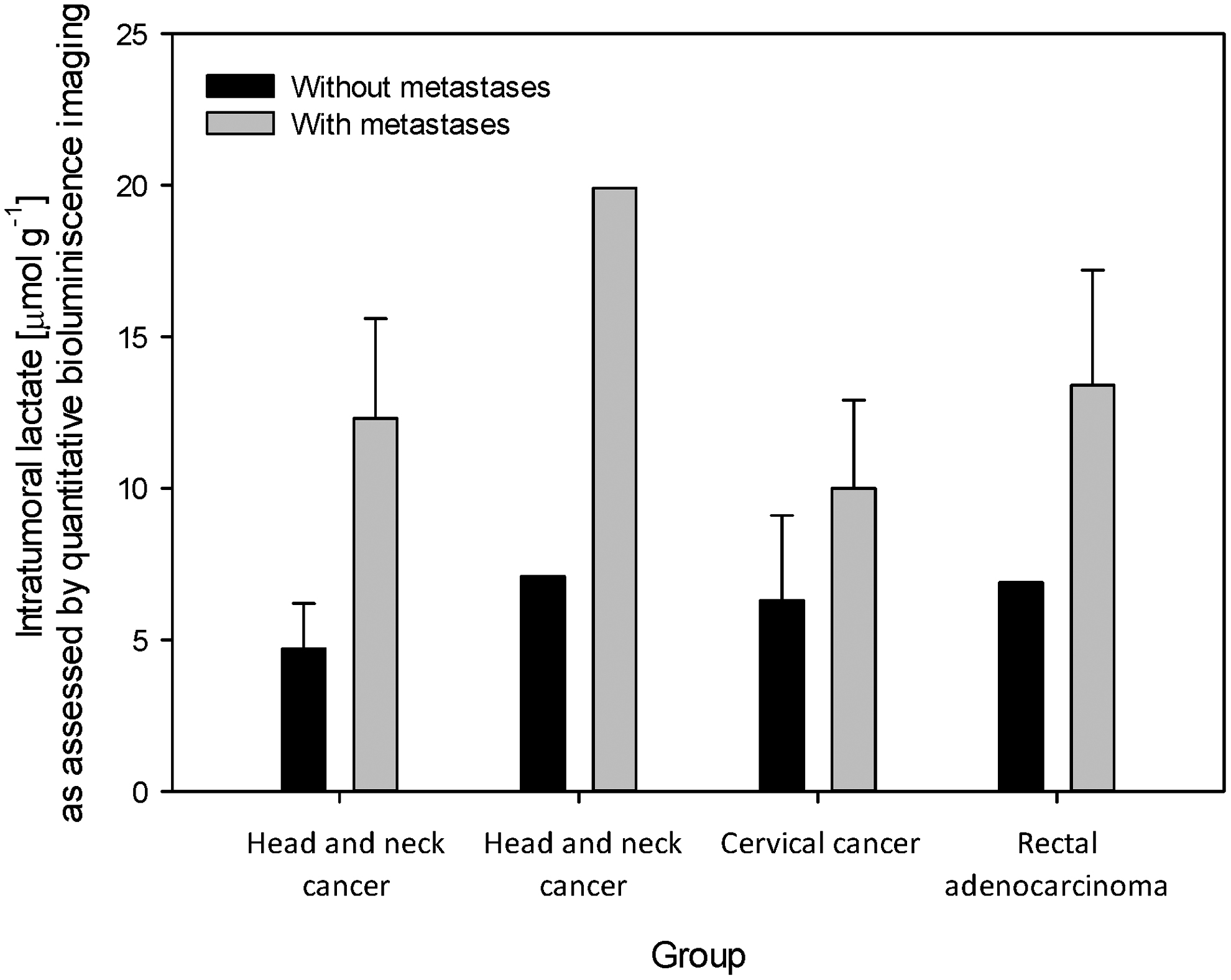
The mechanisms include remodeling of the extracellular matrix (67), angiogenesis stimulation by vascular endothelial growth factor release (240), suppression of T cell activity, and induction of macrophage phenotypic switch toward the M2 phenotype (26, 132, 166). In response to the acidic environment within the tumor, infiltrating T cells suppress interferon-γ translation and can be activated by simply increasing the extracellular pH (166). Cancer cells stressed by acidic environments may adapt by increasing genomic instability (89) and epigenetic changes (123).
