
Abstract
Significance:
Oxygen deprivation (hypoxia) is a common feature at sites of inflammation. Immune cells and all other cells present at the inflamed site have to adapt to these conditions. They do so by stabilization and activation of hypoxia-inducible factor subunit α (HIF-1α and HIF-2α, respectively), enabling constant generation of adenosine triphosphate (ATP) under these austere conditions by the induction of, for example, glycolytic pathways.
Recent Advances:
During recent years, it has become evident that HIFs play a far more important role than initially believed because they shape the inflammatory phenotype of immune cells. They are indispensable for migration, phagocytosis, and the induction of inflammatory cytokines by innate immune cells and thereby enable a crosstalk between innate and adaptive immunity. In short, they ensure the survival and function of immune cells under critical conditions.
Critical Issues:
Up to now, there are still open questions regarding the individual roles of HIF-1 and HIF-2 for the different cell types. In particular, the loss of both HIF-1 and HIF-2 in myeloid cells led to unexpected and contradictory results in the mouse models analyzed so far. Similarly, the role of HIF-1 in dendritic cell maturation is unclear due to inconsistent results from in vitro experiments.
Future Directions:
The HIFs are indispensable for immune cell survival and action under inflammatory conditions, but they might also trigger over-activation of immune cells. Therefore, they might be excellent setscrews to adjust the inflammatory response by pharmaceuticals. China and Japan and very recently (August 2021) Europe have approved prolyl hydroxylase inhibitors (PHIs) to stabilize HIF such as roxadustat for clinical use to treat anemia by increasing the production of erythropoietin, the classical HIF target gene. Nonetheless, we need further work regarding the use of PHIs under inflammatory conditions, because HIFs show specific activation and distinct expression patterns in innate immune cells. The extent to which HIF-1 or HIF-2 as a transcription factor regulates the adaptation of immune cells to inflammatory hypoxia differs not only by the cell type but also with the inflammatory challenge and the surrounding tissue. Therefore, we urgently need isoform- and cell type-specific modulators of the HIF pathway. Antioxid. Redox Signal. 37, 956–971.
Classical HIF Regulation and Oxygen Sensing
Hypoxia is the condition that arises when oxygen demand exceeds its supply. It is a feature that forces every cell to adapt its metabolism. The cell needs oxygen to generate adenosine triphosphate (ATP), with the biological energy equivalent keeping all cells alive. Thus, all cells have specialized means to ensure an enduring generation of ATP when oxygen levels fall. The transcription factor family of hypoxia-inducible factors (HIFs) largely coordinates the cellular answer to hypoxia by the induction of a distinct set of target genes that regulate glycolysis, angiogenesis, metabolism, as well as cell proliferation and survival.
The HIFs are heterodimeric proteins and consist of an oxygen labile α-subunit (HIF-1α, HIF-2α, HIF-3α) and a constitutively expressed, oxygen-independent β-subunit (also known as aryl hydrocarbon nuclear translocator, ARNT). To ensure an instantaneous reaction toward hypoxia, the cell makes the effort to continuously produce the α-subunit but immediately degrade it through specialized enzymes, the prolyl hydroxylase domain (PHD) proteins (26, 62). The PHD enzymes belong to the Fe(II) and 2-oxoglutarate-dependent dioxygenase superfamily, and PHD activity directly depends on the availability of oxygen. The apparent KM value for oxygen (the concentration of oxygen providing half of enzyme maximal activity) ranges from 60 to 220 μM for different constructs of PHDs and HIF-1α/HIF-2α proteins.
The values for the longest (and therefore most physiological) constructs are ∼100 μM (23, 46, 47). Since the intracellular oxygen level (10–30 μM) is significantly lower, oxygen concentration limits PHD activity when other co-factors are abundant. The PHDs incorporate one atom of molecular oxygen into the hydroxyl group at the prolyl residue (Fig. 1), whereas the other oxygen atom enables the oxidative decarboxylation of 2-oxoglutarate yielding succinate and CO2 (59). The von Hippel-Lindau (VHL) protein immediately recognizes the hydroxylated HIF-α and binds to it.

Subsequently, VHL recruits a ubiquitin E3 ligase that targets HIF-1α for proteasomal degradation by poly-ubiquitination (16). Factor inhibiting HIF (FIH)-1 is another Fe(II) and 2-oxoglutarate-dependent oxygenase that hydroxylates a conserved carboxy-terminal asparagine of HIF-α under normoxia. This leads to steric hindrance preventing the recruitment of the coactivators p300 and cyclic adenosine monophosphate response element binding protein (CREB) binding protein (CBP) [for reviews see Kaelin (42), Schofield and Ratcliffe (72)].
There is evidence that very low oxygen levels completely inhibit the PHDs whereas FIH-1 is still active. It then suppresses the activity of HIFα proteins that have escaped degradation (20, 46). HIF-1α is more sensitive to FIH-1-mediated inactivation compared with HIF-2α (10, 104).
Consequently, HIF-responsive genes feature a correspondence to the depth of hypoxia and a distinct relative dependency on HIF-1 and/or HIF-2.
(How) Does Inflammation Cause Hypoxia?
