
Abstract
Significance:
Immune cell therapy involves the administration of immune cells into patients, and it has emerged as one of the most common type of immunotherapy for cancer treatment. Knowledge on the biology and metabolism of the adoptively transferred immune cells and the metabolic requirements of different cell types in the tumor is fundamental for the development of immune cell therapy with higher efficacy.
Recent Advances:
Adoptive T cell therapy has been shown to be effective in limited types of cancer. Different types and generations of adoptive T cell therapies have evolved in the recent decade. This review covers the basic principles and development of these therapies in cancer treatment.
Critical Issues:
Our review provides an overview on the basic concepts on T cell metabolism and highlights the metabolic requirements of T and adoptively transferred T cells.
Future Directions:
Integrating the knowledge just cited will facilitate the development of strategies to maximize the expansion of adoptively transferred T cells ex vivo and in vivo and to promote their durability and antitumor effects. Antioxid. Redox Signal. 37, 1303–1324.
Introduction
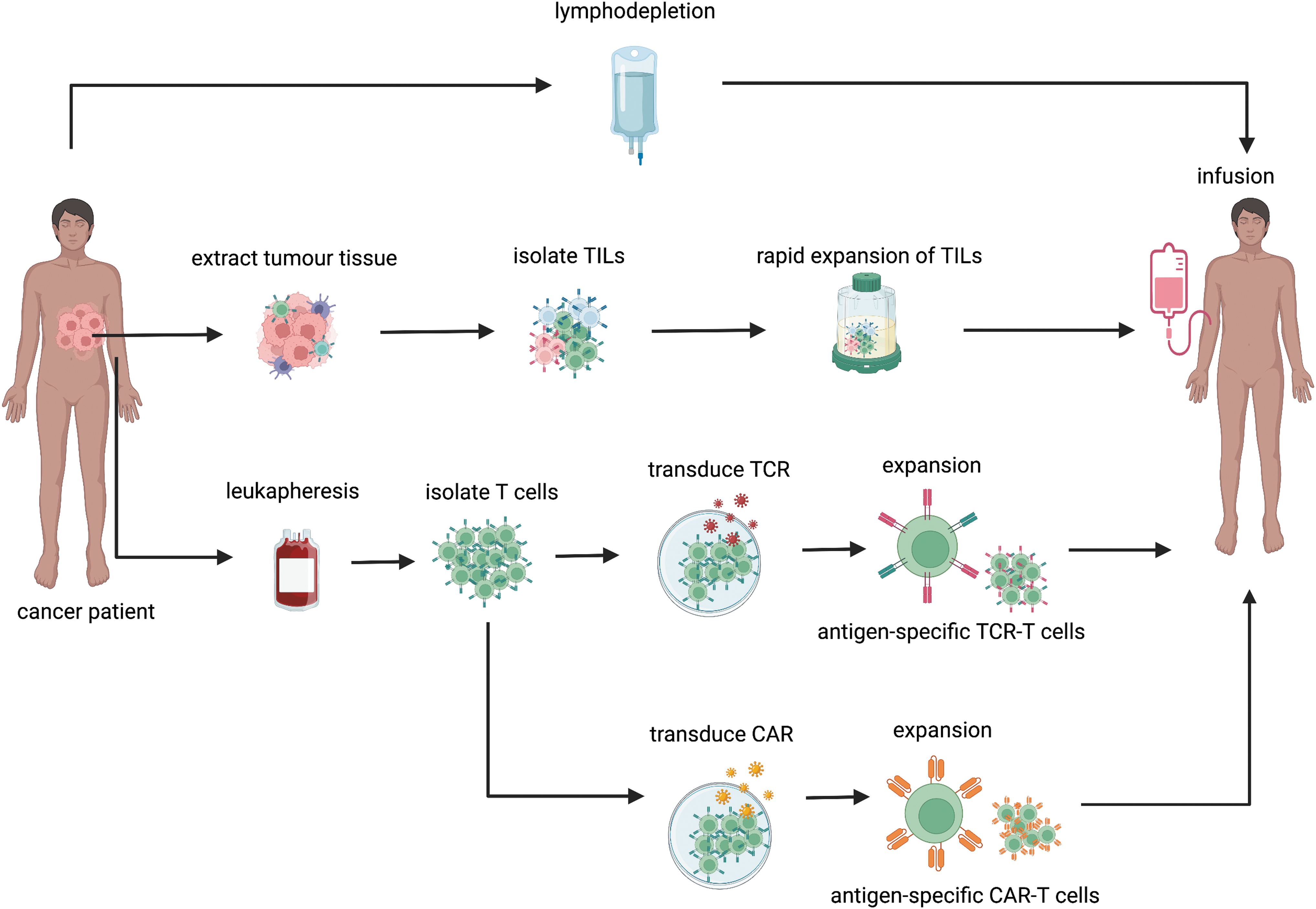
Adoptive cell therapy (ACT), also known as immune cell therapies, is a form of treatment that involves the transfer of antitumor lymphocytes into cancer patients. Lymphocytes are activated and expanded ex vivo prior to infusion into patients. With the advancement of genetic engineering, these lymphocytes can be modified with improved functionality prior to transfer. There are currently three different types of ACT. These are tumor infiltrating lymphocytes (TIL)-based therapy, engineered T cells receptor (TCR)-based therapy, and chimeric antigen receptors (CAR) T therapy, which differ by their production process and mechanism of actions (June et al, 2018).
The potential of ACT as cancer therapy has been demonstrated as early as the year 1964 by Delorme in an experiment that involves transferring allogeneic lymphocytes in rats. This experiment demonstrated that transferred lymphocytes could delay the growth of fibrosarcoma (Delorme and Alexander, 1964). Later, clinical evidence showed that stem cell transplants or infusion of immune cells in hematological cancer patients can result in cure in some patients (Collins et al, 1997; Thomas et al, 1957). These experiments do not only highlight the critical role of immune cells in tumor control, but they also open up new avenues for ACT development.
It is now known that tumor elimination is mediated primarily by cytotoxic immune cells, specifically CD8+ T cells, which kill tumor cells via cytotoxic proteins and induced cell death (Vesely et al, 2011). In hematological cancer, clinical studies have shown that infusion of allogeneic T cells can induce complete remissions in some patients with chronic myeloid leukemia (Collins et al, 1997). In solid tumors, a high level of tumor infiltrated CD8+ T functions as a prognostic biomarker for some cancer types (Fridman et al, 2017; Idos et al, 2020; James et al, 2017; Kmiecik et al, 2013; Kolberg-Liedtke et al, 2020; Piersma et al, 2007; Zhang et al, 2019). Bioinformatics and histology studies further revealed the infiltration tumor antigen-reactive T cells and their association with clinical outcomes (Fridman et al, 2017; McGranahan et al, 2016).
The level of intratumoral CD8+ T cells was shown to be associated with positive clinical outcomes in cancer patients (e.g., breast cancer (Zgura et al, 2018), colorectal cancer (Galon et al, 2006), and ovarian cancer (Hwang et al, 2012; Zhang et al, 2003). In addition, high infiltration of CD4+ T cells, γδ T cells, and natural killer (NK) cells also correlates with favorable outcomes in some cancer types (Hwang et al, 2012; Zgura et al, 2018). Together, these findings highlight two important ideas that formed the principles of ACT. First, tumor tissues consist of immune cells with tumor-killing abilities. Second, repopulation of the tumor microenvironment (TME) with tumor reactive immune cells can potentially result in tumor elimination.
The ACT is the most rapidly expanding sector of cancer immunotherapy. More than 600 clinical trials for ACT were reported in the year 2020 (Yu et al, 2020). Among the different types of ACT, CAR T therapy showed the greatest number of registered trials, followed by TCR therapy and TIL-based therapy (Yu et al, 2020). Remarkable results have been reported for lymphoma. However, the efficacy of ACT in solid tumors remains disappointing (Yu et al, 2020). Many factors have been shown to contribute to the lack of ACT efficacies in solid tumors. This includes tumor heterogeneity (Jamal-Hanjani et al, 2017; Rosenthal et al, 2019), immunosuppressive TME (Fridman et al, 2017; Thorsson et al, 2018), and disorganized blood vessel networks in solid TME.
A significant research effort is currently underway to unravel the factors contributing to the immunosuppressive TME. The TME is a complex network of cells with dynamic interaction that involve growth factors, cytokines, metabolites, and receptor-ligand binding. Many studies have shown that tumor metabolism is involved in the regulation of TME (Elia and Haigis, 2021; Kouidhi et al, 2017; Molon et al, 2016). Tumors harbor dysregulated metabolic pathways that do not only promote tumor development but also promote immune evasions (Hanahan and Weinberg, 2011). Hence, understanding how tumor metabolism contributes to immunosuppressive TME can potentially elucidate strategies to improve ACT.