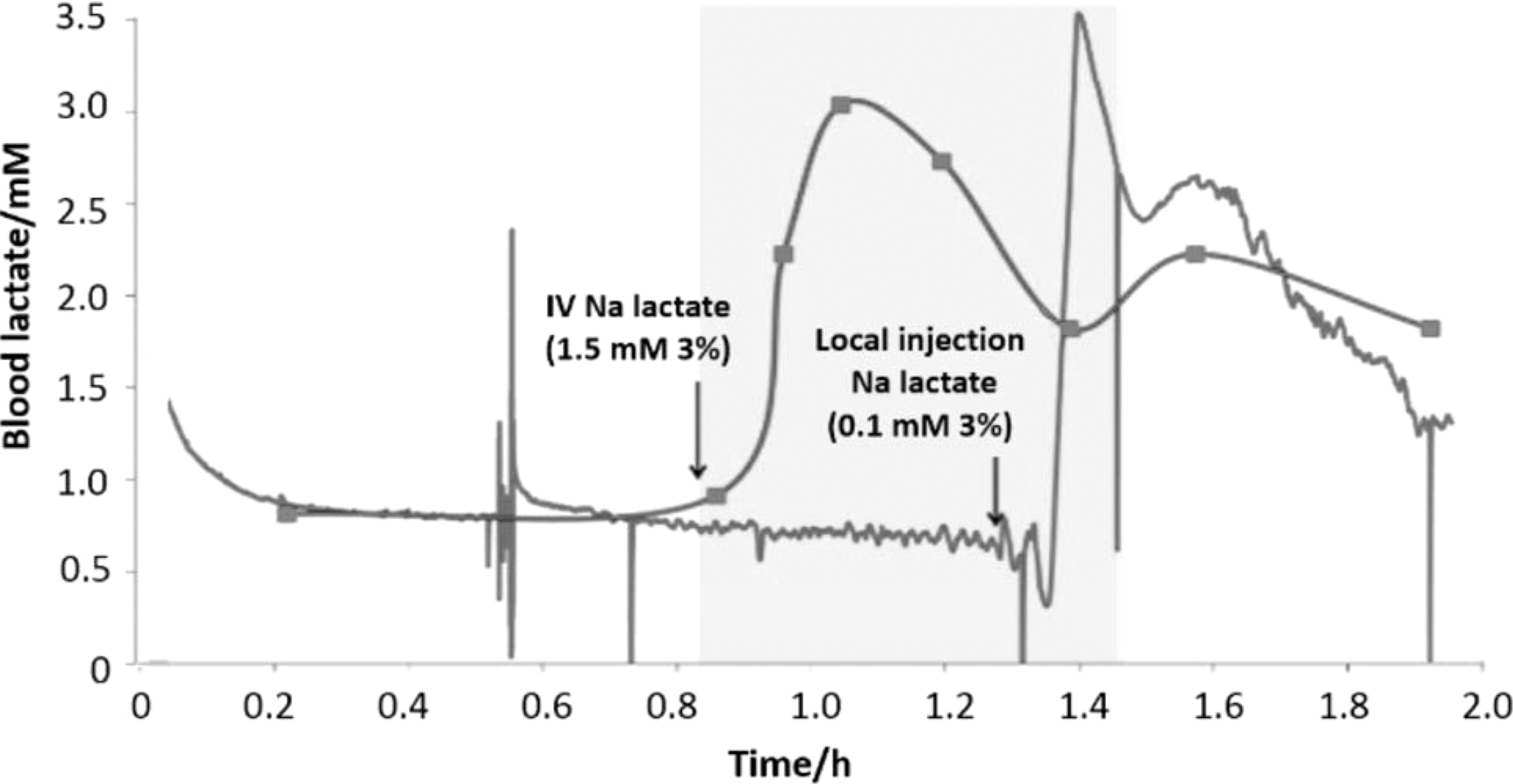
The difficulty of treating tissue-specific lactic acidosis can be illustrated by the fact that the increase in systemic lactate is not projected within hours in the interstitial fluid (Fig. 4) (205). Therefore, the organism is capable of compensating for large-scale systemic changes in lactate, and these changes minimally affect tissue-specific lactate levels.
Treatment of Solid Cancer-Associated Systemic Lactic Acidosis
The treatment of lactic acidosis associated with solid tumors often consists of
This finding corresponds to the fact that treatment with sodium bicarbonate is considered controversial since it may induce CO2 accumulation in tissues and thus further promote acidosis and worsen treatment outcomes (120). In addition, lactate production itself can be stimulated by sodium bicarbonate therapy (76), which corresponds to the results from experimental animals with phenformin-induced lactic acidosis that were treated with bicarbonate. In this model, blood lactate increased, liver and erythrocyte pH declined, and mortality did not change (7).
The administration of bicarbonate represents the most common form of direct neutralization of lactic acidosis. Although sodium bicarbonate administration has limited prosurvival effects when treating systemic lactic acidosis, the results are more promising when targeting tumor pH and metastasis formation. In experimental studies in animal models, orally administered sodium bicarbonate effectively suppressed tumor metastatic activity and increased tumor pH, whereas it did not affect systemic pH (142, 143, 170, 182, 199).
