
Abstract
Significance:
Evidence for a role for the oxytosis/ferroptosis regulated cell death pathway in aging and neurodegenerative diseases has been growing over the past few years. Because of this, there is an increasing necessity to identify endogenous signaling pathways that can be modulated to protect cells from this form of cell death.
Recent Advances:
Recently, several studies have identified a protective role for the AMP-activated protein kinase (AMPK)/acetyl CoA carboxylase 1 (ACC1) pathway in oxytosis/ferroptosis. However, there are also a number of studies suggesting that this pathway contributes to cell death initiated by various inducers of oxytosis/ferroptosis.
Critical Issues:
The goals of this review are to provide an overview and analysis of the published studies and highlight specific areas where more research is needed.
Future Directions:
Much remains to be learned about AMPK signaling in oxytosis/ferroptosis, especially the conditions where it is protective. Furthermore, the role of AMPK signaling in the brain and especially the aging brain needs further investigation.
Introduction
A variety of different causes can lead to cell death. Over the past few decades, it has become apparent that although cell death can be initiated by a large number of conditions ranging from aging to acute toxicities, the process of death generally involves a series of biochemical and sometimes transcriptional changes. Thus, this process has been named regulated cell death. Multiple variations of regulated cell death have been identified by using genetics, small-molecule inhibitors or, frequently, a combination of the two (105).
After the description of apoptosis in 1972 (55), one of the next regulated cell death pathways to be described in detail was oxytosis (104). The early characterization of the distinct steps of this cell death pathway relied on small-molecule inhibitors of enzymes and specific cellular pathways, which could be considered an early application of a chemical biology approach to understanding a natural process. In 2012, this pathway was rediscovered and renamed ferroptosis (23, 62, 72).
Although some have argued that oxytosis and ferroptosis are distinct cell death pathways (e.g., 23), the observations that (i) both are initiated by inhibition of system xc − (62), (ii) both share the same steps (38, 62, 72), and (iii) both are blocked by exactly the same group of chemical inhibitors (99) strongly support the idea that they are the same regulated cell death pathway. Since 2012, there have been a growing number of papers indicating a role for this pathway in a variety of diseases, including Alzheimer's disease (AD) (60, 72, 89, 91, 108, 122) as well as Parkinson's disease (24), hemorrhagic stroke (128), kidney injury (30, 67), and cardiomyopathy (29). This, in turn, has led to a search for endogenous molecular pathways that can be regulated to protect cells from oxytosis/ferroptosis.
In this review, we will begin by discussing the basic pathway of oxytosis/ferroptosis and describe how distinct components of this pathway are related to aging and neurodegenerative disease. We will then provide background information on AMP-activated protein kinase (AMPK), as AMPK is a recently identified novel target whose activation we and others have shown to protect against oxytosis/ferroptosis (20, 59, 64). The potential mechanisms underlying this AMPK-mediated protection will be discussed. Because there is some controversy about the protective effects of AMPK against oxytosis/ferroptosis (42, 98, 109, 114, 123, 125), these studies will be covered as well. The overall goal is to stimulate further research on this important topic.
Relevance of Oxytosis/Ferroptosis to Aging and AD
The oxytosis/ferroptosis regulated cell death pathway is a non-apoptotic form of cell death that is characterized by glutathione (GSH) loss and enhanced production of reactive oxygen species (ROS) from mitochondria, as well as other sources, that result in lethal lipid peroxidation and dysregulation of calcium homeostasis (62, 72, 103, 104) (Fig. 1).

At the cellular level, oxytosis/ferroptosis can be initiated by the inhibition of cystine uptake via system xc − by using either glutamate or the synthetic chemical erastin, which results in the depletion of intracellular GSH (62, 72, 103, 104). System xc − is a heterodimeric amino acid antiporter composed of two subunits, the 4F2 heavy chain (SLC3A2) that is common to multiple amino acid transporters, and xCT (SLC7A11), the antiporter-specific light chain. System xc − can transport glutamate and cystine in both directions.
However, since the intracellular pool of cystine is negligibly small because intracellular cystine is rapidly reduced whereas the intracellular glutamate concentration is generally higher than in the extracellular space, it generally imports cystine while exporting glutamate in a 1:1 ratio (63). Within the cell, cystine is reduced to cysteine, which is the rate-limiting amino acid for GSH synthesis. Consistent with the inhibition of system xc − leading to cystine loss, oxytosis/ferroptosis can also be initiated by culture in cystine-free medium (82).
GSH depletion leads to a reduction in the activity of a number of GSH-dependent antioxidant enzymes, including GSH peroxidase 4 (GPX4), and to the activation of lipoxygenases (LOXs). GPX4 can also be directly inhibited by the chemical RSL3 (116). Regardless of the initiating stimulus (glutamate, erastin, cystine deprivation, RSL3), ROS and lipid peroxides accumulate in the cells (33, 103), eventually leading to the potentiation of intracellular calcium influx through store-operated calcium entry (SOCE) and cell death (38, 47, 65).
