
Abstract
Significance:
Type 2 diabetes mellitus, which is related to oxidative stress and mitochondrial dysfunction, is one of the most prevalent diseases in the world. In the past decade, alterations in autophagy have been shown to play a fundamental role in the development and control of type 2 diabetes. Further, mitophagy has been recognized as a key player in eliminating dysfunctional mitochondria in this disease.
Recent Advances:
Recently, much progress has been made in understanding the molecular events associated with oxidative stress, mitochondrial dysfunction, and alterations in autophagy and mitophagy in type 2 diabetes.
Critical Issues:
Despite increasing evidence of a relationship between mitochondrial dysfunction, oxidative stress, and alterations of autophagy and mitophagy and their role in the pathophysiolology of type 2 diabetes, effective therapeutic strategies to combat the disease through targeting mitochondria, autophagy, and mitophagy are yet to be implemented.
Future Directions:
This review provides a wide perspective of the existing literature concerning the complicated interplay between autophagy, mitophagy, and mitochondrial dysfunction in type 2 diabetes. Further, potential therapeutic targets based on these molecular mechanisms are explored. Antioxid. Redox Signal. 39, 278–320.

I. Introduction
Diabetes mellitus, commonly known as diabetes, is undoubtedly the most prevalent metabolic disease globally. There are two main types of diabetes: type 1 and type 2. Type 1 diabetes, an autoimmune disease whose development has been related to exposure to viruses or other environmental factors, causes the destruction of the insulin-producing β cells in the pancreas, preventing the body from producing enough insulin to adequately regulate blood glucose levels. Type 2 diabetes is a metabolic condition of unbalanced hyperglycemia related to impaired pancreatic β cell function and defects in glucose metabolism and/or insulin action (reviewed in DeFronzo et al, 2015).
Type 2 diabetes currently affects around 537 million people worldwide; in other words, around 10.5% of the world's population, and this number is predicted to rise to 783 million (12.2%) by 2045 (Sun et al, 2022). This rising prevalence is especially relevant in developing countries, where diabetes is reaching epidemic proportions; with 1 in 10 people currently diagnosed, ∼90% of whom have type 2 diabetes (Khan et al, 2020). Moreover, concerns have been raised that more than 1 million deaths per year can be attributed to diabetes alone, making it the ninth leading cause of mortality (Khan et al, 2020). In addition to genetic predisposition, the rising prevalence of type 2 diabetes (6059 cases/100,000 in 2017) can be attributed to multiple factors, mainly (1) improved medical care and higher survival rates among those who suffer it, and (2) increases in risk factors, particularly obesity, a condition that appears due to unhealthy eating habits and a sedentary lifestyle (Cho et al, 2018).
Importantly, this chronic condition affects individuals' quality of life and their functional capacities, leading to premature mortality and significant morbidity, the latter of which is a result of the development of multiple type 2 diabetes-related pathologies, among which dyslipidemia, obesity, depression, and hypertension are the most prevalent (reviewed in Trikkalinou et al, 2017 and in Zheng et al, 2018).
One of the most characteristic effects in the development of type 2 diabetes is the defective signaling downstream of the insulin receptor (INR), as well as a functional deficit and/or early destruction of insulin during its synthesis. It is well known that pancreatic β cells release insulin when the concentration of glucose in the blood increases, thus regulating its uptake in target tissues such as adipose tissue, and skeletal muscle. In this way, when there is a balance between insulin release and glucose levels, there is generally an adequate control of anabolic metabolism.
Defects in insulin secretion or insulin signaling/action can lead to impaired metabolic homeostasis, causing diseases such as obesity and atherosclerosis (reviewed in Semenkovich, 2006; Taylor, 2012). The relative deficiency of insulin secretion from β cells and alterations in glucose, protein, and lipid metabolism, as well as inflammation and oxidative stress due to excess energy intake and obesity, are typically combined with, or often preceded by, varying degrees of peripheral insulin resistance (Fig. 1).
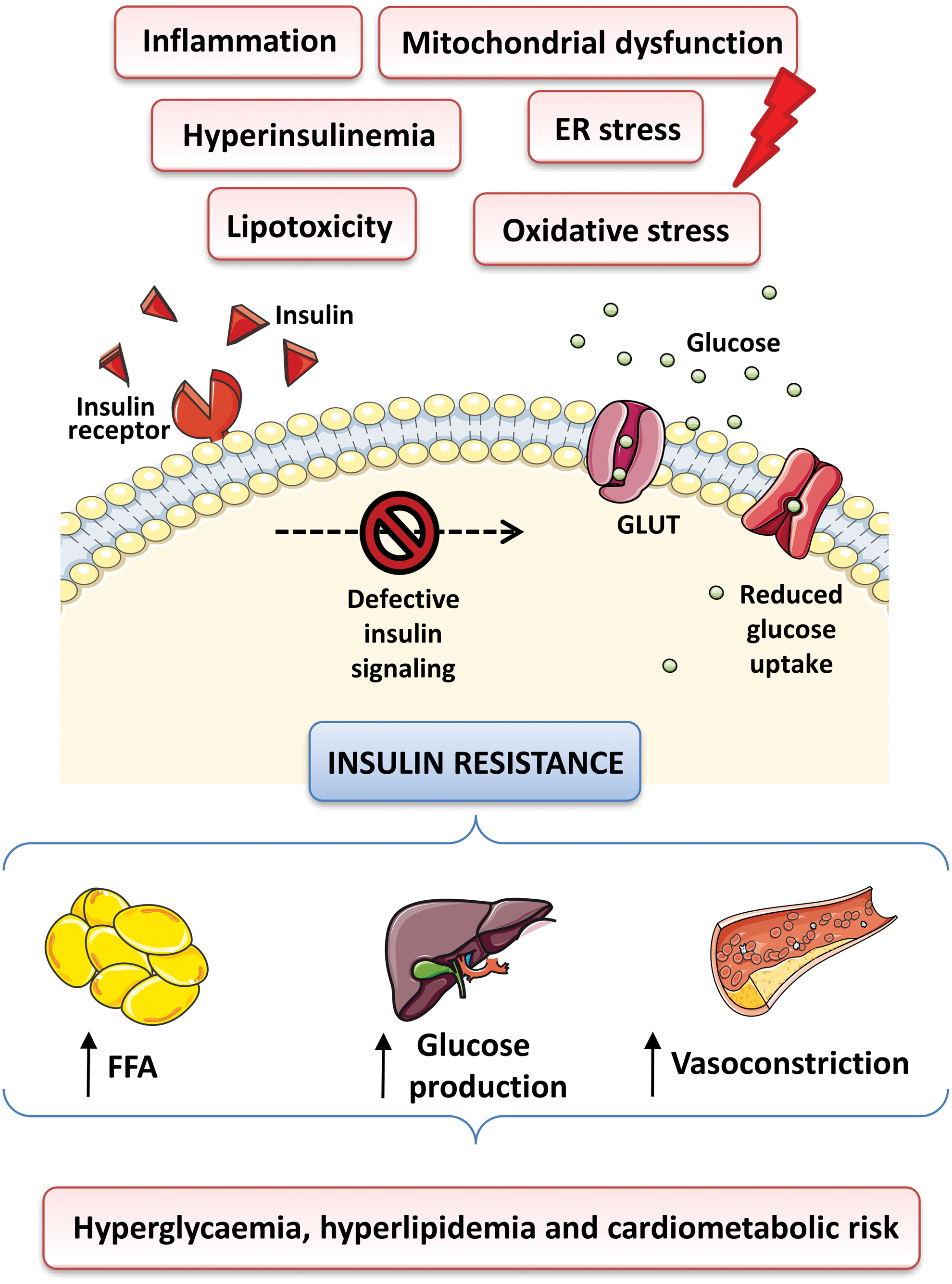
In this pathological condition, target tissues are unable to respond appropriately to insulin levels due to an impaired or reduced insulin signal transmission and decreased cellular sensitivity to the action of this hormone (reviewed in Freeman and Pennings, 2021). Alterations in insulin and glucose levels are also accompanied by chronic inflammation and alterations of the blood lipid profile, both of which progressively lead to the appearance of micro- and macrovascular alterations, which, together with other cellular disturbances, lead to numerous complications (such as coronary artery disease, retinopathy, cardiomyopathy, nephropathy, neuropathy, and peripheral vascular disease) (Figs. 1 and 2) (reviewed in DeFronzo et al, 2015). Thus, type 2 diabetes can be considered not merely a metabolic disease but also a vascular one.

Type 2 diabetes is generally related to hyperglycemia (high levels of glucose, related to the development of insulin resistance), and to dyslipidemia (characterized by elevated fasting and postprandial triglycerides, low high-density lipoprotein cholesterol [HDL-C], elevated low-density lipoprotein [LDL]-cholesterol, and the predominance of small dense LDL particles). These lipid changes represent the major link between diabetes and its increased cardiovascular risk. Diabetic individuals also present a long list of comorbidities, defined as the occurrence of one or more chronic conditions in the same person.
Semantically, diabetic complications and comorbidities are difficult to distinguish between, as the former participate in the pathogenesis of these diseases. Diabetes-associated comorbidities include not only established pathologies, such as chronic renal disease, heart disease, obesity, metabolic syndrome, dyslipidemia, or arterial hypertension, but also periodontal disease or vascular dementia and mild cognitive impairment. In addition, diabetics are at a greater risk of suffering from asthma or infections, as a result of the altered function of the immune system. This growing list of conditions is the most important concern once a patient is diagnosed with diabetes.
Diabetic cardiomyopathy is characterized by the presence of cardiac dysfunction in diabetic individuals who do not have a vascular condition (i.e., arterial hypertension, atherosclerosis, and coronary artery disease) or other risk factors that can cause structural heart disease (Bonora and DeFronzo, 2018). Myocardial ischemia-reperfusion (I/R) injury is also closely related to diabetes mellitus, and hyperglycemia exacerbates myocardial injury during I/R. Retinopathy is one of the most frequent complications in type 2 diabetes and one of the main causes of acquired blindness in adults. It can be defined as a neurovascular disorder since it can affect both the neuroglia and blood vessels.
Diabetic nephropathy is the main cause of chronic kidney disease (24-h urinary protein excretion >0.5 g) in subjects initiating renal replacement therapy (Saran et al, 2020) and is related to a significant increase in cardiovascular mortality (Valmadrid et al, 2000). According to a fundamental study for the knowledge and development of diabetes (UK Prospective Diabetes study), the cumulative incidence of microalbuminuria in people with type 2 diabetes (study population of 3982 women and 3250 men) was 12.6% over 7.3 years, the incidence of microalbuminuria was 2% per year, and the prevalence 10 years after diagnosis was 25% (Adler et al, 2003). Proteinuria in type 2 diabetes is extremely variable, ranging between 5% and 20% (Fava et al, 2001).
Over the past two decades, there has been a notable advance in knowledge of the mechanisms and risk factors of diabetic nephropathy, and in developing therapies to prevent the disease or halt its development. Ideally, primary prevention is based on the prompt detection of diabetic nephropathy, the use of multifactorial therapies targeting the central risk factors (dyslipidemia, hyperglycemia, hypertension, sedentary lifestyle, and/or smoking), and the use of drugs with a renoprotective effect (angiotensin-converting enzyme [ACE] inhibitors and/or angiotensin II receptor blockers [ARBs]) that greatly decrease the progression of kidney disorder. Thus, the treatment of arterial hypertension is one of the main strategies to prevent and control the development of the disease.
II. Pathogenesis of Type 2 Diabetes: General Aspects
Current individualized therapeutic approaches (non-pharmacological and pharmacological) seek to normalize blood glucose levels, blood pressure, and plasma lipid profiles (Borse et al, 2021; Carrasco-Sanchez et al, 2021). Nevertheless, a better understanding of the modulation of the molecular pathways that underlie the pathogenesis and progression of type 2 diabetes could prevent or minimize the deleterious effects and complications of this condition. Importantly, its pathogenesis involves two types of cells: pancreatic β cells, which are insulin-producing, and cells that are targets of insulin's action, among which skeletal muscle cells and adipocytes are particularly relevant.
Some of the effects of type 2 diabetes involve both cell types, whereas others are specific. However, it is very difficult to establish a timeline and a cause-consequence relation in the pathogenic process, as the alterations in the functions of these different cell types are strongly interconnected. During the progression of the disease, the insulin-resistant state forces β cells to compensate for the lack of insulin by elevating its synthesis to restore the normal blood glucose level. However, despite fairly normal β cell replication and islet formation, the number of β cells declines due to enhanced apoptosis; thus, in advanced diabetic patients, β cell exhaustion occurs, which leads to subsequent persistent hyperglycemia.
Indeed, many authors point to β cell reprogramming or even dedifferentiation/transdifferentiation as a core phenomenon contributing to the development of type 2 diabetes (Remedi and Emfinger, 2016; Wang and Zhang, 2021). Chronic hyperglycemia impairs the normal functions of the circulatory system (the endothelium), the immune system, the nervous system, the adipose tissue, the gastrointestinal tract, and the liver, and these alterations lead to further progression of the disease while generating the pathophysiological milieu that underlies the development of many chronic comorbidities (Fig. 3).
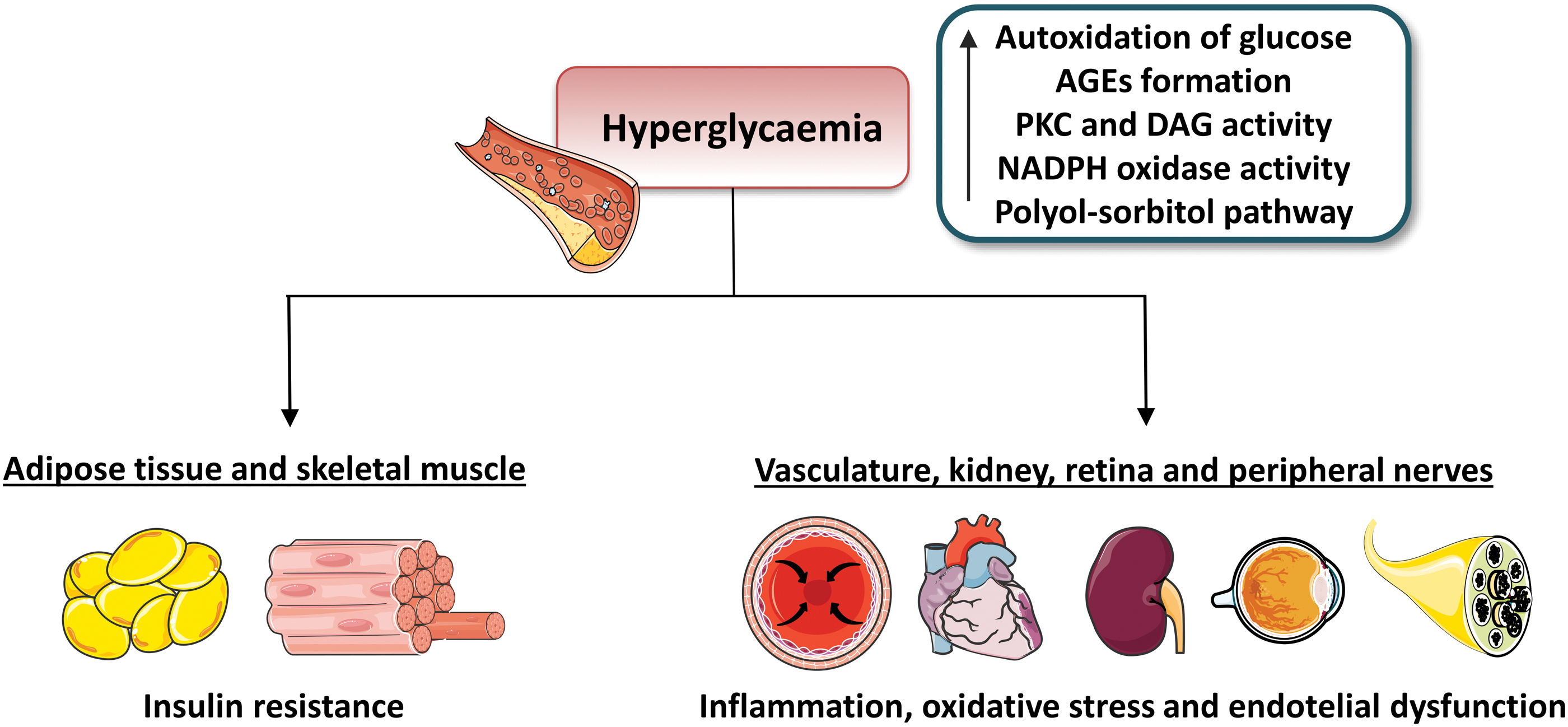
Type 2 diabetes is understood as a metabolic imbalance in which redox signaling and adenosine triphosphate (ATP) levels are compromised, and in which mitochondria act as critical players (reviewed in Patti and Corvera, 2010). In this sense, redox imbalance can activate different stress pathways involving serine/threonine (Ser/Thr) kinases, which eventually cause impairment of insulin signaling (reviewed in Rains and Jain, 2011). In addition, hyperglycemia can increase membrane lipid peroxidation and oxidative damage of lipoproteins (which affects cellular membranes and other structures containing lipids), and this is known to be directly proportional to blood glucose concentrations in diabetic patients (Jain et al, 1989 ) and/or to glucose concentrations in vitro (Jain, 1989 ).
Mitochondria are essential double-membrane cellular organelles involved in multiple important processes, including energy supply, metabolism, redox homeostasis, cell differentiation, and ion homeostasis, and they are thus the main regulators of both the life and death of cells (Cooper, 2000). They constantly generate reactive oxygen species (ROS), highly reactive derivatives of molecular oxygen that act as cell signaling molecules in multiple regulatory processes involved in cell survival, mitochondrial function, and insulin sensitivity.
In this way, altered mitochondrial function is one of the hallmarks of diabetes and insulin resistance (reviewed in Kim et al, 2008). In this regard, defects in mitochondrial fatty acid oxidation are related to insulin resistance, which eventually lead to the impairment of insulin signaling as a result of the intracellular accumulation of fatty acid derivatives (fatty acyl CoA and diacylglycerol [DAG]) as seen in the skeletal muscle (reviewed in Park and Seo, 2020) (Fig. 4).

Further, it is important to consider that many type 2 diabetic patients suffer from comorbidities such as metabolic syndrome and obesity, which also contribute to altered lipid levels.
It is well known that a deterioration in the processes that maintain cellular homeostasis can lead to the accumulation of damaged organelles and can consequently contribute to high levels of ROS and the alteration of the redox state, together with or leading to mitochondrial dysfunction. These events can activate alternative signaling pathways that cause different pathological effects, including pancreatic β cell dysfunction and insulin resistance, features commonly related to type 2 diabetes (reviewed in Petersen et al, 2004).
In this regard, a number of studies performed in rodents have suggested that insufficient autophagy in β cells makes them susceptible to endoplasmic reticulum (ER) stress through unfolded protein response (UPR) activation (Quan et al, 2012 and reviewed in Herbert and Laybutt, 2016; Pandey et al, 2019), to which multiple diabetes-related events are attributed, such as an increase in ROS generation and in cell death.
Experimental evidence has also shown that ER stress in β cells can be caused by elevated levels of proinflammatory cytokines, glucose, and/or free fatty acids (FFAs), all of which are common features of insulin resistance, metabolic syndrome, and type 2 diabetes (Cunha et al, 2008; Hasnain et al, 2014 and reviewed in Bensellam et al, 2012). Interestingly, deregulated mitophagy has been associated with the accumulation of dysfunctional mitochondria and increased ROS generation in peripheral blood mononuclear cells of individuals with type 2 diabetes (Bhansali et al, 2017).
Given the important interrelationship among mitochondria, oxidative stress, and metabolism and the clear role of autophagy/mitophagy in the regulation of type 2 diabetes, it is not surprising that the modulation of these processes has generated substantial interest as a potential therapeutic approach. In recent years, different natural compounds of animal and plant origin (including urolithin A, actinonin, resveratrol, and melatonin) (reviewed in Shakeri et al, 2020; Su et al, 2021), as well as other compounds that include synthetic agents (such as elamipretide, niclosamide, and metformin) (Barini et al, 2018; Bhansali et al, 2020; Escribano-Lopez et al, 2019), have been the subject of investigation to test their ability to modulate mitochondrial function, mitochondrial dynamics, and mitophagy.