Consistent with the earlier findings, orally administered alkaline solution Basenpulver (Pascoe, Germany), which is a mixture of sodium bicarbonate and carbonate salts, controlled melanoma growth in a mouse model (10). This effect is likely mediated by pH neutralization and is not connected to lactate itself, as it can be achieved by buffers lacking sodium bicarbonate (109, 176). Of high clinical interest are, however, the effects of pH neutralization on metastasis formation, as invasion and metastasis account for greater than 90% of cancer-associated mortality. Classical migrastatics usually target actin polymerization and contractility (80). However, in this case, pH neutralization likely plays a role. In addition to the examples mentioned earlier, the administration of neutralization buffers did not affect the already established primary tumor but both prevented cancer onset (108) and
In addition to bicarbonate,
Another alternative to sodium bicarbonate treatment involves the use of
Perhaps closer to practical use could be the employment of dichloroacetate as a radiosensitizer, as recently reported in high-grade gliomas (42). Moreover, it is necessary to keep in mind that dichloroacetate is metabolized differently by adults and children. Age-dependent differences in dichloroacetate metabolism and elimination cause its lower toxicity in children (197).
Previous proposals for the treatment of cancer-associated systemic lactic acidosis include the reduction of tumor burden (99), hemodialysis (77, 169), or thiamine supplementation (214). The suggestion for thiamine supplementation stems from the fact that thiamine deficiency is an uncommon but well-documented cause of hyperlactatemia in beriberi disease (124, 201). Among the patients with solid tumors listed in Supplementary Table S1, six were treated with thiamine. Two died within a week, two survived up to 10 weeks, and the other two survived over 10 weeks after lactic acidosis onset. Therefore, there is insufficient evidence of the benefits of thiamine supplementation to resolve fatalities in patients with cancer-associated systemic lactic acidosis.
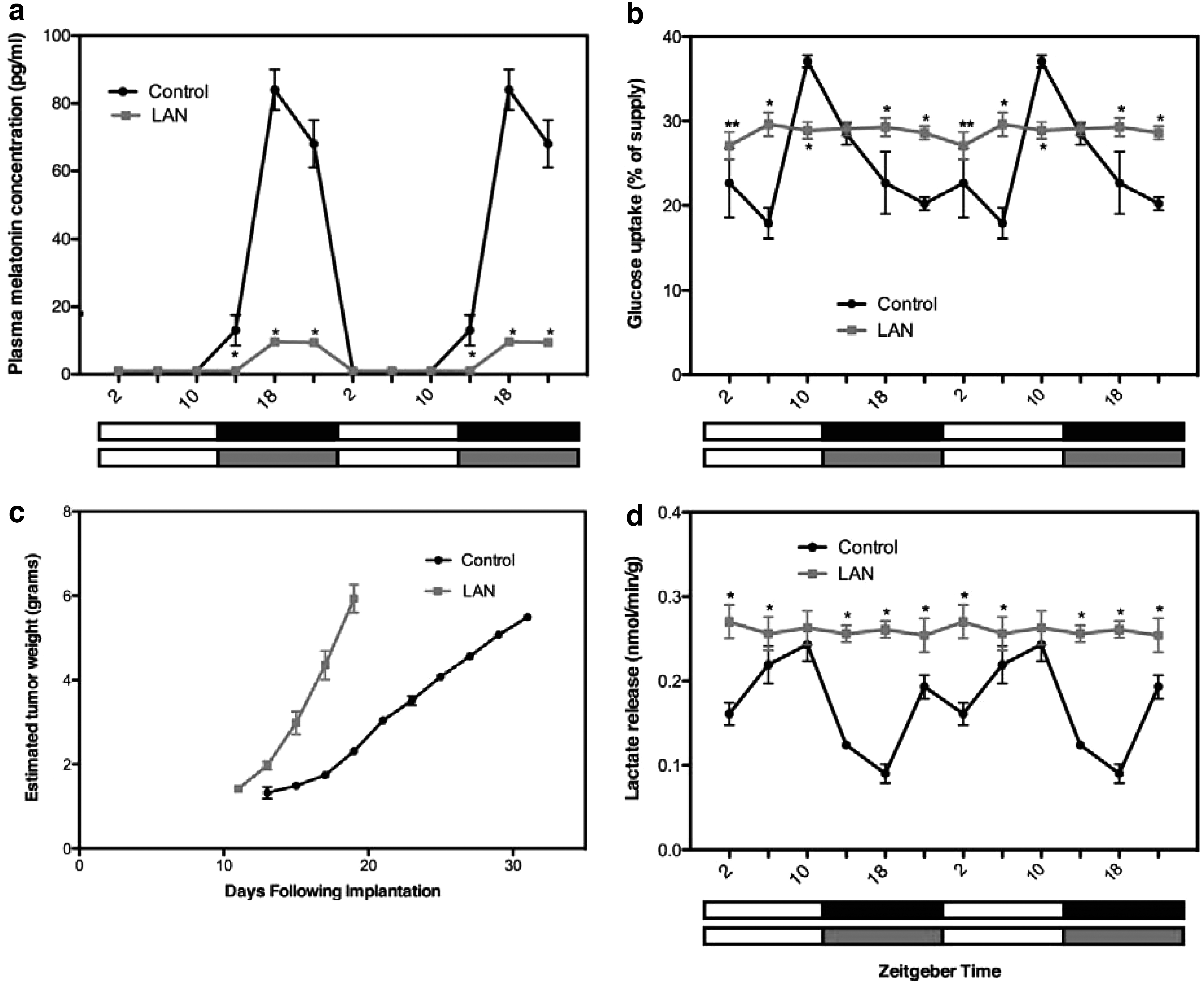
Several compounds known as glycolytic agents stimulate the shift of cancer cells from aerobic glycolysis to oxidative phosphorylation. Melatonin, the hormone responsible for the synchronization of circadian rhythms, also functions as an inhibitor of aerobic glycolysis in cancer cells. The mechanism involves the downregulation of pyruvate dehydrogenase kinase. Melatonin suppresses the proliferative activity of cancer cells, stimulates apoptosis, and has migrastatic activity. Under a standard light:dark cycle, lactate levels are subject to circadian fluctuations.