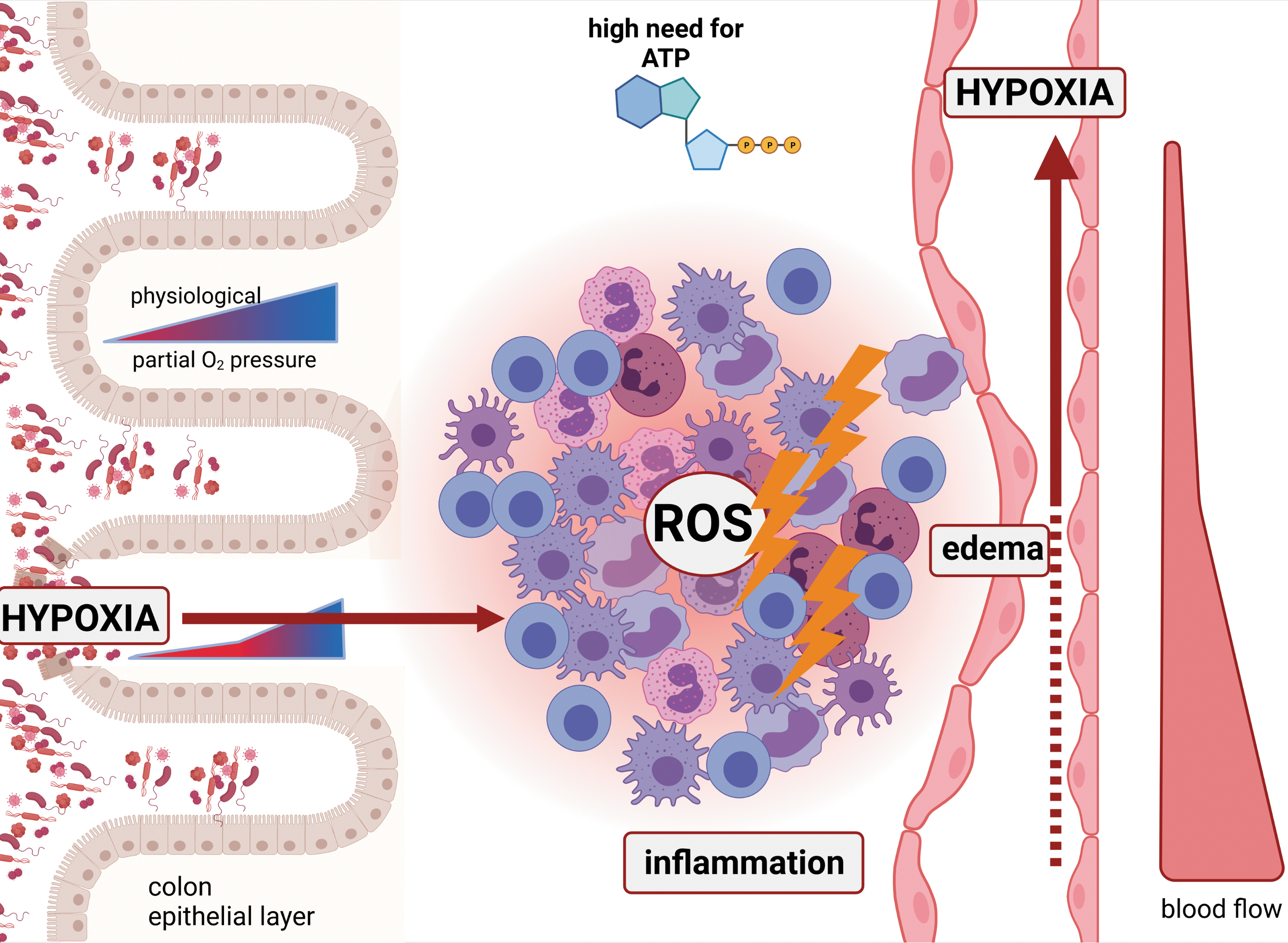
Sites of inflammation are hypoxic. There are several reasons that favor hypoxia under inflammatory conditions.
Inflammation can suppress oxygen delivery: Although blood flow may increase in acute inflammation, the formation of edema [most likely due to disassembly of endothelial cell junctions (51)] can diminish the delivery of blood and oxygen toward the site of inflammation. In turn, hypoxia itself can induce edema formation not only in the brain but also in peripheral microcirculation (61, 76) further aggravating oxygen and nutrient supply. Especially chronic inflammation exhibits a reduced blood flow, because it can lead to vascular damage and tissue infarction, which is to a high degree driven by platelet activation favoring the formation of microthromboses (93).
Inflammation recruits huge amounts of metabolically active cells: Neutrophils, eosinophils, and macrophages consume oxygen to produce reactive oxygen species (ROS) during oxidative burst (12, 56, 92).
Inflammation costs energy: All cells in the inflamed area are highly active in terms of metabolism. The production of cytokines, chemokines, and reactive oxygen or nitrogen (ROS or reactive nitrogen species [RNS], respectively), and antibodies as well as the activation of enzymes catalyzing the formation of these substances demands high amounts of cellular ATP. Elevated ATP production, in turn, needs an elevated oxygen consumption, which further deepens tissue hypoxia.
Inflammation can expand (physiological) hypoxic areas toward the surrounding tissue (left part of Fig. 2). This is especially true for inflammation of the colon (but can surely be transferred to other organs that show physiologically hypoxic areas as e.g., the bladder as well), where physiological hypoxia that exists in the epithelial tip layer of the crypts spreads toward deeper layers with disruption of the mucosa (6, 43). Interestingly, it was shown that the expansion of epithelial hypoxia is a very early feature of acute colitis as endothelial damage, increased colonic vascular permeability, perivascular edema, and epithelial hypoxia precede the epithelial barrier dysfunction whereas erosions, ulceration, and inflammation are following the features of ulcerative colitis (87).
Figure 2 summarizes all mechanisms, highlighting the need to consider hypoxia as a common feature of inflammatory conditions.
Activation of HIFs by Inflammatory Stimuli
The HIFs accumulate in inflamed areas (6, 28, 43, 44, 87). Interestingly, hypoxia is not a precedent condition for HIF-α stabilization as many inflammatory stimuli such as bacterial lipopolysaccharides (LPS) or non-methylated cytosine-phosphatidyl-guanine (CpG) islands, but also viruses [ranging from double-stranded desoxyribonucleic acid (DNA) viruses to single-stranded ribonucleic acid (RNA) viruses, excellently reviewed in Reyes et al. (68)] can also induce HIF-1α in the presence of oxygen (29, 100). The mechanisms that contribute to increased HIF are hereby multifaceted and include a stabilization of either protein or the messenger RNA (mRNA) of HIF1A/HIF2A—a phenomenon that is exclusive to inflammatory stimulation and does not occur under sole hypoxia (Fig. 3).