In this review, we will outline three main modalities of ACT, which are TIL-based therapy, TCR-based therapy, and CAR T therapy. We will discuss their mechanism of actions and clinical efficacies in solid tumors. We focus particularly on T cells as they are the most widely used immune cell type being developed for ACT treatment. We will provide an overview of T cell metabolism and discuss how it is dysregulated in the solid TME. We also discuss the promising strategies that are being investigated to ACT efficacies through metabolic modulation.
ACT Modalities for Solid Tumors
TIL-based therapy
TIL-based therapy is a form of ACT that involves the infusion of autologous or allogeneic TIL isolated from tumor tissue fragments into cancer patients (Topalian et al, 1987) (Fig. 1). The TILs are a heterogeneous population of immune cells that are present in the TME. It consists of mainly CD8+ T cells, CD4+ T cells, and NK cells. Among them, CD8+ T cells are the most abundant (Fridman et al, 2017). In preparation of TIL-based therapy, patients-derived TILs are activated, expanded, and selected for their tumor reactivity ex vivo (Rosenberg et al, 1994; Topalian et al, 1987; Yossef et al, 2018). It is anticipated that transferred TILs that consist of tumor reactive T cells would then migrate to the tumor sites and eliminate tumor cells.
The efficacies of TIL-based therapy are dependent on the ability of transferred T cells to recognize and mount antitumor response on encountering tumor cells. T cells recognize human leukocyte antigen (HLA)-bound tumor antigens via TCR. It is a heterodimeric protein composed of an α-chain and a β-chain and is noncovalently associated with the CD3 signaling complex (Hwang et al, 2020). The activation of TCR complex and co-stimulatory molecules leads to T cell activation, which is mediated by a range of downstream signaling pathways such as extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), mammalian target of rapamycin (mTOR), and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). This results in clonal expansion and activation of the effector function in T cells (Hwang et al, 2020).
The first positive clinical response of TIL-based therapy for solid cancer was observed in melanoma in a study conducted by Rosenberg and colleagues. The clinical trial reported an objective response rate (ORR) of up to 72% in melanoma patients who received the non-myeloablative lymphodepletion regime followed by TIL therapy and high-dose interleukin (IL)-2. Overall, 10%–20% of these patients achieved complete remission and 40% of the patients achieved durable clinical responses (Rosenberg et al, 2011). In line with this, a meta-analysis has also reported an estimated ORR of 41% in advanced cutaneous melanoma patients obtained from 13 studies (n = 410) (Dafni et al, 2019). In addition to melanoma, other solid tumors such as renal, breast, and cervical cancer and cholangiocarcinoma also showed clinical benefits from TIL-based therapy (Andersen et al, 2018; Lee et al, 2017). However, they reported a significantly lower ORR and durability as compared with cutaneous melanoma patients.
For instance, a phase II TIL-based therapy study in 21 uveal melanoma patients reported an ORR of 35% (Chandran et al, 2017). In metastatic cervical cancer, three out of nine TIL-based therapy-treated patients showed tumor regression; among these, two showed a durable response (Stevanović et al, 2015).
It is unclear why TIL-based therapy is significantly more effective in melanoma as compared with other form of solid tumors. One of the hypotheses is that melanoma tends to harbor higher tumor mutation burden (TMB) as compared with other tumor types. High TMB can lead to higher expression of tumor antigens, which can be recognized by T cells, and potentially drive the infiltration and expansion of T cells of unique tumor antigen specificity (Chalmers et al, 2017; Schumacher and Schreiber, 2015). Given that the presence of tumor-reactive T cells is a prerequisite for TIL-based therapy, it is hypothesized that tumors with higher TMB and immunogenicity will show a better response to the treatment as compared with tumors that are poorly immunogenic.
Tumor immunogenicity and antitumor activities can be influenced by the level of tumor antigens. Many tumor antigens have been identified to date. For instance, the melanoma antigen family (MAGE)A-1-4 is highly expressed on metastatic melanoma (∼70%) (Valmori et al, 1997), carcinoembryonic antigen (CEA), and New York-esophageal squamous cell carcinoma-1 (NY-ESO-1) are highly expressed in colorectal cancer and synovial sarcoma, respectively (Lai et al, 2012; Parkhurst et al, 2011). In addition, oncoviral antigens are also commonly expressed by virally associated tumors (Stevanović et al, 2015). Recognition of tumor antigens leads to the clonal expansion of antigen-specific T cells.
Hence, tumor antigen-specific T cells are detectable in cancer patients, which is indicative of the presence of antitumor response (Abd Hamid et al, 2020). However, a study also showed that high antigen exposure can drive T cells exhaustion, which then undermines the function of these antitumor T cells (Ghorani et al, 2020).
The efficacy of ACT is limited in tumors that are poorly immunogenic. For instance, breast, pancreatic, and renal cancer tumor tissues were shown to express low levels of tumor antigens, which are then likely to contain a low level of tumor-specific TILs (Vareki, 2018). Even for immunogenic tissues, only a small fraction of TILs (∼30%) are tumor reactive (Kumar et al, 2021). This poses a challenge to TIL-based therapy, as it requires sufficient TILs to be isolated from a patient's tumor tissues. Although the ex vivo selection process was able to enrich the tumor-reactive T cells population, the process remains too time-consuming for routine clinical application. The generation of tumor antigen-specific TCR and CAR T cells using the genetic engineering method aims at filling this gap. This will be discussed in greater detail in the subsequent sections.
TCR-based therapy
Engineered TCR was first performed in mice in early 1986. A murine exogenous alpha and beta gene pair was successfully transferred into other murine cytotoxic T cells, and it equipped the recipient cells with new TCR specificity (Dembić et al, 1986). This was later tested on human immortalized T cells and subsequently on primary human T cells with exogenous melanoma antigen recognized by T cells (MART)-1-specific TCR that was cloned from melanoma TILs. The expression of exogeneous tumor antigen-specific TCR T cells was shown to confer cytolytic activities in preclinical studies, which is suggestive of their potential as cancer treatment (Orentas et al, 2001; Stanislawski et al, 2001).
The first TCR-based therapy was used in the clinic in metastatic melanoma patients (Morgan et al, 2006). Peripheral blood lymphocytes collected from melanoma patients were modified to express transgenic TCR that targets HLA2-A2-restricted MART-1. MART-1 is a lineage-specific antigen expressed on melanoma cells. These cells were then infused into patients with metastatic melanoma after lymphodepletion. Although the response rate was low (2 out of 15 patients or 13%), MART-1-specific T cells were shown to persist for at least 2 months. Remarkably, two responding patients showed long-term engraftment of MART-1-specific T cells for up to 2 years (Morgan et al, 2006). To improve the efficacy of TCR-based therapy, MART-1-specific TCR of a higher avidity was generated. This results in an improved response rate from 14% to 30% in 36 treated metastatic melanoma patients in subsequent clinical trials (Johnson et al, 2009).
Apart from MART-1-specific TCR, modified TCR toward various tumor-associated antigens (TAA) have also been developed. As of June 2021, more than a hundred clinical trials of TCR therapy of various antigen specificities have been identified in
Melanoma is the most prevalent studied entity for TCR-based therapy and was reported to achieve an ORR of 33% (Oppermans et al, 2020). Clinical trials in other solid tumors report varying results. For instance, NY-ESO-1-specific T cells in 42 patients with synovial sarcoma reported an ORR of 35.7%. Moreover, T cell products with engineered TCR targeting human papillomavirus (HPV)-16 E7 were also shown to induce extensive tumor regression in 6 out of 12 patients (50%) with metastatic HPV-associated epithelial cancers (Nagarsheth et al, 2021).
The benefits reported for TCR-based therapy in melanoma is considerably less than TIL-based therapy (72% vs. 30%) (Johnson et al, 2009; Rosenberg et al, 2011). This could be due to the presence of polyclonal TIL populations obtained from tumor tissues of patients, which allows diverse tumor antigens to be targeted. In TCR-based therapy, tumor-specific T cells are largely homogeneous as they are expanded from a T cell clone that have been engineered with tumor-specific TCR. However, as our knowledge on the antitumor response of T cells improves, modification can be made accordingly to improve the efficacy of TCR-based therapy.