The influx of calcium and mitochondrial ROS production appear to be tightly coupled (103), but the precise mechanisms underlying this relationship have not yet been fully elucidated. Mitochondria may also promote oxytosis/ferroptosis through their bioenergetic and biosynthetic activities (31). Intracellular labile free iron also plays an important role in oxytosis/ferroptosis, as iron chelators have been shown to prevent oxytosis/ferroptosis-mediated cell death (23, 72). Evidence suggests that iron can not only generate ROS and lipid peroxides via the Fenton reaction but also activate the non-heme iron-containing LOX enzymes (13). In addition, iron can induce neuronal redox-sensitive ryanodine receptor (RyR)-mediated calcium release from the endoplasmic reticulum (ER) and subsequent mitochondrial fragmentation that might also contribute to the promotion of oxytosis/ferroptosis (37, 92).
Although mammalian cells in culture die within 24 h after the induction of oxytosis/ferroptosis, it is not clear that this would necessarily be the case in vivo (72). As outlined earlier, oxytosis/ferroptosis follows a series of discreet steps that, in vivo, could take place over a lengthy time frame, thereby leading to a slow and progressive loss of basic nerve cell functions before any overt cell death. Thus, in the context of aging and age-associated neurodegenerative diseases, oxytosis/ferroptosis could become a chronic process rather than an acute event such that its inhibition would be relevant throughout the entire course of the disease rather than only at its point of initiation or termination (72).
Importantly, there is strong evidence indicating that all of the cellular pathophysiological changes that are associated with the oxytosis/ferroptosis pathway are also seen in the aging brain and are exacerbated in age-associated neurodegenerative diseases (Fig. 1). GSH plays a central role in maintaining cellular redox homeostasis (93). Importantly, recent studies using double-edited proton (1H) magnetic resonance spectroscopy have shown that GSH levels decrease with age in the human brain and this decrease is exacerbated in AD (1, 4, 21, 28, 73). The loss of GSH in the aging brain correlates with multiple pathological features of AD, including increases in cognitive impairment, microglial activation, and endothelial dysfunction (1, 4, 21, 73). Moreover, age-dependent decreases in blood GSH levels in humans have been shown in multiple studies (25, 36, 71, 95) and these decreases were also found to be exacerbated in mild cognitive impairment and AD (5, 87).
Lipid peroxidation also has been correlated with AD and is postulated to be an early feature of the disease since post-mortem brain samples from subjects with mild cognitive impairment already show increased levels of lipid peroxidation (6, 35, 90, 101). Furthermore, by-products of lipid peroxidation, including malondialdehyde, 4-hydroxy-2-nonenal (4HNE), acrolein, prostanes and isoprostanes in plasma, urine and cerebrospinal fluid, are increased throughout the course of the disease (6, 35, 90, 101). In addition, several different LOXs have been suggested to play a role in AD (53, 54, 58, 97, 115). Early in the course of the disease, 12/15LOX is upregulated in the most vulnerable brain regions (53) whereas 5LOX expression shows an age-dependent increase in the brains of both mice and humans, which is further enhanced in AD (54).
Biometal dyshomeostasis is another important stress that could contribute to both the onset and the progression of AD (41, 44). Iron is the most abundant transition metal in the brain and participates in a variety of critical brain functions, including neurotransmitter synthesis, myelination, and mitochondrial function (44). However, there is an increased accumulation of iron in the aging brain that is exacerbated in AD and some other neurodegenerative diseases and is believed to contribute to the neuropathophysiology of the diseases by increasing oxidative stress through Fenton chemistry (41), in a process similar to that seen during oxytosis/ferroptosis.
Mitochondrial dysfunction and associated ROS production is another critical aspect of oxytosis/ferroptosis (62, 72, 103, 104) and is also one of the hallmarks of aging (70) that has been directly implicated in AD (18). The pathology and symptoms of AD are preceded by a decline in cerebral energy metabolism that is more severe than that observed in normal aging (15, 17, 18, 118). This is at least partly due to the observations that most of the energy derived from glucose oxidation is produced in mitochondria but multiple mitochondrial-dependent functions are impaired in aging and AD. Indeed, there are multiple lines of evidence that mitochondrial dysfunction occurs early and directly contributes to the development of AD, including declines in electron transport chain (ETC) activity and respiration, significant changes in the mitochondrial metabolite landscape, increases in mitochondrially derived oxidative stress, and oxidative damage to mitochondrial DNA (mtDNA), RNA, lipid and protein species, as well as an accumulation of mtDNA mutations (18, 66, 83, 102, 119).
The requirement for calcium influx for the execution of cell death in the oxytosis/ferroptosis pathway was noted in the very first studies on this cell death pathway (81). Calcium influx occurs downstream of both ROS production and LOX activation (65). The movement of calcium both between intracellular organelles, including the ER and mitochondria, and across the plasma membrane plays a key role in many of the fundamental functions of neurons. Thus, it is not surprising that disruptions in calcium homeostasis are associated with multiple neurological disorders, including AD (37, 130). These disruptions include changes in calcium buffering capacity, dysregulation of calcium channel functions, and alterations in other calcium-regulated proteins (37, 130). Several studies have shown that SOCE is activated in oxytosis/ferroptosis (38, 47). Interestingly, dysregulation of SOCE is observed in aging (11) and imbalances in SOCE components may contribute to AD pathogenesis (106). In summary, many of the pathophysiological changes that are seen in cells after activation of the oxytosis/ferroptosis pathway also occur in the aging brain and are exacerbated in AD.