mRNA induction of HIF-α: the role of nuclear factor-κB
Inflammatory stimuli such as bacterial LPS that activate the nuclear factor-κB (NF-κB) pathway can also stabilize HIF-1α protein. Moreover, observations showed an increase of HIF1A mRNA as well (29). van Uden et al. identified a potential NF-κB binding site −197/188 bp upstream from the initiation site of the HIF1A promoter (91). The authors crucially analyzed the impact of the different NF-κB family members and found that all of them could activate the HIF1A promoter, although to various extents. In addition, they found that NF-κB family members control the basal mRNA expression of HIF1A in U2OS cells, resulting in the fact that hypoxic stabilization of the HIF-1α protein is no longer possible under completely blocked NF-κB activation (91).
Javan et al. observed a significant reduction of the HIF-1α protein as well as the Hif1a mRNA expression in a cardiomyocyte-specific knockout of the NF-κB family member p65 even under control conditions (37). This was further evaluated in in vivo experiments by Rius et al.: Mice lacking the NF-κB activating kinase IKK (inhibitor of κB kinase)-β in hepatic and Kupffer cells showed a reduced expression of Hif1a and Vegfa mRNA and a diminished hypoxia-induced accumulation of the HIF-1α protein in these cells (69).
These data nicely fit to the findings of Greten et al., who linked the deletion of the IKK-β enzyme to reduced inflammation and inflammation-associated cancer development in the colon (32).
HIF and NF-κB crosstalk
During the past almost 20 years, there has been an intensive study of the interference between HIFs and NF-κB. The results show that the interdependence of NF-κB and especially HIF-1 occurs in multiple ways and is not restricted to immune cells. Jung et al. detected a simultaneous induction of NF-κB dependent transcription and HIF-1α protein stabilization in A549 (adenocarcinomic human alveolar basal epithelial cells), Jurkat (immortalized human T lymphocyte cells), MCF-7 (human breast cancer cells), and NIH-3T3 (murine embryonic fibroblasts) cells (40). HIF-1, in turn, can induce NF-κB in hypoxic neutrophils, which is highlighted in more detail in the section about neutrophils in inflammation given next (95).
Other studies highlighted the role of the prolyl hydroxylases that regulate both HIF-αs and members of the NF-κB pathway: Homozygous deficiency for PHD1 protected mice in a model of dextran sodium sulfate (DSS)-induced colitis (81) whereas PHD2-deficient heterozygotes and PHD3-deficient homozygotes were equally as susceptible as wild-type controls. The PHD1 is proven to hydroxylate the IKK-β (19, 65), and a knockdown of the enzyme inhibited “anoxia” (0.1% O2) induced NF-κB activation in leukocytes (99).
In addition, the von Hippel Lindau protein can favor NF-κB activation as a loss of VHL, hypoxia, or PHD inactivation decreases vascular cell adhesion molecule (VCAM)-1 levels in clear cell renal cell carcinoma (ccRCC) cell lines. This occurred through a transcriptional mechanism that was independent of HIFs but dependent on the nuclear factor κB signaling pathway (50).
Up to date, we know much less about the interaction of NF-κB and HIF-2(α): The hypoxia associated factor (HAF) mediates a switch from HIF-1α to HIF-2α through activation of the NF-κB pathway, which contributes to the malignancy of T24 bladder cancer cells and is accompanied by the maintenance of stem-cell markers (33). The HIF-2α protein accumulates at sites of inflammation, and a conditional knockout of HIF-2α in colon epithelial cells protects mice from damage in an experimental model of acute colitis (103).
HIF-2α and NF-κB colocalize in calcified aortic valves (2), and bacterial stimuli such as LPS in combination with nicotine induce HIF-2α protein and mRNA expression in human periodontal ligament cells. The authors of these findings also observed a dramatic reduction of NF-κB p65 after a small interfering RNA (siRNA) mediated knockdown of EPAS1 but did not investigate the necessity of NF-κB activation for gene or protein expression of HIF-2α (7). A stimulation of macrophages with LPS/interferon (IFN)-γ also accumulated HIF-2α protein above the normoxic and hypoxic control, respectively (34).
The interdependency of NF-κB and the HIFs is complex. It includes a direct regulation of the HIF1A gene in humans and mice (then termed Hif1a) by NF-κB family members as well as translational modifications of both HIFs and NF-κB family members by the prolyl hydroxylases. An interdependence of NF-κB with HIF-2α seems likely but is still under investigation.
Induction of HIFs by inflammatory cytokines
It is a well-established fact for more than a decade that inflammatory cytokines can shape the expression of HIF-1α. Depending on the cell type and the protocol, the data differ with respect to a limitation of the effect to protein expression or involvement of transcriptional changes in HIF1A mRNA as well. Kuo et al. found that tumor necrosis factor (TNF)-α enhances HIF-1α protein expression in various breast cancer cell lines under normoxic or hypoxia-mimicking conditions, but it has little effect on the HIF1A mRNA level.
Herein, a depletion of IKKβ consistently reduced the amount of HIF-1α protein, indicating a role of NF-κB in the TNF-α mediated increase of HIF-1 (49). In contrast to that, TNF-α enhanced normoxic and hypoxic HIF-1α protein and mRNA levels in airway smooth muscle cells. Inhibition of the NF-κB pathway suppressed the TNF-α-mediated induction of HIF-1 gene transcription and despite the upregulation of HIF-1α protein the transcription of HIF-1 target genes remained low in the presence of TNF-α at normoxia and was even reduced under hypoxia (89).
The secretion of TNF-α is a common feature of tumor-associated macrophages (TAMs), and Jeong et al. have illustrated that this favors glycolysis in tumor cells and therefore directly shapes tumor cell metabolism (38). The observed effects are likely to be HIF-dependent, although the authors did not show HIF-1α protein stabilization in the tumors. Nonetheless, they observed a highly significant positive correlation of CD68 (a gene characteristically expressed by human macrophages) and HIF1A.
Tashiro et al., in turn, observed that an inactivation of prolyl hydroxylases by either hypoxia or hypoxia-mimicking conditions was involved in the repression of TNF-α-induced TSLP (thymic stromal lymphopoietin) expression in keratinocytes. The TSLP is a cytokine that is involved in allergic reactions and is upregulated under chronic inflammation and TNF-α induces its expression. Hypoxia and hypoxia-mimicking agents, therefore, did not inhibit the mRNA expression of TNFA, IL6, IL8, MCP1, and VEGFA and treatment with an HIF-2α antagonist could significantly reverse the inhibition of TSLP induction whereas an HIF-1α inhibitor did not show any effect (85).