Due to the effects of melatonin on pyruvate dehydrogenase kinase, irregularities in circadian rhythms cause constitutive elevation of lactate levels (14, 174) (Fig. 5). The effects of melatonin on lactic acidosis and its reactive oxygen species (ROS) scavenging function could contribute to the effects of melatonin treatment of patients who undergo chemotherapy with tamoxifen or doxorubicin (50, 238).
Use of Tissue Lactic Acidosis for Ion Trapping
Under physiological conditions, the extracellular interstitial pH is ∼7.3, and the intracellular pH of the cellular cytoplasm is ∼7.2. When acidosis is induced locally, the pH can be reduced to 6.7 in the interstitial fluid, whereas it remains at 7.0–7.1 in the cytoplasm in vivo (236) (Fig. 6 for experimentally induced acidosis of the cell growth medium [164, 204]). Therefore, the pH gradient is reversed from −0.1 to +0.3–0.4. Importantly, some chemotherapeutics are attracted by the less acidic pH of the cells in acidic environments. The mechanism consists of the entrapment of a nonionized membrane-permeable form of the compound, which is then converted intracellularly to a charged compound that cannot cross biological membranes.
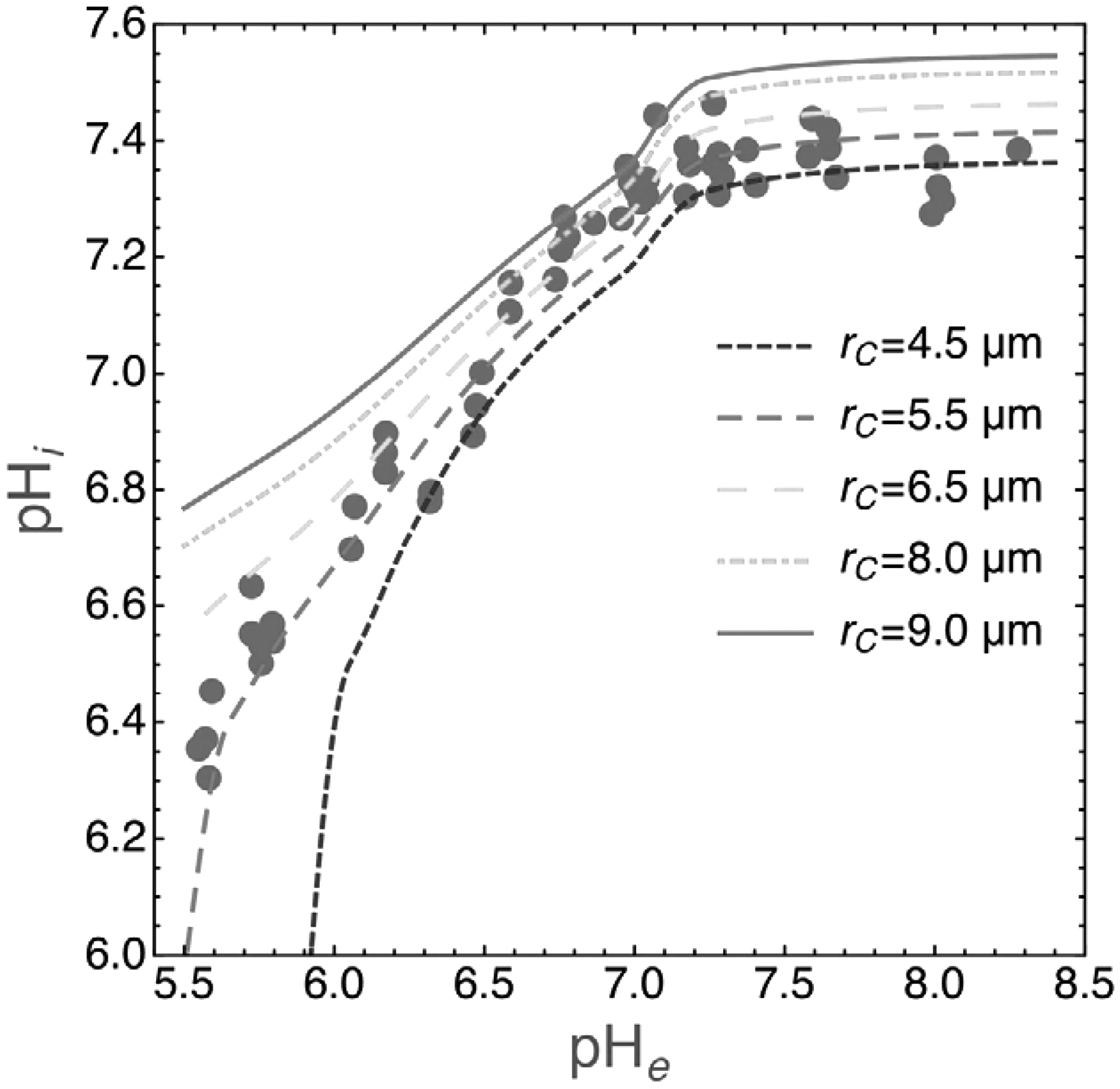
This phenomenon leads to the exclusion of weak base chemotherapeutics from more alkaline environments and the sequestration of weak acids (236). The majority of anticancer compounds fulfill the earlier criteria and have ionizable species under physiological conditions; importantly, these include many commonly used chemotherapeutic agents (Table 1). When tumor acidity is targeted in parallel to the standard treatment regimen, the therapeutic efficacy of chemotherapeutics that are subject to ion trapping may substantially change. Typically, weakly basic compounds enhance their efficacy with a buffer-induced increase in tumor pH, whereas weakly acidic compounds display decreased activity (87, 142, 170, 222).
Examples of Chemotherapeutic Agents That Are Subject to Ion Trapping
The groups of compounds that are sorted according to their dissociation in aqueous solutions are indicated. The effect (shown in brackets) denotes the effects of the higher acidity of the tumor environment. Note that the therapy designed to neutralize tumor acidity would completely reverse these effects. Adapted from Gillies et al. (88).
Use of Tissue Lactic Acidosis for Compound Activation
Several groups of compounds are activated only in acidic environments, that is, typically in the localized areas of tumor-induced lactic acidosis. These include nanoparticles that release their cargo in acidic environments, proton pump inhibitors, and carbonic anhydrase IX inhibitors.