AMPK Signaling
AMPK is a serine/threonine kinase that is ubiquitously expressed in eukaryotic cells (for reviews see 34, 43, 51, 77, 88) (Fig. 2). AMPK was first described in 1987 as an enzyme that was allosterically activated by AMP and by phosphorylation by an unknown kinase (43). Since then, it has become clear that AMPK is a critical regulator of energy homeostasis in cells, both promoting catabolic reactions and inhibiting anabolic reactions under conditions of energetic stress.

AMPK is composed of a heterotrimeric complex consisting of one catalytic subunit (α) and two regulatory subunits (β and γ). In mammals, there are two different α and β isoforms and three different γ isoforms, leading to a possible 12 different complexes. The expression of the different subunit isoforms varies among distinct cell types, and it is not yet clear whether the different complexes are functionally redundant.
The α subunit contains the catalytic domain and Thr172 in the activation loop, which is phosphorylated by upstream kinases to activate the enzyme (46). The β subunit contains a domain that allows AMPK to sense glycogen and also a myristoylation site, which helps to target AMPK to membranes (49). The γ subunit binds adenine nucleotides, thereby allowing the enzyme to respond to changes in the AMP/ATP or ADP/ATP ratio.
Phosphorylation of Thr172 in the α subunit of AMPK can be carried out by three distinct kinases: liver kinase B1 (LKB1), calcium/calmodulin-dependent kinase 2 (CaMKK2), and TGFβ-activated kinase 1 (TAK1) (for reviews see 34, 43, 51). LKB1 is the kinase responsible for Thr172 phosphorylation in response to energetic stress, which is promoted by the binding of AMP or ADP to the γ subunit (45, 96, 113). Binding of AMP or ADP to the γ subunit also induces a conformational change in AMPK, which protects Thr172 from dephosphorylation by protein phosphatases.
The other two kinases phosphorylate AMPK under conditions that are not directly dependent on energetic stress. CaMKK2 is activated by increases in intracellular calcium levels whereas the conditions that result in AMPK activation by TAK1 are still being elucidated, although a recent paper suggested that one of these conditions is damage to lysosomal membranes (52).
AMPK activation leads to a restoration of energy levels by differentially modulating the activity of ATP-consuming and ATP-generating pathways and by altering the levels of transcription factors that play key roles in regulating cellular metabolism (for reviews see 34, 43, 51, 77). Specifically, AMPK both inhibits the synthesis of lipids and glucose and promotes their breakdown. With respect to lipids, AMPK directly phosphorylates and thereby inhibits ACC1 and ACC2, which is the first enzyme in fatty acid synthesis converting acetyl-CoA to malonyl-CoA. The inhibition of malonyl-CoA synthesis by AMPK also promotes fatty acid oxidation because malonyl-CoA blocks the uptake of the substrates of fatty acid oxidation into mitochondria where the process occurs (76). This combination of effects on fatty acid synthesis and breakdown can also enhance NADPH and GSH levels because although fatty acid synthesis uses NADPH (7), fatty acid oxidation generates it and NADPH is required for the reduction of oxidized GSH by glutathione reductase (16). In addition, AMPK phosphorylates and inhibits sterol regulatory element binding protein 1 (SREBP1), a key transcriptional regulator of lipid synthesis. AMPK also phosphorylates and inhibits 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), leading to a decrease in steroid synthesis (10).
In addition to these effects on glucose and lipids, AMPK inhibits cap-dependent protein synthesis by multiple mechanisms (for reviews see 34, 43, 51, 88). AMPK inhibits mammalian target of rapamycin 1 (mTORC1) activity, which is required for cap-dependent protein synthesis, both by phosphorylating and activating tuberous sclerosis complex subunit 2 (TSC2) (50), a negative regulator of mTORC1, and by phosphorylating and inhibiting Raptor (40), a subunit of the mTORC1 complex. AMPK also phosphorylates and activates elongation factor 2 kinase, (eEF2K), an inhibitor of protein elongation.
Another way that AMPK activation increases the levels of molecules that can be used to generate energy is by activating autophagy, a process for the digestion of cellular components, via several mechanisms (for reviews see 34, 43, 51). These mechanisms include (i) the phosphorylation and activation of unc51-like autophagy activating kinase 1 (ULK1) (26, 56), which is a protein required for the initiation of autophagy, (ii) the inhibition of mTORC1 (40, 50),whose activity blocks autophagy, and (iii) the phosphorylation and activation of Beclin-1 (121), a component of the class III phosphoinositide 3-kinase (PI3K) complex I that plays a key role in the assembly of the autophagosome.
Not only does AMPK promote general autophagy but it also coordinates mitochondrial fission/fusion and stimulates mitophagy, a process for the digestion and removal of damaged mitochondria (for reviews see 34, 43, 48, 51). In addition, AMPK promotes mitochondrial biogenesis by upregulating the activity of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α), a key transcriptional regulator of mitochondrial biogenesis (for review see 48). Thus, by both stimulating the removal of damaged mitochondria and upregulating the synthesis of new mitochondria, AMPK promotes the health of mitochondria.