This review takes an in-depth look at the contribution of oxidative stress and mitochondrial damage to the onset and progression of type 2 diabetes, with a special focus on autophagy and mitophagy, and the mechanisms that could be key to their use as potential therapeutic strategies in the management and prevention of type 2 diabetes.
A. Insulin signaling
Insulin signaling modulates blood glucose levels via different mechanisms that enable cellular glucose uptake, thereby promoting cell growth and proliferation as well as regulating protein, lipid, and carbohydrate metabolism (reviewed in Wilcox, 2005). Insulin mediates its actions by binding to the extracellular subunit of INR, a well-known ligand-activated tyrosine kinase, triggering its autophosphorylation, which leads to tyrosine phosphorylation of downstream insulin receptor substrates (IRS), particularly IRS-1 or IRS-2, though a total of six isoforms have been described.
After IRS phosphorylation, two pathways are activated: the mitogen-activated protein (MAP) kinase and the phosphoinositide 3-kinases (PI3K)-protein kinase B (Akt) pathways (reviewed in De Meyts, 2016; Świderska et al, 2018) (Fig. 5). Mitogen-activated protein kinase (MAPK) and PI3K/Akt signaling systems are known to interact through different pathways in the context of diabetes and can be viewed as a common target for the treatment of this disease.

The PI3K pathway is responsible for most metabolic effects of insulin and is connected exclusively through IRS. Insulin signaling in this pathway is followed by activation of PI3K, Akt (Akt1, Akt2, and Akt3), protein kinase C (PKC; of which more than 20 isoforms have been recognized), phosphoinositide-dependent kinase-1 (PDPK1), ribosomal protein S6 kinase beta 1 (S6K1), and mammalian target of rapamycin (mTOR) (Scott et al, 1998 and reviewed in Janus et al, 2016). This cascade of events eventually results in increased translocation of glucose transporter type 4 (GLUT4) to the membrane, thus enhancing glucose uptake and regulating blood glucose levels (reviewed in Bogan, 2012).
There are different stimuli that enhance the trafficking of GLUT4 storage vesicles, including the activation of canonical insulin signaling cascade in adipose tissue and skeletal muscle, and through physical exercise, which provides muscle contraction. Several studies have attempted to clarify these mechanisms, demonstrating that the signaling cascades involve remodeling of the cytoskeleton, a process that facilitates the movement of GLUT4-containing vesicles and, consequently, the uptake of glucose by the tissues (reviewed in Tunduguru and Thurmond, 2017).
After uptake, glucose is promptly phosphorylated to glucose 6-phosphate (G6P), which can be metabolized by different metabolic pathways (Fig. 5). The MAPK pathway (RAF/Ras/MEK/MAPK pathway, also described as an extracellular signal-regulated kinase [ERK] pathway) emanates from both IRS and Src-homology-2-containing (Shc) protein and is involved in the regulation of gene expression and, in cooperation with the PI3K pathway, in the control of cell growth (“mitogenesis”) and differentiation.
This pathway mediates several non-metabolic actions of insulin, including pro-atherogenic, pro-inflammatory, mitogenic, and proliferative effects. It involves the binding of other essential molecules, such as growth factor receptor-bound protein (Grb2), insulin-like growth factor 1 (IGF1), and the binding of insulin to its specific receptor on a target cell membrane, with the subsequent phosphorylation of specific substrates such as IRS-1, IRS-2, and Shc. These are associated with proline-rich sequences in son of Sevenless (SOS), which, once activated, can promote the shift of membrane-bound Ras from an inactive form (Ras-GDP) to an active form (Ras-GTP).
Ras-GTP binds to the protein kinase RAF, favoring its activation, which, in turn, directly activates MEK1 and MEK2. Consequently, these serine/tyrosine/threonine kinases phosphorylate and activate the MAPK ERK1/2. Finally, activated ERK1/2 regulates different transcription factors and INR signaling by phosphorylating several substrates and producing an appropriate biological response (reviewed in De Meyts, 2016). Of note, high levels of IGF1 activate Akt, which then phosphorylates RAF at serine residue Ser259 and thus suppresses the activity of the RAF-MEK-ERK signaling pathway (Moelling et al, 2002).
Finally, in the context of insulin signaling, ROS have a dual role, as both facilitators and inhibitors of the insulin signaling cascade. The ROS mediate these effects through redox modifications of cysteine residues, affecting the activity of phosphatases, stress-sensitive kinases, and metabolic sensors (Besse-Patin and Estall, 2014).
III. Mitochondrial Function and Oxidative Stress, and Their Link to Inflammation
Mitochondrial O2 consumption is linked to the primary role of these organelles, which is to generate most of the cell's energy supply by breaking down molecules that include carbohydrates and fatty acids, therefore forming ATP through a key energetic process known as oxidative phosphorylation (OXPHOS) (reviewed in Zhao et al, 2019). This process is mediated by the transfer of electrons from donor to acceptor molecules through the mitochondrial electron transport chain (ETC), which includes four different transmembrane protein complexes (I–IV) coupled to the ATP synthase, also named complex V (Fig. 6). Three of the ETC complexes—complexes I, III, and IV—exhibit proton-pumping activity; through these complexes, an electrochemical proton gradient (or proton-motive force) is generated between the mitochondrial matrix and the mitochondrial intermembrane space, while complex V drives the synthesis of ATP via chemiosmosis.
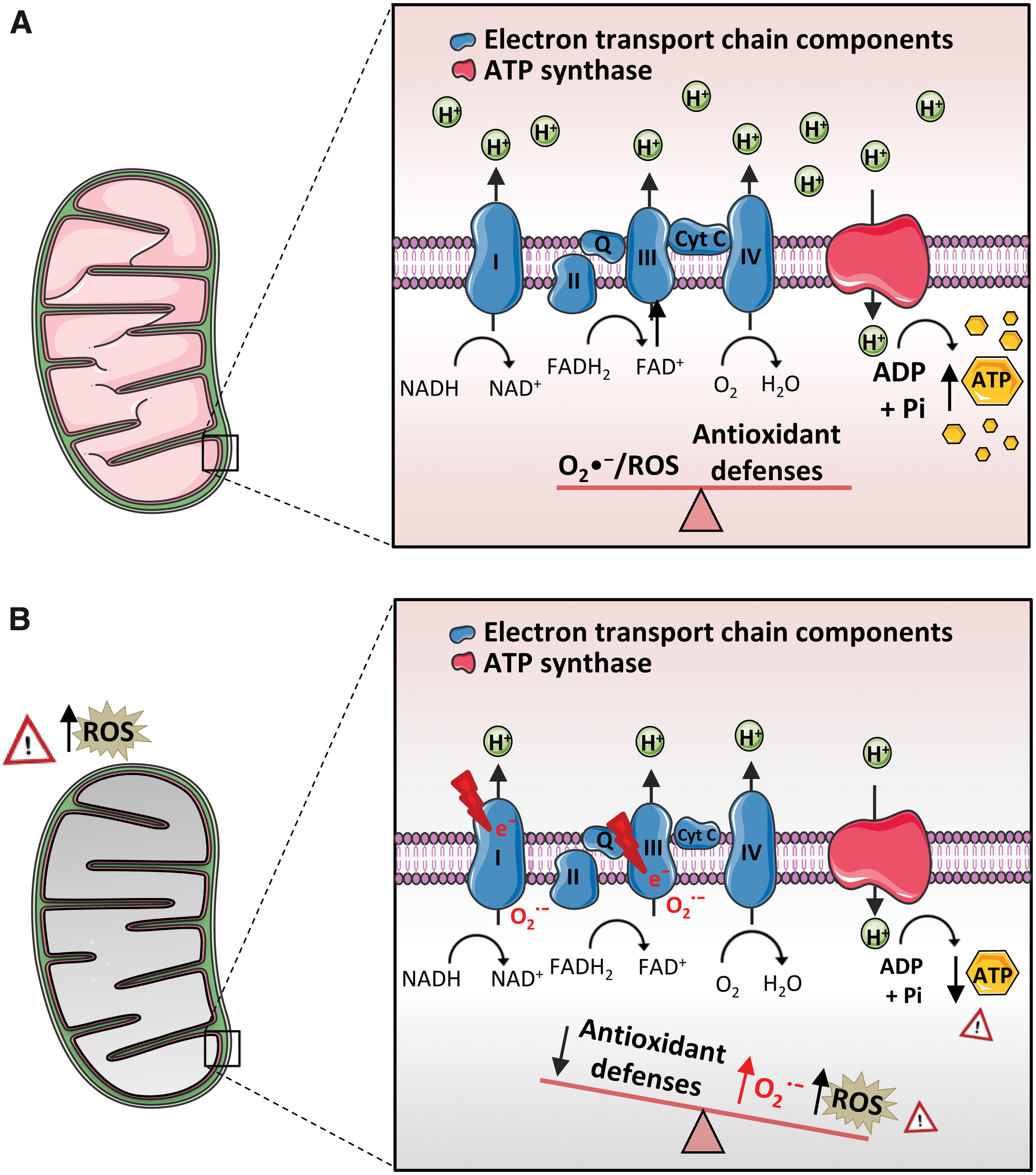
The electron transfer at ETC is not completely efficient, and some electrons “escape” or “leak” during the process and are transferred directly to O2, leading to ROS production (reviewed in Zhao et al, 2019) (Fig. 6). It is important to highlight that ROS include superoxide anion (O2 •−), hydroxyl radical (•OH), hydrogen peroxide (H2O2), and singlet oxygen, as well as a high variety of reactive molecules, such as alkoxyl radical (RO•), peroxyl radical (•OOH), lipid hydroperoxides (LOOH), and sulfate radical (SO4 •−).
The ROS act as modulators of ion channels, membrane receptors, and transcription factors, thus modulating inflammation, cell differentiation, cell survival, and death (reviewed in D'Autreaux and Toledano, 2007). The ROS vary greatly in their duration and site of action; for example, while •OH and O2 •− act only within a small molecular distance from their origin, H2O2 is longer-acting and can diffuse into more distant areas of the cells. Of note, modulation of the redox state of biomolecules by ROS is involved in many cellular processes (Sies, 2014), and redox biology has been established as a research field of its own.
There is growing evidence that physiological levels of ROS are fundamental for insulin signaling and glucose homeostasis. For instance, glucose-stimulated insulin secretion (GSIS) has been shown to require H2O2 signaling by nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) (Plecita-Hlavata et al, 2020). This physiological role of ROS as signaling molecules is highly concentration-dependent. For example, H2O2, an oxidative molecule that regulates signaling pathways through the oxidation of cysteinyl thiols in multiple proteins, can either enhance or impair insulin signaling depending on its concentration, as seen in H4IIEC hepatocytes (Iwakami et al, 2011).
This dual effect can be attributed to the different sensitivity of protein-tyrosine phosphatase 1B (PTP1B) inhibition and c-Jun N-terminal kinase (JNK) activation, kinases that stimulate and inhibit insulin signaling, respectively. Under homeostatic conditions, excess of ROS can be neutralized by different compensatory antioxidant defenses (enzymatic and non-enzymatic), such as paraxonases (PON), catalase (CAT), superoxide dismutases (SOD), glutathione (GSH) and its related enzymes glutaredoxin 2 (GRX2), glutathione peroxidases (GPX) and glutathione reductase (GR), thioredoxin 2 (Trx2), thioredoxin reductases (TRXR), and thioredoxin peroxidases (TRXP; also known as peroxiredoxins [PRDX]), as well as coenzyme Q, uric acid, lipoic acid, and vitamins (vitamin C and vitamin E) (reviewed in Rabilloud et al, 2001; Zhang et al, 2016), by which mitochondria and the cell, in general, are able to maintain their homeostatic activity.
However, dysregulation or inadequacy of the antioxidant response, combined with a gradual increase in both ROS and reactive nitrogen species (RNS; such as peroxynitrite [ONOO−] and nitric oxide [NO], as well as metal-oxygen complexes), can eventually lead to oxidative stress. In general terms, prolonged oxidative stress is related to the pathogenesis of many conditions, such as heart diseases, diabetes, autoimmune diseases, cancer, and neurological disorders.
This oxidative stress status results in redox modifications of nucleic acids, proteins, and membranes, as well as damage (and permanent defects) to mitochondrial DNA [mtDNA]-encoded subunits of the OXPHOS complexes, thus resulting in the reduction of ATP generation by mitochondria, and cellular and organism failure as a consequence. Indeed, when levels are excessive, ROS cannot provide controlled regulation of the biochemical reactions needed for the cells to stay healthy (reviewed in Zorov et al, 2014).
Remarkably, on ROS-mediated DNA damage, an ADP-ribosylating enzyme essential for DNA repair named poly-ADP-ribose polymerase-1 (PARP1) (reviewed in Groslambert et al, 2021) is modified, leading to ATP depletion. The absence of PARP1 undermines AMP-activated protein kinase (AMPK) activation and prevents the complete loss of mTOR activity, leading to deficient autophagy (Rodriguez-Vargas et al, 2012). mtDNA is more susceptible to oxidative damage than nuclear DNA (nDNA) because it is not compacted around histones and is localized near the ETC, which is a major source of ROS.
In addition, mtDNA possesses at most a few noncoding regions and sometimes none, thus increasing the probability of mutagenic alterations in coding regions. In addition, mitochondria are highly enriched in iron microenvironments, thus favoring the formation of •OH, which, due to its short half-life, preferentially reacts with mitochondrial components, including mtDNA. Due to these effects, it has been reported that mtDNA damage is 10- to 50-fold higher and persists longer when compared with that of nDNA (Stowe and Camara, 2009). In comparison to nDNA repair, knowledge regarding mtDNA repair is limited; however, base excision repair (BER) is the primary nuclear and mitochondrial repair pathway for oxidative DNA damage. Several other mechanisms have also been suggested, such as mismatch repair, homologous recombination, and non-homologous end-joining.
In most textbooks, mitochondria are depicted as small bean-shaped structures, which is how they appear in electron microscopy images. However, visualization of live cells has revealed that mitochondria exist as an active reticulum that moves to different regions of the cell by interacting with the cytoskeleton. The mitochondria within this reticulum are in continuous communication through dynamic fusion and fission events (a process called mitochondrial dynamics) (Tilokani et al, 2018).
Fusion helps to alleviate stress by combining the contents of partially damaged mitochondria, whereas fission is necessary to create new mitochondria and contributes to quality control by enabling the removal of damaged mitochondria and promoting apoptosis. Mitochondrial dynamics not only regulate intrinsic mitochondrial processes such as oxygen consumption and ATP generation, but they also contribute to multiple other functions, including cellular Ca2+ handling, metabolic control, and ROS production.
Moreover, the number of mitochondria per cell varies greatly according to physiological context and in a tissue-specific manner. The term mitochondrial biogenesis is used to describe the multiplication of mitochondria in response to increased energy demands, or for cell growth and division. Transcription of the mitochondrial genome is under the control of a single nDNA-encoded transcription factor called mitochondrial transcription factor A (TFAM), which, in turn, is regulated by the transcription factors NRF (nuclear respiratory factor)-1 and NRF-2, which transactivate numerous nuclear-encoded genes involved in mitochondrial respiration.
Importantly, ROS and mitochondria have been nominated as the active modulators of pro-inflammatory cytokine production (reviewed in Gilkerson and Materon, 2014). Similarly, pro-inflammatory signaling may also alter mitochondrial function, thus fueling a vicious cycle (reviewed in Escames et al, 2012; Lopez-Armada et al, 2013). Indeed, a great number of human and animal studies show that NO, as well as proinflammatory cytokines such as interleukin (IL)-1β and tumor necrosis factor alpha (TNF-α), can promote mitochondrial impairment through an increase in ROS levels, thus leading to a reduction of mitochondrial membrane potential and ATP synthesis.
In this sense, it has been demonstrated that IL-1β can contribute to the postprandial stimulation of insulin secretion. Accordingly, lack of endogenous IL-1β signaling in mice during refeeding and obesity diminished the concentration of insulin in plasma. IL-1β and insulin increased the uptake of glucose into macrophages, and insulin reinforced a pro-inflammatory pattern via INR, glucose metabolism, production of ROS, and secretion of IL-1β mediated by nucleotide oligomerization domain (NOD), leucine-rich repeat (LRR), and pyrin domain (PYD) (NLRP3) inflammasome (Dror et al, 2017). Further, postprandial inflammation would appear to be limited by normalization of glycemia, since it was prevented by inhibition of the sodium-glucose cotransporter 2 (SGLT2).
Mitochondria have other important functions, including the modulation of immune response, both innate and that of acquired immunity. Among the described mechanisms, redox-sensitive inflammatory pathways, and direct activation of the NLRP3 inflammasome have been described, including the activation of NRLP3 by released mtDNA (reviewed in Qiu et al, 2022). NLRP3 inflammasome is a multiprotein complex whose activation results in increased levels of caspase-1, thereby allowing for cleavage and the subsequent activation of the inactive precursors pro-IL-18 and pro-IL-1β into IL-18 and IL-1β (reviewed in Yang et al, 2019), two cytokines that can promote inflammation and insulin resistance (reviewed in Wieser et al, 2013).
NLRP3 plays an important role in the pro-inflammatory response and is the most studied inflammasome complex. Its activation is the result of several steps that generally depend heavily on pro-inflammatory cytokines and/or pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) and increases the expression of the different components of the inflammasome (reviewed in Kelley et al, 2019). Altered mitochondrial dynamics with concomitant mitochondrial ROS overproduction is a fundamental factor in triggering NLRP3-mediated inflammation.
This is relevant to the development of type 2 diabetes mellitus. Using a trans-mitochondrial cybrid cell model harboring mitochondrial haplogroup B4, which is more likely to develop type 2 diabetes, it was demonstrated that mitochondria and specifically altered mitochondrial dynamics take part in the pathological process of insulin resistance and pro-inflammatory phenotype (activation of NLRP3) (Chang et al, 2020). Putti et al (2015) showed that long-chain saturated fatty acids promote skeletal muscle inflammation and insulin resistance by impairing mitochondrial bioenergetics and promoting their fission phenotype, whereas omega-3 polyunsaturated fatty acids improve insulin sensitivity in the skeletal muscle by modulating mitochondrial dynamics and function. Considering the available knowledge, it is safe to nominate mitochondrial dynamics as a potential link between nutrient excess and insulin resistance. NLRP3 inflammasome activation is also specifically related to mitophagy (Kim et al, 2016).
Clearly, NLRP3 plays an important role in type 2 diabetes, and its levels are increased in diabetic pancreatic tissue. In fact, inhibiting activation of the NLRP3/caspase-1/gesdermin-d (GSDMD) pathway in β cells confers protection to diabetic pancreatic tissue in experimental animals (Liu et al, 2021a). It has been described that chronic treatment of 3T3-L1 cells (a cell line derived from mouse 3T3 cells and used in biological research on adipose tissue) and primary human adipocytes with IL-1β inhibits GLUT4 translocation to the plasma membrane in response to insulin (Jager et al, 2007).
Likewise, evidence obtained in human adipocytes shows that IL-18 inhibits insulin action via phosphorylation of PKB/Akt, a key step in the initiation of insulin's metabolic effects, therefore impairing insulin signaling and sensitivity (Kobashi et al, 2009). Consistent with these data, high plasma levels of IL-18 have been observed in pre-diabetic subjects with impaired glucose tolerance (Esposito et al, 2002). In fact, the study in question demonstrated that high levels of glucose acutely enhance circulating pro-inflammatory cytokine concentrations through an oxidative mechanism, an effect that was more pronounced in pre-diabetic patients with impaired glucose tolerance. These results highlight a causal role for hyperglycemia in the altered immune response in type 2 diabetes.
Although mitochondria are undoubtedly the main source of ROS, there are many other non-mitochondrial sources of ROS, including, but not limited to, lipoxygenase (LOX), cytochrome P450 reductase, monoamine oxidase (MAO), cyclooxygenase (COX), hemoxygenase (HO), xanthine oxidase (XO), endothelial nitric oxide synthase (eNOS), and NOX (reviewed in Kayama et al, 2015; Kietzmann et al, 2017; Rivera et al, 2010). There is extensive evidence that type 2 diabetes-associated hyperglycemia activates an integrated network of non-mitochondrial sources of ROS involving NOX, PKC, sorbitol (polyol), DAG, glucose auto-oxidation, and formation of advanced glycation end products (AGEs) (Volpe et al, 2018).