A pH gradient can be employed to deliver
Other functional pH-responsive nanoparticles were designed to encapsulate doxorubicin into poly-L-histidine-based polymeric micelles that are destabilized in an acidic cancer cell environment (57, 118). Another example is the pH-sensitive conjugates of doxorubicin with methacrylamide copolymers (37). As a last example, nanoparticles of polyethylene glycol-conjugated doxorubicin that also encapsulated curcumin are stable at neutral pH but release both doxorubicin and curcumin in an acidic environment (248). Regarding acid-based linkers (e.g., hydrazine), examples include the conjugation of trastuzumab via acid-cleavable thiomaleamic acid (29). Regarding the pHLIP-MMAE conjugates and the AuNS-pHLIP-cargo particles, these were extensively reviewed (e.g., 116, 178). pHLIPs are 35- to 37-amino acid peptides that can facilitate the internalization of nanomaterials at low pH; when conjugated to the microtubule inhibitor MMAE, they show potent anticancer activity in vivo (22). The AuNS have a core-shell structure consisting of a silica core surrounded by a thin gold shell and are then conjugated to pHLIP and a cargo, such as the Ce6 photosensitizer. Although pHLIP is responsible for the delivery of the cargo into the cell, laser irradiation is used to unload and dequench from AuNS. This reaction uses the photothermal effect of AuNS and leads to enhanced photodynamic ablation of target cancer cells (244). pH-mediated intracellular cargo delivery has been subject to rapid development in recent years and is definitively of use in multiple future clinical applications. However, this topic exceeds the scope of this review.
Cells growing in acidic medium can be selectively targeted with
Another phase III open-label trial of devimistat in combination with modified folfirinox in patients with metastatic adenocarcinoma of the pancreas failed to meet the primary end point of overall survival (NCT03504423; AVENGER 500) (185). Further research should identify the conditions that limit devimistat action in vivo.
Treatment of Solid Cancer-Associated Tissue-Specific Lactic Acidosis
Tissue-specific lactate levels can be affected by targeting
Multiple LDH inhibitors are available, but most of them suffer from low potency and high off-target effects. When
Representative Anticancer Drugs That Target Glycolysis, Mitochondrial Metabolism, Lipid Synthesis, and Nucleic Acid Synthesis
Adapted from Schmidt et al. (193), updated. The data on the status of clinical studies as published at
Tissue-specific lactate levels can also be affected by targeting monocarboxylate transporters (MCT)1 (SLC16A1) and MCT4.