The authors concluded that the hypoxic binding of HIF-2 to a hypoxia responsive element in the TSLP promoter region repressed the TSLP expression in keratinocytes.
Other inflammatory cytokines such as TNF-α are able to modulate HIF activation; most of them induce the accumulation of the HIF-α protein. Recent data for the modulation of HIFs by interleukin (IL)-6 points toward an induction of both HIF-1α and HIF-2α in different cell types. Xu et al. have analyzed the linkage of IL-6 and HIF-1 in ovarian cancer cell lines and found that IL-6 induced the HIF-1α expression via the signal transducer and activator of transcription 3 (STAT3) signaling under hypoxic conditions.
Additional experiments revealed an induced nuclear translocation of HIF-1α and a direct enhancement the transcriptional activity of HIF-1 by IL-6 via the STAT3 signaling pathway (102). The enhancement of this signaling axis, in turn, was already known to be elevated in ovarian clear cell carcinoma (OCCA), which is refractory to platinum-based chemotherapy and, therefore, difficult to treat (5). Endothelial cell derived IL-6 drives macrophage alternative activation via the induction of HIF-2 and, consequently, a robust arginase-1 expression in tumor-associated macrophages in a glioblastoma model (96).
IL-1β also induces HIF-1α in various cells, for example, dependent on reticular activating system (Ras)-induced NF-κB activation. Sharma et al. observed this in glioma cells under normoxic conditions, and IL-1β failed to induce NF-κB and HIF-1α activity in cells transfected with dominant negative RasN17 (75). Although this increased Ras expression and induced an increased phosphorylation of AKT, ERK (extracellular-signal regulated kinases), JNK (C-Jun-N-terminal kinase), and p38MAPK (mitogen activated protein kinase), the individual inhibition of these Ras effectors failed to hinder HIF-1α induction.
The co-inhibition of both AKT and ERK, in turn, resulted in a significant decrease in IL-1β-induced HIF-1α activation. In A549 cells, IL-1β upregulates functional HIF-1α protein through NF-κB and cyclooxygenase (COX)-2 activation, with a subsequent upregulation of vascular endothelial growth factor (VEGF) (41).
Inflammatory cytokines such as TNF-α, IL-6, or IL-1β affect HIF-1α on the protein level, by either an induced protein accumulation or an induced transcriptional activity. TNF-α and IL-6 are able to stabilize transcriptionally active HIF-2(α).
Table 1 summarizes the effects of inflammatory cytokines on HIFs and compares regulations in rodents and humans.
Effects of Inflammatory Cytokines on Hypoxia-Inducible Factors
COPD, chronic obstructive pulmonary disease; HIF, hypoxia inducible factor; mRNA, messenger RNA.
Ways of Hypoxia-Inducible Factor-1 Stabilization via Reactive Oxygen Species
Induction of HIFs by ROS
A long discussion exists of the role of ROS for the regulation of HIFs. There is no doubt anymore that ROS such as hydrogen peroxide (H2O2), O2 −•, or NO• can inhibit the prolyl hydroxylases under normoxic conditions, which allows the accumulation of HIFα (25, 90). In this article, we will not discuss the mechanisms of ROS generation in immune cells but will rather try to highlight the most recent data on the crosstalk of ROS and HIF stabilization under inflammatory conditions.
Box 1 gives a very brief overview on the mechanisms used by ROS to stabilize HIF-1. Infection with Helicobacter pylori affected the polarization of macrophages by intracellular induction of ROS and stabilization of HIF-1α (55). The amount of bacteria hereby regulated the polarization of the macrophages: The more bacteria, the more HIF-1 activation and the higher the polarization toward a proinflammatory phenotype. Stabilization of HIF-1 via the mitochondrial ROS axis also enabled elevated glucose levels in monocytes after infection with severe acute respiratory distress syndrome-corona virus 2 (SARS-CoV-2).
This, in turn, favored viral replication and cytokine production in these cells (17). Another recent publication highlighted that ROS generated under hypoxia enable macrophages to eliminate the intracellular protozoan parasite Leishmania amazonensis more effectively than under normoxic conditions. Herein, hypoxia triggered the expression of nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) and thereby the production of ROS in a murine macrophage cell line.
Either the silencing of HIF-1α, or the inhibition of Nox2 or antagonizing macrophage migration inhibitory factor (MIF) reversed the hypoxic killing efficiency of these cells (3).
In short, the generation of ROS stabilizes HIF-1α and thereby establishes a proinflammatory innate immune cell, which can render the cells more attractive for viral replication. In turn, HIF-1α can induce cellular ROS under hypoxia via Nox, thereby improving the parasite killing functions of macrophages (Box 1).
Upregulation of HIF induced by either bacterial or viral compounds represents a signal for the migration of immune cells toward hypoxic and/or inflamed tissues. Once they have arrived the site of action, the cells critically depend on HIFs to perpetuate their cellular functions and support or defeat inflammation. The following chapters will deal with the role of HIFs in the different innate immune cell types.