The ROS have been shown to alter ETC function, primarily via dysregulation of O-GlcNAcylation of mitochondrial proteins (Hu et al, 2009), which contributes to further oxidative stress and can eventually lead to a significant decline in mitochondrial function and mitochondrial membrane potential (reviewed in Darley-Usmar et al, 2012).
Of note, the sources of ROS and their mechanisms of action vary with respect to different tissue and cellular sites. Further, each type of oxidant species (RNS or ROS) plays specific pathological roles depending on the impaired organ. All these specificities regarding the types and sources of ROS and other oxidant molecules need to be carefully considered in the search to discover drugs (reviewed in Elbatreek et al, 2019).
Accordingly, clinical approaches targeting specific sources and types of cytotoxic oxidants in type 2 diabetes are ongoing or likely to multiply in the near future. In addition, it is important to highlight that lipotoxicity effects, such as excessive fatty acids, can substantially enhance the matrix pro-oxidative state.
IV. Oxidative Stress and Mitochondrial Impairment in Type 2 Diabetes and Insulin Resistance
Mitochondrial dysfunction and, specifically, mitochondrial ROS play a critical role in the pathophysiology of type 2 diabetes and its comorbidities. Prolonged exposure to high blood glucose levels has been implicated in the deregulation of antioxidant pathways and the disruption of ROS homeostasis (reviewed in Ighodaro, 2018). In fact, several studies have reported that antioxidant enzymes are altered in type 2 diabetic subjects, with both increases and decreases in their presence and activity having been described in blood samples.
In one study, subjects with metabolic syndrome presented higher activity of CuZnSOD and GR, lower activity of CAT and PON1, and lower concentrations of GSH when compared with controls (Vavrova et al, 2013). In another study, CAT activity and malondialdehyde (MDA) levels were found to be considerably increased, whereas the activity of the antioxidant enzyme GPX and GR and levels of GSH were lower in blood samples of type 2 diabetic patients (with or without nephropathy) than in those of controls.
These differences have also been described in type 2 diabetic patients with nephropathy versus those without nephropathy (Kumawat et al, 2013). In another study, leukocytes from type 2 diabetic patients exhibited high levels of mitochondrial ROS and reduced messenger RNA (mRNA) levels of antioxidant enzymes and redox-regulating mediators with respect to controls, including GPX1 and sirtuin 3 (SIRT3), respectively; and metformin was shown to reverse these effects (Diaz-Morales et al, 2017).
Along with mitochondrial production of O2 •−, NOX enzymes seem to be critical sources of ROS in type 2 diabetes (Palicz et al, 2001 and reviewed in Kaneto et al, 2007) and have been implicated in the pathogenesis of micro- and macrovascular disease in type 2 diabetes (reviewed in Forbes et al, 2008). The NOX are tissue-specific complexes that generate ROS as secondary messengers. Unlike other enzymatic sources, which have other important functions, NOX are the only enzyme family whose sole known action is the generation of ROS (reviewed in Casas et al, 2015).
Further, it is widely accepted that the general oxidative stress status typical in type 2 diabetic patients can lead to alterations in cell signaling and metabolic imbalances (reviewed in Roberts and Sindhu, 2009). For example, insulin resistance and type 2 diabetes have been related to increased plasma oxidative stress and decreased antioxidant defenses (including SOD, GR, CAT, or PON1), which positively correlate with anthropometric parameters, including waist circumference or blood pressure, and clinical parameters, including HDL-C and triglycerides (reviewed in Holvoet, 2008; Tangvarasittichai, 2015).
Altered mitochondrial function is one of the main characteristics of type 2 diabetes and insulin resistance. In this sense, it has been demonstrated that in INS-1 cells, a β cell line, cultured under hyperglycemia conditions, displays upregulation of glycolytic enzymes, reduced glucose-stimulated O2 consumption, and downregulation of mitochondrial proteins (Haythorne et al, 2019). The authors hypothesized that impaired mitochondrial function directly reduces equivalents (such as NADH and flavin adenine dinucleotide [FADH2]) that drive ATP synthesis and ATP-dependent processes, thus contributing to insulin-resistant glucose metabolism (Haythorne et al, 2019).
Mitochondrial alterations related to type 2 diabetes also involve changes in mitochondrial biogenesis, and pharmacological induction of mitochondrial biogenesis has been proposed as a therapeutic strategy for this disease (Zamora et al, 2015). Fewer and smaller-sized mitochondria have been reported in skeletal muscle biopsy samples from individuals with type 2 diabetes in whom insulin-mediated muscle glucose uptake was about 60% lower than in control subjects (Morino et al, 2005). Therefore, the reduced number and density of functional mitochondria in the skeletal muscle can modify the expression of essential proteins involved in redox balance, including the peroxisome proliferator-activated receptor coactivator 1α (PGC-1α) and PGC-1β (St-Pierre et al, 2003), considered a master regulator of mitochondrial functionality.
Thus, exacerbating stress signals result in reduced OXPHOS (reviewed in Kim et al, 2008). Insulin signaling directly regulates mitochondrial biogenesis through the insulin/IRS/Akt pathway's regulation of mTOR and FOXO transcription factors and activation of PGC-1α. PGC-1α-responsive genes involved in OXPHOS are downregulated in human diabetic muscle (Mootha et al, 2003). The altered insulin signaling in the skeletal muscle of diabetic patients is also reflected by the fact that high physiological insulin levels enhance mitochondrial oxidative capacity (related to an increased expression of mitochondrial proteins encoded by both nuclear and mtDNA) in non-diabetics, but not in insulin-resistant type 2 diabetics (Stump et al, 2003). Diabetes and insulin resistance have also been related to altered mitochondrial dynamics (Jheng et al, 2012; Lin et al, 2018 and reviewed in Yoon et al, 2011).
Moreover, under conditions of insulin resistance, the PI3K-Akt pathway is impaired, whereas the MAPK pathway is not, thus promoting a significant imbalance between these two signaling pathways (Fig. 7). In this way, endothelial NO production is decreased, with the consequent impairment of endothelium-dependent relaxation and reduction of GLUT4 trafficking (reviewed in Bogan, 2012; Mueckler, 2001), which leads to lower fat and skeletal muscle glucose uptake, as well as a compensatory increase in insulin production.
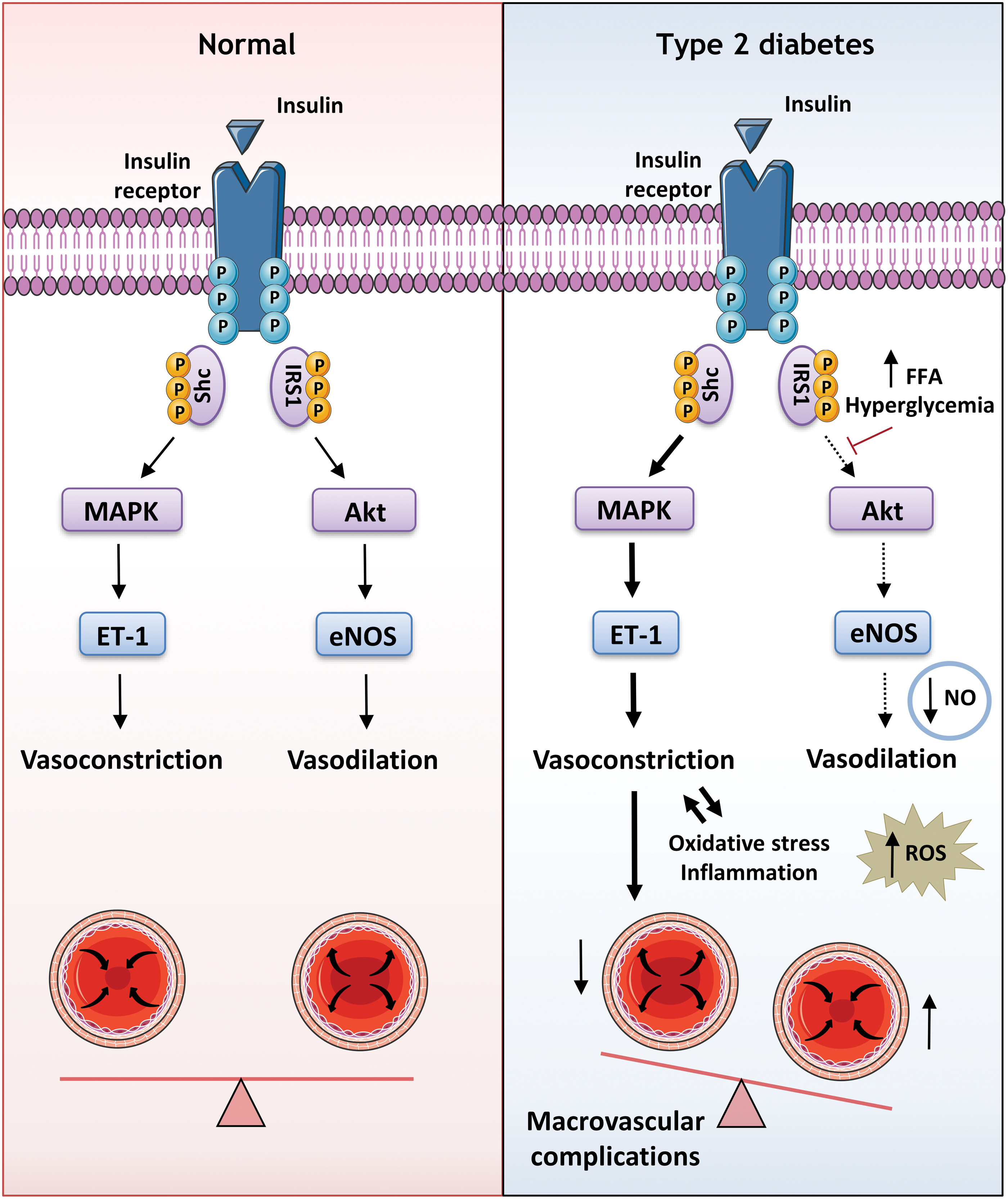
However, as the MAPK pathway is unaltered, there is a high production of endothelin-1 (ET-1), a potent vasoactive peptide involved in vascular lesion development (Fig. 7). As a result of these alterations, insulin resistance can lead to the cardiovascular complications that predispose individuals to atherosclerosis.
Importantly, different studies in experimental models and humans have shed light on the direct involvement of metabolism-associated ROS production and inflammation in the etiology of insulin resistance and type 2 diabetes, which occurs through multiple mechanisms (Tripathy et al, 2003 and reviewed in Wieser et al, 2013). In this context, elevated circulating levels of FFAs, commonly seen in diabetic individuals and mainly associated with excess nutrition, can increase ROS levels as well as affect nuclear factor-kappa B (NF-κB) and mTOR signaling (reviewed in Rogero and Calder, 2018). In isolated mitochondria from kidney cortical tubules of diabetic rats, increased amounts and activities of selective fatty acid oxidation enzymes were found to be associated with increased OXPHOS and net ROS production, whereas pyruvate oxidation was decreased, and pyruvate-supported ROS production was unaltered.
The study suggested that mitochondrial fatty acid oxidation was the source of the increased net ROS production, and the site of electron leakage was proximal to coenzyme Q at the flavoprotein that shuttles electrons from acyl coenzyme A (acyl-CoA) dehydrogenases to coenzyme Q (Rosca et al, 2012). Non-mitochondrial sources of ROS associated with lipotoxicity have also been described. Among them, NOX enzymes stand out; in one study, rat islets exposed to palmitate displayed increased protein expression of the NOX subunit p47phox and increased NOX-derived superoxide, and this effect was reversed by NOX inhibition (Morgan et al, 2007).
Fatty acid-induced oxidative stress is among the main mechanisms responsible for disrupting insulin signaling, and this occurs through a kinase cascade (involving PKC and JNK1), which phosphorylates IRS-1, on serine307, inhibiting IRS-1 binding of PI3K and thus impairing the action of insulin on target cells such as muscle cells, including insulin-stimulated GLUT translocation to the membrane (Yu et al, 2002 and reviewed in Chang et al, 2004).
Concurrently, FFA can activate different pattern-recognition receptors, such as toll-like receptors (TLR), and facilitate pro-inflammatory and pro-atherogenic signals through inhibition of I-kappa B kinase subunit beta (IKKβ) or activation of JNK pathways (reviewed in Yin et al, 2014; Yung and Giacca, 2020), by which IRS can be phosphorylated and insulin signaling damaged. Data from Boden and Shulman (2002) revealed that elevated plasma FFA concentrations induce defects involving muscle glucose transport and/or translocation in type 2 diabetic patients, resulting in reduced glycogen synthesis and glycolysis, along with the inhibition of insulin-stimulated glucose uptake.
Further, UPR and ER stress are highly relevant to the pathogenesis of type 2 diabetes (reviewed in Thomas et al, 2010). They can activate different transcription factors such as activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase R-like endoplasmic reticulum kinase (PERK), which are involved in protein synthesis, the transcriptional regulation of UPR, and the interplay between apoptosis and inflammation. Oxidative stress profoundly affects redox homeostasis in the ER, thereby causing alterations in the protein-folding environment, increasing ROS accumulation, and generating mitochondrial dysfunction and inflammation (reviewed in Cao and Kaufman, 2014).
In this sense, ER stress activates different networks that mediate the inflammatory response, such as JNK kinases (reviewed in Kaneto et al, 2005). This molecular mechanism, once activated, is involved in the reduction of insulin synthesis and secretion induced by high levels of ROS and/or oxidative stress. Indeed, studies performed in isolated rat islets have shown that JNK overexpression is associated with a decrease in the DNA-binding activity of the pancreatic duodenal homeobox 1 (PDX1) (Kaneto et al, 2002), an essential transcription factor that regulates β cell function and pancreas development, as well as insulin gene expression.
Likewise, adult mice with β cell–specific deletion of Pdx1 exhibit severe hyperglycemia accompanied by a rapid loss of cell identity and subsequent acquiring of islet α-like features (Gao et al, 2014b). Further, a recent in vitro study indicated that ER stress can interfere with the transport of newly synthesized insulin proreceptors from the ER to the plasma membrane, therefore inhibiting their proteolytic maturation and thus promoting insulin resistance (Brown et al, 2020).
The ER and mitochondria communicate physically and functionally in a well-regulated manner, forming specific microdomains named mitochondria-associated membranes (MAMs) (reviewed in Herrera-Cruz and Simmen, 2017). These specialized contact sites are key actors in cellular Ca2+ homeostasis and they have also been reported to regulate lipid transfer, ROS production, mitochondrial dynamics and bioenergetics, inflammation, and mitochondria-mediated apoptosis (reviewed in Janikiewicz et al, 2018; Kerkhofs et al, 2018; Missiroli et al, 2018).
It is important to highlight that MAMs consist of membrane fractions from both the outer mitochondrial membrane (OMM) and the ER, and they contain many cell-specific molecular components involved in the tethering complex. Abnormal ER-mitochondria interaction and alterations in MAMs composition are related to several pathological disorders, including type 2 diabetes, where organelle miscommunication has been proposed to trigger ROS production, cell death, β cell inflammation, and altered metabolic function (reviewed in Rieusset, 2018).
One of the primary hypotheses of insulin resistance development is that excessive generation of ROS, which occurs mainly due to defects in mitochondrial function or NOX activation, can lead to the phosphorylation of IRS proteins via activation of serine kinases, as well as undiminishing insulin signaling and glucose levels (reviewed in Rains and Jain, 2011). However, the primary molecular events by which this occurs are yet to be clarified. It is important to stress that the skeletal muscle is the major tissue of insulin-stimulated glucose uptake and is therefore one of the main locations of insulin resistance in type 2 diabetic and obese individuals.
Multiple studies have recognized mitochondrial abnormalities (which regard function and/or morphology) in the skeletal muscle as a major link between ROS overproduction and insulin resistance (Kelley et al, 2002; Petersen et al, 2004; Simoneau et al, 1999 and reviewed in Hojlund et al, 2008) (Fig. 8). In this sense, insulin resistance in the human skeletal muscle is characterized by decreased content and functional capacity of mitochondria, decreased insulin-stimulated glucose metabolism and disposal, and triacylglycerol accumulation, which can eventually lead to mitochondrial impairment.
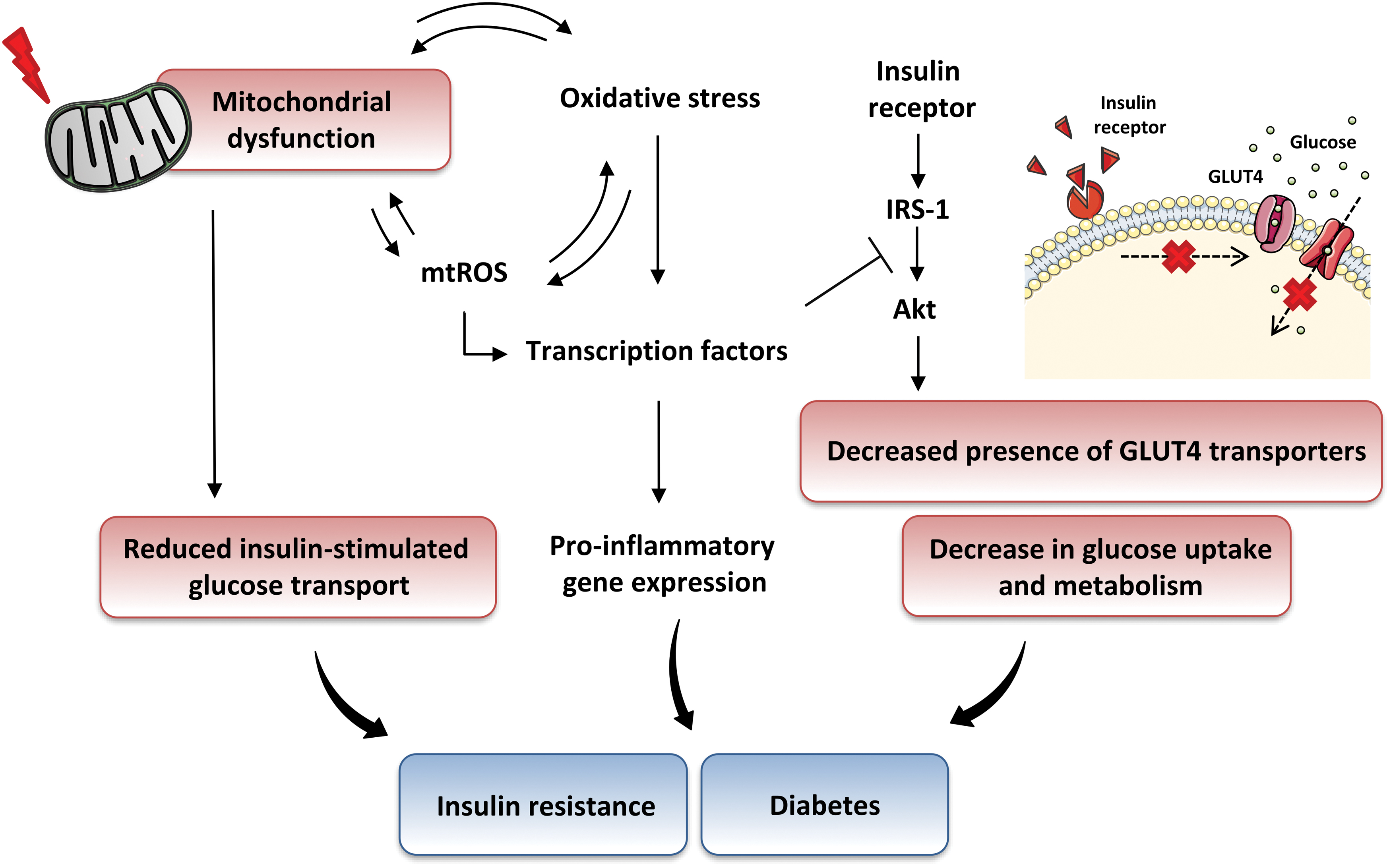
In addition, patients with type 2 diabetes display a decreased expression of genes involved in OXPHOS, which is regulated by factors that stimulate mitochondrial biogenesis, such as PGC-1α and NRF, thus suggesting altered mitochondrial biogenesis and function (Mootha et al, 2003).
Moreover, reductions in mitochondrial density have been shown to result in decreased mitochondrial activity in muscle biopsy samples obtained from type 2 diabetic patients (Morino et al, 2005). As a result, the intramyocellular content of fatty acid levels increases and phosphorylation of IRS-1 is suppressed, thus leading to changes, including a reduction in muscle glycogen synthesis and insulin-stimulated glucose transport translocation (Dresner et al, 1999) (Fig. 8).