This includes the improvement of TCR avidity, as discussed earlier (Johnson et al, 2009). Unlike TIL-based therapy that obtained T cells from tumor tissues, T cells are usually obtained from the peripheral blood of patients for TCR-based therapy. This allows patients with a low TIL count to benefit from the treatment (Oppermans et al, 2020).
One of the major drawbacks of TIL and TCR-based therapy is the need to identify and generate antigen-specific TCR T cells for different HLA. As mentioned, TCR recognition is HLA-restricted. Given the polymorphism of this gene in the human population, the efforts to produce TIL and TCR-based therapy will have to be customised for patients' HLA, which will be costly and labour intensive. Today, the most common HLA being investigated for TCR-based treatment in clinical trials is HLA-A*0201, which is found in 50% of the Caucasian population (Gaissmaier et al, 2020).
This shows that the majority of patients who do not harbor this specific HLA haplotype are being excluded from the treatment. Moreover, the downregulation of HLA also appeared to be a common tumor resistance mechanism. A study showed that HLA class 1 is downregulated or deficient in 40%–90% of patients (Cornel et al, 2020). Genomic studies of tumors from patients with HPV-associated epithelial cancer had also identified antigen presentation as a primary resistance mechanism of TCR-based therapy (Nagarsheth et al, 2021). In addition, loss of heterozygosity of HLA also appeared to be a common immune evasion mechanism (Rosenthal et al, 2019).
CAR-T therapy
Since the first conception of CARs in the year 1989 by Zelig Eshhar, the design of CAR constructs has expanded dramatically (June et al, 2018). A typical CAR construct consists of four essential components. This includes the extracellular antigen-recognizing domain, hinge, transmembrane domain, and intracellular signaling domain (Larson and Maus, 2021; Stoiber et al, 2019). The antigen-binding domain is composed of a single-chain variable fragment (scFv), which is derived from antibodies against tumor targets of interest. The ScFv consists of a variable light (VL) and variable heavy (VH) chain joined by a linker that is commonly derived from repeated glycine and serine residues (Stoiber et al, 2019).
Other engineered and artificial domains composed of binding moieties such as natural ligands or artificial protein-binding constructs have also been developed (Larson and Maus, 2021). Unlike TCR, CAR recognizes tumor antigen directly, bypassing the requirements of peptide processing and HLA presentation of tumor cells. It can recognize proteins, glycolipids, glycoproteins, and carbohydrates that are expressed extracellularly. A flexible hinge connects scFv to the transmembrane domain that spans across the lipid bilayer and increases the conformational range of the antigen-binding domain (Stoiber et al, 2019).
The hinge is then connected to the intracellular domain that mediates CAR signaling. The intracellular domain consists of TCR-derived CD3ζ and the costimulatory domain (Stoiber et al, 2019). The activation of CD3ζ leads to the recruitment of zap-70, subsequently phosphorylating the transmembrane protein LAT. Phosphorylated LAT then initiates the activation of pathways involved in T cell activation, differentiation, proliferation, and cytokine release.
To date, four generations of CAR design have been developed that differ mainly by the composition of costimulatory domains (e.g., 41BB/CD28) (Fig. 2) (Stoiber et al, 2019). Initial clinical trials using first-generation CAR designs in patients were disappointing. The lack of the co-stimulatory domain in the first-generation CAR construct endows CAR-transduced T cells with limited persistence and function in vivo (June et al, 2018). This observation has led to the design of a second-generation CAR that incorporates 41BB or the CD28 co-stimulatory domain and thus results in the remarkable success of CD19-targeting CAR-T cell therapy (Stoiber et al, 2019). CD19 is a lineage marker of B cells that is also highly expressed in B cell malignancies.
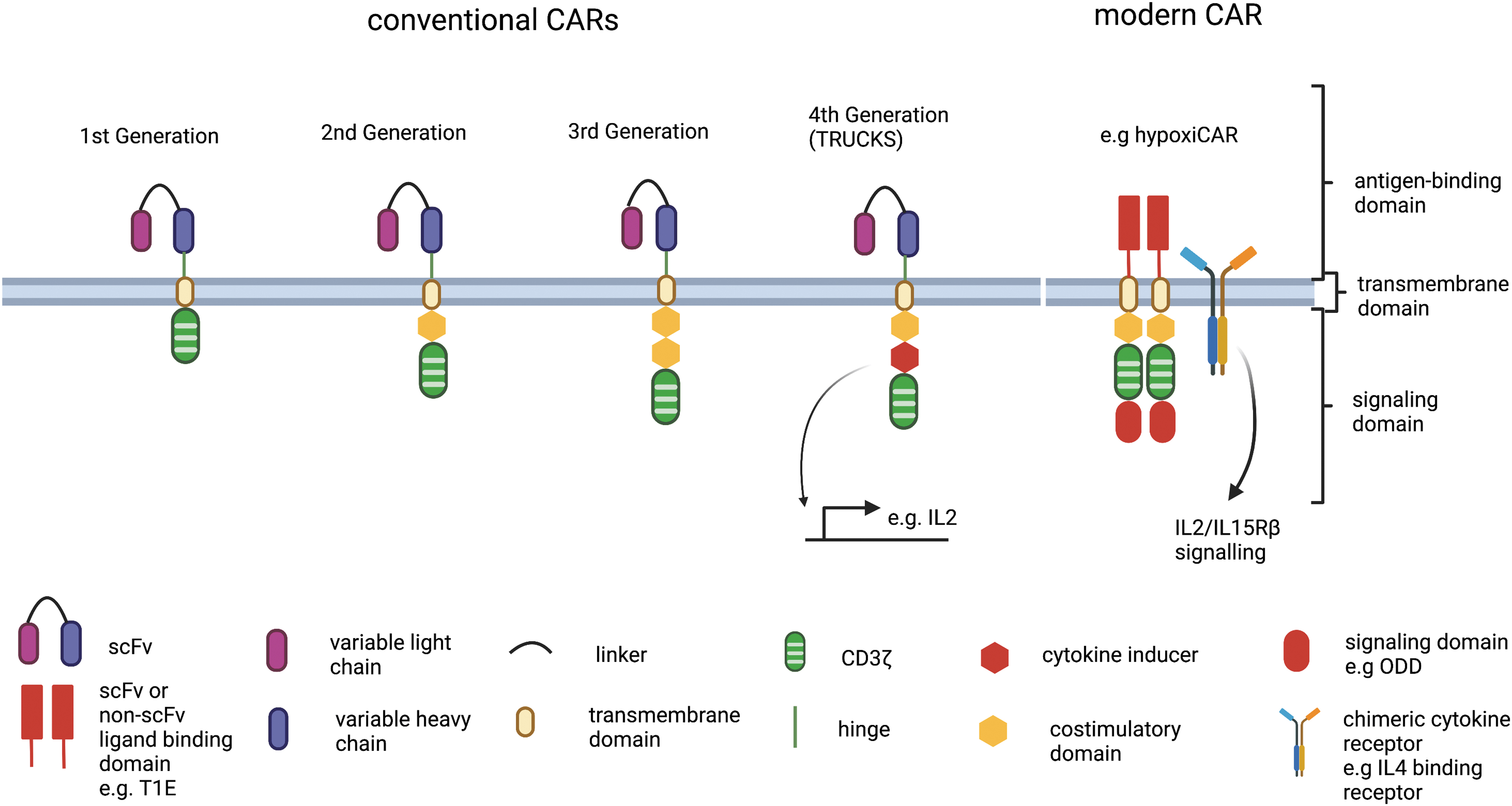
Patients with B cell malignancies have been successfully treated with anti-CD19 CAR-T therapy (Brentjens et al, 2013; Grupp et al, 2013; Kochenderfer et al, 2010). This leads to FDA approval of two CAR therapies that target CD19 for leukemia: KYMRIAH and YERCARTA. The third generation of CARs consists of additional co-stimulatory domains in the construct, potentially leading to enhance signaling. The fourth generation of CARs, also known as T cells redirected for universal cytokine killing (TRUCKS), are engineered with enhanced function to produce additional protein molecules such as cytokines and receptors (Stoiber et al, 2019). Despite the improvements, the second generation of CAR remains the most popular design in clinical trials.
It remains challenging to assess the efficacies of CAR-T therapy in solid tumors given the wide range of CAR designs and different protocols that are used in the preparation of CAR T cells for clinical trials. A comprehensive analysis of clinical trials on CAR-T therapy conducted by Schaft (2020) showed that clinical benefits remained low in patients with solid tumors. Of the 42 clinical trials registered at clinicaltrials.gov, 375, patients with solid tumors were treated and clinical benefits were observed in less than 10% of these patients. They are treated mainly with autologous T cells (Schaft, 2020).