In theory, AMPK can be indirectly activated by any compound that causes AMP or calcium accumulation (for reviews see 43, 48, 51, 57, 77, 88). One of the most widely employed indirect AMPK activators is metformin. Several studies in the early 2000s showed that metformin increases AMP levels and activates AMPK by partially inhibiting Complex I of the mitochondrial ETC (27, 84). However, these studies used supra-pharmacological concentrations of metformin (27, 84), calling into question the relevance of this Complex I inhibition to the in vivo effects of metformin. Importantly, a more recent study showed that although at pharmacological concentrations metformin does activate AMPK, it does not do so by inhibiting Complex I (112). Metformin has also been reported to have AMPK-independent effects that need to be taken into consideration when evaluating the results of any study (43, 57).
Several direct AMPK activators are also widely available. These fall into two classes: AMP mimetics and AMPK interactors. The first direct activator to be described was 5-aminoimidazole-4-carboxamide riboside (AICAR), which is an adenosine analog that can be phosphorylated by adenosine kinase to generate an AMP mimetic (57, 77). However, since AICAR will also activate other AMP-dependent kinases, additional experimental support is needed for any study to attribute its effects specifically to the activation of AMPK (43, 57). Two more recently described direct activators are A769662 and compound 911. Both activate AMPK in a highly specific manner but do so independently of Thr172 phosphorylation (57, 77, 88) and appear to be specific to the β1 subunit isoform.
The only AMPK inhibitor that is widely available is compound C (dorsomorphin). Unfortunately, it is not very specific for AMPK and so any results obtained with this compound need to be viewed with caution (43, 77).
AMPK in aging and AD
Many of the actions of AMPK overlap with canonical anti-aging pathways activated by treatments such as dietary restriction (94). Furthermore, the activation of AMPK can extend lifespan in worms and flies whereas knockdown reduces lifespan (8). Whether this is also true in mammals is not clear. Although metformin was reported to extend lifespan and health span in C57BL/6 mice (74), the same dose fed to genetically heterogeneous mice failed to do so in a very large study carried out at three separate locations (100).
While AMPK loss of function results in neurodegeneration in flies (79), the role of AMPK in neuroprotection in the mammalian brain is unclear (79, 88, 111). Since a decline in cerebral energy metabolism occurs during aging and is exacerbated in AD, it might be expected that AMPK, an enzyme that upregulates energy metabolism, would be beneficial under these conditions. Evidence for this is only beginning to be generated. For example, a recent study (80) showed that AMPK regulates neuronal survival in a non-cell autonomous manner by promoting astroglial glycolysis and function. Loss of AMPK in neurons also stimulated neuronal hyperexcitability and enhanced seizure susceptibility (80).
Since metformin has a long history of clinical use for the treatment of type 2 diabetes and is considered safe, there have been a number of studies that have looked at its effects on aging and AD (for reviews see 8, 9, 78). However, the results with respect to both outcomes are not entirely clear. Given metformin's use for the treatment of diabetes, there is a large amount of both epidemiological data and results from a few randomized clinical trials that examined its effects on cognitive function in the context of aging and the development of AD. Several recent meta-analyses (9, 78) of these studies concluded that although metformin did seem to reduce the risk of cognitive impairment and AD in aging diabetic patients, there were no clear benefits in non-diabetic subjects. One of the analyses (9) also suggested that the studies where no or negative effects of metformin were seen in diabetic patients correlated with metformin-induced B12 deficiency since vitamin B12 deficiency has been linked to cognitive impairment.
Similarly, since an increase in cancer risk is also associated with aging (3), the effects of metformin on cancer have been examined in a number of epidemiological studies, especially in the context of type 2 diabetes (61, 117). For many types of cancer, metformin appears to reduce cancer mortality and improve the efficacy of first-line cancer drugs in patients with type 2 diabetes (61, 117). However, consistent with the studies on cognitive impairment and AD, the benefits of metformin in the context of cancer appear less clear in non-diabetic patients (61).
Relevance of AMPK targets to oxytosis/ferroptosis
As described earlier, AMPK has multiple targets that could contribute to the protection of cells against both endogenous and exogenous inducers of oxytosis/ferroptosis and thereby preserve cellular function in the context of both aging and neurodegenerative diseases (Fig. 3). A key target of AMPK relevant to oxytosis/ferroptosis is ACC1, whose inhibition leads to a decrease in free fatty acids and particularly polyunsaturated fatty acids (PUFAs), which are believed to be particularly susceptible to peroxidation prior to integration in the cellular membranes (20, 59). Furthermore, since ACC1 inhibition also promotes fatty acid oxidation, this can lead to a further decrease in oxidizable fatty acid levels. As noted earlier, this combination of effects on fatty acid synthesis and breakdown also enhances NADPH and GSH levels and the maintenance of both are important for protection against oxytosis/ferroptosis.