Abundant evidence supports the hypothesis that insulin sensitivity is closely interconnected to mitochondrial function and that impairment of these organelles contributes directly or indirectly to a diminished action of insulin and to the development of diabetes-related complications and comorbidities.
A. Oxidative stress and mitochondrial impairment in diabetes-related complications and comorbidities: the example of diabetic cardiomyopathy and retinopathy
As previously described, diabetes is related with different complications and comorbidities. Here, we focused on diabetic retinopathy and cardiac disease due to the major role that mitochondria/oxidative stress play in their pathogenesis. Diabetic retinopathy is the most common microvascular complication. The retina is particularly susceptible to oxidative stress being one of the highest oxygen-consuming tissues in the human body (the retina oxygen tension is over 70 mmHg). This environment with abundant photosensitizers, visible light exposure, and a high energy demand supports a highly oxidative milieu.
Cardiovascular disease is the most frequent cause of death in diabetic individuals. It is important to highlight that one-third of the heart's volume is composed of mitochondria, and an adequate mitochondrial function is essential for its proper functioning, as this organ requires a high-energy supply. In this sense, it has been described that mitochondrial dysfunction plays a critical role in the onset, progression, and development of diabetic cardiomyopathy (reviewed in Jubaidi et al, 2020). Indeed, glucose utilization in the diabetic heart is significantly decreased due to insulin resistance (because of a deficiency in insulin release and/or its action).
In addition, there is an alteration in pyruvate dehydrogenase activity and low levels of GLUTs (especially GLUT4). Consequently, the use of glucose as a source of energy is decreased in mitochondria in diabetic cardiomyocytes, in which ATP production relies mainly on fatty acid. This process generates more ROS and disrupts OXPHOS and mitochondrial Ca2+ handling, which leads to cell death. In addition, this metabolic switch can eventually result in intracellular lipid accumulation, as mitochondria are unable to metabolize all incoming fatty acids.
It is important to highlight that there are different antioxidants and/or ROS scavenger molecules that have been shown to have beneficial effects in diabetic animal models with diabetic cardiopathy, by decreasing cardiomyocyte death and modulating diabetic cardiac injury (Fiordaliso et al, 2004; Shen et al, 2006). However, the literature includes examples in which antioxidant-based therapies have proved to be unsuccessful in type 2 diabetes or its complications (Heart Outcomes Prevention Evaluation Study Investigators et al, 2000; Lonn et al, 2005), suggesting that simply scavenging ROS through antioxidants is not sufficient.
A treatment strategy that is likely to be more effective is to improve mitochondrial quality control through adequate mitochondrial dynamics and thus induce a correct mitochondrial turnover by mitophagy. In this way, a population of healthy mitochondria can be preserved, and a normal cardiac contractile function maintained.
In diabetic retinopathy, hyperglycemia is considered the main pathogenic factor and the efficient control of glucose levels can prevent/delay the development of this condition (Diabetes Control and Complications Trial Research Group et al, 1993). However, several studies have failed to detect improvements in diabetic retinopathy after normalizing glucose levels; consequently, oxidative stress and mitochondrial dysfunction persist in type 2 diabetic subjects, thus driving the progression of retinopathy (Bixler et al, 2011; Chew et al, 2014).
This can be attributed to the possible epigenetic alterations induced by a high glucose environment, which causes changes in the expression of genes involved in protection against oxidative stress, among other effects (Bixler et al, 2011). As a result, oxidative damage leads to altered transcription of mtDNA-encoded ETC subunits—key actors in OXPHOS—and additional mitochondrial dysfunction (Madsen-Bouterse et al, 2010; Mishra and Kowluru, 2019 and reviewed in Kowluru and Mishra, 2018; Kowluru and Mishra, 2015).
Recent research has revealed that mtDNA copy numbers are reduced in type 2 diabetes, with an increase in damaged, inflamed, and generally non-functional mitochondria, which accumulate in the retina, thus leading to partial christolysis and a decrease in mitochondrial oxygen consumption (Kowluru and Abbas, 2003 and reviewed in Bek, 2017; Kowluru and Mishra, 2018; Kowluru et al, 2015). In this sense, it seems that the susceptibility of the retina to oxidative damage could be associated with its lipid composition, which makes it particularly prone to oxidation. In summary, retinal mitochondria have been shown to be dysfunctional in type 2 diabetes, but the mechanism by which this occurs has not been fully clarified.
Obesity and type 2 diabetes are intrinsically connected, and obesity can be viewed as both a diabetes risk factor and a comorbidity. Further, it is important to highlight that obesity also takes part in diabetes-related pathologies and one of the major links among them is insulin resistance. Clinical manifestations of obesity-driven insulin resistance initiate when insulin-target cells, such as adipocytes, inadequately respond to insulin, and type 2 diabetes pathology emerges over time (Ahima, 2011). Some of the key factors contributing to insulin resistance in these patients are excessive nutrient supply to adipocytes, which leads to ROS production, pro-inflammatory processes ER stress, cell aging, and altered mitochondrial dynamics (Gao et al, 2014a; Patti and Corvera, 2010; Petersen et al, 2003).
Thus, mitochondrion is both the origin and target of multiple metabolic signals whose integration maintains insulin sensitivity. Importantly, it has been suggested that mitochondrial redox signaling plays a key role in white adipose tissue (WAT), regulating different processes (such as adipocyte differentiation or adiponectin secretion) through the modulation of redox-sensitive transcriptional factors (Wang and Hai, 2015). Not surprisingly, given that WAT is the largest endocrine organ in the human body, any alteration in adipocyte mitochondria could result in significant homeostatic disturbances.
B. Targeting oxidative stress and mitochondrial dysfunction as a therapeutic strategy in diabetes
Numerous studies performed in animal models affirm the beneficial effects of antioxidants in diabetes and its related disorders (reviewed in Byrne et al, 2021; Oyenihi et al, 2015; Thakur et al, 2018). The pharmacological approach in most of these studies was non-enzymatic and involved the use of common antioxidant compounds such as tocopherol, ascorbic acid, β-carotene, selenium, N-acetyl cysteine, and α-lipoic acid. In addition, a plethora of molecules of plant origin have been assayed, and these include polyphenols from green tea, proanthocynidins from grape seeds and other flavonoids, such as hesperetin, fisetin, rutin, quercetin, and curcumin.
Nevertheless, the results obtained in humans have been less encouraging; even though many antioxidants have been tested in clinical trials (Balbi et al, 2018), only a few have shown sufficient efficacy and safety to be formally approved in some countries; among these is α-lipoic acid, which is used in diabetic polyneuropathy (Ziegler et al, 2011).
The mitochondrion houses the highest concentration of intracellular antioxidants and is the major site of the production of ROS, thus emphasizing its importance to the overall cellular redox status. In this context, it would be logical to treat diseases that display increased oxidative stress and altered mitochondrial function by increasing the antioxidant capacity of these organelles. However, low bioavailability of exogenous antioxidant agents in mitochondria in vivo is a limitation. To overcome this challenge, antioxidants have been developed to target the mitochondria (Apostolova and Victor, 2015); for example, by conjugation to lipophilic cations to exploit the negative membrane potential (about −140 mV) of the organelle.
This strategy has proved successful when triphenylmethylphosphonium (TPMP) is conjugated with coenzyme Q10, vitamin E, the SOD mimetic TEMPO and TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxy radical) or PBN (alpha-phenyl N-tertiary-butyl nitrone), as MitoQ10, MitovitE, MitoTEMPO, MitoTEMPOL, or MitoPBN, respectively. There is evidence of the beneficial effects of these targeted compounds in animal models of diabetes and associated complications, such as the case of MitoQ in diabetic kidney disease (Chacko et al, 2010; Ward et al, 2017) or that of peripheral neuropathy (Fink et al, 2020), and of their capacity to modulate oxidative stress, inflammation, and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients (Escribano-Lopez et al, 2016).
In addition, MitoQ has been tested in the context of different pathologies in several clinical trials, including one pilot study of endothelial dysfunction in type 2 diabetics, in which supplementation with MitoQ did not significantly improve brachial artery flow-mediated dilatation or reduce vascular adhesion molecules and p-selectin (Alsweiler, 2018). MitoTEMPO has shown therapeutic inhibition of mitochondrial ROS in experimental animals with diabetic cardiomyopathy (Ni et al, 2016) and has also demonstrated beneficial effects by inhibiting ROS production, CD36 overexpression, and NLRP3 activation in renal tubular epithelial cells (TEC) of diabetic kidneys (Hou et al, 2021).
Other approaches include Szeto-Schiller (SS) peptides, molecules that have the capacity to selectively cross the inner mitochondrial membrane and exert antioxidant properties. The beneficial effects of SS-31 peptides in diabetes have been reviewed (Ding et al, 2021). For instance, SS-31 has demonstrated beneficial effects by ameliorating diabetes-induced alterations in mitochondrial function in the pancreas of diabetic male TallyHO/JngJ mice (Bhatti et al, 2021).
The administration of agents that induce the activities of mitochondrial uncoupling proteins (UCP) or of artificial uncouplers has also shown potential as therapeutic agents against mitochondrial ROS. The UCP are usually lipophilic weak acids that can lower the membrane potential gradient and can reduce mitochondrial ROS production. In this regard, interference with the activity of UCP would appear to be an attractive strategy for diabetes and diabetes-related diseases (reviewed in Dludla et al, 2018).
Genipin, a natural compound from Gardenia jasminoides used in Chinese medicine to treat diabetes (among other illnesses), is an inhibitor of proton transport mediated by UCP2 (Kreiter et al, 2019). Genipin can protect against mitochondrial damage of ARPE-19 cells (retinal pigment epithelia) under hyperglycemic conditions through the Akt pathway mediated by the miR-4429/JAK2 signaling axis (Xu et al, 2022).
In addition, mitochondrial oxidative damage can be attenuated by supplementation with tetrahydropterin (BH4), a coenzyme required for full functioning of eNOS. In diabetes, BH4 is oxidized to BH2, and low BH4 levels result in decreased eNOS activity and increased superoxide production by eNOS. Given the proximity of this enzyme to the OMM, eNOS-derived ROS may cause mitochondrial damage. Chronic coadministration of sepiapterin, a BH4 precursor, with L-citrulline, an L-arginine precursor, has been shown to prevent cardiac dysfunction in diabetic db/db mice (Baumgardt et al, 2016).
In addition, obesity and obesity-associated pathologies have been related to mitochondrial dysfunction (James et al, 2012). Detrimental accumulation of visceral adipose tissue (VAT), the major intra-abdominal fat depot, significantly increases the risk of metabolic comorbidities, insulin resistance, and type 2 diabetes (Neeland et al, 2012).
Due to the relationship between mitochondrial dysfunction in type 2 diabetes and changes in autophagy, we will describe their relationship, relevance, and signaling in the next section.
V. Autophagy
A. General aspects
Autophagy plays a crucial role in recycling intracellular components, including damaged organelles, specific proteins, and protein aggregates, and in clearing pathogens. The three known forms of autophagy are microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA) (reviewed in Yang and Klionsky, 2010) (Fig. 9). Among them, macroautophagy (also typically called autophagy) is probably the most common and studied of these processes. In general, a hallmark of autophagy is the generation of a large double-membrane vesicle named the autophagosome, which envelops the cytosolic materials destined for degradation.
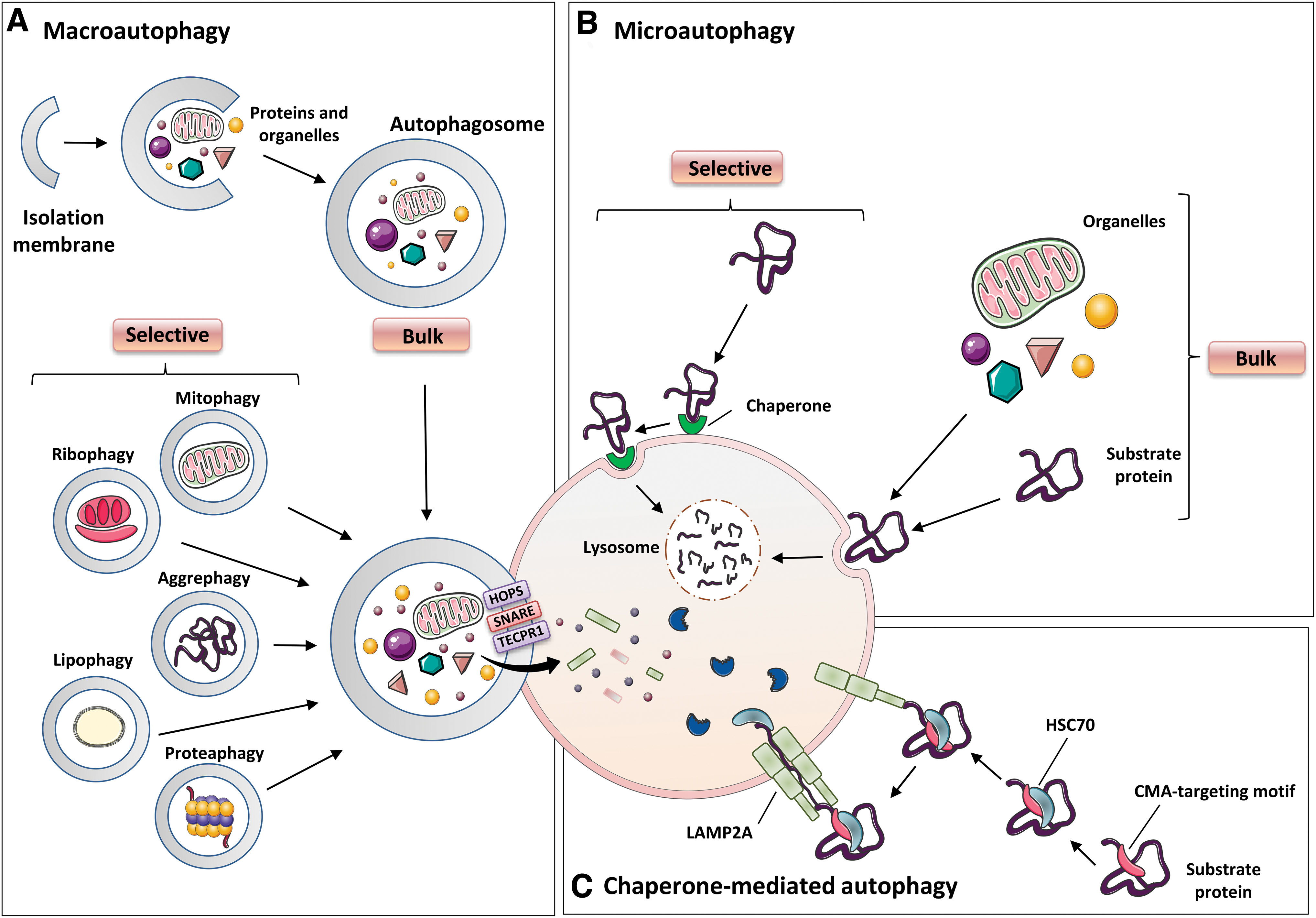
Subsequently, the outer membrane of the autophagosome matures and fuses with the membrane of the endosome or the lysosome, and the contents, along with the inner membrane, are degraded by exposure to acidic hydrolases (reviewed in Oku and Sakai, 2018). The degraded products are then exported to the cytosol and reused for synthesis of new cellular components and to obtain energy. Autophagy is stimulated by a large number of factors, such as alterations in the lipid metabolism (reviewed in Xie et al, 2020), amino acid starvation (Sutton et al, 2019), caloric restriction (reviewed in Chung and Chung, 2019), exercise (Brandt et al, 2018), impaired intracellular cholesterol transport, hypoxia, ER stress, damaged or surplus organelles, and infectious pathogens (reviewed in Xiong et al, 2019).
Microautophagy involves the direct invagination or deformation of the lysosomal membrane, which then engulfs the contents of the cell (reviewed in Oku and Sakai, 2018; Parzych and Klionsky, 2014). To date, three types of microautophagy have been identified depending on the organelles involved in the process and the architecture of the membranes: Type 1 mainly involves protrusion of the lysosomal membrane to sequester the cytoplasmic contents, whereas invagination of the lysosomal or endosomal surface is typical of type 2 and type 3 microautophagy, respectively (reviewed in Oku and Sakai, 2018).
The CMA does not lead to the generation of the autolysosome or autophagosome, and the protein cargo is administered directly to the lumen of the lysosome (Itakura and Mizushima, 2010).
B. Key players and events in autophagy
Autophagy is a complex process consisting of multiple sequential steps, including initiation, phagophore nucleation, autophagosome formation, and finally lysosome fusion and degradation (reviewed in Parzych and Klionsky, 2014; Pyo et al, 2012). Autophagosome biogenesis occurs through the sequential action of autophagy-related proteins (ATG) (reviewed in Mizushima et al, 2011). The initiation of autophagy includes two protein complexes: Unc-51-like kinase 1 (ULK1) complex, which is composed of different proteins, including ATG13, FAK family–interacting protein (FIP) 200, ATG101, and ULK1/2; and the class III PI3K complex, consisting of Beclin1, vacuolar protein sorting (VPS) 34, ATG14L, and p150/VPS15. PI3K (or PtdIns3K) generate PtdIns3P, which is critical for phagophore nucleation. Further, the ULK1 complex is activated by ULK1 kinase, which, in turn, is controlled by mTOR complex 1 (mTORC1) and AMPK, the major bioenergetic sensor of the cell (Fig. 10).
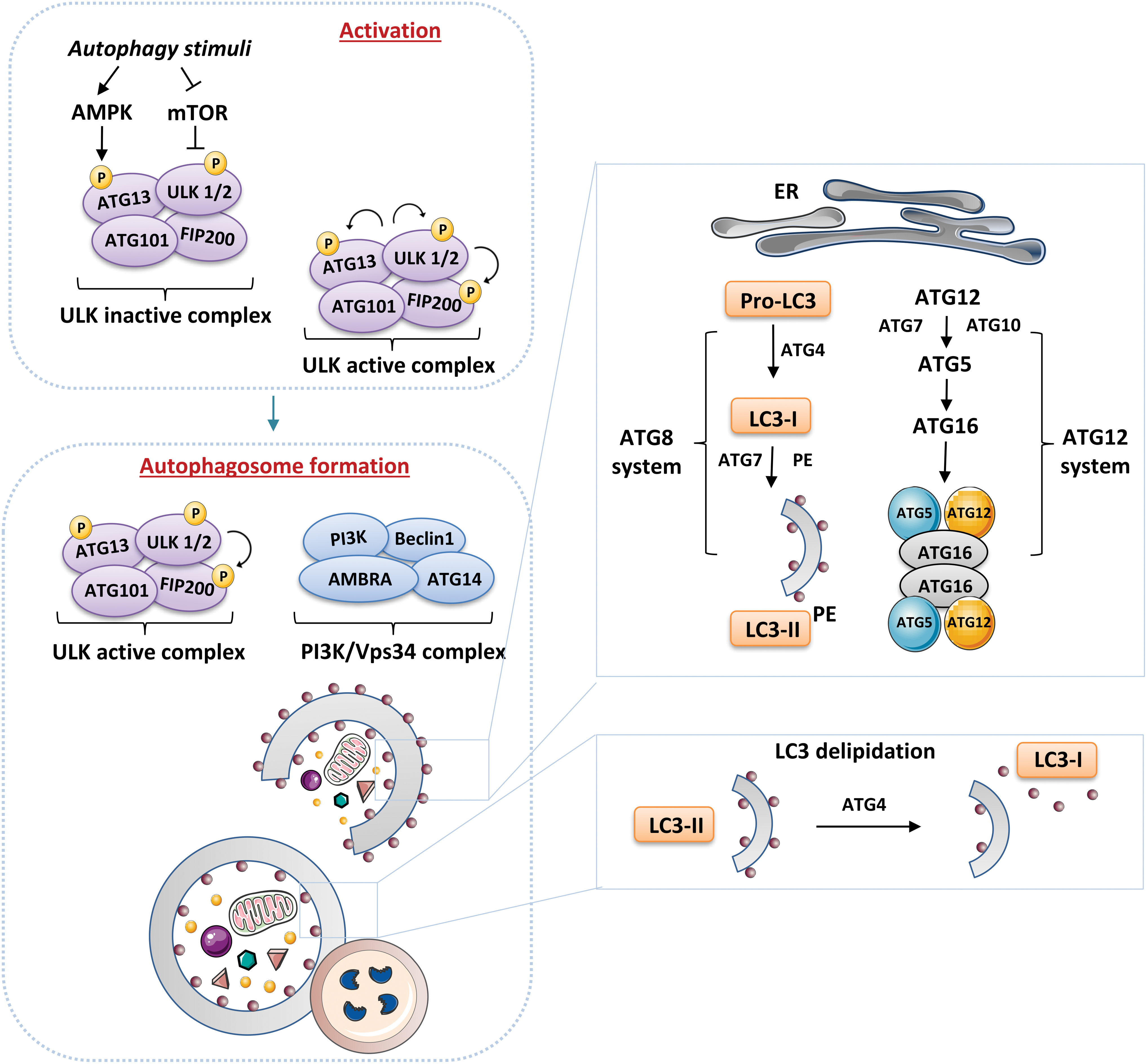
This regulation allows the ULK1-ATG13-FIP200-ATG101 complex to sense the nutritional and energetic status of the cell, which is one of the primary determinants of autophagy. When activated, ULK1 also phosphorylates other essential autophagy proteins, such as VPS34 and Beclin1. In addition, ATG14-class III PI3K complex can initiate classic macroautophagy by associating with ATG14 or facilitating the stimulation of other molecular pathways (e.g., the endocytic pathway) by associating with UVRAG (reviewed in Burman and Ktistakis, 2010). In addition, autophagy is regulated in a positive or a negative way by many different proteins. Some are proteins that cooperate with Beclin1; for example, the anti-apoptotic protein B cell leukemia/lymphoma 2 (BCL2), which interacts with Beclin1 to prevent its interaction with PIK3C3 and thus suppress autophagy; or AMBRA1, which binds to Beclin1, thereby promoting binding between Beclin1 and its target kinase VPS34.