The common targets of CARs include epidermal growth factor receptor (EGFR), natural killer group 2D receptor (NKG2D) ligands, human epidermal growth factor receptor 2 (HER2), B7-H3, mucin 1 (MUC1), and CEA. These targets are not only commonly expressed at a low level on the cell surface but also significantly enriched and sometimes mutated in tumor cells (Schaft, 2020). For instance, EGFR and HER2 belong to epidermal growth factor family receptor tyrosine kinases and are known to play a role in cell division. It is highly expressed in many cancers such as non-small cell lung cancer (NSCLC), colorectal cancer, and breast cancer. Clinical benefits were observed in 15 out of 19 patients with advanced biliary tract cancers treated with EGFR-specific CAR T therapy (Guo et al, 2018). Objective response and persistence of EGFR-CAR was also observed in 2 out of 11 patients with NSCLC (Feng et al, 2016). However, CAR T cells that target these antigens can also result in on-target off-tumor toxicities, which can be fatal (Schaft, 2020).
The design of CAR formats continues to expand as our knowledge about tumor and T cell biology improves. A new generation of CARs is being developed with the aim of improving antitumor response, persistence, and reducing toxicities. These CARs incorporate distinct signaling domains, in addition to the conventional CD3ζ and co-stimulatory domains. For instance, the incorporation of hypoxia-responsive element and the oxygen-dependent degradation domain of hypoxia-inducible factor (HIF)-1α in the CAR construct permits the control of CAR expression (Kosti et al, 2021). Under normoxic condition, CARs would be degraded through ubiquitination whereas under hypoxic condition, activation of HIF-1α leads to the expression of CARs. These CAR T cells do not only show improved ability to kill but also reduced off-tumor toxicities in vivo (Kosti et al, 2021).
There is also a focus on creating universal CAR that can potentially be used to treat different cancer types and patients. The strategies include the use of allogeneic T cells, γδ T cells, or NK cells and also the identification of tumor-specific antigen that is widely expressed across different patients and cancer types (Depil et al, 2020; Reindl et al, 2020). For instance, one of the promising universal targets for CAR is NKG2D-ligands, which are commonly upregulated on stressed, infected, and transformed cells but absent from healthy tissues (Liu et al, 2019). Many NKG2D CARs are being developed and tested in preclinical and clinical studies (Curio et al, 2021; Prenen et al, 2021).
Combinations of NKG2D CAR with chemotherapy were shown to result in a partial response in three out of 15 patients with advanced colorectal cancer and progression-free survival of 3.9 months (Prenen et al, 2021). Its efficacies in other solid tumors such as liver metastasis, nasopharyngeal cancer, prostate and gastric cancer, and glioblastoma are also being investigated (Curio et al, 2021).
Unlike hematological cancer, the clinical efficacies of CAR T cells and other form of ACT in the treatment of solid cancer remain poor. One of the major roadblocks for ACT in treating solid tumors is the lack of tumor-specific antigens. Many of the CARs and TCRs that have been developed so far target TAA, which are also expressed at a low level on healthy tissues (Morgan et al, 2013; Neelapu et al, 2018). This leads to the issues of on-target off-tumor toxicities (Morgan et al, 2013; Neelapu et al, 2018). In comparison to TCR-based therapy, the use of antibody's antigen-recognition machinery in CARs limits its targets to extracellular tumor antigens. It is estimated that only 10% of tumor antigens expressed extracellularly that are targetable by CARs (Garber, 2018).
Hence, TCR-based therapy appeared to be a more promising therapy for solid tumors as it is able to recognize both surface and intracellular peptides when presented on HLA (Harris and Kranz, 2016). In addition, it has also been shown that the amount of antigens required to activate T cells via TCR is also significantly lower as compared with CAR-T cells (1–50/cell vs. >103/cell) and indicates that TCR-based therapy will be beneficial for solid tumors that express a low level of tumor antigens (Gaissmaier et al, 2020; Harris and Kranz, 2016) (Table 1). Another important challenge to the efficacies of ACT is the immunosuppressive TME in solid tumors.
Comparison Between T Cells' Receptor-Based Therapy and Chimeric Antigen Receptor T Therapy
scFv, single-chain variable fragment; HLA, human leukocyte antigen; ITAM, immunoreceptor tyrosine-based activation motif; TCR, T cell receptor; CAR, chimeric antigen receptor.
Immunosuppressive factors and cells in the TME can hamper the infiltration and antitumor response of transferred T cells and thus leads to ACT resistance. One of the resistant mechanisms is through the metabolic modulation of T cells. This will be discussed in greater detail in the subsequent sections.
T cell metabolism in a nutshell
Cellular metabolism involves the production of energy and macromolecules through complex catabolic and anabolic processes that support cellular function, proliferation, and growth. In T cells, metabolic rewiring occurs as they progress through different developmental stage and differentiation (Zhang and Romero, 2018) (Fig. 3). Normal cells process glucose into pyruvate, which is converted into acetyl-CoA, which is metabolized by tricarboxylic acid (TCA) cycle in the mitochondria. NADH, generated by glycolysis and TCA cycle, donates electrons to start the electron transfer at the electron transport chain (ETC) and finally to oxygen molecules.

This process generates energy, ATP. This metabolic mode is referred to as oxidative phosphorylation (OXPHOS). Reactive oxygen species (ROS) are generated during the electron transfer as side products. When oxygen level declines, due to the lack of electron recipient at the ETC, normal cells would alternatively convert pyruvate into lactate by lactate dehydrogenase. Highly proliferating cells such as cancer cells metabolize glucose into lactate, despite the presence of oxygen to maximize the channeling of metabolites into anabolic reactions, a process called aerobic glycolysis. OXPHOS is an efficient ATP-generating metabolic route whereas aerobic glycolysis is relatively a less efficient ATP-generating metabolic route; however, aerobic glycolysis allows the branching of glycolytic metabolites into the pentose phosphate pathway and serine synthesis pathway or one carbon metabolism through which generate a great number of metabolic intermediates to sustain the nutrient demand that supports rapid cell division and proliferation.
Interestingly, growing studies confirmed that almost all actively proliferating cells, including activated immune cells such as activated T cells that need to be expanded quickly, adopt aerobic glycolysis. Aerobic glycolysis is coupled with increased glutamine uptake and metabolism. Aerobic glycolysis allows the generation of macromolecules, including lipid, nucleotides, amino acids, and antioxidants (Buck et al, 2017; Zhang and Romero, 2018).
Naive T cells (Tn) require a relatively low level of nutrient demand to maintain their quiescent state. They mostly utilize OXPHOS to generate ATP (MacIver et al, 2013). As T cells differentiate into effector T cells (Teff), nutrient and energy are required to promote rapid proliferation and secretion of cytotoxic molecules and cytokines. Increases in nutrient demand are met by enhanced glycolysis, which allows glucose to be metabolized by aerobic glycolysis to generate anabolic precursors and antioxidants. After antigen clearance, a small fraction of Teff differentiates into memory T cells (Tmem). Similar to Tn, Tmem has a metabolically low state, which is maintained by OXPHOS and fatty acid oxidation (FAO) (Zhang and Romero, 2018). This shows that metabolism is intricately linked to the differentiation process of T cells.
Three signals are known to drive the differentiation of Tn into Teff. This includes the activation of the TCR complex, costimulatory molecules, and cytokine receptors (Fig. 4). Together, they upregulate anabolic activities through the activation of phosphatidylinositol 3-kinase (Pl3K)/AKT/mTOR and rat sarcoma protein (Ras)/MAPK/ERK, as well as downregulate catabolic activities through 5’ adenosine monophosphate-activated protein kinase (AMPK) signaling (Shyer et al, 2020). MTOR functions as an energy sensor and a key regulator of T cell metabolic programs. The activation of mTOR1 is required for the expression of glucose transporter 1 (GLUT1) and glycolytic enzymes. It mediates the activation of transcription factors (TFs): c-MYC and HIF-1α, which are known to regulate metabolic genes (Finlay et al, 2012; Shi et al, 2011). In addition, mTOR activation also opposes catabolic activities through inhibiting AMPK signaling (Herzig and Shaw, 2018). Overall, these enhance glucose import through increased GLUT1 expression, glycolysis, amino acid metabolism, and fatty acid synthesis (FAS) (Shyer et al, 2020). T cells with mTOR1 deficiency have been shown to exhibit reduced glycolysis, defective lipid synthesis, and OXPHOS in vitro, which are associated with poor cytotoxic function in effector T cells (Yang et al, 2013).