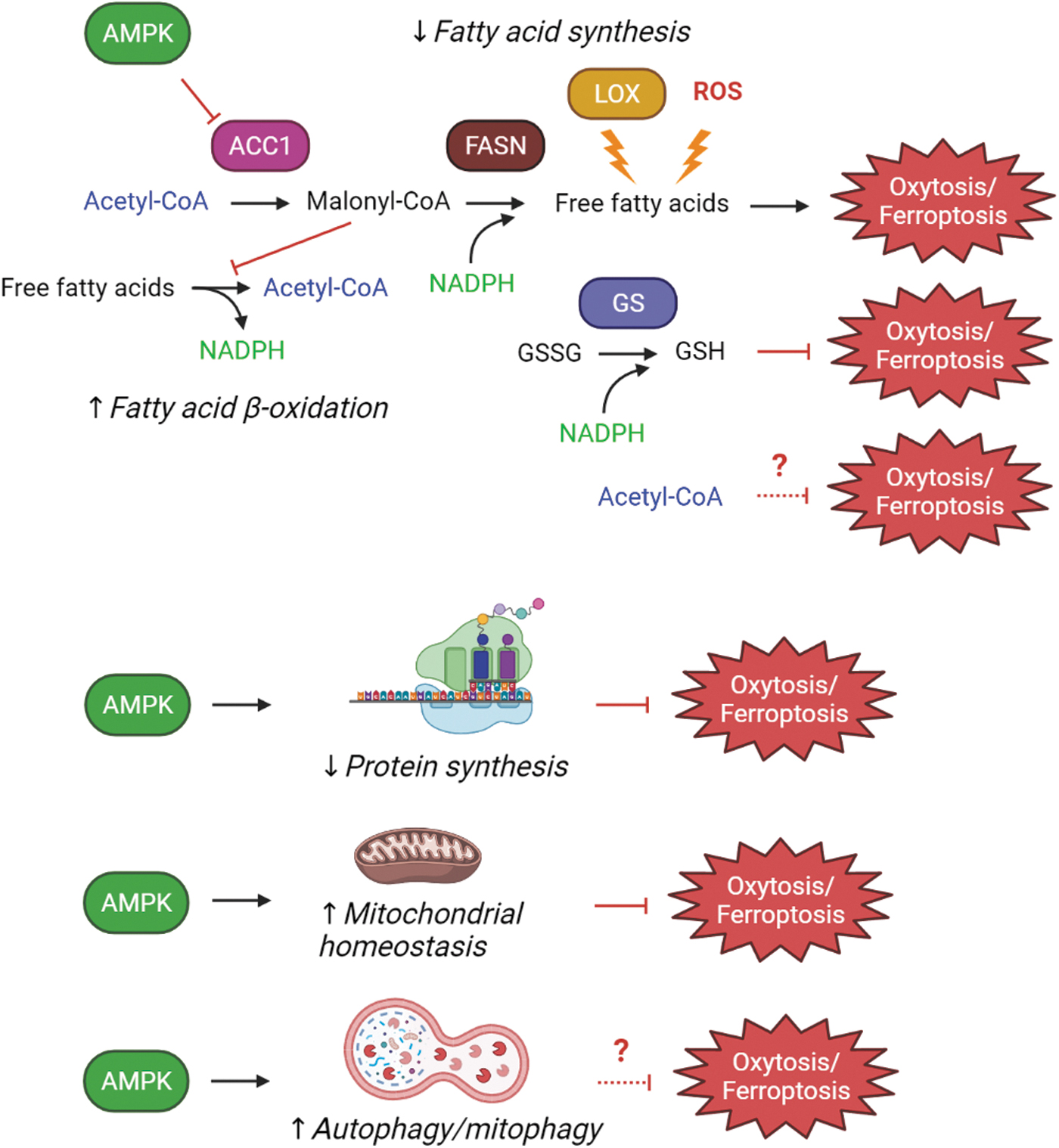
In addition to the actions of AMPK described earlier, AMPK activation also leads to a decrease in protein synthesis and protein synthesis inhibitors have been shown to block oxytosis/ferroptosis induced by system xc − inhibitors as well as other treatments that reduce intracellular GSH levels (23, 62). Although the role of protein synthesis inhibition in protection against oxytosis/ferroptosis has not been explored extensively, it might be tied to p53, which has been shown to induce oxytosis/ferroptosis (69). Among the transcriptional targets of p53 are several LOXs (69), which are known to promote this cell death pathway. Thus, an AMPK-mediated decrease in protein synthesis could also contribute to the protective effects of AMPK activation. However, it was recently shown that the activation of mTORC1 via a pathway independent of TSC2 protects from oxytosis/ferroptosis by upregulating the synthesis of GPx4 (124). In addition, the mTORC1 inhibitor rapamycin does not protect from erastin-induced cell death (59). Together, these results suggest that any protective effects of a reduction in protein synthesis via AMPK activation must target a very specific group of proteins.