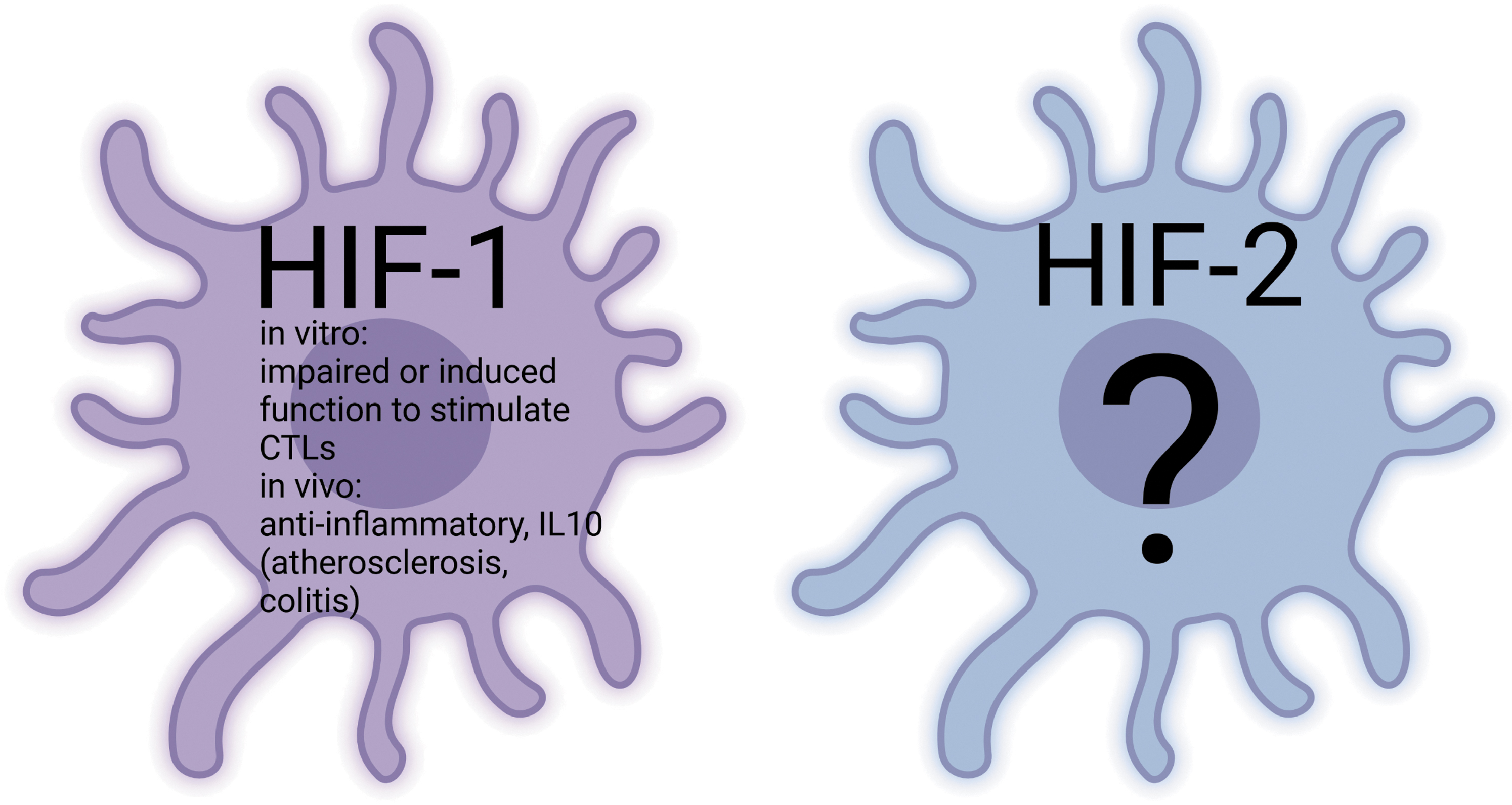
Role of Neutrophils in Inflammatory Settings
Neutrophil granulocytes are the first cells to arrive at a site of inflammation. Once activated, their “oxidative burst,” the generation of ROS, will directly increase their oxygen consumption. For the oxidative burst, neutrophils assemble the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase complex that generates ROS and, along with the enzymes superoxide dismutase and myeloperoxidase (MPO), H2O2 and hypochlorous acid (HClO•) (71).
All these reactions deplete local oxygen. Thereby, infiltrating neutrophils leave their mark while they cross the epithelial border: They promote microenvironmental hypoxia, they induce a transcriptional imprinting in epithelial cells while passing the epithelial border, and finally they effectively resolve inflammation (12). The stabilization of HIFs is hereby essential for the function of neutrophils: HIF-1α enables a prolonged neutrophil survival under hypoxic conditions.
In this context, NF-κB is hypoxia- and HIF-1-dependently induced as the deletion of HIF-1α in murine neutrophils resulted in a reduction in NF-κB signaling and anoxia-stimulated cell death (95). Later work of the same group further highlighted the underlying mechanisms and identified prolyl hydroxylase 3 as pivotal for neutrophil survival (94). PHD3 is a direct target of HIF-1 and was elevated in neutrophils under both hypoxic and inflammatory conditions. A loss of PHD3 hereby caused a reduction of the expression of B-cell lymphoma-extra large (BcL-xL), a protein that is crucial for the prolonged neutrophil survival under hypoxia.
Although Schuster et al. already stated in 2007 that bacterial LPS stimulate the neutrophils' uptake of glucose in an HIF-1-dependent manner (73), the HIF-dependent regulation of neutrophil metabolism has attracted special attention during the acute corona virus induced disease (COVID)-19 pandemic. Neutrophils undergo immune-metabolic reprogramming in severe COVID-19 illness with increased expression of cytosolic PKM2 (pyruvate kinase muscle type 2), phosphorylated PKM2, HIF-1α, and lactate (57).
The role of HIF-2 for the function of neutrophils is less clear than for HIF-1, but there are data describing a stabilization of HIF-2α in human and murine peripheral blood neutrophils by both acute and chronic inflammatory stimuli. HIF-2α-deficient murine inflammatory neutrophils, in turn, displayed an increased sensitivity to apoptosis induced by nitrosative stress ex vivo. Neutrophil apoptosis in vivo was increased, too, which reduced neutrophilic inflammation and tissue injury (86). In contrast to this, we recently found that a myeloid knockdown of HIF-2α resulted in a more severe inflammation in a murine model of colitis due to increased numbers of MPO-expressing neutrophils in the colon (44).
These neutrophils were likely to produce more inflammatory cyto- and chemokines recruiting other proinflammatory immune cells to the site of inflammation, such as dendritic cells (DCs) and CD4 T cells.
Neutrophils are special cells in terms of bacterial killing, as they can kill hostile microorganisms not only by pathogen uptake but also extracellularly by neutrophil extracellular traps (NETs). Although this release of cytotoxic granules to the extracellular environment is for a protective purpose, it can have tissue-damaging effects as well, which is why it appears to be difficult to find simple ways of pharmacological interference here (54). The HIF activation under hypoxia drives both the synthesis of degranulation proteins needed for the intracellular bacterial killing and the formation of NETs (11).
