
Abstract
Significance:
Glioblastomas (GBMs) are among the most lethal tumors despite the almost exclusive localization to the brain. This is largely due to therapeutic resistance. Radiation and chemotherapy significantly increase the survival for GBM patients, however, GBMs always recur, and the median overall survival is just over a year. Proposed reasons for such intractable resistance to therapy are numerous and include tumor metabolism, in particular, the ability of tumor cells to reconfigure metabolic fluxes on demand (metabolic plasticity). Understanding how the hard-wired, oncogene-driven metabolic tendencies of GBMs intersect with flexible, context-induced metabolic rewiring promises to reveal novel approaches for combating therapy resistance.
Recent Advances:
Personalized genome-scale metabolic flux models have recently provided evidence that metabolic flexibility promotes radiation resistance in cancer and identified tumor redox metabolism as a major predictor for resistance to radiation therapy (RT). It was demonstrated that radioresistant tumors, including GBM, reroute metabolic fluxes to boost the levels of reducing factors of the cell, thus enhancing clearance of reactive oxygen species that are generated during RT and promoting survival.
Critical Issues:
The current body of knowledge from published studies strongly supports the notion that robust metabolic plasticity can act as a (flexible) shield against the cytotoxic effects of standard GBM therapies, thus driving therapy resistance. The limited understanding of the critical drivers of such metabolic plasticity hampers the rational design of effective combination therapies.
Future Directions:
Identifying and targeting regulators of metabolic plasticity, rather than specific metabolic pathways, in combination with standard-of-care treatments have the potential to improve therapeutic outcomes in GBM. Antioxid. Redox Signal. 39, 957–979.
Introduction
Glioblastoma, previously known as glioblastoma multiforme (GBM), is a grade IV astrocytoma, the most common and most lethal primary brain tumor with a current 5-year survival rate of ∼5% in the United States. Without prompt treatment, the survival of GBM patients is only about 3 months (Schapira, 2007). Maximal safe resection is the first attempt at controlling the tumor, followed by radiation therapy (RT) given in 2 Gy fractions over 6 weeks for a total dose of 60 Gy. Ever since Walker et al. (1979, 1978) demonstrated in the late 1970s the effectiveness of postsurgical RT in significantly increasing the survival in GBM patients, RT has remained an indispensable treatment for GBM, extending the median survival to about 1 year (Stupp et al., 2009).
Despite extensive efforts to further improve survival with RT, only one alkylating agent, temozolomide (TMZ), has increased GBM median survival to ∼15 months (Stupp et al., 2009; Stupp et al., 2005) in combination with RT. Therefore, current standard-of-care for treating GBM remains postsurgical RT with concurrent and adjuvant TMZ. Recently, tumor treating fields have shown some promise as an alternative strategy for treating GBM (Fabian et al., 2019), but this approach is yet to enter clinical practice.
GBM can be highly invasive, but metastasis outside the brain is extremely rare (Sullivan et al., 2014). RT can shrink or even cure most localized tumors, but GBM recurrence is almost universal, and often occurs within the irradiated field, pointing to a remarkable resistance to RT. Proposed reasons for such resistance are numerous, and include tumor metabolism. A comprehensive metabolic profiling of gliomas has revealed that distinct metabolic profiles associate with tumor grades in GBM (Chinnaiyan et al., 2012) pointing to a key role for tumor metabolism in GBM progression. GBMs are a heterogeneous group of tumors classified into four different subtypes, mesenchymal, classical, proneural, and neural, based on gene expression profiles (Brennan et al., 2013; Verhaak et al., 2010).
Another important classification is based on the mutational status of isocitrate dehydrogenase (IDH1/2), which occurs in ∼10% of GBM patients (Louis et al., 2016). The implications of IDH mutations in metabolism and therapy response have been recently reviewed (Zhou and Wahl, 2019) and are therefore out of the scope of this review. The dynamic nature of GBM metabolism and its interactions with the tumor microenvironment (TME) have been recently reviewed by Morrow et al. (2021) and we have recently reviewed the extent of radiation effects on tumor metabolism in general, elsewhere (Read et al., 2022). In this study, we explore the role of tumor metabolism in protecting GBM from the cytotoxic effects of the current standard-of-care therapies and attempt to provide some insight into the potential of combining existing therapies with metabolic regulators to improve GBM outcomes.