T cells with MYC deficiency further showed reduced proliferative capacity, which was associated with low GLUT1 expression, reduced nutrient uptake, glycolytic activities, and glutaminolysis (Gao et al, 2009; Wang et al, 2011). Moreover, the lack of HIF-1α activities also impairs the development of Th17 and CD8+ T cells whereas they promote the development of regulatory T cells (Tregs) (Finlay et al, 2012; Shi et al, 2011). Tregs display a distinct metabolic profile. In addition to glycolysis, Tregs utilize FAS and FAO to meet their metabolic demands. This allows them to prevail in the oxygen and nutrient-limiting environment (Pacella et al, 2018).
Although glycolysis is known to promote Teff differentiation, heightened glycolysis can promote differentiation of terminal Teff (Kawalekar et al, 2016; Sukumar et al, 2013). This is presumably a mechanism to limit T cell response and prevent excessive T cell effector function that can potentially cause host damage. By contrast, heightened glycolysis is required for the differentiation of CD4 T cells into helper T cells (Th)-1, Th2, Th9, Th17, and follicular T helper cells (Tfh) and moderate glycolytic activities are required to promote memory T cell differentiation (Pacella et al, 2018).
This shows that T cell differentiation is finely regulated by the level of glycolytic activities. Although the mechanisms remain unclear, mitochondria fitness might contribute to the regulation of metabolic activities in T cells. Buck et al (2016) showed that effector T cells underwent mitochondrial fission. This led to fragmented architecture of mitochondria with reduced metabolic fitness and increased ROS production. By contrast, memory T cells were shown to maintain a fused morphology with tight cristae, which was important to facilitate TCA cycle activities. By altering the mitochondria cristae morphology, the study showed that the state of T cells could also be modulated (Sukumar et al, 2013).
Tmem can persist in the long term and be reactivated into effector T cells on subsequent encounter of specific antigens. They are metabolically primed but rely on mitochondrial oxidation and FAO to maintain its quiescent state (O'Sullivan et al, 2014). A study has shown that these cells harbor substantial mitochondrial mass and spare respiratory capacity (SRC) (van der Windt et al, 2012). Moreover, AMPK is also known to play a key role in promoting Tmem differentiation. It regulates nutrient import, glycolysis, glutamine and lipid metabolism, and ATP production, which are critical in Tmem formation (Blagih et al, 2015).
It also functions as the energy sensor and is activated in response to low cellular energy store in the cells (Blagih et al, 2015). The activation of AMPK inhibits mTOR1, leading to increased catabolism and reduced anabolism in the cell, which allows cellular energy to be restored (Herzig and Shaw, 2018). It is also critical in dampening intracellular ROS and protects T cells from ROS-mediated apoptosis, which might contribute to the persistence of Tmem. A study demonstrates that AMPKα1-deficient mice harbor disability to generate central memory T cells. They show comparatively less effector memory T cells as compared with the wild-type mice (Lepez et al, 2020). This highlights that memory T cells exhibit a distinct metabolic profile as compared with effector T cells.
Metabolism of T cells can also be modulated by nutrients and oxygen in the external environment. Nutrients such as glucose, lipid, glutamine, methionine, serine, glycine, leucine, tryptophan, and arginine are known to play a critical role in T cell effector functions and differentiation (Geiger et al, 2016; Jacobs et al, 2008; Maciver et al, 2013; Maciver et al, 2008; Munn et al, 1999; Newsholme et al, 1985). Glucose and glutamine are generally needed in different types of T cells at all status (Araujo et al, 2017; Klysz et al, 2015; Swamy et al, 2016; Zhao et al, 2016). Especially glucose that enters glycolysis fuels T cells to produce interferon (IFN) (Chang et al, 2015).
Serine and glycine (one carbon metabolism) have been demonstrated to be crucial to support the activation, proliferation, and cytotoxic functions of T cells (Ma et al, 2017; Ron-Harel et al, 2016). Methionine has been shown to promote histone methylation (H3K79me2) through providing methyl donor SAM, thereby promoting signal transducer and activator of transcription 5 (STAT5) expression to promote T cell activity and cytotoxicity such as IFN-γ, tumor necrosis factor (TNF)-α, and Granzyme B production (Bian et al, 2020). Branched chain amino acids, including leucine, isoleucine, and valine, also support cytotoxicity of T cells (Ren et al, 2017). Alanine has also been shown to support protein synthesis during the activation of naive and memory T cells (Ron-Harel et al, 2019).
Different amino acids potentiate mTOR1 signaling through interaction with Rag GTPases (Sancak et al, 2008; Zhang et al, 2021b). CD8+ T cells with Rag deficiency are dysfunctional and showed reduced proliferative capability and cytokine secretion. Lipid also functions as an important signaling mediator. On activation of TCR and co-stimulation, T cells activate lipid synthesis pathways to support proliferation (Howie et al, 2017; Shyer et al, 2020). Given the intricate link between metabolism and T cell function, the antitumor response of T cells can potentially be improved through modulation of T cells' metabolic pathways.
Although T cells rely on glutamine, a recent study demonstrated that T cells have the adaptability to rewire their metabolic program by using acetate to replenish the TCA cycle when glutamine metabolism is blocked (Leone et al, 2019). Further, c-MYC is activated and AMPK is inactivated to enhance glycolysis, which channels metabolites into the pentose phosphate pathway to generate NADPH in glutamine antagonist-treated T cells (Leone et al, 2019). Interestingly, cancer cells lack this metabolic adaptability; therefore, they are more vulnerable to glutamine blockade (Leone et al, 2019).
This interesting study suggested that although the proliferating cancer and immune cells generally use similar metabolic machineries to maximize anabolism and to achieve REDOX homeostasis, their abilities or speed to adapt to different nutrient stress or metabolic blockade vary, and through understanding their divergences introduce new therapeutic windows or modalities to more specifically target cancer cells while maximizing the longevity and cytotoxicity of antitumor T cells, including inherent T cells or adoptively transferred T cells.
How TME Affects T Cells Metabolism and ACT Efficacies
The TME is a hostile environment that is often characterized by having a disorganized blood vessels network, high interstitial fluid pressure, and hypoxic acidosis. Such an environment poses significant metabolic challenges to immune cells, which can undermine the local immune response and thus limit the efficacies of ACT (Kouidhi et al, 2017).
Rapidly proliferating tumor cells exhibit a metabolic profile that is different from non-proliferating somatic cells. To fulfill the metabolic requirements for tumor growth and proliferation, tumors undergo metabolic reprogramming. They showed heightened aerobic glycolysis, also known as the Warburg effect, in which glucose is metabolized into lactate even in the presence of oxygen (Singer et al, 2011). They also exhibit increase lipid metabolism and decomposition of glutamine (Elia and Haigis, 2021). Many known oncogenes or tumor suppressor genes such as Ras, mTOR1, HIF-1, c-MYC, and p53 have been shown to drive metabolic reprogramming of tumor cells. For instance, the activation of P13K/AKT/mTOR1 signaling was also shown to increase glucose uptake from the environment and enhance catabolic metabolites extraction from mitochondria (Ward and Thompson, 2012).
The downstream signaling of the mTOR pathway leads to the activation of TFs HIF-1 and c-MYC. These TFs act in concert to divert the cells from OXPHOS and toward aerobic glycolysis to support de novo synthesis of nucleic acids, fatty acids, and amino acids (Li et al, 2020; Semenza, 2000). In addition, tumor cells with mutated p53—a known tumor suppressor gene—also showed enhanced glucose import to support aerobic glycolysis (Zhang et al, 2013). These show that metabolic reprograming is critically involved in oncogenesis.
Aberrant expression of the metabolic proteins does not only provide the tumor cells with metabolic advantage to thrive in the hostile microenvironment, but it also supports immune evasion. As discussed, the function and survival of effector T cells is nutrient-dependent. Given the enhanced metabolic activities of tumors, nutrient availability in the TME becomes limited. This leads to the suppression of the antitumor function of TILs (Chang et al, 2015). In a mouse model experiment, T cells obtained from tumors with a high glucose concentration in the extracellular milieu were shown to exhibit higher glycolytic activity and IFN-y production as compared with T cells derived from glucose-restricted TME (Chang et al, 2015).