While AMPK activation also leads to the induction of autophagy, the role of autophagy in oxytosis/ferroptosis is not clear. Although there is substantial evidence from multiple studies that inducers of oxytosis/ferroptosis activate autophagy (for review see 68), whether or not autophagy induction contributes to cell death or is a protective response still needs to be determined. For example, in human HT1080 cells, we found no protection against glutamate, erastin, or RSL3 by either the autophagy inhibitor bafilomycin A or knockdown of autophagy related 5 (ATG5), a key protein involved in autophagy (99). This is in agreement with one other report (23) but in contrast to others (32, 107, 129) and is likely related to both different levels of baseline cell death and the time point after toxin treatment at which cell death was measured.
AMPK activation also promotes healthy mitochondria (48), and mitochondrial dysfunction is a key feature of oxytosis/ferroptosis as well as age-associated neurodegenerative diseases. Thus, this is yet another way that AMPK activation could contribute to protection against oxytosis/ferroptosis.
Identification of AMPK in Protection against Oxytosis/Ferroptosis Using Chemical Biology
Because of the strong evidence indicating that all of the cellular pathophysiological changes that are associated with the oxytosis/ferroptosis pathway are also seen in the aging brain and are exacerbated in age-associated neurodegenerative diseases, we have used protection from this form of cell death as our primary screen to identify potential AD drug candidates (86). For most of these screens, we have used HT22 cells, a mouse cell line derived from hippocampal neurons that we developed in the laboratory in combination with millimolar concentrations of extracellular glutamate to inhibit system xc-. While the in vivo relevance of system xc- inhibition to neurodegeneration is not clear, what is apparent is that a disturbance in the cellular redox balance is a central event in neurodegeneration and can be initiated by a variety of pathophysiologically relevant insults.
Two of the AD drug candidates that we identified using inhibition of oxytosis/ferroptosis as the primary phenotypic screen were subsequently shown to modulate AMPK activity, which appears to play an important role in their neuroprotective activity (Table 1). One of these drug candidates, CMS121, is a derivative of the flavonoid fisetin with improved medicinal chemical properties that are more consistent with those of known central nervous system drugs and significantly enhanced neuroprotective activity against oxytosis/ferroptosis (14). CMS121 recently received IND approval and we plan to start a phase 1 clinical trial for the treatment of AD in 2022.
Effects of AMPK Modulation on Oxytosis/Ferroptosis
N/A indicates that cell death was not examined in the context of AMPK manipulation.
BSO, buthionine sulfoximine; IRI, ischemia reperfusion injury; IMCA, 2-imino-6-methoxy-2H-chromene-3-carbothioamide; ko, knockout; SOR, sorafenib; SSA, sulfosalacylic acid.
A second compound is based on the natural product curcumin. This compound, called J147, has a low nanomolar EC50 in the oxytosis/ferroptosis assay (12). The target was identified as ATP5A, a catalytic subunit of the mitochondrial ATP synthase complex (39). J147 is currently in a phase 1 clinical trial for the treatment of AD (NCT03838185).
After their identification and characterization in vitro, both CMS121 and J147 were tested in multiple models of AD, including the senescence accelerated prone mouse (SAMP8) (20). SAMP8 mice are an excellent animal model for the most common, sporadic form (>97% of cases) of AD because this is one of the few mouse AD models that is not biased toward the rare, dominant genetic forms of the disease, is age-dependent, and shows pathology (Aβ, tau phosphorylation, memory loss, vascular damage) and disease progression that is similar to what is seen in AD in humans (19, 85).
We aged three cohorts of mice to 9 months and then fed them with control, CMS121, or J147 diet for an additional 4 months (13 month-old mice). A fourth cohort of 9 month-old mice was used as a control (20). Although we did see improvements in cognitive function with both compounds as compared with the control mice, the goal of this study was to use these oxytosis/ferroptosis inhibitors in a chemical biology approach to identify specific, age-related molecular pathways with therapeutic potential for AD.
To achieve this goal, we carried out large data multi-omic analyses and integrated multiple different types of molecular parameters (genes, metabolites, and proteins) (19, 20). Using this approach, acetyl-CoA was identified as a central metabolite that appeared to underlie the neuroprotective actions of CMS121 and J147. The modulation of acetyl-CoA metabolism by the compounds correlated with the preservation of mitochondrial homeostasis and histone acetylation at an epitope that is required for memory and is downregulated in the brains of AD patients (20).
Most relevantly, the in vivo effects of the compounds on acetyl-CoA metabolism appeared to result from the inhibition of ACC1 via the activation of AMPK (20). As noted earlier, AMPK phosphorylates ACC1, thereby causing an inhibition in its activity. A decrease in the brain and plasma levels of free PUFAs, the targets of lipid peroxidation, after treatment of the mice with both compounds was observed in agreement with the role of ACC1 in fatty acid synthesis (20). Consistent with the in vivo results, both compounds also induced AMPK activation and ACC1 inhibition in the HT22 cells. Moreover, using mouse embryonic fibroblasts (MEFs) that lack AMPK, we showed that both the protective effects of the two compounds, as well as their ability to increase acetyl-CoA levels, were significantly reduced as compared with wild-type MEFs. J147 was found to activate AMPK via its ability to partially inhibit the mitochondrial ATP synthase, which leads to the activation of CaMKK2 (39). Although it is not yet clear how CMS121 activates AMPK, it is likely through its action on the lipid biosynthetic enzyme, fatty acid synthase (FASN) (127), as described next, although this possibility requires further study.