Metabolic reprogramming is an emerging hallmark of cancer (Hanahan and Weinberg, 2011) and is largely driven by oncogenic mutations (Pavlova and Thompson, 2016). “Metabolic reprogramming” generally refers to sustained, long-term changes in the overall cellular metabolic state to support a specific function. In the case of tumors, during transformation, cancer cells reprogram their metabolism to sustain a rapid proliferation rate while surviving in a nutrient-poor TME. Over the last decade it has been appreciated that, once “reprogrammed,” tumor metabolism is not a rigid metabolic state. Rather, tumors exhibit remarkable metabolic plasticity or flexibility, generally defined as the capacity of a cell to reroute metabolic fluxes on-demand, based on its needs and nutrient availability. It is a dynamic process that allows cancer cells to adapt to an ever-changing TME or to therapy-induced stress.
Such metabolic changes are thought to be of a transient nature aimed at surviving a finite stress, for example, adapt to restrictions in the bioavailability of a nutrient or oxygen or respond to the cytotoxic effects of an anticancer therapy, that is, potentially lethal oxidative stress or DNA damage generated during RT, the backbone treatment for GBM. Some of the same metabolic pathways that generate antioxidants in cells (see: Glycolytic Branches: A Reservoir of Building Blocks and Antioxidant Defenses That Can Promote Therapy Resistance) are also the main producers of precursors for nucleotide synthesis, which would further support DNA repair in radioresistant tumors following damage by oxidative stress. The metabolic redox changes that perpetuate oxidative stress induced by radiation in cells in general are thoughtfully reviewed by Spitz et al. (2004). In this study, we focus on studies that shed light into prosurvival metabolic reprogramming following radiation of cancer cells and the impact of specific oncogenic mutations, with emphasis on GBM.
The Impact of Oncogenic Mutations on GBM Glucose Metabolism and Implications for Therapeutic Resistance
Based on extensive evidence, it is now largely accepted that most malignant cells enhance their glucose consumption to sustain the nutrient and bioenergetic needs of rapid proliferation, and GBM is no different. This is reflected in the “Warburg effect,” characterized by enhanced oxidation of glucose to lactate via glycolysis even when oxygen is plentiful (aerobic glycolysis) (DeBerardinis and Chandel, 2020; Warburg, 1924). In GBM, increased glucose uptake correlates with enhanced expression of glucose transporters (GLUTs), particularly GLUT1 (solute carrier family 2 member 1, SLC2A1) and GLUT3 (solute carrier family 2 member 3, SLC2A3) (Boado et al., 1994; Cosset et al., 2017; Flavahan et al., 2013), which are the predominant isoforms expressed in GBM and the normal brain.
The expression levels of all glycolytic genes are increased in GBM relative to low-grade gliomas (LGG) (Stanke et al., 2021), and a higher expression of certain glycolytic genes, including SLC2A3, hexokinase 2 (HK2), and the M2 isoform of pyruvate kinase (PKM2), is associated with poorer survival in GBM (Flavahan et al., 2013; Stanke et al., 2021). Moreover, higher lactate levels in the cerebrospinal fluid of GBM patients are associated with worse survival (Nakamizo et al., 2013). Together, these findings point to a supportive role for enhanced glycolysis in GBM progression.
The most common mutations in GBM are implicated in modifying glucose metabolism in a way that benefits tumor growth, as well as promotes therapy resistance. GBMs are characterized by activating mutations in the epidermal growth factor receptor (EGFR) gene, inactivation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), or mutations in the TP53 gene that result in loss of its tumor suppressor functions. Below, we summarize findings that shed light into the effects that these mutational events have on glucose metabolism. Where possible, we highlight GBM-specific studies, but in the lack thereof, we include discussions of other tumor models to glean insight into the potential consequences for GBM metabolism and response to therapy.