Neutrophil phagocytosis, in contrast, is enhanced under short-term hypoxia but this occurs most likely via HIF-independent but NF-κB dependent effects as the concentration of HIF-1α increased in the cytosol but not in the nucleus (30). Bacterial killing, instead, is possibly impaired under hypoxia, as the decreased abundance of oxygen results in an inhibition of the capacity to form ROS (58).
The HIF-1 prolongs neutrophil survival and thereby enables neutrophils to kill pathogens via the oxidative burst. In addition, it favors NET formation and therefore improves the extracellular killing of pathogens. There are few and conflicting data on neutrophilic HIF-2α, revealing either an increase or a decrease in neutrophil-mediated inflammation on loss of HIF-2 activity (Fig. 4).

Role of Macrophages in Inflammatory Settings
Macrophages are facing hypoxia after recruitment toward sites of inflammation or to the microenvironment of solid tumors. The activation of HIFs in macrophages ensures function and viability under these austere conditions by the maintenance of cellular ATP levels.
During the past years, our understanding of the mechanisms enabling macrophage functions under hypoxia steadily increased, although it is still far from complete. Starting with the observation that hypoxia significantly increased the phagocytosis rate of particles by macrophages in vitro, we gained detailed insights into the importance of HIFs for macrophage functions (18). In 2007, Anand et al. observed that hypoxic RAW264.7 cells and primary peritoneal macrophages exhibited an increased phagocytosis of labeled bacteria in vivo when the mice were hypoxic. Herein, hypoxia significantly increased the phosphorylation of p38MAPK and the inhibition of p38 reversed the hypoxic effects on phagocytosis, supporting a role for p38 in the hypoxic induction of phagocytosis.
Further, the authors suggested a link between p38 activation and HIF-1α expression under hypoxia. The siRNA knockdown of Hif1a prohibited the hypoxic induction of phagocytosis, demonstrating a specific role for HIF-1α in the regulation of phagocytosis (4). In addition to and somewhat contradictory to that, Cramer et al. convincingly showed that efficient bacterial killing in macrophages requires the abundance of HIF-1α because macrophages with an HIF-1α knockdown showed seven times more replicable bacteria than wild-type cells, although bacterial uptake between the cells was not different (18).
The activation of Toll-like receptor (TLR) 4 shifts macrophage metabolism from oxidative phosphorylation toward glycolysis. Many glycolytic enzymes get induced (including 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 [PFKFB3], pyruvate dehydrogenase kinase 1 [PDK1], and phosphoglycerate kinase 1 [PGK1]) (70, 82). The HIF-1 activation drives the majority of these changes, and the enhanced transcription of glycolytic enzymes results in a polarization of macrophages toward the proinflammatory phenotype (called “M1 macrophages”).
The overexpression of HIF-1α in macrophages leads to a hyper-inflammatory state with an enhancement of M1 markers and a diminished rate of oxygen consumption. Thus, HIF-1 induces M1 polarization of macrophages (64, 98). In inflammatory macrophages, the decrease of oxidative phosphorylation accompanies the increased glycolysis rate. Anti-inflammatory macrophages (M2) show the opposite phenotype: They change from glycolysis to oxidative metabolism. This switch is most likely induced by adenosine monophosphate (AMP)-activated protein kinase (AMPK) activation through a high ratio of AMP/ATP (22).
Interestingly, HIF-2 might be involved in the polarization of macrophages toward an anti-inflammatory phenotype, as it induces the expression of arginase 1 (79), a key player in anti-inflammatory cells.
The increase in the transcriptional activity of HIF-1 after inflammatory stimulation allows the inhibition of apoptosis and thereby increases the lifespan of phagocytes. In addition, it promotes phagocytosis and enhances antimicrobial peptide as well as pro-inflammatory cytokine release (TNF-α, IL-1β, IFN-γ, and IL-12) (1). Further, there are reports that hypoxia and HIF-1 activity induce TLR4 expression on macrophages (45). The inducible nitric oxide synthase (iNOS) is a target gene of HIF-1 in various species and is crucial for the production of nitric oxide (31, 48, 79).
Nitric oxide in turn—aside from its bactericidal activity—ablates HIF degradation via inhibition of the PHDs (90), thereby multiplying the fast activation of phagocytosis [reviewed in Dzik (22) and Nizet and Johnson (64)]. Only recently, Talwar et al. postulated that macrophages activated by LPS exhibit a metabolic reprogramming (80). Herein, HIF-1α drove glucose transporter 1 (GLUT1) expression, which increased glucose uptake. The abundance of glucose triggered proinflammatory cytokine production, especially that of IL-1β.
Mitogen activated protein (MAP) kinases tightly control this process, including the MEK (mitogen activated protein kinase kinase)/ERK pathway as well as HIF-1α. MEK-2 deficiency herein did not significantly affect the secretion of proinflammatory mediators such as NO•, TNF-α, and IL-12 after LPS stimulation of murine bone marrow derived macrophages (BMDM), but the cells exhibited a reduced IL-10 production.
This came along with higher HIF-1α and GLUT1 protein expression, and it significantly increased IL-1β as well as IL-6 production in response to LPS stimulation. Knockdown of HIF-1α expression via short interference RNA decreased the Il1b mRNA induction and IL-1β production in response to LPS treatment, indicating an important interplay of MEK-2 and HIF-1 in the inflammatory induction of IL-1β in murine BMDM (80). This glucose metabolism pathway is also attractive for viral invaders: for example, human immunodeficiency virus (HIV)-1 is able to exploit it. Barrero et al. transduced Macrophage-like differentiated human U937 cells with an adenovirus construct harboring the viral protein (Vpr) gene.
This changed the expression of more than 130 genes in these cells, among them being numerous enzymes of the glycolytic pathway. Vpr stabilized HIF-1α, which, in turn, induced the expression of, for example, hexokinase (HK), glucose-6-phosphate dehydrogenase (G6PD), and PKM2. The induction of HIF-1 thereby ensured a long-term survival of the macrophages with simultaneous facilitation of viral replication and biogenesis, which makes it a perfect virus supporter under these conditions (8). Pharmacologic treatment also affects the inflammatory regulation of HIF-1: Propofol suppressed the LPS-induced HIF-1α protein accumulation in monocytic THP-1 cells in a dose-dependent manner by inhibiting the neo-synthesis of HIF-1α protein (83).