Depletion of glutamine in the culture medium is also shown to block the proliferation and cytokine production of T cells (Berod et al, 2014). These T cells exhibit reduced mitochondrial function along with increased expression of exhaustion markers and co-inhibitory molecules (Scharping et al, 2016). These show that nutrient competition in TME serves as a major barrier to effective tumor control for ACT.
Local acidosis is prevalent in TME. Enhanced aerobic glycolysis of tumors leads to increased secretion of lactic acid, which has been shown to suppress the effector functions of CD8+ T cells (Brand et al, 2016). In line with this, an in vitro study showed that mouse CD8+ T cells cultured in medium with low pH exhibited decreased cytotoxic activities against tumor cells. Further, the reactivation of antigen-specific T cells from memory cells also appeared to be inhibited. This could be rescued by neutralizing the pH of culture (Nakagawa et al, 2015).
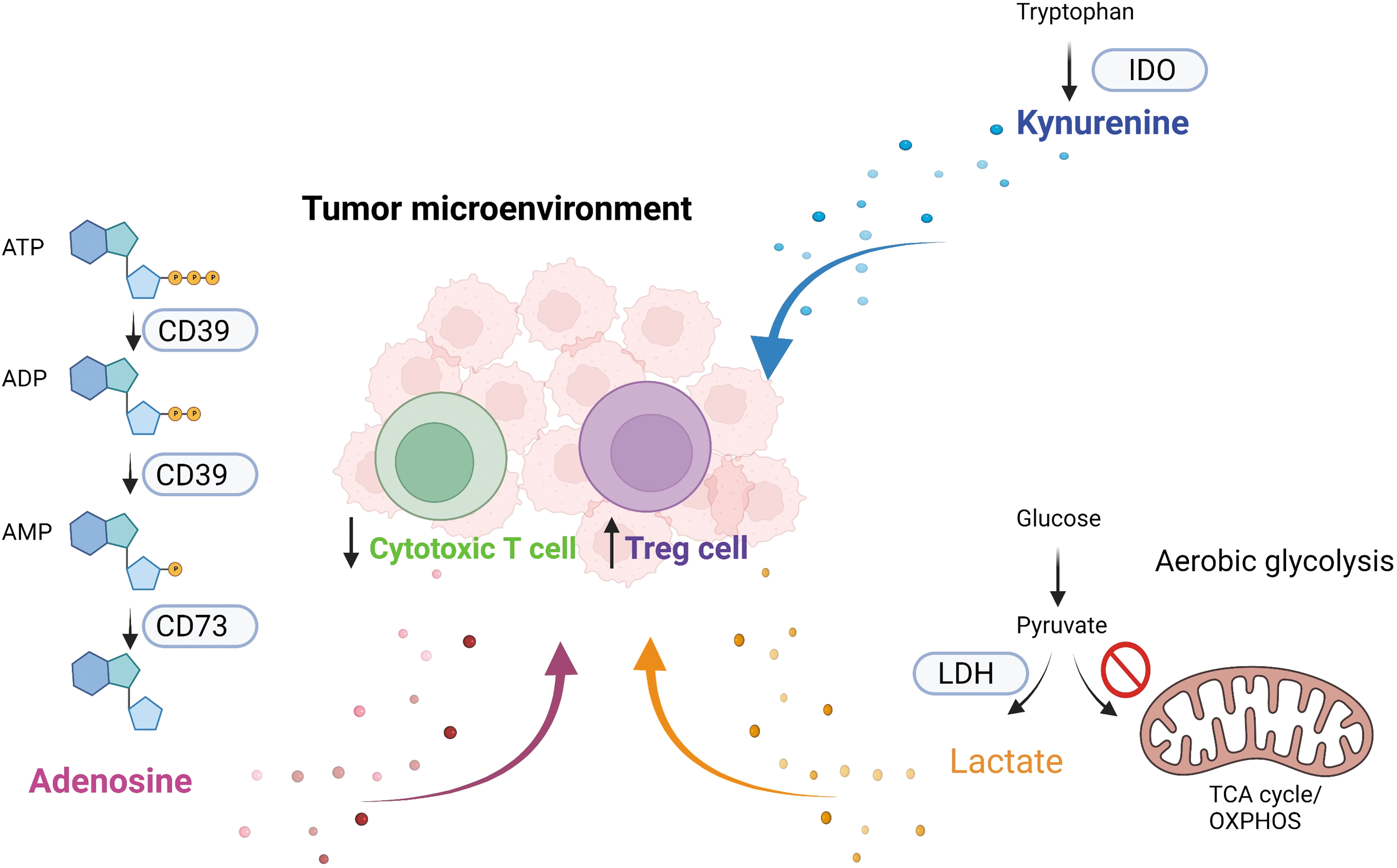
Tumors also inhibit the function of TILs by depleting important amino acids such as glutamine and tryptophan from the TME through their increased uptake. Tumor cells also highly express indoleamine 2,3 dioxygenase (IDO), which can metabolize tryptophan to kynurenine (KYN), thus further reducing the availability of tryptophan in TME. Meanwhile, increased KYN in TME can also suppress antitumor immunity through binding to aryl hydrocarbon receptor (AHR) on T cells. The binding of AHR induces immunosuppressive phenotypes in T cells and favors the differentiation of Treg but not Th17 cells (Cheong and Sun, 2018; Triplett et al, 2018).
In the colorectal cancer mouse model, inhibition of IDO was shown to inhibit KYN-mediated Treg differentiation (Zhang et al, 2021a). It was also shown to harbor a higher level of TILs with superior cytotoxic capabilities and proliferation as compared with the wild-type mice (Zhang et al, 2021a). These show that the antitumor response of transferred T cells during ACT can be undermined by the presence of immunosuppressive metabolites. Moreover, there is also a risk that transferred T cells might acquire Treg phenotypes in response to the high level of KYN in TME.
Adenosine is often increased in the tumor microenvironment due to the enhanced expression of ectonucleotidases CD39 and CD73, which sequentially carries the conversion of ATP ADP AMP and then AMP adenosine (Longhi et al, 2021) (Fig. 5). Adenosine is immunosuppressive and has been shown to suppress effector functions of T cells and favor the development of Treg cells (Longhi et al, 2021). Adenosine interacts with adenosine receptors on different immune cells to elicit their immunosuppressive functions. CD39 and CD73 as well as adenosine receptors could be induced by hypoxia (HIF) and chronic inflammation (transforming growth factor-beta [TGF]-β) and other stressors or stimuli (Allard et al, 2020; Clambey et al, 2012).
Long-term exposure to antigenic tumors drives exhaustion in T cells. This could be due to chronic activation of AKT signaling, leading to a sustained reliance on glycolysis of infiltrating T cells in TME where glucose levels are low (Siska and Rathmell, 2015). Activated T cells with chronic AKT stimulation tend to differentiate toward terminal exhaustion (Scharping et al, 2016). Man et al further showed that high amounts of TCR activation induce the expression of TFs interferon regulatory factor (IRF)4, basic leucine zipper ATF-like transcription factor (BATF), and nuclear factor of activated T cell complex 1 (NFAT1) (Man et al, 2017).
These TFs were shown to impair T cell metabolism and repress TCF1—a TF critical for memory T cell differentiation. In particular, T cells with IRF4 haploinsufficiency were also shown to highly express genes involved in OXPHOS, amino acid metabolism, and FAO (Man et al, 2017). Together, these show that metabolic reprogramming of tumor cells can serve as a mechanism of immune evasion and resistance for ACT.
Modulate T Cell Metabolism to Improve ACT Efficacies
Efficacy of ACT depends on the ability of T cells to mount strong antitumor response and form long-lived memory T cells that can respond to future antigens to prevent recurrence. Given that the function of T cells is intricately linked to their metabolic status, strategies that improve the metabolic fitness of effector T cells and promote memory T cell formation can potentially enhance the effectiveness of ACT therapy. Many strategies have been investigated so far and they can be broadly categorized into two main approaches: ex vivo modulation and systemic modulation.
Ex vivo modulation
The preparation of ACT ex vivo provides the opportunity to modulate and improve the metabolic fitness of T cells prior to infusion. The preparation process involves three key steps. First, allogeneic or autologous T cells are isolated from donors or cancer patients. Second, ex vivo activation, expansion, and modification are conducted. Third, infusion is conducted in cancer patients (Rohaan et al, 2018; Tsimberidou et al, 2021) (Fig. 1).
T cells isolated from patient or donors are cultured in a medium that consists of growth factors and nutrients. They commonly include soluble anti-CD3 antibody, IL-2, and glutamine (Jackson et al, 2020; Levine et al, 2017; Sabatino et al, 2016). Current methods of culturing T cells tend to drive the cells toward terminal differentiation, which might undermine its antitumor function (Jackson et al, 2020; Levine et al, 2017; Sabatino et al, 2016).