The elongation/expansion of the phagophore consists of two ubiquitin-like conjugation systems. The first one, the ATG12-ATG5-ATG16L1 complex, associates with the phagophore membrane, but it disassociates after autophagosome completion. The second, ATG8/microtubule-associated protein 1 light chain 3 (LC3) system (Kabeya et al, 2000 and reviewed in Geng and Klionsky, 2008) is activated by ATG4, which cleaves pro-LC3 to form LC3-I, which, in turn, is lipidated to form LC3-II, thus generating the phagophore membrane, which subsequently becomes elongated (Fig. 10). This elongation process includes ATG9, a key protein located in the late endosome and the trans-Golgi network during nutrient-rich conditions, and at the autophagosome under starvation conditions (Young et al, 2006).
The fusion of autophagosomes with the lysosome is the least understood step of the autophagic process, despite being crucial in maintaining autophagy flux (extensively discussed [reviewed by Cabukusta and Neefjes, 2018; Lorincz and Juhasz, 2020]). It is well known that it depends on the cytoplasmic microtubule network, since autophagosomes must be delivered to the perinuclear region of the cell where they can fuse with lysosomes.
C. Selective versus bulk autophagy
Autophagy was originally described as a bulk and relatively non-selective degradation pathway induced by nutrient deficiency, and whose function was to recycle building blocks to compensate for lack of nutrients. Over the decades, however, it has become evident that autophagy can act on intracellular homeostasis in non-starved cells through a specific degradation of the cargo material. Multiple selective forms of autophagy have been recognized, including mitophagy (mitochondria), lipophagy (lipids), ribophagy (ribosomes), ER-phagy (ER), pexophagy (peroxisomes), aggrephagy (aggregated proteins), xenophagy (microorganisms), proteaphagy (proteasome), lysophagy (lysosomes), crinophagy (secretory vesicles), and ferritinophagy (degradation of specific proteins).
The molecular mechanism of selective autophagy should provide efficient charge recognition and ensure its tethering to a nascent autophagosome (usually performed by simultaneously binding to ATG8 family proteins and to the cargo on the autophagosomal membrane). All these processes are mediated by specific proteins called autophagic cargo receptors (reviewed in Burman and Ktistakis, 2010; Xiong et al, 2019), which can either bind directly to their cargo or identify polyubiquitin chains attached to the surface of the cargo, as occurs in mammalian cells.
Despite abundant evidence of the cytoprotective role of autophagy in β cells, the importance of non-selective versus selective autophagy in β cells is yet to be determined. This shortfall is related to a limitation in many of the studies carried out to explore this aspect; namely, they involved the deletion and deterioration of proteins that, in general, can impair both non-selective and selective autophagy. However, non-selective macroautophagy is generally induced by nutrient deficiency, whereas type 2 diabetes is a disease characterized by high blood glucose/nutrients, thus suggesting that the role of other forms of autophagy in β cell survival and function has been underestimated. The present review refers to one type of selective autophagy: mitophagy, which can be ubiquitin-dependent or -independent, depending on the pathway involved.
D. Autophagy signaling pathways relevant to type 2 diabetes
Regulation of autophagy is a complex process that is closely associated with critical signaling pathways, including mTOR, p53, MAPK/ERK1/2, PI3K/Akt, AMPK, HIF-1α/PDK4, and β-catenin (Colella et al, 2019). Particularly relevant for type 2 diabetes is the AMPK/mTOR pathway (Jia et al, 2019 and reviewed in Shi et al, 2019). AMPK signaling is considered fundamental for insulin resistance, and the pathogenesis of diabetes and its complications (reviewed in Entezari et al, 2022).
Under nutrient-rich conditions, such as those often present in diabetic patients, together with the metabolic alterations related to hyperglycemia and dyslipidemia, mTORC1 can associate with the ULK1 complex and phosphorylate ULK1 and ATG13; on the other hand, during nutrient deprivation, mTORC1 dissociates, resulting in dephosphorylation and induction of autophagy. Generally, AMPK is activated by a high AMP/ATP ratio and then suppresses mTORC1 through phosphorylated tuberous sclerosis complex 2 (TSC2). Further, AMPK can directly phosphorylate ULK1 at other sites to activate the autophagy pathway.
Thus, the inhibition of mTORC1 by tizoxanide (Shou et al, 2020) or the widely employed autophagic inducer rapamycin (Civiletto et al, 2018) triggers autophagy, whereas the inhibition of AMPK blocks the initiation of ULK1-mediated autophagy (Liu et al, 2019). In this sense, evidence shows that autophagy regulation is limited to mTORC1, since ULK1 does not bind to the mTORC2-specific component Rictor (rapamycin-insensitive companion of mTOR) (Ganley et al, 2009; Jung et al, 2009). mTORC1 also controls autophagy by activating the autophagy suppressor death-associated protein 1 (DAP1) (Koren et al, 2010) or the phosphorylation of WD-repeat protein interacting with phosphoinositide 2 (WIPI2), a keypositive regulator of autophagosome formation that is prone to proteasomal degradation on mTORC1 phosphorylation (Wan and Liu, 2019). mTOR-dependent autophagy is also influenced by the activation of PI3K/Akt signaling.
Another important pathway that requires further exploration involves ROS. Although many different mechanisms are involved in ROS-induced autophagy, a few of them directly implicate the role of ROS signaling in autophagy. The Ser/Thr kinase ATM (ataxia-telangiectasia mutated) is one of the few candidate proteins and is involved in glucose transport as a major upstream activator of Akt. It contributes to the translocation of GLUT4 to the cell membrane, and so its deficiency can cause insulin resistance (Halaby et al, 2008).
ATM also acts as an ROS sensor, triggering autophagy to maintain physiological cell homeostasis (reviewed in Stagni et al, 2020). Finally, the molecular relationship between inflammation and autophagy also merits consideration. NLRP3 inflammasome activity is enhanced in type 2 diabetes, and this activation is mediated through the effects of both hyperglycemia and dyslipidemia. Several works reveal an intriguing interplay between the NLRP3 inflammasome and autophagy in various conditions, including metabolic disorders such as diabetes.
Notably, NLRP3 has been shown to be a binding partner of mTOR (Cosin-Roger et al, 2017), and mTOR may constitute a fundamental axis connecting the NLRP3 inflammasome and autophagy.
E. Evidence of alterations in autophagy in type 2 diabetes
It is widely accepted that autophagy contributes to the maintenance of β cell function (including insulin secretion), mass, and survival under both normal and stress conditions (Blandino-Rosano et al, 2017; Hayes et al, 2017; Riahi et al, 2016 and reviewed in Watada and Fujitani, 2015). Various stress stimuli are well known to upregulate (and to be alleviated by) autophagy in β cells, including oxidative stress, mitochondrial dysfunction, glucolipotoxicity, and ER stress (Fig. 11). Insufficient autophagy compromises β cell function; for example, it has been associated with UPR, which promotes type 2 diabetes (Quan et al, 2012).
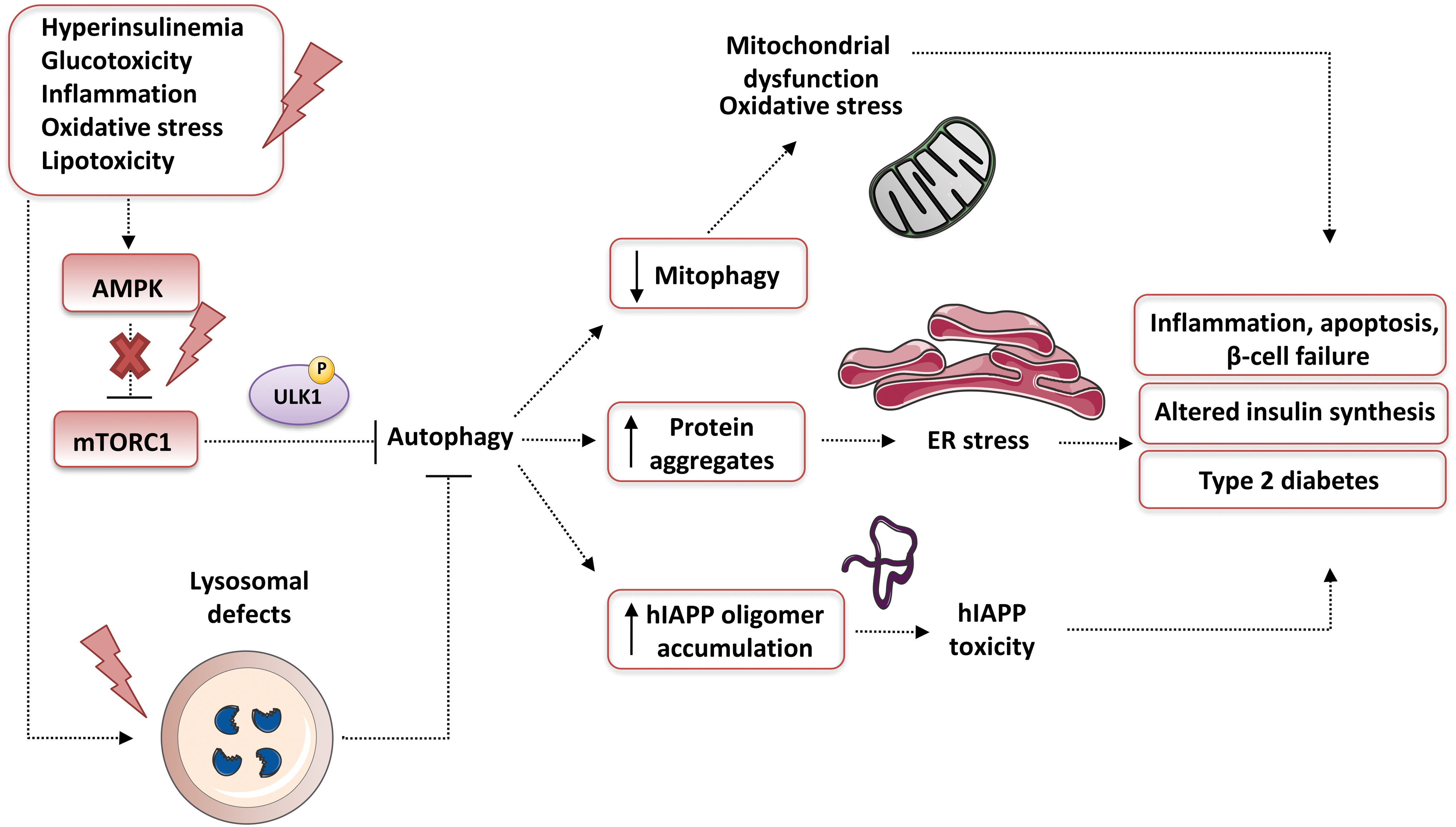
In this sense, increased ROS levels are an alarm signal that activates autophagy under a variety of metabolic stresses, including those present in type 2 diabetes. In this context, autophagy is vital for the elimination of damaged/oxidized biological macromolecules and organelles (such as mitochondria). β cells are extremely susceptible to oxidative stress, principally of the hyperglycemia-induced type, which leads to altered abnormal insulin secretory responses and induced apoptosis (reviewed in Robertson, 2004).
Importantly, stimulation of the antioxidant nuclear factor erythroid 2-related factor 2 (Nrf2) in these cells can prevent apoptosis and cellular damage by the transcription of autophagy genes, whereas its knockout results in diminished β cell mass (Yagishita et al, 2014). Of note, autophagy can also activate Nrf2 through sequestosome-1 (p62/SQSTM1)-mediated disruption of its interaction with Kelch-like ECH-associated protein 1 (KEAP1) (Jain et al, 2010), the primary negative regulator of Nrf2, which promotes its proteasomal degradation via the ubiquitin-proteasome pathway.
β cells are highly sensitive to changes in nutritional status, and autophagy has been extensively reported as a pivotal metabolic regulator of these cells. It is noteworthy that a short-term starvation diet can induce a specific autophagic process of insulin-containing vesicles known as β cell crinophagy, whereas prolonged fasting can stimulate classic autophagy (reviewed in Watada and Fujitani, 2015). Moreover, β cell autophagy is stimulated by cholesterol (Wu et al, 2017), omega-3 fatty acids (Hwang et al, 2015), vitamin D (Wang et al, 2016), and FFA, including palmitate (Martino et al, 2012), therefore playing a protective role by preventing the activation of the apoptotic process.
Finally, a dipeptidyl peptidase-4 (DPP-4) inhibitor (MK-626) and glucagon-like peptide 1 (GLP-1) receptor agonists (liraglutide and exendin-4) have been shown to stimulate β cell autophagy and improve β cell function in both in vivo and in vitro studies, thus protecting β cells against tacrolimus-induced glycolipotoxicity and apoptosis (Wang et al, 2016; Zummo et al, 2017).
In light of all of this evidence, it is apparent that impaired/dysregulated autophagy is an essential contributing factor to the pathogenesis of type 2 diabetes (reviewed in Marasco and Linnemann, 2018). Enhanced vacuole overload and the presence of autophagosomes have been related to β cell loss in human type 2 diabetic samples (Masini et al, 2009). Nevertheless, it is not easy to distinguish whether enhanced autophagosomal content is a protective response to stress or a marker of pathogenic dysregulation of autophagy.
Generally, it seems that, although diabetes is associated with augmented autophagosome generation, the overall autophagic clearance under diabetic conditions is malfunctional. The underlying mechanism of the blockage of β cell autophagic flux may include lysosomal deficiencies or impairment in response to chronic glucolipotoxicity conditions (Trudeau et al, 2016). Another phenomenon associated with reduced autophagic clearance in β cells is human islet amyloid polypeptide (hIAPP) aggregation (Fig. 11), which is the principal contributor to pancreatic dysfunction during type 2 diabetes in humans, but not in animals (Rivera et al, 2014).
In summary, autophagy is vital for the maintenance of β cells survival and function, whereas type 2 diabetes is associated with diminished pancreatic autophagy. Indeed, the protective effects of the anti-diabetic drug metformin against β cell death are related to the enhancement of autophagy that results from mTORC1 inhibition, with or without the implication of AMPK activation, and other pathways (Li et al, 2019a).
Besides the pancreas, autophagy may be deregulated in different important organs involved in the pathogenesis of type 2 diabetes and its associated comorbidities. First, subjects with type 2 diabetes often display metabolic disturbances, including obesity and metabolic syndrome. In this sense, adipogenesis, a highly controlled process in which primary fibroblasts develop into mature adipocytes, entails profound cytoplasmic reorganization, which comes about through massive autophagy.
Adipose tissue from type 2 diabetic and obese subjects has been shown to exhibit increased autophagic flux compared with that from lean individuals without type 2 diabetes (Kosacka et al, 2015). Similarly, studies in genetically modified rodents have revealed that autophagic damage is closely linked to a defective function of insulin-sensitive tissues, including skeletal muscle (reviewed in Barlow and Thomas, 2015). In addition, autophagy-deficient skeletal muscle shares most of the characteristics of insulin-resistant muscle, including mitochondrial dysfunction. To date, it seems that autophagy signaling is undermined by the onset of insulin resistance, but it is unclear whether this is a response to hyperinsulinemia or an intrinsic defect in autophagy.
Further, the emerging regulatory network for autophagy-glycemia-insulinemia is hugely complicated, with much data requiring clarification. Insulin is thought to inhibit autophagy in human skeletal muscle; however, there are conflicting data that suggest that autophagy continues to respond to the suppressive effects of insulin in obese and insulin-resistant mice (Ehrlicher et al, 2018). Moreover, infusion with physiological insulin concentrations has been shown to reduce the skeletal muscle protein content of LC3B-II, an important marker of autophagosome formation, in both lean and obese people.
However, this response was absent in diabetic subjects under conditions of euglycemia and became normalized when fasting glucose levels rose (Kruse et al, 2015). A possible mechanism by which insulin resistance suppresses autophagy is the activation of mTORC1 by phosphorylation at Ser2448 (Timmerman et al, 2010). It is noteworthy that attempts to evaluate whether basal autophagy is dysregulated in the skeletal muscle of type 2 diabetic individuals have yielded contradictory rather than conclusive data, with both downregulation (Henriksen et al, 2019) and a lack of difference being reported with respect to healthy controls (Kruse et al, 2015).
There is abundant evidence to support a relationship between altered autophagy and the development of diabetic nephropathy; indeed, the regulation of autophagy has been proposed as a priority therapeutic goal in the treatment and follow-up of diabetic nephropathy (reviewed in Gonzalez et al, 2021; Kitada et al, 2017). More specifically, a dearth of autophagy in renal cells, including TEC and podocytes, has been described in diabetic nephropathy. Several studies have reported a decreased autophagic flux in kidney biopsy samples from patients with diabetes nephropathy, manifested as an accumulation of p62/SQSTM1 protein in the glomeruli (Tagawa et al, 2016), and the same was observed in a diabetes nephropathy rat model with severe proteinuria (Tagawa et al, 2016), and in the kidneys of diabetic Wistar fatty (fa/fa) rats (Kitada et al, 2011).
It has been described that lysosome dysfunction and impaired autophagy occur in cultured podocytes treated with sera from rats and patients with type 2 diabetes and high levels of proteinuria, circumstances that eventually lead to cell death (Kitada et al, 2017). In fact, altered autophagic activity in podocytes results in podocyte loss, which ultimately promotes proteinuria and the development of diabetic nephropathy (Dong et al, 2019; Zhao and Fan, 2020). In this sense, one of the main mechanisms involved in autophagic malfunction in diabetic nephropathy is high AGE levels. AGE can disrupt the autophagy-lysosome pathway in renal TEC (Liu et al, 2015).
More specifically, they suppress autophagic flow in different cell types, including podocytes, by activating mTOR and inhibiting the nuclear translocation of the transcription factor EB (TFEB) and, consequently, the biogenesis of lysosomes (Zhao et al, 2018). Of note, TFEB is a master regulator of autophagy, lysosomal biogenesis, exocytosis, and lipid catabolism; on starvation or lysosomal stress, mTOR inhibition induces TFEB dephosphorylation, resulting in nuclear localization and increased activity of this transcription factor.
In addition, another study demonstrated that the activation of Smad3 (mothers against decapentaplegic homolog 3) can regulate blockage of AGE-induced autophagic flow, since Smad3 is able to bind to the 3′-untranslated region (UTR) of the TFEB gene, thus inhibiting its transcription and leading to deficient lysosome biogenesis (Yang et al, 2021a).