These observations led us to further explore the role of AMPK activation and subsequent ACC1 inhibition in protection against oxytosis/ferroptosis using the HT22 cells. Interestingly, we found that direct inhibition of ACC1 with 5-(tetradecyloxy)-2-furoic acid (TOFA) or knockdown of ACC1 in the cells not only led to increased acetyl-CoA levels but was also highly protective against oxytosis/ferroptosis (20). In agreement with these results, high concentrations of acetate, a precursor of acetyl-CoA, could also protect against oxytosis/ferroptosis. Thus, these studies identified the AMPK/ACC1 axis as a new molecular regulator of oxytosis/ferroptosis. Whether the protective effect is due to directly decreasing the levels of free PUFAs, increasing the levels of acetyl-CoA, or some combination of the consequences of AMPK activation warrants further investigation.
Additional support that the protective effects of AMPK activation/ACC1 inhibition against oxytosis/ferroptosis are due to decreases in free PUFAs is provided by our recent study on the effects of CMS121 in the APPswe/PS1ΔE9 transgenic mouse model of AD (2). Similar to the SAMP8 study, these mice were aged to 9 months, a time when they already showed cognitive impairment (19, 22), and then treated for 3 months with CMS121. CMS121 treatment alleviated the loss of cognitive function, as assessed in multiple behavioral tests, while at the same time it reduced lipid peroxidation and neuroinflammation in the brains of the mice. Importantly, we showed that the beneficial effects of CMS121 in this model correlated with its ability to partially inhibit FASN, thereby reducing the levels of PUFAs. Further, using HT22 cells and mouse BV2 microglial cells, we showed that FASN knockdown phenocopied the protective effects of CMS121 against oxytosis/ferroptosis and inflammation, respectively.
Thus, these studies provided the first evidence that activation of AMPK could provide protection against oxytosis/ferroptosis induced by multiple stimuli, including both system xc − inhibitors and RSL3. Whether the protective effects of AMPK activation are mediated solely by its inhibition of ACC1 or also through other downstream targets as described earlier still remains to be determined.
Further evidence for protection by the AMPK/ACC axis against oxytosis/ferroptosis
Similar results regarding the protective role of AMPK activation were obtained by Lee et al. using a very different approach (59) (Table 1). In this study, the authors found that when MEFs were exposed to multiple inducers of oxytosis/ferroptosis (erastin, cystine deprivation, RSL3, GPX4 knockout) under conditions of glucose starvation, cell death was prevented. By using different concentrations of glucose in the medium, they showed that ATP depletion correlated directly with the protective effects of glucose starvation. Since ATP depletion can activate AMPK (see AMPK Signaling), further studies focused on this kinase. Known AMPK activators (AICAR, A769662) were found to protect cells from oxytosis/ferroptosis, whereas AMPK knockdown greatly reduced the protective effects of both glucose starvation and the AMPK activator A769662. Consistent with our studies, these authors identified ACC1 as the key target of AMPK that regulates oxytosis/ferroptosis. Comparative lipidomic analyses on cells exposed to the AMPK activator A769662, the ACC1 inhibitor TOFA, or lacking AMPK suggested that AMPK activation suppresses whereas AMPK loss enhances PUFA-containing lipid biosynthesis, thereby altering the cellular sensitivity to oxytosis/ferroptosis (59). These studies were extended to an in vivo model of ferroptosis, renal ischemia reperfusion injury (IRI). Previous studies have shown that the cell death induced by renal IRI has many of the characteristics of ferroptosis, including alterations in renal epithelial cell mitochondrial morphology and protection by the ferroptosis inhibitor, ferrostatin 1 (30, 75). In this study (59), the AMPK activator AICAR reduced IRI-induced renal tubular injury and blood urea nitrogen, a marker of kidney injury, as well as 4-HNE staining, a marker of lipid peroxidation.
Very similar results regarding the protective effects of both AMPK activation and ACC1 inhibition against ferroptosis were obtained in another study (64) (Table 1). In addition, these authors found that in non-small cell lung cancer cells, LKB1 levels and/or activity correlated directly with protection of the cells against ferroptosis. Interestingly, they also showed that in both MEFs and H1299 cells, erastin itself induced AMPK phosphorylation whereas the knockdown of AMPK enhanced the sensitivity of the cells to ferroptosis, suggesting that the activation of AMPK might be a protective response to treatment with ferroptosis inducers and is perhaps reflective of the ∼40% of cells that go on to survive. Further, consistent with our results, these authors found that FASN inhibition or knockdown also protected MEFs from ferroptosis.