MCT1 inhibitors with anticancer activity in vitro and in vivo include α-cyano-4-hydroxycinnamic acid analogs (27) and 7-aminocarboxycoumarine derivatives (61). Another MCT1 inhibitor, AZD3965, has potent anticancer properties in small cell lung cancer cell lines and their xenografts (167). The outcomes of the AZD3965 phase I clinical trial were reported at the most recent ASCO meeting. AZD3965 can be safely administered at 10 mg twice a day, but monotherapy led to complete response in only one patient and stable disease in another patient out of a total of six examined patients (96).
MCT1 facilitates lactate uptake, whereas
The MCT4 inhibitors include bindarit (78) and diclofenac (which was already mentioned earlier given its anti-LDH activity) (190). Simultaneous inhibition of MCT4 and MCT1 was reported for AZ93 (146), syrosingopine (12), and the clinically approved indazole lonidamine (154).
Lactate-sensing receptor G-protein coupled receptor 81 (
Other less explored treatment options consist of the inhibition of proton transport through the plasma membrane. The best examples include inhibitors of the Na+/H+ exchanger (see also the chapter on the Use of tissue lactic acidosis for compound activation). For example, omeprazole reduced the expression of MMP-9 and invasiveness of the tumor and reduced the formation of lung metastases after i.v. injection of pretreated cancer cells (117).
In addition, the molecular pathways that are activated by low pH are considered therapeutic targets. For example, the inhibition of Ca2+- or proton-activated TRPM5 channels by acidic pH decreased MMP-9 expression and reduced metastatic potential in an in vivo melanoma model (140). In addition, the blockade of the pH-dependent voltage-gated sodium channel Nav1.5 by phenytoin has inhibitory effects on the migration and invasion of cancer cells (241).
Hypoxia-inducible factor 1α (
Hyperlactemia and Acidity in Diabetes
Many patients with cancer manifest type 2 diabetes as a comorbidity. Diabetes is well known as a factor that contributes to cancer onset and progression, but it appears unclear as to whether the diabetes-associated increase in hyperlactemia could play a pathogenic role in cancer onset and progression. Patients with type 1 and type 2 diabetes mellitus have lactate levels that are 0.3–1.0 mM higher than those of individuals without diabetes (98). Hyperlactemia is particularly prominent when diabetic ketoacidosis is manifested, particularly due to the combination of hypovolemia and altered carbohydrate metabolism. The lactate levels in diabetic ketoacidosis reach 3.5 mM (95% CI: 1.2–8.3 mM) (46). The decrease in pH of the interstitial fluid in patients with type 2 diabetes explains part of diabetes-causing insulin resistance. The insulin receptor has a lower affinity for insulin under low values of interstitial fluid pH (101). In contrast, the effects of diabetes on acidity are much better understood. Type 2 diabetes manifests as mitochondrial dysfunction, which affects the distribution of acid load. The three main mechanisms of effects on acid load consist of the overproduction of lactate due to the impaired tricarboxylic acid (TCA) cycle, increased renal acid load and net endogenous acid production due to intake of high amounts of protein (175), and production of ketone bodies that are consumed in muscle and brain tissues for ATP synthesis (90).
Effects of Acidosis on Cancer Cells
Cancer cells display
The cells that face acidosis increase lysosomogenesis and redistribute lysosomes from a perinuclear space to the plasma membrane, where they can release their contents, including proteases and protons, to the extracellular space. The redistribution of lysosomes prevents lysosome-associated membrane protein 2 from acid hydrolysis (48), induces autophagy (235), and separates the mammalian target of rapamycin complex from the Ras homolog enriched in the brain and other parts of its regulatory complex (229). The
Acidosis-exposed cells need to reprogram their metabolic pathways. Glyceraldehyde phosphate dehydrogenase (GAPDH) and glucose-6-phosphate dehydrogenase (G6PDH) are enzymes that are crucial for survival at low pH (163). As a rate-limiting glycolysis enzyme, GAPDH can be targeted by a number of inhibitors, such as arsenate, arsenic trioxide, 3-brompyruvate, iodoacetate, koningic acid, and other compounds (165). G6PDH is a rate-limiting enzyme allowing the entry of glucose-6-phosphate into the PPP pathway. G6PDH can be inhibited by the natural glucoside polydatin (resveratrol precursor), which is well tolerated and has already succeeded in a phase II clinical trial for the treatment of nonneoplastic pathologies (47).
Lactate derived from tumors potently induces
Cancer cells that have already adapted to acidic environments are more sensitive to
High rates of glycolysis and high levels of lactate contribute to
Studies That Have Exploited Lactate Metabolism to Improve Cancer Radiotherapy Outcomes
Adapted from Liu et al. (137).
Lactate derived from tumors was hypothesized to undergird the existence of tumors that are responsive and nonresponsive to uprosertib (GSK2141795), an ATP-competitive pan-Akt inhibitor. Uprosertib has already been assessed in phase II clinical trials (63), but disease regression was observed only in a small subset of patients (2). Uprosertib treatment led to reduced glucose uptake (93). Its action was independent of the presence of lactate, but the inhibition of lactate transport potentiated uprosertib-induced apoptosis. In addition, lactate acidosis conferred uprosertib resistance (11). Akt phosphorylation at Ser473 is changed in cancer cells subjected to acidosis, but additional research is needed to reach a consensus on the direction and extent of changes in Ser473 phosphorylation.