Dysfunctional autophagy is also involved in diabetic myocardial I/R injury (Yu et al, 2018), and multiple mechanisms have been proposed. It has previously been highlighted that the NLRP3 inflammasome is a key player in the development of diabetic myocardial I/R injury (Qiu et al, 2019); however, whether there exists an interplay between the NLRP3 inflammasome and autophagy in the development of diabetic myocardial I/R injury remains to be clarified. In rat diabetic myocardial I/R injury, autophagy was found to be inhibited and the NLRP3 inflammasome activated, whereas rapamycin (an autophagy activator) improved the injury by decreasing the blood levels of creatine kinase and LDH, and diminishing the infarct size (Zhang et al, 2020).
Similar data were obtained in high glucose-treated H9C2 cells (rat cardiomyoblast cell line) with I/R injury (Zhang et al, 2020). In contrast, the activation of autophagy aggravated I/R-induced injury to the myocardium in an animal model of streptozotocin-induced diabetes (Ma et al, 2018a). These discrepancies could be due to evaluation of different tissues and/or stages of the disease, and therefore a more concerted effort to investigate is required.
Moreover, altered autophagy has been described in the central nervous system in relation to diabetes. In streptozotocin-treated rats, hyperglycemia worsens cerebral I/R-induced neuronal damage via ERK1/2-activated cell autophagy and mitochondrial fission (Liu et al, 2022), whereas in db/db mice, a model of type 2 diabetes, characterized by a deletion of the leptin receptor gene and thus displaying obesity and insulin resistance, treatment with the autophagy inducer trehalose alleviated memory and neuroinflammatory brain processes.
Another, in vitro study by Xue et al (2022) suggests that high glucose and palmitic acid induce neuronal senescence by RE1-silencing transcription factor (REST), also known as neuron-restrictive silencer factor (NRSF/REST) elevation and the subsequent suppression of mTOR-related autophagy.
F. Anti-diabetic drugs and autophagy
There is abundant evidence that different anti-diabetic medications can alter autophagy both positively and negatively, in a direct or indirect way. For example, metformin, the most widely used oral anti-diabetic drug, has been shown to influence autophagy in many different models thanks to its capacity to affect multiple signaling pathways (reviewed in Lu et al, 2021). Metformin inhibits elevated autophagy in leukocytes (Diaz-Morales et al, 2018) and restores decreased autophagy in blood mononuclear cells (Bhansali et al, 2020) of type 2 diabetic patients. SGLT2 inhibitors (empagliflozin, dapagliflozin, canagliflozin, and ertugliflozin) act by inhibiting glucose reabsorption in the proximal tubules, thus reducing blood glucose and body weight.
Many of the effects of SGLT2 inhibitors can produce important changes in the autophagic flux, but there is also evidence of these molecules directly enhancing autophagy in diabetic experimental rodents (Aragon-Herrera et al, 2019; Korbut et al, 2020). GLP-1 receptor agonists (exenatide, liraglutide, dulaglutide, and semaglutide) are highly effective anti-diabetic agents with renoprotective effects that are mediated by GLP-1 receptor signaling.
Liraglutide has been shown to stimulate autophagy flux positively through the AMPK-mTOR signaling pathway in experimental diabetic kidney disease (Yang et al, 2020). Finally, DPP-4 inhibitor vildagliptin reduces the type 2 diabetes-induced increase in post-myocardial infarction acute mortality in Otsuka-Long-Evans-Tokushima Fatty rats (OLETF), seemingly by restoring the autophagic response through attenuation of BCL2-Beclin1 interaction (Murase et al, 2015).
VI. Mitophagy
A. Types and significance
The term mitophagy was coined in 2005 to refer to an adaptive mechanism that is triggered under both pathological and physiological conditions (reviewed in Lemasters, 2005). It involves the elimination of damaged or excess mitochondria, thus acting as a sort of mitochondrial quality control (Fig. 12). It was initially described in starved yeast, where the presence of a mutated Uth1p in the OMM was found to inhibit autophagy (Kissova et al, 2004).
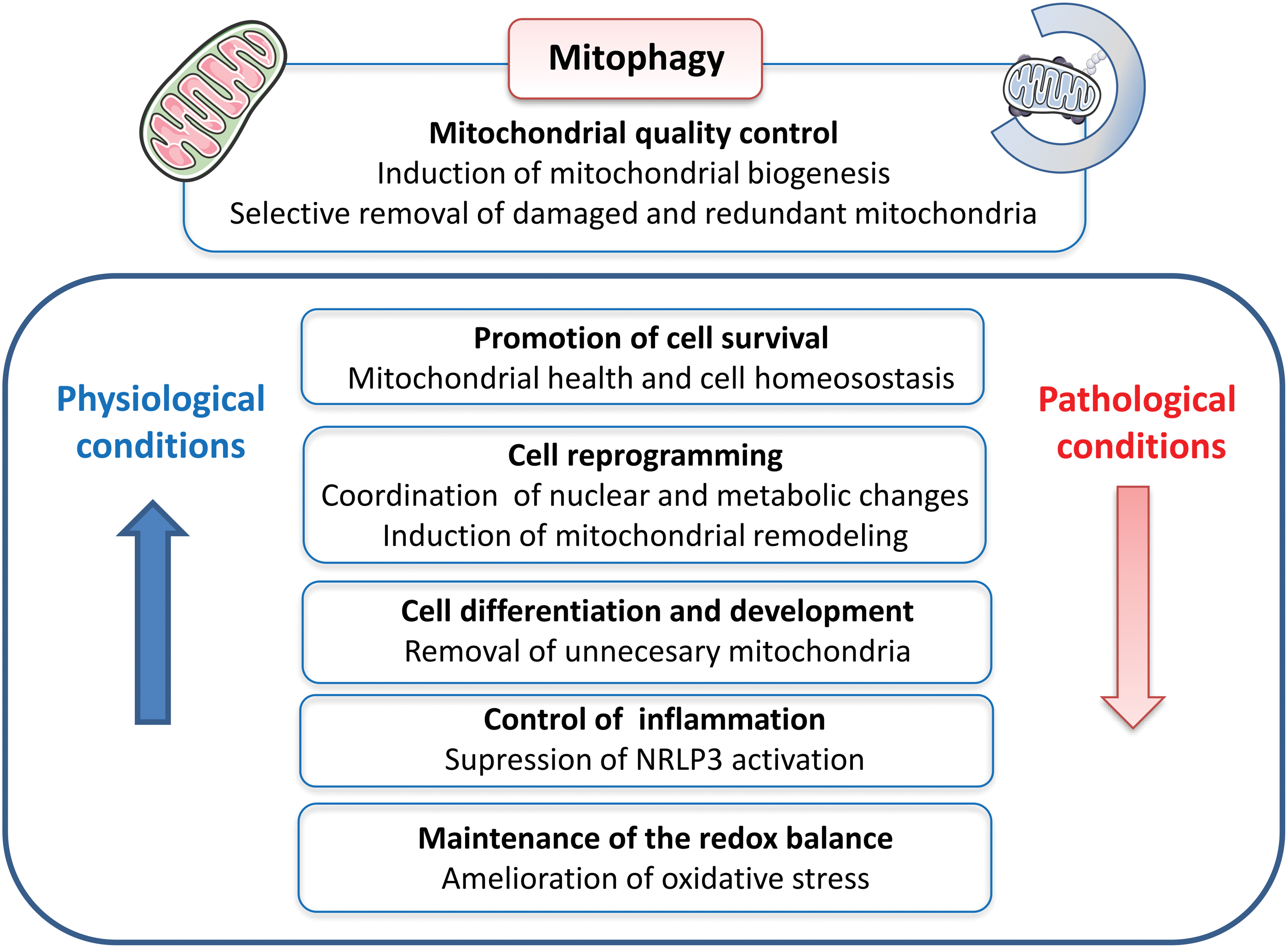
Similarly, the elimination of damaged mitochondria was reported in cultured hepatocytes exposed to oxidative damage. Nowadays, the existence of three types of mitophagy has been reported: type I and II, which are induced by nutrient deprivation and mitochondrial damage, respectively, and type III, also known as micromitophagy, and linked to mitochondria-derived vesicles (MDV) (reviewed in Lemasters, 2014; Zachari and Ktistakis, 2020) (Fig. 13).
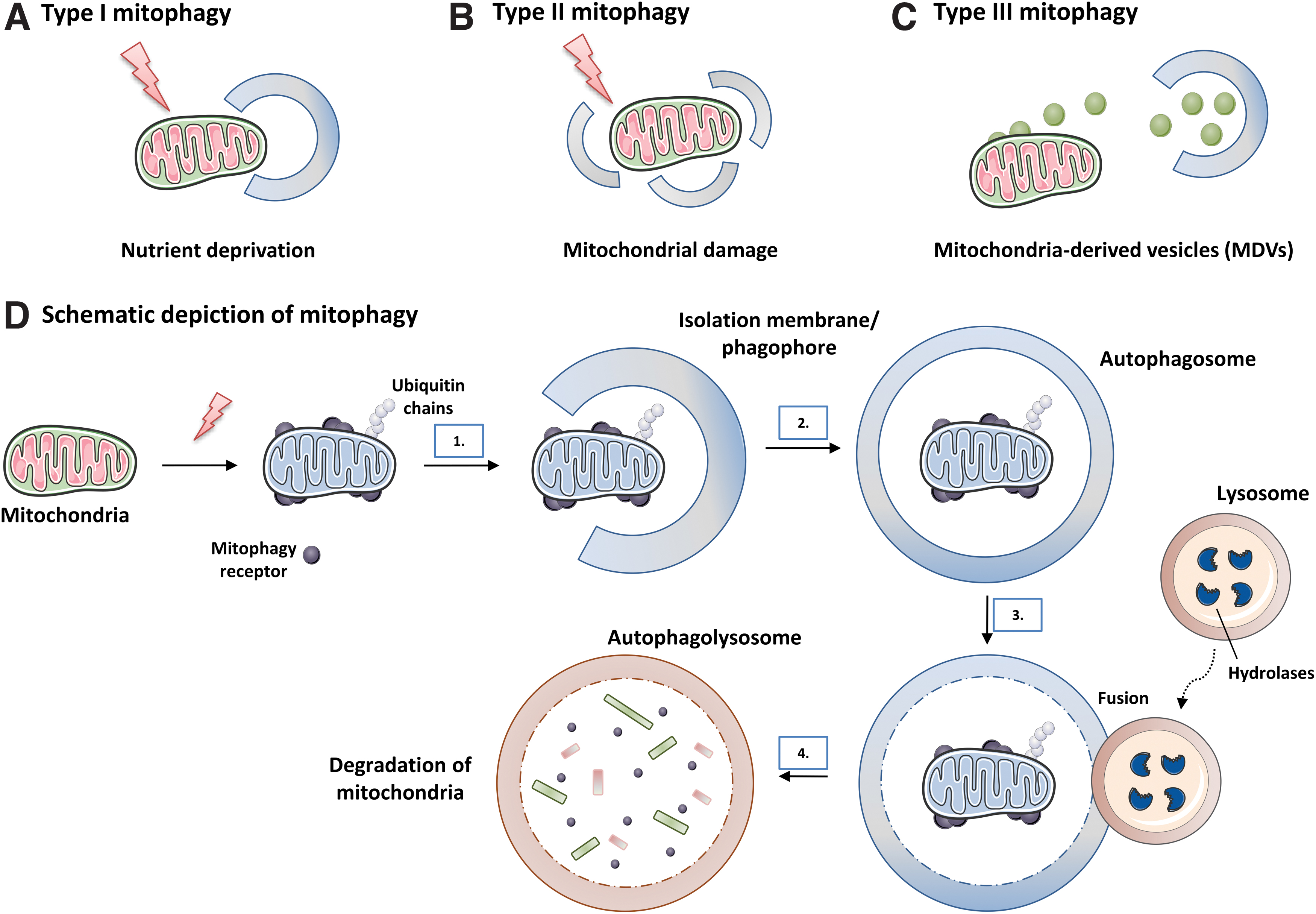
Type I and type II involve autophagosome-encircling mitochondria, which fuse with a lysosome. In type I mitophagy, pre-autophagic structures (PAS) can elongate into surrounding cup-shaped phagophores and thus sequester individual mitochondria in mitophagosomes. This process requires PI3K, which plays a key role in the classic Beclin1/VPS34 autophagic pathway (class III PI3K), and usually occurs simultaneously with the process of mitochondrial fission.
When the selective degradation of mitochondria is stimulated by nutritional deficit, mitochondria can maintain their membrane potential during sequestration, and depolarization does not occur until sequestration is complete. Therefore, and unlike type I mitophagy, type II mitophagy is not affected by PI3K inhibition and is not related to mitochondrial fission or phagophore formation. In type II mitophagy, which is exemplified primarily by photodamage as a prototype stimulus, LC3 is localized on the surface of individual depolarized mitochondria.
These LC3-containing aggregates fuse into complete ring structures, thus inserting each damaged/aged organelle into a mitophagosome. Phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) and Parkin are thought to be crucial for type II but may not be involved in type I mitophagy. In type III mitophagy, the fission event required for this process is different from that associated with binary fission of mitochondria, since MDV formation does not depend on dynamin-related protein 1 (DRP1), a critical GTPase that mediates division of mitochondria (reviewed in Lemasters, 2014).
Subsequently, MDV are internalized into multivesicular bodies in a PINK1/Parkin-dependent manner. These multivesicular bodies fuse with the lysosomes, and mitochondrial components are then degraded by hydrolytic enzymes. Oxidative stress can stimulate the formation of MDV, and MDV themselves are reported to be enriched in oxidized mitochondrial proteins (Soubannier et al, 2012). However, unlike type II mitophagy, the formation of MDV does not require a decrease in mitochondrial membrane potential, and the transit of MDV to lysosomes generally occurs independently of the autophagic proteins LC3 and ATG5.
In this way, micromitophagy can be regarded as a mechanism of mitochondrial quality control by which selective elimination of oxidized and/or damaged mitochondrial components takes place. Further, this process is carried out without depolarization of the mitochondrial membrane or deterioration and damage of mitochondrial function. Finally, it should be stated that micromitophagy in yeast differs from that in mammalian cells; whereas the multivesicular bodies of mammalian cells swallow small mitochondrial fragments of MDV, the digestive vacuoles (lysosomes) of yeast engulf entire mitochondria.
How exactly the three types of mitophagy are regulated in vivo is still not fully understood, and the experimental models to induce mitophagy depending on physiological context and timeframe do not selectively activate specific types of mitophagy. For example, oxidative stress can induce all three types of mitophagy, mainly depending on its severity; type III mitophagy arises with mild oxidative stress and types I and II mitophagy are related to more severe oxidative stress, which eventually causes a decrease in ATP and mitochondrial membrane depolarization.
It is important to highlight that mitophagy plays essential roles during normal organ development, as demonstrated both in vitro and in vivo in fluorescent transgenic mouse models (e.g., mito-QC or mt-Keima) (Sun et al, 2015; Um et al, 2018). It can also be viewed as a fundamental ubiquitous biological mechanism in response to the changing energetic requirements of the cell. Other regulatory pathways have been suggested, such as the mitophagic removal of mitochondria based on their subcellular location or topology. In this sense, serum-starved U2OS osteosarcoma cells have been shown to form “donut” mitochondria that exhibit normal mitochondrial membrane potential and are resistant to mitophagy, whereas mitochondria with low potential are eliminated (reviewed in Zhou et al, 2020b).
Mitochondrial biogenesis is especially linked to mitophagy. In this sense, some tissues, such as skeletal muscle, the nervous system, and the heart, liver, and kidney, display a high activity of basal mitophagy, whereas others present low levels of mitophagy, as occurs in the thymus and the spleen (Sun et al, 2015), with significant differences depending on the tissue under study. In fact, although cell proliferation is slowed down in many adult organs, individual mitochondria change with a half-life of between 10 and 25 days, since mitophagy can eliminate worn out mitochondria in strict synergy with the biogenesis of new mitochondria (Lemasters, 2014).
In summary, mitophagy plays a fundamental role in cellular homeostasis in what is a complex process. It protects against pro-apoptotic protein release, high ROS production, and the inefficient ATP metabolism that occurs in depolarized, damaged, or aged mitochondria, as well as disposing of mitochondria during cytoplasmic remodeling and mtDNA degradation, including mutated and damaged mtDNA (Kim and Lemasters, 2011; Rodriguez-Enriquez et al, 2009; Suen et al, 2010 and reviewed in Kim et al, 2007; Lemasters, 2014; Mizushima et al, 2011; Yin et al, 2008; Youle and Narendra, 2011).
B. Molecular mechanisms involved in mitophagy
The most widely studied mechanism of mammalian mitophagy is PINK1/Parkin-mediated mitophagy. Under normal conditions, Parkin, an E3 ubiquitin ligase, is localized in the cytosol and its E3 activity is inhibited while PINK1, a mitochondrial Ser/Thr kinase, is transported to the intermembrane space and constitutively degraded (reviewed in Shirihai et al, 2015; Yamano et al, 2016). On reduction of the mitochondrial membrane potential, Parkin E3 activity is triggered and full-length PINK1 is exposed in OMM, which subsequently recruits Parkin by phosphorylating it at Ser65 (Heo et al, 2015; McLelland et al, 2018).
Ubiquitin is directly phosphorylated by PINK1 and represents Parkin's receptor in the OMM, which permits its translocation (Kazlauskaite et al, 2014; Koyano et al, 2014). Before mitophagy, PINK1/Parkin must arrest mitochondrial movement; once Parkin is activated, it initiates the ubiquitylation of various proteins at the OMM, preparing the defective mitochondrion for envelopment by autophagosomes (Fig. 14). This includes K11- and K48-associated ubiquitination of mitofusin (MFN)1/2 and of mitochondrial Rho (Miro), an OMM GTPase required for mitochondrial trafficking to various cellular locations (Reis et al, 2009) and which serves as a mitochondrial docking site for Parkin.

These modifications induced by Parkin rapidly trigger Miro's proteasomal degradation, thus avoiding fusion of damaged mitochondria with healthy ones and mitochondrial trafficking (Tanaka et al, 2010; Wang et al, 2011), and Miro-mediated mitochondrial movement prevents mitophagy. Together, PINK1 and Parkin regulate mitophagy at multiple levels, including mitochondrial fragmentation (via MFN1/2), mitochondrial mobility (via Miro), autophagy receptor proteins (via p62/SQSTM1 or optineurin [OPTN]), and autophagy machinery (via Ambra1).
PINK1 also directly regulates mitochondrial dynamics by tightly regulating the phosphorylation of DRP1, the direct antagonist of MFNs and a fundamental promoter of the activation of the mitochondrial fission process, at Ser616 (Han et al, 2020). In parallel, K63-linked ubiquitin chains are recognized by a family of proteins called autophagy receptors, which bind to LC3/GABARAP members attached to the autophagosomal membranes, leading to the recruitment of the autophagy machinery at locations where there are mitochondria with low membrane potential.
Of note, PINK1/Parkin-mediated mitophagy is mainly dependent on the sequestasome (p62/SQSTM1) (Geisler et al, 2010; Iguchi et al, 2013) and the activities of the voltage-dependent anion channel (VDAC) 1. Further, p62/SQSTM1 interacts directly with LC3 to recruit autophagosomes, which allows the exchange and transport of damaged mitochondria to lysosomes (Villa et al, 2017 and reviewed in Manley et al, 2013).
One of the critical processes of selective autophagy is the ubiquitination of the cargo (reviewed in Ney, 2015). In fact, in damaged mitochondria, after OMM remodeling arbitrated mainly by proteasomal degradation of ubiquitinated proteins, adapter molecules are drafted to transport depolarized mitochondria to the perinuclear zone through a microtubule-dependent system (Lazarou et al, 2015). To date, the most widely established hypothesis is that cargo-bound adapter and/or receptor proteins recruit LC3 through an LC3-interacting region (LIR) and bind cargoes with a membrane generated by autophagy, thus promoting mitochondrial sequestration into autophagosomes.
Notably, autophagy receptors are recruited by ubiquitination or are an integral part of the cargo. It has also been reported that a scaffold protein that recruits additional ATG may be involved (reviewed in Johansen and Lamark, 2020). Five cargo-linked mitochondrial receptors (LC3 adapters) that contain an LIR motif have been described (reviewed in Yoshii and Mizushima, 2015) that are recruited by polyubiquitinated substrates into mitochondria through their ubiquitin-binding domain and include neighbouring gene 1 of breast cancer 1 (BRCA1; NBR1), protein 52 of the nuclear domain 10 (NDP52), p62/SQSTM1, HTLV-1 (TAX1) binding protein 1 (TAX1BP1) transcriptional regulator transcriptional regulatory protein (TAX1), and OPTN.