Several factors such as the culturing time, nutrition level, reagents, growth factors, and the state of T cells were shown to play a role (Fig. 6). For instance, shorter culture time of TILs is associated with better clinical response for TIL-based therapy (Itzhaki et al, 2011). Exhausted TILs obtained from metabolically deranged TME were shown to exhibit poor antitumor response even after ex vivo activation and modulation (Thommen and Schumacher, 2018). This indicates that the impaired state of T cells is sometimes irreversible. These processes have been a focus of optimization for improving ACT efficacies.

Less-differentiated Tn, T memory stem cells (Tscm) and central memory T cells (Tcm) appear to be a better candidate for ACT as compared with Teff. They showed longer persistence and better antitumor response when infused into patients and are correlated with objective clinical responses (Klebanoff et al, 2012). Given our understanding on how the metabolic program is linked with T cell differentiation, the use of metabolic modulators has been explored as a strategy to promote the formation of memory T cells ex vivo (Table 2). A study by Sukumar et al (2013) demonstrated that inhibiting glycolytic activities with analog 2-deoxyglucose (2DG), which functions as a hexokinase 2 (HK2) inhibitor in T cells during ex vivo expansion, could enhance the formation of memory CD8 T cells.
Metabolic Modulators Used to Promote T Cells' Function and Antitumor Abilities
2-DG, 2-deoxyglucose; HK2, hexokinase 2; OXPHOS, oxidative phosphorylation; AMPK, 5’ adenosine monophosphate-activated protein kinase; Eomes, eomesodermin; Sirt, silent information regulator; Foxo1, forkhead box O1; ACAT, acetyl-CoA acetyltransferase; Phx, PhysiologixTM xeno-free (XF) hGFC; TIL, tumor infiltrating lymphocytes; mTOR, mammalian target of rapamycin; γc, gamma chain; FAO, fatty acid oxidation; NAD, nicotinamide adenine dinucleotide; JAK; Janus kinase; STAT, signal transducer and activator of transcription; Pl3K, phosphatidylinositol 3-kinase; IL, interleukin; TME, tumor microenvironment.
T cells treated with HK2 inhibitor showed increased expression of TFs (e.g., Tcf7, Lef1, Bcl6) and surface marker CD62L, which was implicated in early memory T cell populations. Notably, when infused in the mouse model, 2DG pre-treated T cells showed improved glycolytic ability, increased IFN-γ and TNF-α production, and prolonged survival as compared with non-treated T cells (Sukumar et al, 2013).
In addition to 2DG, other metabolic modulators have also been used in ACT preparation to promote Tmem differentiation. This includes pharmacological inhibition of mTOR during ex vivo activation of T cells. They were shown to induce the formation of memory T cells, evidenced by increased expression of memory-associated genes that are associated with enhanced FAO and mitochondrial SRC. This suggests that the formation of Tmem could be due to the rewiring of metabolic profile in T cells (Crompton et al, 2015). In addition, inhibiting cholesterol-esterification enzyme acetyl-CoA acetyltransferase 1 (ACAT1) in T cells during ex vivo culture was shown to improve their antitumor function in the melanoma and glioblastoma mouse model.
A similar effect was also observed when ACAT1 inhibitor was delivered systematically. Moreover, exhausted T cells treated with ACAT1 inhibitor also displayed improved cytotoxic function (Hao et al, 2020). These suggest that the metabolic modulators can potentially be used to reprogram the metabolic profile of T cells to improve their antitumor function in vivo.
IL-2 is commonly added in T cells culture as growth factor to promote T cell expansion and activation (Dudley et al, 2003). It is a cytokine that belongs to γc family and is known to be indispensable for T cell survival, development, and activation. IL-2 plays a key role in promoting Tn differentiation into Tmem and Teff by regulating metabolic (e.g., AKT/mTOR) and epigenetic profile through STAT5 (Bevington et al, 2020). Ablation of IL-2Rα in CAR-T cells showed decreased proliferative capability in vitro (Zhang et al, 2018). Despite the important role of IL-2, T cells cultured with IL-2 during ACT preparation were shown to harbor phenotypes of terminal differentiated effector cells (Zhang et al, 2018). Hence, this prompted studies to investigate the potential of other cytokines.
Studies have now demonstrated IL-7, IL-15, and IL-21 as potential candidates to promote and sustain T cell function during ACT preparation (Ding et al, 2017; Dwyer et al, 2019; Murphy et al, 1993; Reif et al, 1997; Richer et al, 2015; Wofford et al, 2008). The CAR T cells expanded with IL-15 were shown to be less differentiated and exhibit superior antitumor activities in vivo as compared with IL-2 treated CAR T cells. IL-15 reduces mTOR activities and helps preserve Tscm (CD62L+CD45RA+ CCR7+) phenotype of CAR T cells (Alizadeh et al, 2019). Consistent with this finding, other studies also showed that a combination of IL-2 with IL-7 or IL-15 supports the homeostasis and maintenance of Tmem through supporting mitochondrial biogenesis in T cells (van der Windt et al, 2012).
In addition, IL-21 has also been shown to enhance the expansion and cytotoxicity of less-differentiated T cells (Du et al, 2021). Transgenic expression of IL-15 and IL-21 in Glypican-3-specific CAR T cells showed robust antitumor activity and superior expansion in the hepatocellular carcinoma mouse model (Batra et al, 2020). This indicates the potential of using multiple cytokines to improve the metabolic fitness of ACT cells prior to infusion.
In addition to cytokine modulation, (Ghassemi et al., 2020) further showed that culturing CAR T cells in a serum-free medium enriched with a nutrient such as carnosine results in enhanced CAR T cell transduction and increased therapeutic potency in xenograft models of neuroblastoma as compared with CAR T cell culture in human serum. Carnosine is a biogenic amine. It was shown to enhance the metabolic fitness of T cells by limiting glycolytic metabolism that gives rise to terminally differentiated effector cells (Ghassemi et al, 2020).
It also rewiews the metabolic profile of T cells toward oxidative state. CAR T cells cultured in carnosine-rich medium also showed superior engraftment and survival after transfusion. Although the mechanism remains unclear, optimizing culture medium through inhibition of glycolysis or addition of carnosine might be a strategy to improve ACT efficacies (Ghassemi et al, 2020).
Another emerging approach of modulating T cell metabolic fitness is through genetic editing. In CAR T cells, the choice of the costimulatory domain was shown to affect the metabolic activities and thus their function in vivo. Kawalekar et al (2016) demonstrated that the addition of CD28 in the CAR construct tends to drive CAR T cells toward aerobic glycolysis with increased expression of the pentose phosphate pathway and glycolytic enzymes such as glucose-6-phosphate dehydrogenase (G6PD) and phosphoglycerate kinase (PGK).
On the other hand, the activation of 41BB tends to drive CAR T cells toward FAO with increased mRNA expression of cartinine palmitoyltransferase 1A (CPT1A)—a metabolic enzyme involved in mitochondrial FAO and biogenesis, and fatty acid binding protein (FABP5) that is involved in fatty acid uptake, transport, and metabolism. The metabolic pathways mediated by 41BB support central memory differentiation and T cell persistence (Kawalekar et al, 2016). 41BB-mediated Tcm showed enhanced mitochondrial SRC and an expression marker of CAR T cells whereas CD28 was shown to promote the differentiation of effector cells and effector memory (Kawalekar et al, 2016).
The metabolic signaling pathway in T cells can also be genetically modified to improve their metabolic fitness and thus their function. For instance, genetically enforced expression of proteins involved in mitochondrial function such as peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1α) and mitochondrial dynamin such as GTPase 1 (OPA1) cells in T cells were shown to promote metabolic shift from aerobic glycolysis to FAO and OXPHOS. This leads to enhanced antitumor responses and persistence in vivo (Buck et al, 2016; Scharping et al, 2016).
After the discovery that extracellular potassium concentration in TME can potentially impair T cell function, potassium channel KV1.3 was genetically introduced into T cells. This leads to improved tumor rejection in the melanoma-bearing mouse (Eil et al, 2016). Moreover, engineered expression of BATF in CAR T cells was also shown to improve function and antitumor response in vivo. This is evidenced by increased proliferation, production of effector cytokines, and reduced thymocyte selection-associated high-mobility group box (TOX) TF in T cells.