Alterations in EGFR-PTEN axis impact GBM glucose metabolism
EGFR pathway alterations significantly correlate with inferior prognosis in GBM (Brennan et al., 2013; Cancer Genome Atlas Research Network, 2008; Huang et al., 2009) and arise from gene amplification, protein overexpression, autocrine signaling loops, and/or mutations resulting in constitutive activation of EGFR, such as EGFR variant III (EGFR-vIII) (Huang et al., 2009). EGFR amplification and/or activating mutations are present in ∼57% of GBM tumors (Brennan et al., 2013), whereas PTEN homozygous deletion occurs in ∼36% (Cancer Genome Atlas Research Network, 2008), across all the different GBM subtypes (Verhaak et al., 2010). PTEN antagonizes EGFR signaling by preventing the activation of AKT and downstream signaling, and therefore, loss of PTEN would be expected to enhance the metabolic consequences of hyperactive EGFR signaling. EGFR mutations and PTEN loss may co-occur, but no significant association between these genomic alterations has been identified (Yan et al., 2020).
EGFR signaling regulates glucose uptake
In other EGFR-mutated cancers, such as lung cancers, EGFR signaling regulates glucose consumption and expression of GLUTs (Kim et al., 2018; Makinoshima et al., 2014). In GBM, there is evidence that mutant EGFR-vIII upregulates the Myc-binding protein Delta Max, which activates Myc (Babic et al., 2013), subsequently increasing the expression of its target genes, including the glycolytic genes, SLC2A1, SLC2A3 (GLUTs GLUT1/3), and HK2 (Fig. 1). This enhances fluorodeoxyglucose (FDG) uptake in vivo (Babic et al., 2013) and induces a dependency on glycolysis in GBM tumors (Tateishi et al., 2016). Further supporting a link between EGFR signaling and glucose metabolism in GBM, a study by Mai et al. shows that inhibition of EGFR signaling via erlotinib decreases glucose consumption in a subset of human GBM tumors in mouse models of patient-derived xenografts. However, no specific EGFR mutation or other pathway alterations could predict this metabolic response to EGFR inhibition (Mai et al., 2017).
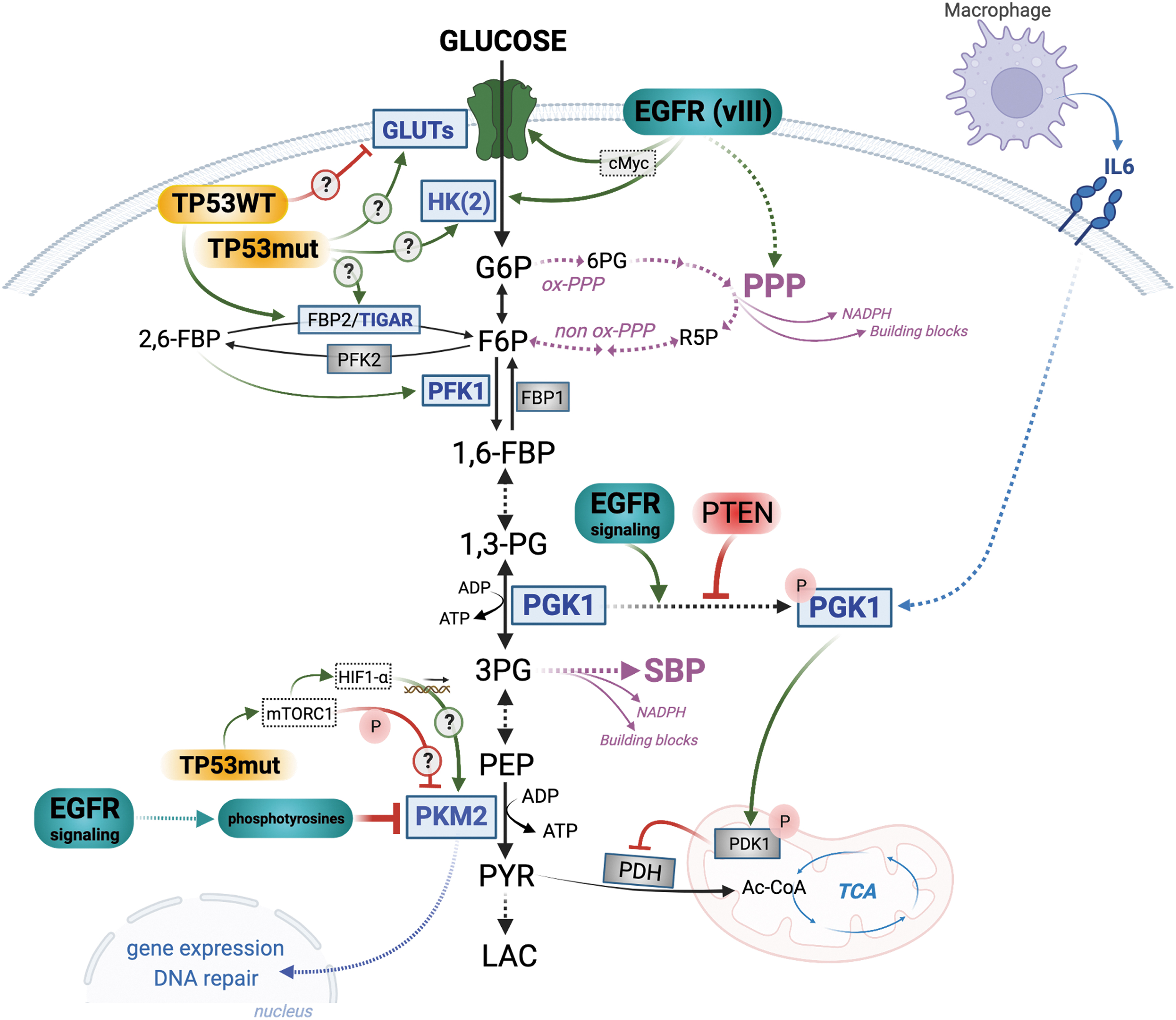