In recent years, evidence has been mounting concerning Parkin-independent mitophagy pathways that include, but are not restricted to, recruitment of alternative E3 ligases to mitochondria, cardiolipin-mediated mitophagy, and transmembrane-receptor mediated mitophagy (Fig. 14). In this context, LC3 adapters can also recognize damaged mitochondria in a ubiquitin-independent manner. These proteins detect mitochondrial injury and then change their subcellular location or the proteins with which they interact, aiding the transfer of impaired mitochondria to the autophagosome. This event is referred to as transmembrane receptor–mediated mitophagy.
The best described systems are involved in programmed mitochondrial clearance during cell and tissue development and include transmembrane OMM proteins B-Cell Leukemia/Lymphoma 2 (BCL2)/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) and its homolog BNIP3-like or Nip-like protein X (NIX/BNIP3L) pathway (reviewed in Zhang and Ney, 2009). There are several studies supporting the critical role of BNIP3 and NIX in hypoxia-induced autophagy, by which they promote the decrease of mitochondrial membrane potential and the depolarization of mitochondria, as well as their fusion with cellular membranes.
Their N-terminal cytoplasmic portion can interact with different molecules that have been related to LC3, thus targeting mitochondria for autophagic degradation. Similarly, BNIP3 can interact directly with PINK1, stabilizing it and promoting Parkin recruitment through DRP1-mediated mitochondrial fission (Tang et al, 2019). BNIP3 can also modulate autophagy by decreasing mTOR activity and promoting LC3 expression (reviewed in Chourasia et al, 2015).
Another mitochondrial protein that operates in a Parkin-independent way and is sensitive to hypoxia is the OMM protein FUN14 domain-containing protein 1 (FUNDC1) (Liu et al, 2012) (Fig. 14). In fact, the list of mitochondrial proteins that detect mitochondrial damage and trigger the process of mitophagy is growing. In short, certain mitochondria-damaging stimuli cause these adaptor proteins to translocate to the OMM, where they interact with LC3 (via LIR) or other autophagic proteins. For example, choline dehydrogenase (CHDH), which is typically anchored at both the IMM and OMM, can accumulate in OMM when there are changes in the mitochondrial membrane potential, where it interacts with p62/SQSTM1 through its domain.
This PB1 (Phox and Bem1) domain eventually leads to the formation of the CHDH-p62-LC3 complex that mediates mitophagy (Park et al, 2014). Further, a protein activating the mitochondrial Rab GTPase and known as member 15 of the TBC1 domain family (TBC1D15) forms a complex with TBC1D17 and migrates to the OMM on interacting with Fis1. This TBC1D15/17 complex subsequently cooperates with LC3 (Yamano et al, 2014).
Another protein that can interact with LC3 is the FK506 binding protein 8 (FKBP8), located in the OMM (Bhujabal et al, 2017). Similarly, BCL2 like 13 (BCL2L13), the mammalian homolog of yeast ATG32 (Xia et al, 2018), spans the OMM and binds to LC3. Notably, certain IMM components have also been revealed to mediate mitophagy, and prohibitin 2 (PHB2) and cardiolipin are the most representative of them.
PHB2 is an IMM protein (Wei et al, 2017) that is exposed to LC3 after OMM rupture is arbitrated by Parkin. Cardiolipin is a membrane lipid localized in the IMM and that plays a central role in mitophagy by maintaining its normal functions. It acts as an LC3 receptor when translocated from the IMM to the OMM in different situations, such as the presence of external depolarizing toxins (reviewed in Li et al, 2015). How cardiolipin modulates the activity of this process is unknown and requires explanation, thus representing an intriguing area for future investigation.
In addition, the involvement of cardiolipin in mitophagy is also supported by the fact that it binds directly to the conserved domain of Beclin1 on the OMM (Huang et al, 2012). Also, AMPK induces Parkin-independent mitophagy by activating and phosphorylating the TNF receptor–associated family member–associated NF-κB activator (TANK)-binding kinase 1 (TBK1) (Seabright et al, 2020), a multi-functional kinase involved in the selective clearance of impaired mitochondria.
It is important to highlight that an emerging potential target of mitophagy is activation of the novel E3 ubiquitin ligase CLEC16A (C-type lectin domain family 16, member A), which is particularly relevant in diabetes, as will be discussed further on.
Recently, there has been great interest in the link between mitophagy and ER stress. A fully functional ER is paramount for the physiological functioning of β cells, since these cells are under a constant burden of high protein folding by which they synthetize proinsulin, converting it into its mature form and secreting it. Various stimuli (dysregulation of Ca2+ homeostasis, oxidative stress, changes in mitochondrial membrane potential among others) can induce ER stress in β cells, therefore leading to protein translation interruption, molecular chaperone synthesis, unfolded/misfolded protein overload, and disruption of cellular homeostasis.
ER stress in β cells has been defined as a stimulus of autophagy and, thus, a promoter of homeostasis (Kong et al, 2017). In relation to the ER, MAMs are key regulators of mitophagy, since many proteins directly involved in autophagy are located in these regions and it has been proposed that damaged mitochondria are ubiquitinated and dynamically encased in ER, providing platforms for formation of the mitophagosomes (Zachari et al, 2019). Indeed, in response to multiple stimuli-inducing mitophagy, Beclin1 and PINK1 relocate in the MAMs, where they promote the association of mitochondria with ER and, in turn, the formation of autophagosomes (Gelmetti et al, 2017).
Moreover, PINK1 controls mitochondrial Ca2+ efflux (Gandhi et al, 2009; Heeman et al, 2011), and PINK1 expression is sensitive to alterations in Ca2+ fluxes (Gomez-Sanchez et al, 2014), suggesting that Ca2+ levels must be highly regulated for the proper functioning mitophagy. Of note, the alternative FUNDC1-mediated pathway of mitophagy takes place at the MAMs (Wu et al, 2016; Wu et al, 2014). In addition, some ER-stress regulators stimulate mitophagy; for example, ER-stress-inducible transcription factor ATF4 can promote mitophagy by increasing Parkin expression (Zhang et al, 2014).
Finally, it is paramount to mention the abundant evidence that mitophagy is induced by oxidative stress and is the primary mechanism by which cells dispose of mitochondria affected by oxidative damage. Oxidative modifications in redox-sensitive amino acids (cysteine and methionine) regulate mitophagy and many other mitochondrial processes. In HeLa cells, Parkin/PINK1-mediated mitophagy requires superoxide production, which occurs through activation of the p38 signaling pathway (Xiao et al, 2017a).
Moreover, direct generation of mitochondrial ROS using a mitochondria-targeted photosensitizer causes mitochondrial depolarization and induces mitophagy, which can be reversed by overexpression of SOD2 (Wang et al, 2012). A shift of the mitochondrial environment toward oxidative conditions promotes mitochondrial fission, and mild oxidative stress triggers mitophagy in a mitochondrial fission-dependent manner (Frank et al, 2012) and through downregulation of the mitophagy-associated proteins OPTN, NDP52, and MFN2 (Zhang et al, 2021).
Considering these and many other similar findings, it is safe to state that mitophagy, altered mitochondrial dynamics, and oxidative stress (redox modification) are closely interlinked. Although an in vitro experimental approach has been used in most of the studies in question making it possible to establish a cause-consequence relation, real-life clinical settings including diabetes mellitus are subject of the chicken-and-egg paradox regarding the pathogenic cause and the clinical effects.
C. Evidence of alterations in mitophagy in type 2 diabetes
The progression to type 2 diabetes is characterized by hyperglycemia and insulin resistance, processes that are intrinsically related to mitochondrial damage, alteration of mitochondrial morphology, dysmetabolism, and oxidative stress. Therefore, it is very likely that these phenomena are the result of a vicious cycle provoked by prolonged exposure to high levels of FFA and glucose, giving rise to glycolipotoxicity, oxidative stress, and high release of mitochondrial ROS, all of which ultimately cause damage to the cells and tissues.
Diabetics are also prone to lipid and lipoprotein abnormalities as an indirect effect of insulin resistance on key metabolic enzymes. However, recent studies imply that lipid changes are not only a consequence of impaired glucose metabolism, but also a causative factor (reviewed in Shetty and Kumari, 2021). Fatty acids influence translocation of GLUTs and INR binding and signaling, in addition to cell membrane fluidity and permeability.
Thus, it has been suggested that FA play an essential role in the development of insulin resistance and type 2 diabetes. Specific combinations of FA within phospholipids and triglycerides have been shown to exhibit the strongest associations with the risk of type 2 diabetes (reviewed in Shetty and Kumari, 2021).
Therefore, maintaining adequate mitochondrial quality control through mitophagy is essential for cellular homeostasis. However, damage and cellular stress overwhelm mitochondrial homeostatic mechanisms, altering mitophagy and leading to the accumulation of damaged and dysfunctional mitochondria (Fig. 15). Importantly, mutations in genes related to mitophagy, including PARKIN, PINK1, PDX1, and CLEC16A, have been associated with the development of diabetes in humans, both type 1 and type 2 (Jin et al, 2014; Qu et al, 2011; Soleimanpour et al, 2015; Soleimanpour et al, 2014).
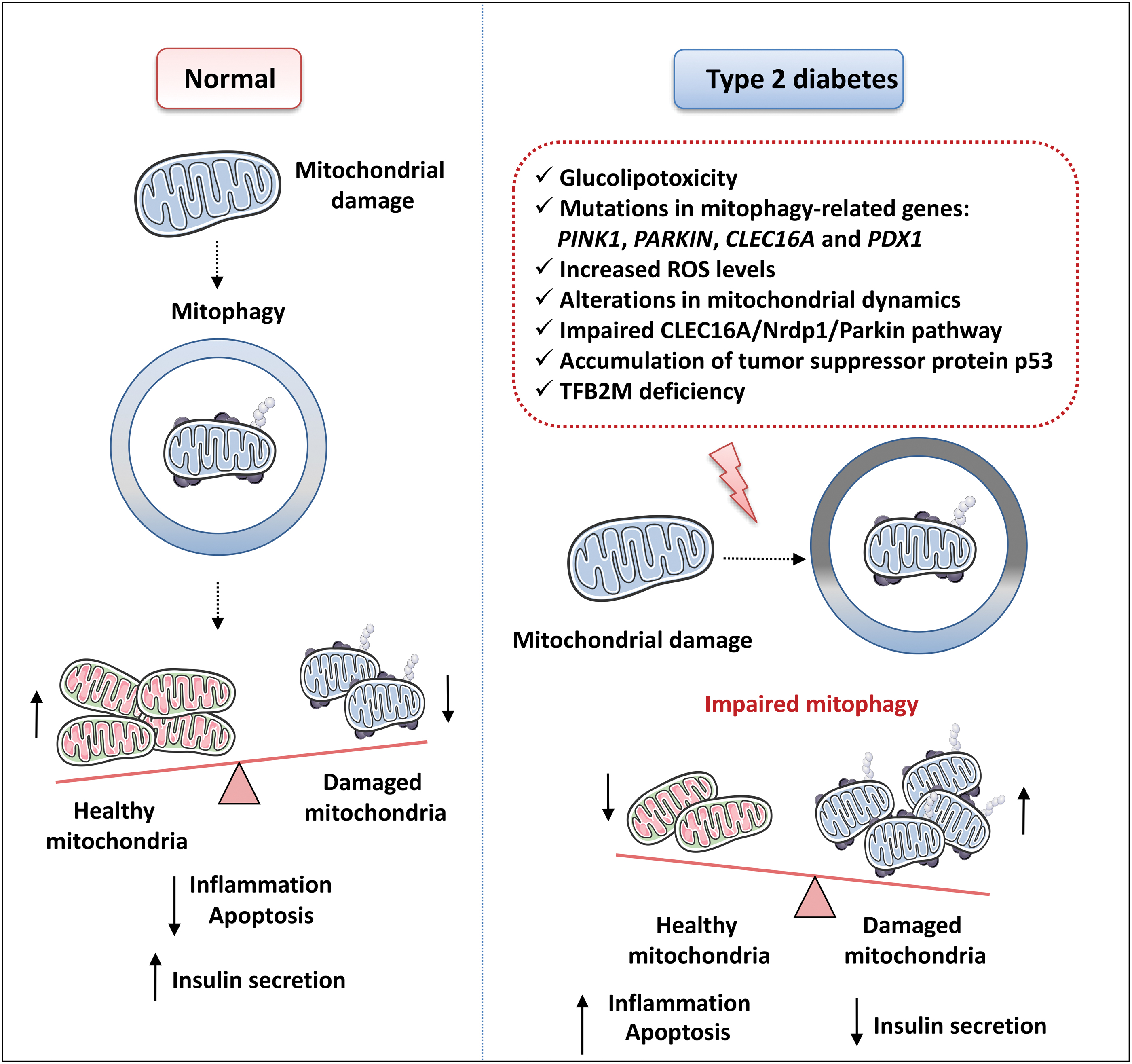
Impaired mitophagy in diabetes has been associated with decreased insulin release from β cells, even when glucose concentrations are high. This occurs because of the inhibition of the OMM protein Miro1, which inhibits mitophagy and interferes with insulin secretion through IRS-Akt-Foxo1 (Chen et al, 2017). Notably, type 2 diabetic patients display impaired mitophagy flux and decreased expression of mitophagy genes, unlike pre-diabetic patients with mild hyperglycemia who exhibit an increase in the expression levels of different mitophagy-related genes, such as Parkin, PINK1, and NIX (Bhansali et al, 2017).
In light of these findings, it is believed that mitophagy is enhanced in subjects with pre-diabetes as an adaptive mechanism by which dysfunctional mitochondria are disposed of, thereby modulating mitochondrial oxidative stress and delaying or preventing the onset and progression of type 2 diabetes. In subjects with established type 2 diabetes, this protective mechanism is overwhelmed, and higher ROS levels not only aggravate the existing mitochondrial damage but also suppress mitophagy, thereby resulting in enhanced accumulation of damaged mitochondria.
Multiple cellular effects of type 2 diabetes have been implicated in mitophagy regulation. For example, hyperglycemia can promote mitochondrial fission (which is involved in mitophagy) and inhibit mitochondrial fusion through OPA1/MFN degradation and DRP1 recruitment, thus altering mitochondrial dynamics (reviewed in Liesa and Shirihai, 2013).
As a result of these alterations, type 2 diabetic patients have smaller mitochondria than healthy controls (Bhansali et al, 2017). In addition, glycolipotoxicity, which occurs during the development of type 2 diabetes, also causes the accumulation of cytoplasmic p53, which disturbs the process of mitophagy by blocking the mitochondrial translocation of Parkin (Hoshino et al, 2014). A link with obesity has also been demonstrated, as the obesity-induced regulator of calcineurin 1 (RCAN1) overexpression in mice leads to β cell failure through Miro1-mediated mitophagy deficiency (Li et al, 2020b).
Abundant evidence points to the necessity of the fine-tuning of Parkin content and activity in β cells for their proper function. For example, one study showed excessive Parkin levels caused by overexpression of the OMM protein mito-NEET to be detrimental to β cells, as manifested by undermined mitochondrial oxidative capacity, increased glucose intolerance due to aberrant Parkin-mediated mitophagy, and reduced GSIS (Kusminski et al, 2016).
A similar finding has been reported with another Parkin-regulating protein, CLEC16A, a pivotal β cell mitophagy regulator, and one of the pathways to have been implicated is CLEC16A/neuregulin receptor degradation protein-1 (Nrdp1)/Parkin-mediated mitophagy. CLEC16A expression, which is required for maintaining normal glucose levels and β cell function in mice, is decreased in individuals with diabetogenic single nucleotide polymorphism (SNP), in whom the production of insulin is insufficient. Consequently, CLEC16A gene locus has been related to a high risk of developing type 1 diabetes.
A role of CLEC16A in β cells has become apparent, as it forms a ubiquitin-dependent complex, together with Nrdp1 and the deubiquitinase enzyme USP8, which acts as a key mitophagy modulator. Under metabolically healthy conditions, CLEC16A stabilizes RNF41 via nondegradative ubiquitination, leading to the proteasomal degradation of Parkin.
When severe mitochondrial damage occurs, dissolution of the CLEC16A-RNF41-USP8 complex can lead to low levels of RNF41 and a notable improvement in the way USP8 deubiquitinates Parkin, thus allowing Parkin's to initiate mitophagy due to its mitochondrial localization, which, in turn, maintains mitochondrial quality control (Pearson et al, 2018). CLEC16A also modulates cytokine-induced mitophagy in β cells and the overexpression of CLEC16A ameliorated cytokine-induced β cell death (Sidarala et al, 2020).
In addition, results obtained in murine models suggest that reducing the levels of the suppressor p53 in the cytosol compartment of pancreatic β cells is a protective mechanism against the development of diabetes, since cytosolic p53 can inhibit Parkin-mediated mitophagy and induce mitochondrial dysfunction, thereby decreasing ATP levels and increasing mitochondrial ROS levels (Hoshino et al, 2014). The proposed mechanism involves the stimulation of NOX by glucolipotoxicity through the activation of the receptor for AGE and TLR4.
The high production of mitochondrial ROS and the oxidative stress generated as a result induce the accumulation of p53 in the cytosol, altering the mitophagy process through an inhibitory interaction with Parkin.
The type of mitophagy that is active under different conditions in β cells remains to be elucidated. In this regard, although Parkin is a widely known modulator of mitophagy, it has been shown to be dispensable for glucose homeostasis (islet architecture, β cell formation, and insulin secretion) in both β cells and adipocytes during diet-induced insulin resistance in mice (Corsa et al, 2019).
In recent years, significant effort has been made to understand the up-stream regulatory pathways, including transcription regulators of mitophagy in β cells. In this regard, PDX1, a master regulator of β cell development and function, was found to modulate CLEC16A-mediated mitophagy through transcriptional regulation of CLEC16A (Soleimanpour et al, 2015).
Mitochondrial transcription factor B2 (TFB2M) is an RNA methyltransferase and a component of the mitochondrial transcription initiation complex necessary to maintain β cell mtDNA content, mitochondrial respiration, and GSIS. TFB2M deficiency in β cells causes an accumulation of mitophagy intermediates, probably because there is impaired mitophagy (Nicholas et al, 2017). Nor1/NR4A3, an orphan nuclear receptor that functions as a transcriptional activator, also plays a pro-apoptotic extranuclear role, and it is upregulated in pancreatic islets in type 2 diabetes.
In INS1 832/13 cells exposed to inflammatory cytokines, Nor1/NR4A3 translocates to mitochondria, which aberrantly enhances mitophagy and reduces mitochondrial function and GSIS (Close et al, 2020; Close et al, 2019). Similarly, in the insulinoma cell line RIN-m5F, treatment with N ɛ-(carboxymethyl) lysine-conjugated bovine serum albumin (CML-BSA), a major component of AGE, was found to induce mitochondrial dysfunction and undermine insulin production, while enhancing mitophagy (Lo et al, 2015).
Of note, treatment with CML-BSA has been shown to reduce the viability of RIN-m5F cells, an effect that is partially reversed by concomitant treatment with 5-aminolevulinic acid (ALA) or the autophagy inhibitor 3-methyladenine (3-MA). These results support the idea that excessive activation of excessive autophagy (and therefore mitophagy) can cause alterations in β cell function and apoptosis.
Mitophagy has been found to be altered in different organs and cell types in relation to various diabetic complications. In the context of diabetic nephropathy, patients with this condition display decreased renal expression of OPTN, a key receptor of mitophagy, and their OPTN levels correlate negatively with NLRP3 inflammasome activation and renal interstitial inflammation. Moreover, mitophagy inhibits high-glucose-induced NLRP3 inflammasome activation in cultured renal TEC (Chen et al, 2019).
In cultured podocytes exposed to high glucose levels, the glycoprotein progranulin (PGRN) has been shown to play a protective role by improving mitochondrial homeostasis (restoring both mitochondrial biogenesis and mitophagy) via PGRN-SIRT1-PGC-1α/FoxO1 signaling. In addition, data from the study in question indicated that the administration of recombinant human PGRN significantly protected against podocyte injury in rodents with diabetic nephropathy, whereas the expression levels of PINK1 were recovered in their glomeruli (Zhou et al, 2019a).