Lactic Acidosis Caused by Anticancer Drugs
Other biguanides, such as phenformin, have similar or even stronger effects on lactate production. Multiple events of severe lactic acidosis in patients with diabetes were behind the withdrawal of phenformin from the U.S. market in 1977 and the delay of approval of metformin until 1995. In contrast to phenformin, lactic acidosis associated with therapeutic doses of metformin is much less frequent, and some authors concluded that the observed association could be coincidental (189) and that lactate levels are similar among patients with type 2 diabetes regardless of the use of metformin therapy (54).
In addition to metformin, a number of different drugs have been reported as putative causative agents of elevated lactate levels. As summarized by Smith et al. (203), there are a number of drugs that rarely but repeatedly induce hyperlactatemia or lactic acidosis. These include
Research Gaps
Previous research has addressed pH gradients in the serum, interstitial fluid, and cytoplasm. However, whether changes in nuclear pH play a role has not been assessed. Under physiological conditions, the nucleus has a slightly more alkaline pH than the cytoplasm, and various studies agree on the range of 7.55–7.88 (39, 147, 195). The activity of transcription factors, the DNA replication apparatus, and DNA-dependent protein kinases that are responsible for nonhomologous end joining repair are pH sensitive (106, 223), and many pH-sensitive chemotherapeutics act in the nucleus. Therefore, it remains to be tested whether changes in nuclear pH affect chemotherapy outcomes and whether the nuclear pH of cancer cells differs from the nuclear pH of nontransformed cells.
Another pH-dependent process, which is minimally understood in the context of cancer-associated lactate acidosis, is O-glycosylation in the Golgi apparatus. Glycosylation is pH sensitive (141, 181), and the more alkaline Golgi pH in cancer cells compared with nontransformed cells (180, 181) could explain the repeatedly reported pH-dependent glycosylation changes in cancer cells (95, 129). Altered glycosylation promotes carcinogenesis and metastasis (95), but the effects of lactic acidosis and its treatments on O-glycosylation in cancer cells remain poorly understood.
The next questions stem from the fact that cells, including cancer cells, respond differently to acidosis in the presence and absence of lactate in vitro. It is more difficult for cancer cells to survive in an acidic medium in the absence of lactate, particularly because the pentose phosphate pathway is blocked by the absence of lactate (81). Relevant metabolomic studies from patients or at least from patient-derived xenograft models are lacking.
Many studies, including very recent studies, have based their conclusions on cancer cell lines grown in vitro under standard cell culture conditions. However, this methodology requires the cells to be grown for a long time in alkaline growth media. When such cells are switched to acidic medium, they typically slow their growth. This does not reflect the situation in vivo, where the cancer cells have already adapted to acidic conditions or even contribute to their maintenance (165). Therefore, any reports on growth inhibition by acidic conditions in vitro must be taken with extreme caution.
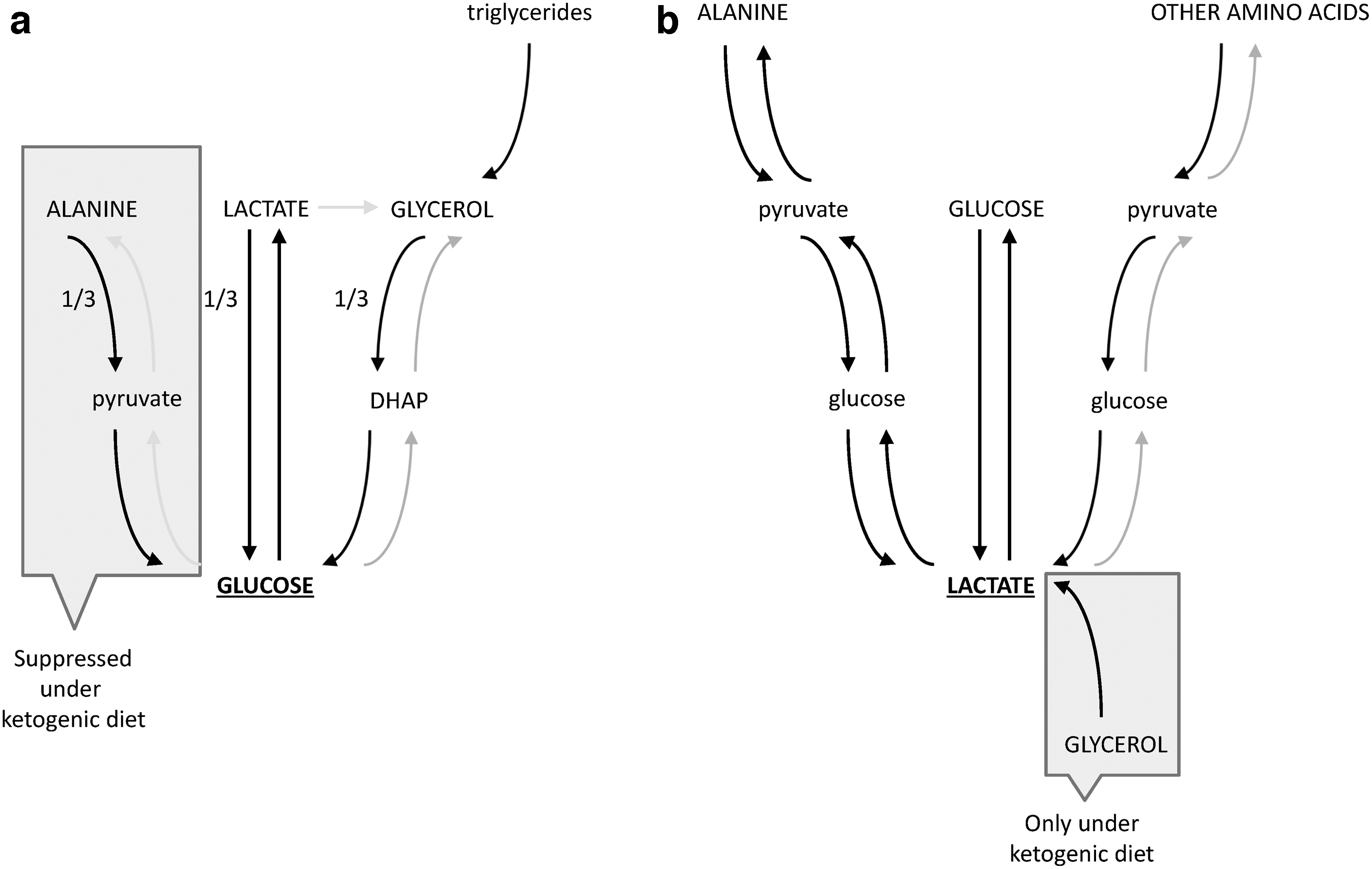