Hypoxic stabilization, in turn, was unchanged. Propofol also diminished LPS-induction of HIF-1 gene expression of glycolytic enzymes and VEGFA (vascular endothelial growth factor).
Overall, HIF-1 is indispensable for macrophage activation under inflammatory conditions. Cramer et al. were the first who systematically analyzed the importance of HIF-1 for the function of myeloid cells. They used murine conditional knockout models wherein the mice lacked the expression of functional HIF-1α, its negative regulator VHL, and VEGF, one of the best studied HIF target genes (18). They showed a need of HIF-1 for the glycolytic capacity in macrophages and neutrophils, with a reduced ATP production in the HIF-1α knockout macrophages and neutrophils.
In analogy to this, HIF-1α knockout macrophages showed a significantly reduced release of lactate whereas VHL knockout macrophages (with constitutive HIF-1α stabilization) exhibited an induced lactate release. Because of the diminished glycolytic capacity of HIF-1α-deficient myeloid cells, the authors found a reduced migration and invasion of macrophages. This turned out to be favorable under inflammatory conditions: Mice with a loss of functional HIF-1α in myeloid cells showed a better outcome in models of phorbol ester-induced ear inflammation, chronic cutaneous inflammation, and passively induced arthritis (18).
Several groups have extended these data in the past 15 years. Among them, Settelmeier et al. could underline the role of HIF-1 for the recruitment of myeloid cells to sites of inflammations, as the total number of recruited macrophages was significantly elevated in mice lacking the negative HIF-1α regulator PHD2. After the induction of skeletal muscle trauma, these elevated numbers of macrophages helped to reduce the traumatic area and to accelerate skeletal muscle regeneration (74).
Imtiyaz et al. have crucially analyzed the role of HIF-2 for the function of macrophages (34). They found that a conditional knockout of HIF-2α in myeloid cells protected mice in acute endotoxemia, mainly by a reduced release of proinflammatory cytokines such as IL-β, IL-12, and TNF-α (34). Isolated BMDM lacking HIF-2α manifested this reduction in cytokine mRNA levels and protein release. In a model of acute ear inflammation, they observed less recruitment of immune cells toward the site of inflammation in mice with a myeloid-specific loss of HIF-2α.
Although there is some implication that HIF-1 and HIF-2 might have similar effects in myeloid cells, it has been of great interest whether the two different HIF isoforms expressed by myeloid cells (HIF-1 and HIF-2) have complementary or opposing effects. A study that analyzed the role of either HIF-1 or HIF-2 in hypoxic monocyte-derived human macrophages revealed that hypoxic gene regulation per se is under the control of HIFs. The authors did not present any hypoxic target genes that showed a specificity for either one or the other isoform (27).
A limiting point of this study is that the authors observed only one time point (18 h of hypoxia) and HIFs show a time-dependent and -specific expression pattern (HIF-1α is stabilized after very few hours whereas it takes much longer [16–24 h] to observe HIF-2α accumulation in most cells). Until very recently, in vivo studies analyzing the role of myeloid HIFs in acute inflammation were limited to colitis (only a few weeks ago, this has been extended to myocardial infarction [MI] as well, as referred to at the end of this section).
A study from our group described a protective effect of a myeloid HIF-1α knockout in acute DSS colitis (6). We detected limited numbers of macrophages in the colon after DSS treatment. The expression of inflammatory as well as anti-inflammatory cytokines exhibited a reduced and delayed induction in the myeloid HIF-1α knockout animals. Further, less other proinflammatory cells such as DCs or Th17 T cells migrated toward the site of inflammation whereas the number of regulatory T cells (Tregs) remained unchanged.
All this resulted in less inflammation in the myeloid HIF-1α knockout animals. The deletion of HIF-2α in myeloid cells, in turn, aggravated the symptoms of acute DSS colitis (44). Interestingly, this was not due to obvious changes in the numbers of recruited macrophages, but it depended on HIF-2α-deficient neutrophils. We found significantly elevated numbers of HIF-2α-deficient neutrophils in the inflamed colon, and these cells brought along a couple of other proinflammatory cells such as DCs and CD4 T cells.
Although these animals showed a (most likely compensatory) induction of Tregs in the inflamed colon as well, the aggravating effects outweighed anti-inflammatory effects in the acute inflammation. This was underlined by an increased expression of various proinflammatory cytokines (e.g., Ifng [interferon γ], Tnfa, Il17a, Il6). Of note, a simultaneous and specific knockout of HIF-1α and HIF-2α in these cells did reverse both phenotypes of HIF-1 and HIF-2 and gave results that were not significantly different from the wild-type animals (44).
This is at the first glance contradictory to the results of Lin et al., who analyzed the role of a depletion of HIF-1β as a common dimerization partner of HIF-1α and HIF-2α (52). Loss of HIF-β in myeloid aggravated the inflammation, worsened the outcome of the mice, and preserved the resolution of acute colitis. Although HIF-1β has other dimerization partners than HIF-α, it is currently unclear whether this knockout model is fully comparable with HIF-1α and HIF-2α deletion.
Very recently, DeBerge et al. examined the role of myeloid HIF-1 and HIF-2 in a murine model of MI because tissue sections of acute MI in humans had revealed both elevated HIF-1α and HIF-2α levels in CD68+ macrophages (21). They discovered that a loss of either HIF-1 or HIF-2 activity in myeloid cells resulted in a reduced infarct size and a better cardiac function, but for different reasons. Abundant HIF-2α suppressed the anti-inflammatory mitochondrial metabolism of the macrophages, whereas HIF-1α enforced a glycolytic reprogramming of these cells (21).
In contrast to that, a simultaneous loss of both myeloid HIF-1α and HIF-2α exhibited a catastrophic outcome with macrophage necroptosis, impaired fibrogenesis, and cardiac rupture.