A possible function has been proposed for the ubiquitously expressed long non-coding RNA (lncRNA) nuclear paraspeckle assembly transcript 1 (NEAT1), a lncRNA that is upregulated in the serum of patients with diabetic nephropathy. NEAT1 facilitates the high damage caused by high glucose levels in HK-2 cells, decreasing and, in some cases, suppressing mitophagy through the miR-150-5p-DRP1 axis (Yang et al, 2021b).
The progression of diabetic retinopathy has a negative correlation with the level of mitophagy. A deficient removal of damaged mitochondria can lead to apoptosis of retinal capillary cells (endothelial cells pericytes), and an increase in ROS levels, which contribute to a significant metabolic change by which glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity in glycolysis is altered and the formation of AGE is increased.
These events lead to PKC activation, which is associated with inflammation and a decrease in the number of cells and microvascular dysfunction (Wang et al, 2015 and reviewed in Bek, 2017). Moreover, it has been shown that mitophagy can be inhibited by ROS-mediated inactivation of the PINK1/Parkin signaling pathway (Zhang et al, 2019). This effect depends mainly on the activation (under high glucose conditions) of the thioredoxin-interacting protein (TXNIP), an endogenous regulator of thioredoxin (Trx) that inhibits its antioxidant action, thereby trigging alteration of the redox balance and cellular oxidative stress (Devi et al, 2017 and reviewed in Yoshihara et al, 2014), as observed in cultured retinal endothelial cells and the retinas of diabetic rats (Devi et al, 2017; Perrone et al, 2010; Perrone et al, 2009 and reviewed in Singh, 2013).
Of note, high levels of TXNIP can reduce mitophagy and induce mitochondrial damage in retinal Müller cells, where TXNIP is involved in the targeting of defective mitochondria to lysosomes during mitophagy through Parkin-dependent ubiquitination (Devi et al, 2017). Interestingly, TXNIP enhances DRP1's association with mitochondria, which is relevant in the context of altered mitochondrial dynamics and its close relation to mitophagy.
This protein has also been recognized as one of the links between diabetes and Parkinson's disease, as it can regulate Parkin/PINK1-mediated mitophagy in dopaminergic neurons under hyperglycemic conditions (Su et al, 2020). Finally, it has been shown that retinal ganglion cells (RGCs), bridging neurons that reside within the inner retinal layers that form the optic nerve, also play a fundamental role in the initiation and progression of diabetic retinopathy.
In this sense, several studies have shown that the differentiation of mouse RGCs depends mainly on mitophagy (Deczkowska and Schwartz, 2017; Esteban-Martinez et al, 2017). This process is activated when tissue hypoxia increases NIX expression. Indeed, retinas from NixKO rodents contain a high mitochondrial mass and exhibit decreased expression of glycolytic enzymes and diminished neuronal differentiation (Deczkowska and Schwartz, 2017).
In diabetic retinopathy, alterations in the rate of mitophagy have been associated with the advanced stage of the disease. Post-mortem retinal tissue from human diabetic donors or experimental animals presents a significant reduction in the number of mitochondria during the initial stages of the disease, mainly due to an altered mitochondrial biogenesis and consequent inability to compensate for diabetes-exacerbated mitophagy (Hombrebueno et al, 2019).
However, as type 2 diabetes progresses and symptoms become more severe, PINK1-dependent mitophagy progressively deteriorates and consequently there is an accumulation of mitochondria susceptible to degradation due to their deterioration. In this regard, impaired mitophagy during prolonged diabetic conditions has been linked to the onset of retinal senescence. In addition, given the fact that overactivation of mitophagy may be related to damage of the retinal pigment epithelium or other cells in the retina, mitophagy in the context of diabetic retinopathy needs to be viewed as a dynamic, cell type- and context-dependent process.
Specifically, in vitro, TNF-α released from Müller cells has been shown to activate the EGFR/p38/NF-κB/p62 pathway to enhance mitophagy and mitophagy-related apoptosis in retinal pigment epithelial cells under high glucose conditions (Liu et al, 2021b). Similarly, diabetic rat models and the RGC-5 cell line cultured under diabetic conditions present enhanced mitophagy and an analogue of incretin/GLP-1 can alleviate RGC injury by weakening PINK1/Parkin-associated mitophagy (Zhou et al, 2020a).
There is also evidence that diabetic neuropathy is associated with an impairment of mitophagy. Interestingly, sensory neurons isolated from mice (transgenic Mito-QC mice, which have a global, constitutive knock-in of mCherry-GFP-FIS1 that causes mitochondria to fluoresce both red and green under homeostatic conditions) with 16 weeks of diabetes were found to present increased mitophagy (Rodriguez et al, 2021).
This increase reflects an adaptive response to eliminate defective mitochondria; however, it appears to be insufficient to prevent the decline in mitochondrial bioenergetics in diabetic rodents. Given that the assessment occurred at a single time point, it is likely that mitolysosome degradation was altered or that mitophagy eventually decreased, since prolonged diabetes can reduce the capacity of this autophagic elimination, thus leading to significant mitochondrial fragmentation and dysfunction.
Altered mitophagy has also been related to diabetic cardiomyopathy, an aspect reviewed in detail recently (Zheng et al, 2021). In this regard, data from animal models of type 1 diabetes have demonstrated that mitophagy and autophagy are decreased in different tissues, including the heart. However, controversy arises in the case of type 2 diabetes, as there are varying reports of the status of cardiac autophagy being unchanged, diminished, or increased.
However, functional mitophagy is known to have a protective role, since it is critical for maintaining cardiac function during diabetic cardiomyopathy, which is generally induced by a high-fat diet (HFD), as demonstrated in a murine model (Tong et al, 2019). Among the factors that may account for the aforementioned discrepancies are the different models and methods used to assess the state of autophagy/mitophagy. In addition, as type 2 diabetes is a disease of slow progression, the status of mitophagy can vary at its different stages.
Importantly, autophagic removal of mitochondria needs to be compensated by enhanced mitochondrial biogenesis to avoid an insufficient energy supply to cardiomyocytes. In addition, general autophagy and mitophagy do not always coincide; cardiac autophagic flux in mice fed an HFD was shown to peak at 6 weeks and subsequently decline, whereas mitophagy was activated for 2 months (Tong et al, 2019).
Mitophagy has also been proposed as an important process to be targeted in maladaptive left ventricular remodeling and heart failure after myocardial infarction in diabetes. The study in question, carried out in type 2 diabetic mice, found impaired mitophagy in cardiomyocytes from the peri-infarct zone of the left ventricle, and this was related to an enhanced extracellular and intracellular release of mtDNA-derived DAMP, which led to inflammasome hyperactivation, proinflammatory cytokine secretion (IL-18), and cell death (Durga Devi et al, 2017).
Mechanistically, it has been reported that Mst1/SIRT3/Parkin signaling contributes to the pathogenesis of diabetic cardiomyopathy. Similar to Mst1, mammalian sterile 20-like kinase 1, a key component of the “Hippo” signaling pathway, has been shown to specifically inhibit SIRT3 expression, thereby undermining Parkin-dependent mitophagy in different in vitro models and in experimental animals (Wang et al, 2019).
Another molecule involved in the process is BRD4 (bromodomain-containing protein 4), a member of the bromodomain and extra-terminal domain (BET) family of proteins. This protein binds to transcription factors and acetylated histones through its bromodomain domains. As a result, it can recruit transcriptional regulators, which is evidence of its fundamental role in the process. BRD4 was found to be upregulated in the hearts of mice with high-sugar diet-induced diabetic cardiomyopathy, in which PINK1/Parkin-mediated mitophagy decreased.
Consistent with these data, JQ1 inhibition of BRD4 was shown to prevent HFD-induced diabetic cardiomyopathy by restoring PINK1/Parkin-mediated mitophagy (Mu et al, 2020).
Finally, the negative effect of excess mitophagy has been documented in adipose-specific Trx2 KO mice, which develop hepatic insulin resistance and hyperglycemia. Trx2 deficiency induces excessive mitophagy, mitochondrial loss, and mitochondrial structural injury in WAT. Mechanistically, Trx2 deficiency causes overproduction of ROS, which promotes NF-κB–dependent accumulation of the autophagy receptor p62/SQSTM1, essential for the selective removal of defective mitochondria.
Importantly, in the study in question, an NF-κB inhibitor prevented NF-κB/p62–mediated mitophagy and ameliorated adipose dysmetabolism and the progression of type 2 diabetes. In support of these results, increased gene expression of PINK1, Parkin, and p62/SQSTM1 was observed in the visceral WAT of type 2 diabetic patients with hepatic steatosis compared with non-diabetic controls (He et al, 2021).
Taking into account all the information that we have shown, it should be noted that mitophagy can be a target process for the study of the development of diabetes and its control.
VII. Targeting Mitophagy
Due to the important role of mitophagy in type 2 diabetes, its pharmacological regulation would seem to be essential to both prevention and treatment. A few reports have been published on mitophagy modulators, and most available data come from preclinical studies. It is noteworthy that different natural compounds and pharmacological drugs have been tested, and some have been identified as activators of mitophagy.
This beneficial effect is mainly due to their mechanism of action, which points to their potential in the treatment of disorders in which mitophagy and mitochondrial dysfunction are leading players, such as cardiometabolic diseases and type 2 diabetes (Table 1).
Phytochemicals That Have Been Shown to Exhibit Beneficial Effects in Different Diseases While Showing Mitophagy-Activating Activities In Vivo
AD, Alzheimer's disease; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NLRP3, nucleotide oligomerization domain (NOD), leucine-rich repeat (LRR) and pyrin domain (PYD); Nrf2, nuclear factor erythroid 2-related factor 2.
Although displaying beneficial effects in the experimental context of other pathologies, some mitophagy-stimulating compounds are yet to be assessed in models of diabetes mellitus, as exemplified later. Moreover, these molecules have been studied in vivo after systemic administration, and so their tissue-specific actions have been difficult to determine. There is evidence that the antibiotic actinonin and urolithin A, a small compound generated by gut microflora from ellagitannins present in food sources such as berries and pomegranates, increase the expression of mitophagy proteins, including Parkin and PINK1, in the context of Alzheimer's disease (Fang et al, 2019).
Indeed, urolithin A has been studied in humans in clinical trials in which it was administered to healthy, sedentary elderly individuals (Andreux et al, 2019). It modulated plasma acylcarnitines and skeletal muscle mitochondrial gene expression indicative of improved mitochondrial and cellular health. Moreover, urolithin A increased muscle function in young rats and diminished age-related decline muscle function in mice (Ryu et al, 2016).
The capacity of salidroside, a phenylpropanoid glycoside extracted from Rhodiola to ameliorate intervertebral disk degeneration, could be due, at least in part, to the activation of Parkin-mediated mitophagy. It has been shown to increase Parkin expression, thereby activating mitophagy and lowering ROS levels in vitro (Zhang et al, 2018). Salidroside also protects dopaminergic neurons in a mouse model of Parkinson's disease (Li and Chen, 2019) and spinal cord I/R injury (Gu et al, 2020), in both cases by enhancing PINK1/Parkin-mediated mitophagy.
In addition, other compounds, such as macluraxanthone I and gerontoxanthone, which are isolated from Garcinia bracteata and exert protective effects against myocardial IR injury, have been shown to promote Parkin phosphorylation and PINK1 stabilization, which again promotes the mitophagic process through the PINK1/Parkin pathway in an in vitro model (Xiang et al, 2020). Similarly, iron chelators have also been proposed as mitophagy inducers, and they generally execute this role in highly glycolytic cells that do not require Parkin activation or PINK1 stabilization (Allen et al, 2013).
Recent studies have highlighted SIRT1 activators (such as resveratrol) as another therapeutic approach to modulate mitophagy. In this sense, polydatin, a resveratrol glycoside with a high antioxidant capacity, has been shown to attenuate high-fat-induced endothelial dysfunction and improve BNIP3-related mitophagy in vitro (Li et al, 2020a), and to protect against acute respiratory distress syndrome or sepsis-induced acute kidney injury through Parkin-dependent mitophagy (Gao et al, 2020; Li et al, 2019b).
Further, some studies have demonstrated that nicotinamide adenine dinucleotide (NAD+) precursors (NMN and NAM) stimulate mitophagy (reviewed in Guofeng et al, 2020), an effect that has been related to the control of mitochondrial fission and the SIRT1-dependent deacetylation of ATG8, ATG7, and ATG5.
In a similar vein, treatment with tomatidine extends the lifespan and improves mitochondrial and muscle function against age-associated deterioration in Caenorhabditis elegans. The proposed underlying mechanism is that tomatidine induces mild oxidative stress and activates SKN-1/Nrf2 and other cellular antioxidant responses, which induces mitophagy, an effect that also appears to take place in cultured human cells (Fang et al, 2017).
Melatonin is another promising molecule, as it can promote mitophagy in a murine model of atherosclerosis via activation of the SIRT3/FOXO3a/Parkin pathway (Ma et al, 2018b). This pathway is very important in diabetes, as the downregulation of mitophagy mediated by its suppression plays a crucial role in the development of diabetic cardiomyopathy (Yu et al, 2017). Melatonin also has beneficial effects in zebrafish suffering from Parkinson's-like disease; melatonin activated the flow of mitophagy, thus restoring the PINK1/Parkin/DJ-1/MUL1 network and impeding the development of the said condition (Diaz-Casado et al, 2016).
In the context of diabetes nephropathy, D-glucaric acid, a naturally occurring aldaric acid found in vegetables and fruit and used as a dietary supplement, has shown beneficial effects. D-glucarate can inhibit myoinositol oxygenase (MIOX), an essential enzyme that participates in the pathogenesis and development of diabetic nephropathy, whose activity catabolizes myoinositol to D-glucuronate, which then enters the pentose phosphate pathway.
In diabetic mice, D-glucarate was shown to decrease tubular damage and improve kidney function, while modulating oxidative stress, mitochondrial fragmentation, and apoptosis and restoring autophagy/mitophagy in renal tubular cells (Zhan et al, 2015).
A new type of molecule originally identified as antineoplastic is attracting the scientific community's interest as a potential therapeutic mitophagic tool. The so-called ubiquitin-mediated “autophagy-directed chimera” (AUTAC) enhances mitochondrial turnover and function and facilitates the clearance of fragmented organelle in cultured fibroblasts (Takahashi and Arimoto, 2020; Takahashi et al, 2019). Further, oleanolic and ursolic acids seem to exert potent antitumor activity that is highly dependent on their activation of mitophagy.
Treated cancer cells have been shown to exhibit altered mitochondrial morphology, increased PINK1 levels, and enhanced ROS production. However, in the study in question, only oleanolic acid triggered an increase in Parkin, whereas ursolic acid was also shown to activate mitophagy through the Akt/mTOR pathway (Castrejon-Jimenez et al, 2019). It is noteworthy that the antihelmintic drug niclosamide and its analogue AM85 (dibromsalan) robustly activate PINK1 in HeLa cells and primary cortical neurons, thus triggering the PINK1/Parkin mitophagy pathway.
The effects of these drugs are probably produced by indirect mechanisms such as a reversible reduction of the mitochondrial membrane potential (Barini et al, 2018). Moreover, over the past few years, new molecules that target mitophagy have been investigated, such as the deubiquitinating enzyme (DUB). DUB can preserve mitochondrial ubiquitination by inhibiting deubiquitination or enhancing mitochondrial ubiquitination, even in the absence or loss of Parkin or PINK1 function. It is noteworthy that one of the most promising targets is the USP family, in particular USP30, which is a cysteine protease located in the OMM that opposes Parkin-mediated mitophagy (Bingol et al, 2014). In addition, there are different natural compounds and small molecules that may act as inhibitors, although they are yet to be tested in humans (Kluge et al, 2018).
Regarding type 2 diabetes more specifically, several molecules have been highlighted, both natural and synthetic. In vitro studies have highlighted that Ginseng-Sanqi-Chuanxiong (GSC) extracts improve diabetes-induced endothelial cell senescence through the upregulation of mitophagy via the AMPK pathway (Wang et al, 2020). Similarly, notoginsenoside R1 (NGR1), a saponin extracted from Panax notoginseng, has been described as a potential therapeutic strategy in the prevention and treatment of diabetic complications (including retinopathy), an action based principally on its capacity to increase mitophagy and enhance the levels of Parkin and PINK1, and the ratio of LC3-II/LC3-I in the retinas of db/db mice (Zhou et al, 2019b).
Another molecule that enhances mitophagy while producing beneficial effects in the context of type 2 diabetes is elamipretide (SS-31), an SS tetrapeptide that promotes mitophagosome formation in INS1 β cells (Petcherski et al, 2018). It is known that the main mechanism by which SS-31/elamipretide improves mitochondrial function is the decrease of ROS production and recovery of mitochondrial function and ATP synthesis by direct interaction of SS-31 with cardiolipin.
However, the protective effect of elamipretide goes beyond this action, as it prevents mitochondrial fragmentation and hyperpolarization of INS1 β cells induced on exposure to excess nutrients. In the same line, by acting on mitochondrial ROS production, MitoQ has been shown to ameliorate tubular injury in diabetic kidney disease through mitophagy via Nrf2/PINK1 (Xiao et al, 2017b) and myocardial I/R injury by enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats (Ji et al, 2022).
In addition, the screening of 15,000 small molecules in INS1 β cells stably expressing MitoTimer has led to the discovery of two chemically related benzothiophene derivatives (MWP00839 and SPB08007) that are capable of increasing basal mitochondrial turnover through enhanced mitophagy without triggering the loss of mitochondrial membrane potential (Cerqueira et al, 2020).
VIII. Conclusions
Type 2 diabetes is a highly prevalent condition leading to multiple chronic comorbidities. Therefore, the discovery of targets and new treatments is essential. More specifically, type 2 diabetes is highly related to mitochondrial impairment and damage, and so it is vital to understand the molecular pathways involved. Similarly, it is necessary to develop new therapeutic strategies to modulate mitochondrial homeostasis, including biogenesis, ROS production, recycling, and mitochondrial metabolism.
Autophagy is an essential regulatory signaling pathway in type 2 diabetes and its comorbidities, as it regulates insulin secretion, lipid metabolism, and glucose and affects different tissues and cells, including, besides β cells, adipose tissue, skeletal muscle, and the liver. There is growing evidence that autophagy can promote β cell survival by enabling adaptive responses to mitigate or prevent the deleterious effects of oxidative stress, mitochondrial dysfunction, and ER stress.
Identifying the mechanisms that regulate β cell autophagy in these pathological environments needs to be a major research goal, so as to better understand β cell function and survival and develop strategies that directly target β cells.
Mitochondria are a promising target in the pathogenesis of different metabolic conditions, including type 2 diabetes. Mitophagy is responsible for maintaining a healthy and functional population of mitochondria and inhibiting the mismatching of O2 and mitochondria number. In this way, mitochondrial quality control depends on a stable mitophagy. Indeed, impairment of mitophagy can interfere with basic tissue metabolic patterns. In this sense, regulators such as FUNDC1, BINP3, PINK1, and Parkin have been identified as critical players in the regulation of mitophagy.
Although our knowledge about the underlying mechanisms of type 2 diabetes has greatly improved in recent years, many of the key issues covered in this review require further clarification and are at present the focus of intense research. Restoring mitochondrial homeostasis by controlling these key factors has become a fundamental approach. Drug advances based on the targeting of these candidates is sure to accelerate the prevention and treatment of mitochondria-related metabolic disorders.
Footnotes
Acknowledgments
The authors thank Brian Normanly (University of Valencia-CIBERehd) for his editorial assistance. Servier Medical Art was used to create the high-quality images.
Authors' Contributions
N.A.: Conceptualization; writing—original draft. T.V.: Conceptualization; writing—original draft. J.M.: Conceptualization. M.R.: Writing—review and editing. V.M.V.: Writing—review and editing.
Author Disclosure Statement
The authors have nothing to disclose.
Funding Information
This research was supported by the European Regional Development Fund (ERDF “A way to build Europe”); FISABIO (Foundation for the Promotion of Health and Biomedical Research in the Valencian Region, grants UGP15-220 and UGP-21-236); Generalitat Valenciana (grants PROMETEO/2019/027 and AICO/2021/017); and Spanish Ministry of Science and Innovation (grants CIBERehd CB06/04/0071, PI22/00424, PI22/1009, PI19/0437, and PI19/00838 from Carlos III Health Institute, and grant PID2021-127945OB-I00 funded by MCIN/AEI/10.13.039/501100011033/and by FEDER).