EGFR-PTEN axis regulates phosphoglycerate kinase 1
EGFR signaling can also promote glycolysis by activating phosphoglycerate kinase 1 (PGK1) (Li et al., 2016b) (Fig. 1). While PGK1 is normally autophosphorylated (on Y324), EGFR signaling leads to ERK-dependent phosphorylation of PGK1, although on a different residue (S203) (Li et al., 2016b) (Fig. 1). In addition to its function as a glycolytic enzyme, phosphorylated PGK1 can moonlight as a protein kinase (Lu and Hunter, 2018). It translocates to the mitochondria where it phosphorylates and activates pyruvate dehydrogenase kinase 1 (PDK1) (Li et al., 2016b). PDK1 inhibits pyruvate dehydrogenase complex (PDH), leading to a decrease in the production of acetyl-CoA from pyruvate and attenuation of the tricarboxylic acid (TCA) cycle (Golias et al., 2019) (Fig. 1). There is GBM-specific evidence that PTEN dephosphorylates (on Y324) and inactivates PGK1 (Qian et al., 2019) and that enhanced EGFR signaling dampens mitochondrial metabolism, whereas wild-type (WT)-PTEN GBM cells have a higher mitochondrial respiration capacity (Comelli et al., 2018).
It seems therefore that in GBM, the EGFR-PTEN axis can coordinate an enhanced glycolytic flux with attenuated mitochondrial metabolism, and this may be, in part, regulated by PGK1 (Fig. 1). Adding another layer of regulation, it has been reported that M2 tumor-associated macrophages can also phosphorylate and activate PGK1 in GBM cells via secretion of interleukin 6 (IL6) (Zhang et al., 2018b) (Fig. 1). Taken together with evidence that PGK1 gene expression is upregulated in GBM compared with a normal brain (Stanke et al., 2021) and that phosphorylation levels of PGK1 correlate with GBM grade and survival (Zhang et al., 2018b), PGK1 lends itself as a potential therapeutic target in GBM especially in the context of PTEN loss. Supporting a potential therapeutic impact of PGK1 targeting, Qian et al. (2019) observed decreased glycolytic flux in the context of PTEN loss when a mutated PGK1 (Y324F) was expressed to prevent autophosphorylation, thus mimicking a knockdown of PGK1 glycolytic activity.