Several studies in cancer cells have also shown protective effects of AMPK activation against oxytosis/ferroptosis (110, 126). Zhong et al. explored the mechanism underlying the cell death-promoting effects of a compound called FTY720 against multiple myeloma cells (126). They found that FTY720-induced cell death could be blocked by inhibitors of oxytosis/ferroptosis, including ferrostatin and deferoxamine, and was associated with a decrease in AMPK phosphorylation (126). The decrease in AMPK phosphorylation was dependent on the activation of protein phosphatase 2A by FTY720 (126). Importantly, the AMPK activator AICAR was able to prevent FTY720-induced cell death (126). Very recently, Wang et al. showed that both erastin and RSL3 induced CaMKK2 activation and AMPK phosphorylation in melanoma cells (110). Interestingly, the inhibition of CaMKK2 or knockdown of AMPK greatly potentiated erastin- or RSL3-induced cell death, which could be prevented by ferrostatin (110).
In contrast to these papers, there are a number of studies carried out mainly in cancer cells that suggest that AMPK activation might promote oxytosis/ferroptosis (42, 98, 109, 114, 123, 125) (Table 1). However, while all of these studies do show that oxytosis/ferroptosis inducers activate AMPK, as also reported by Li et al. (64), only one provides evidence that knockdown of AMPK is protective (98). Although some of these studies (42, 98, 109) used the AMPK inhibitor compound C to demonstrate a role for AMPK in promoting oxytosis/ferroptosis, Li et al. (64) showed that compound C protects both wild-type and AMPK knockout MEFs from erastin-induced cell death. Furthermore, other studies have confirmed that compound C is not specific to AMPK (43, 77). Moreover, while one of these studies (114) showed that the AMPK activator metformin could induce ferroptosis in breast cancer cells, the authors found that this was through an AMPK-independent mechanism. The only one of these papers that used AMPK knockdown (98) demonstrated a novel role for AMPK in promoting ferroptosis. In this study, the authors showed that beclin 1, a component of the class III PI3K complex I that is best known for its key role in autophagy, plays an autophagy-independent role in ferroptosis. Specifically, these authors found that beclin 1 bound to the antiporter-specific light chain of system xc −, xCT, in an AMPK-dependent manner, resulting in inhibition of the antiporter. Both compound C and knockdown of AMPK decreased beclin 1 phosphorylation and thereby its interaction with xCT and reduced erastin-mediated cell death. Since the pro-ferroptotic effect of AMPK in this scenario is specific to compounds that induce ferroptosis by inhibiting system xc −, then AMPK knockdown or activation should have no effect on ferroptosis induced by compounds such as RSL3 that act downstream of system xc − inhibition. However, the authors did not test this idea. In addition, although knockdown of both AMPK and compound C also reduced the erastin-induced loss of GSH as well as the increase in lipid peroxidation, neither had any effect on the erastin-induced increase in free Fe+2 seen in this study (98). This finding with regard to the effects of compound C on the induction of free Fe+2 by erastin is in contrast to the report of Han et al. (42), where compound C reduced erastin-induced increases in free Fe+2.
Thus, the bulk of the evidence supporting a pro-oxytotic/ferroptotic role for AMPK is weak. However, it does raise some questions, especially regarding the mechanisms underlying the activation of AMPK by inducers of oxytosis/ferroptosis and whether this induction is related to the cells that survive or is evidence of a pro-death role for AMPK. Careful analysis of percentage of cell death versus levels of AMPK activation will be needed to figure this out. In addition, since AMPK is believed to play a dual role in cancer cells (88,120), it may be that in some contexts its activation does promote ferroptosis. For example, some types of cancer cells rely strongly on fatty acid synthesis for growth; thus, AMPK activation leading to ACC inhibition could put these cells in a metabolically stressful condition such that a further insult such as an oxytosis/ferroptosis inducer might tip them over the edge to death. Again, this possibility is something that will need to be carefully studied in a variety of different cancer cell lines in comparison with normal cells, correlating their metabolic requirements with their sensitivity to oxytosis/ferroptosis inducers as well as their levels of AMPK activity.
Conclusions
Several research groups, including our own, have now identified AMPK as a potential protective enzyme against oxytosis/ferroptosis (Fig. 3). The current data strongly suggest that the key target of AMPK in this context is ACC1, the rate-limiting enzyme for fatty acid synthesis. However, AMPK has a number of other targets that could potentially contribute to its protective effects against oxytosis/ferroptosis and these are worth exploring in future studies (Fig. 3). In addition, the cell type and context-dependent roles of AMPK in protection against oxytosis/ferroptosis need to be examined much more thoroughly. Finally, although there is reasonably strong evidence for a role for AMPK in promoting longevity, whether it also plays a direct protective role in the aging brain and particularly in neurodegenerative diseases requires much more evidence. Since AMPK regulates systemic metabolism and the health of peripheral tissues can play a critical role in brain function, the possible indirect beneficial effects on the brain should also be considered.
Footnotes
Authors' Contribution
A.C., Writing-review & editing (lead), Visualization; D.K., Writing-review & editing (equal); Z.L., Writing-review & editing (equal); P.M., Conceptualization, Funding acquisition, Supervision, Writing-original draft, and review & editing (equal).
Author Disclosure Statement
The authors have nothing to declare and no conflicts of interest. The Salk Institute holds patents on both J147 and CMS121.
Funding Information
This work was supported by grants from NIH to P.M. (AG054714, AG069206, AG061296). D.K. is supported by a grant from the Bundy Foundation.