TOX TF is known to be associated with T cell exhaustion (Seo et al, 2021). Although the mechanism of BATF in T cell regulation was not investigated in this study, another study had previously suggested that BATF plays a role in metabolism (Man et al, 2017). This shows that ACT efficacy can be enhanced through genetically modifying the metabolic pathways of T cells. Other potential targets that warrant further investigations include genes that are known to be involved in the metabolic regulation and transduction such as mTOR, AMPK pathways, and TFs such as MYC-1 and HIF-1α.
Lipid accumulation in T cells could be caused by CD36, a transporter that imports lipid from extracellular space. Blockade of CD36 reduced lipid accumulation in T cells and prevented T cells from ferroptosis, a mode of iron-dependent cell death that is caused by lipid-associated oxidative stress. Interestingly, it was shown that ferroptosis inhibitor (ferrostatin)-treated CD8+ T cells are more sustainable and have more antitumor effects after being adoptively transferred into mice (Ma et al, 2021). Meanwhile, overexpressing glutathione peroxidase 4 (GPX4), an anti-ferroptosis gene that uses glutathione to reduce lipid ROS, in T cells could also improve their antitumor effects in adoptive T cell therapy (Xu et al, 2021).
The NAD has been shown to promote and prolong the antitumor effects of adoptively transferred T cells (Chatterjee et al, 2018). The NAD acts as a co-substrate for many enzymes, including the silent information regulator (SIRT) family. Hybrid Th1/17 cells have more robust NAD salvage metabolism and are more effective in suppressing melanoma in mice than Th1 or Th17 cells when transferred adoptively (Chatterjee et al, 2018). Th1/17 cells relied on intracellular NAD to maintain memory, cytotoxicity, and antitumor effects through promoting SIRT1, which activates deacetylation and expression of forkhead box O1 (Foxo1), which are responsible for the transcription of memory-associated genes (Chatterjee et al, 2018).
The NAD supplements could be a source to replenish intracellular NAD, and a future study testing the effects of NAD supplementation on improving the therapeutic efficacy of Th1/Th17 cells in cancer is awaited.
Systemic Modulation
Lymphodepletion is a common procedure performed in patients prior to ACT. Clinical studies show that lymphodepletion improves therapeutic efficacy (Dafni et al, 2019; Schaft, 2020). Infused T cells or CAR T cells exhibit increased persistence and antitumor response in lymphodepleted patients. A clinical study on melanoma patients showed that patients were treated with lymphodepleting regime (e.g., cyclophosphamide and fludarabine) for 5 days prior to a TIL infusion (ORR = 46%) as compared with patients without lymphodepletion (ORR = 31%) (Dudley et al, 2002). The addition of total body irradiation into the lymphodepleting regimes further improves the ORR in melanoma patients (Dudley et al, 2008).
Although the molecular mechanisms on how lymphodepletion improves TIL persistence and function remain unclear, studies hypothesized that lymphodepletion might induce cytokines IL-7 and IL-15, which are known to promote the expansion of infused T cells (Gattinoni et al, 2005). It also reduces the frequency of immunosuppressive cells such as myeloid-derived suppressor cells and regulatory T cells (Tregs) in mouse studies, thus modulating the immunosuppressive TME (Antony et al, 2005; Gattinoni et al, 2005). The depletion of these cells might also reduce the local competition for nutrients in TME. In addition, it is also speculated that some of the lymphodepleting drugs target the metabolism of tumor cells and thus create a metabolically favorable environment for transferred T cells (Zhang and Romero, 2018).
Some metabolic modulators previously discussed are also being explored as adjuvant therapy for ACT. For instance, mTOR inhibitors (e.g., rapamycin and metformin) and IL-2 are being investigated for their ability to improve ACT efficacies in ACT-treated patients (Nian et al, 2021; Zhang and Romero, 2018; Zhang et al, 2020). Small-molecule selective IDO1 inhibitors are currently being developed for advanced cancer treatment. This inhibitor can potentially deplete KYN and increase the availability of tryptophan for transferred T cells in the TME (Cheong and Sun, 2018). Systemic delivery of pegylated kynureninase that degrades KYN into immunologically inert metabolites can also enhance antitumor response (Triplett et al, 2018).
Many ongoing clinical trials in ACT are also exploring the clinical benefits of combining checkpoint inhibitors such as PD-1 blockades or genetically silencing PDCD1 in T cells with ACT (Chong et al, 2017; Rohaan et al, 2018; Wang et al, 2021). It is well known that PD-1 functions as an exhaustion marker and a co-inhibitory molecule in T cells. In addition, it is also involved in the regulation of T cells' metabolic activities. For instance, the ligation of PD-1 was shown to inhibit glycolysis and thus impair T cell differentiation in vitro (Patsoukis et al, 2015). PD-L1 blockade treatments in the mouse model were also shown to enhance IFNγ production of TILs and partially reverse the suppressive effect of Tregs (Liu et al, 2018).
By contrast, knockdown of PD-1 in CAR T cells was shown to inhibit T cell proliferation and impair antitumor function in vitro, which highlights that long-lasting PD-1 blockade might not be beneficial for cancer patients (Wei et al, 2019). Today, significant antitumor response and expansion of CD19 CAR T cells have been observed in CD19 CAR T cells and PD-1 blockade treated patients with lymphoma (Chong et al, 2017; Song and Zhang, 2020). However, evidence for the additive effect of ACT and PD-1 blockades in solid tumors is still limited (Rohaan et al, 2018). A study showed that in high-grade serous ovarian cancer patients treated with PD-1 blockade prior to TIL-based therapy with low-dose IL2, one out of six patients exhibited partial response and the other five showed disease stabilization (Kverneland et al, 2020).
Challenges and Limitations
Despite the promising potential of adoptive T cell therapies, there are challenges yet to be addressed biologically and clinically, especially in solid cancers. The poor recognition of adoptively transferred T cells to their targeted cancer cells remains the major limitation in the field. The TCRs of the adoptively transferred T cells recognize specific neoantigens that are specifically expressed by cancer but not normal cells. It is difficult to confirm and predict whether mutations in the cancer genome always yield neoantigens.
Further, cancer mutations are heterogeneous. Cancer cells from the same or different patients express different mutations clonally. However, the TCRs of the adoptively transferred T cells could only recognize particular antigens expressed by particular cancer clones. Therefore, the adoptively transferred T cells might only specifically target a subset of cancer cells but fail to eliminate all cancer cells. Moreover, HLAs are highly polymorphic across different patients, especially those with different ethnicities. This greatly makes the design of adoptively transferred therapies even more challenging.
Finally, the accreditation of the laboratories needs to be stringently monitored and controlled to ensure biological safety. Although our current review focuses on highlighting how understanding T cell metabolism could enhance the sustainability and functions of adoptively transferred T cells, solutions to overcome the challenges mentioned earlier are direly needed.
Conclusion
Immunotherapy represents one of the most common cancer treatments in the next decades. ACT is actively being investigated for their efficiency in cancer treatment. Adoptive T cell therapies are especially attractive due to the promising clinical outcome in melanoma. This review aims at providing a concise and timely summary of the most common adoptive T cell therapies, their principles, advancements, and clinical outcomes. Although the challenge in adoptive T cell therapy remains mainly due to the lack of clear understanding of neoantigen presentation, research efforts should be continued to detail our understanding on the basic metabolism of T cells, which is highly transferable to adoptively transferred T cells.
Our review highlights the importance of future exploration to expand the knowledge on the metabolic requirements of adoptively transferred T cells both in vitro and in vivo, the metabolic competition of adoptively transferred T cells with stromal and cancer cells within the tumor niche, and the metabolic switch to maintain the durability and memory of these adoptively transferred T cells. This knowledge will benefit the design experimental conditions to maximize the expansion and activity of the T cells in the ex vivo culture as well as the combination treatments that involve metabolic inhibitors or dietary interventions that specifically suppress cancer cells without disturbing the activity, viability, and durability of adoptive T cells. All these will potentially improve the efficiency of ACT.
Footnotes
Author Disclosure Statement
Ge Hui Tan has no disclosure. Carmen Chak-Lui Wong has no disclosure. Ge Hui Tan wrote the review. Carmen Chak-Lui Wong wrote the review.
Funding Information
This work is funded by the Center for Oncology and Immunology under the Health@InnoHK initiative funded by the Innovation and Technology Commission, The Government of Hong Kong SAR. Ge Tan is the recipient of the Hong Kong Fellowship and HKU Presidential Scholarship funded by the Hong Kong Research Grant Council and the University of Hong Kong, respectively. All figures are created with Biorender.com