Overall, HIFs (and especially HIF-1) are indispensable for macrophage activation. Stabilization of HIF-1α enhances macrophage lifespan and function, although there are contradictory results as to whether it regulates bacterial uptake or not. HIF-1 drives macrophage metabolism toward glycolysis and reduces the cellular oxygen consumption under hypoxic inflammation. Consequently, animal models with a myeloid loss of Hif1a reveal a reduced severity of inflammation. A loss of Hif2a in cells of the myeloid lineage results in more organ-specific phenotypes and can either repress or promote innate immunity (Fig. 5).

Role of DCs in Inflammatory Settings
The DCs are masters of antigen presentation and, therefore, have to function properly under hypoxic and inflammatory conditions. Interestingly, there is evidence that inflammatory hypoxia induces a gene set in bone marrow-derived DCs that is different from that which can be detected after hypoxic incubation alone (36). Current research so far appears to cover only the role of HIF-1 in these cells. However, this is obviously not the full story as illustrated by a report claiming HIF-1 to be important for the maturation of bone marrow-derived DCs by induction of the surface receptors CD80 and CD86 (35).
In contrast, other groups did not see any changes in the expression of these markers in murine DCs that dismiss (101) or overexpress HIF-1α (88), and hypoxia even led to a decrease of CD80 and CD86 expression on human DCs (9). What is indisputable until now is that hypoxia and bacterial stimuli such as LPS and CpGs stabilize HIF-1α and induce HIF-1 target gene expression in DCs (35, 36, 88, 100). There are findings indicating that a loss of DC HIF-1α impairs DC function to stimulate T cell responses such as allogeneic lymphocyte proliferation (35) or the induction of Cd278 or Grzmb mRNA (100) after coculture with bone marrow-derived DCs.
In contrast to this, Tran et al. found that bone marrow-derived DCs that overexpressed HIF-1α exhibited a reduced capacity to drive effector CD8+ T cell functions such as Granzyme B positivity of the T cells and their specific ability to kill targeted and CpG stimulated DCs (88). With respect to murine inflammation models, there are accumulating data pointing toward an anti-inflammatory, immune-mediating role of HIF-1 in DCs in murine in vivo models of inflammation.
A conditional deletion of Hif1a in CD11c+ antigen-presenting cells in a mouse model of atherosclerosis caused an accelerated atherosclerotic plaque formation and increased T cell infiltrates into lesions (15). Further supporting that, Wood et al. described that in a mouse model of obesity the deletion of HIF-1α on conventional DCs (cDCs) resulted in enhanced adipose tissue inflammation and atherosclerotic plaque formation (101). They identified a significantly reduced Treg/CD4 T cell ratio in the adipose tissue of the mice and a significantly reduced expression of Il10 in the atherosclerotic plaques.
The activation of HIF-1, in turn, mediated increased lipid synthesis, accumulation of lipid droplets, and altered synthesis of lipid mediators. They concluded that HIF-1α activation in cDCs is necessary to control vessel wall inflammation. In a model of acute colitis, we also observed this immune-modulating role of HIF-1 in DCs (27). The DC loss of functional HIF-1α (driven by Itgax-Cre expression and thus lacking Hif1a in conventional and plasmacytoid DCs) resulted in an aggravated inflammation of the colon due to a reduced expression of immune regulating IL-10 in the inflamed colon.
HIF-1 was also necessary for proper IL-10 secretion of bone marrow-derived DCs in vitro. Mice lacking HIF-1α in DCs also exhibited a significantly reduced expression of Ccr9 mRNA in the mesenteric lymph nodes. CCR9 is a chemokine recruiting Tregs toward sites of inflammation. Consequently, HIF-1α knockout mice displayed significantly reduced numbers of Tregs in the colon (28). Human monocyte-derived DCs also show an HIF-dependent, immunosuppressive phenotype most likely due to an increased production of extracellular adenosine. These cells also induce the expression of adenosine receptor A2B HIF-1 dependently under hypoxic conditions (105). In addition, human monocyte-derived DCs differentiated under hypoxia showed a significantly decreased antigen uptake (24).
The data on HIFs in DCs are much more limited than that about macrophages until now. Bacterial stimuli are able to accumulate HIF-1α in murine bone marrow-derived DCs and a loss of Hif1a in these cells comes along with a reduced ability to stimulate T cells. DC HIF-1 in vivo seems to have an immune suppressive role, as it is indispensable for the recruitment of Tregs (Fig. 6).
Conclusions
Sites of inflammation exhibit hypoxia and inflammatory mediators (e.g., chemokines, cytokines, ROS/RNS, and many more), shaping the response of the present (immune) cells. These conditions trigger the stabilization and activity of HIFs via mechanisms involving the inhibition of HIF hydroxylases by the deprivation of oxygen or the expression of reactive species and TLR-mediated NF-κB activation. We have highlighted the regulation of the most prominent isoforms HIF-1 and HIF-2 herein.
A caveat is that until now, most publications study their regulation in cell culture experiments using varying experimental conditions that are likely to differ from the reality of in vivo situations. This is why we urgently need more data from conditional knockout animals lacking Hif1a and/or Hif2a in specific cells to verify these observations in a physiological context. This would also portray the fact that HIF-1 and HIF-2 do not only exhibit a differential regulation in neutrophils, macrophages, and DCs but also (fine-) tune the immunological activation of these cells.
Although a global stabilization of HIF-1 by the application of approved PHD inhibitors under inflammatory conditions seems to be a promising treatment approach for DCs, it might turn out to have detrimental effects because of a fulminant activation of neutrophils and macrophages. Moreover, HIF-1 and HIF-2 have defined and by far not identical effects in these cells, which underlines the need for not only cell-type but also target-specific inhibitors in the future. Then, target specificity would enable even personalized treatment approaches.
One potential therapeutic mode could include the identification of innate immune cell type specific surface molecules. (Ab)Using the phagocytic capacity of myeloid cells, an antigen-coupled drug might be delivered easily into the cell. Another possibility might be the genetic modification of specific innate immune cell receptors in the laboratory to enable a modified “CAR (chimeric antigen receptor) myeloid cell therapy.”
Footnotes
Acknowledgment
All figures were created with
Authors' Contributions
S.W. conceptualized, wrote, and edited the article. J.F. reviewed and edited the article.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
There is no funding to declare for this work.