Interestingly, one of the least understood research topics is the sources and sinks of lactate in lactic acidosis. Detailed metabolomic tracing studies are lacking. However, the first data are available from healthy animals under various conditions and for several cancer types. In mice on a carbohydrate diet that were fasted for 8 h, circulating glucose was made from lactate, alanine, and glycerol in an approximately equal balance (Fig. 7a). Glucose serves as the dominant precursor of lactate, and lactate serves as a dominant precursor of alanine. Lactate and glucose serve equally as precursors of circulating glycerol, but glycerol is mainly synthesized from stored triglycerides. Glucose, alanine, and other amino acids serve as lactate precursors, but only glucose is the predominant direct contributor (Fig. 7b). In contrast to glucose, amino acids are first converted to circulating glucose by gluconeogenesis and then only to lactate. These metabolic relationships are only marginally unaltered in fed mice as long as we focus on the sources of lactate, alanine, and glutamine. The Cori cycle and triglyceride cycle are unaltered in the fed state. When the mice are maintained on a ketogenic diet, glycerol emerges as a direct lactate precursor, and alanine is no longer a major gluconeogenic precursor. Other primary sources of the circulating metabolites remain the same, and lactate cycling persists despite the near complete lack of dietary carbohydrates (105). Regarding lactate metabolism in cancer, acetate is used as a metabolic fuel in a number of cancer cell types, including breast cancer (119, 160) and non-small-cell lung cancer (72). However, lactate metabolism exhibits heterogeneity between and within tumors (72). Some other tumors, such as clear cell renal cell carcinomas, display a low labeled lactate to 3-phosphoglycerate ratio after [U-13C]glucose infusion (45), suggesting that clear cell renal cell carcinomas do not prefer lactate as their metabolic source. How lactate metabolism differs among cancer types remains to be elucidated.
Conclusions
The field's understanding of the role of lactate has shifted dramatically since its discovery. Long recognized as only a waste product, lactate was subsequently recognized as an alternative metabolic substrate and a secreted nutrient that is exchanged between the tumor and the environment. Most recently, tissue-specific lactic acidosis has been employed in a number of anticancer drug-targeting strategies. In contrast to tissue-specific lactic acidosis, systemic lactic acidosis is associated with poor prognosis; in the case of patients with solid cancer, it is associated with extremely poor prognosis.
Despite attempts at various treatments for solid cancer-associated systemic lactic acidosis, the currently most frequently used bicarbonate and thiamine therapies are inefficient, and patients usually die within days of solid cancer-associated systemic lactic acidosis onset. The only exceptions are patients with neuroendocrine tumors, particularly pheochromocytomas, in which lactic acidosis usually regresses spontaneously after tumor resection. Numerous metabolism-targeting anticancer compounds induce lactatemia, lactic acidosis, or other types of acidosis. Their potential to cause acidic environments is largely overlooked; however, these agents could explain a substantial portion of the observed clinical effects.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by the Charles University project Cooperatio 39.
Supplementary Material
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.