EGFR-PTEN axis and the PKM2
Another glycolytic enzyme that is regulated by EGFR signaling is PKM2 (Fig. 1). Whereas other isoforms of PK exist in a constitutively active conformation, the M2 isoform is allosterically regulated and can shape-shift between an active (tetramer) or inactive (monomer or dimer) conformation, which can effectively constrain lower glycolysis. Paradoxically, increased glycolysis is often accompanied with the expression of the PKM2 isoform. In GBM, PKM2 gene and protein expression increases with grade (Mukherjee et al., 2013), indicating a supportive role for tumor growth and aggressiveness.
PKM2 activity is inhibited by growth factor signaling (Hitosugi et al., 2009; Li et al., 2016a; Yang et al., 2012; Yang et al., 2011) via binding to phosphotyrosine-marked proteins, such as EGFR target proteins (Christofk et al., 2008). In GBM, ERK1/2 kinases downstream of EGFR phosphorylate PKM2 (S37), thus inhibiting its enzymatic activity and promoting its nuclear translocation, and glycolytic gene expression (Yang et al., 2012). Comprehensive metabolic profiling has revealed increased levels of phosphoenolpyruvate (PEP) in mesenchymal gliomas, which correlates with the decreased enzymatic activity of PKM2 (Chinnaiyan et al., 2012), suggesting that hyperactivated EGFR signaling in GBM might sustain the suppression of PKM2 activity (Fig. 1).
This would be advantageous for GBM cells, as the glycolytic bottleneck created by suppressed PKM2 activity results in accumulation of upstream metabolites that become available for use in anabolic glycolytic branches, such as de novo serine biosynthesis pathway (SBP) or the pentose phosphate pathway (PPP), having consequences for therapy resistance (Fig. 1) (see more below). In line with this, Chinnaiyan et al. (2012) have reported higher levels of PPP metabolites, namely 6-phosphogluconate (6PG) and ribose-5-phosphate (R5P), in the metabolic signature of GBM samples, independently of their subtype, suggesting an increased need for R5P for nucleotide synthesis (Fig. 1).
PKM2, similar to PGK1, also has moonlighting functions distinct from its canonical role as a glycolytic enzyme (İlhan, 2022). Glycolytically inactive PKM2 translocates to the nucleus where it regulates expression of genes (Gao et al., 2012; Lee et al., 2008; Luo et al., 2011; Sizemore et al., 2018; Wang et al., 2014; Yang et al., 2012; Yang et al., 2011) such as β-catenin-target genes (Yang et al., 2011), hypoxia-inducible factor (HIF)1 (Luo et al., 2011), and TP53 (Xia et al., 2016). Although protein kinase activity has been attributed to PKM2, this remains controversial (Hosios et al., 2015). There is evidence that such moonlighting activities of PKM2 might promote therapy resistance. For example, following RT of GBM, PKM2 is phosphorylated by ataxia telangiectasia mutated (ATM) and accumulates in the nucleus where it promotes homologous recombination repair mechanisms that can promote radiation resistance (Sizemore et al., 2018) (Fig. 1).
The potential for TP53 pathway alterations in rewiring (GBM) glucose metabolism
Mutations in the tumor suppressor gene TP53, and its various pathway regulators, are found in more than 80% of GBM tumors, although they do not correlate with GBM survival (Zhang et al., 2018a). The frequency of direct mutations in the TP53 gene varies between the different GBM molecular subtypes, with the proneural subtype having the highest (∼50%), while the classical ones lack TP53 mutations (Verhaak et al., 2010). Whereas TP53 deletions are common in other tumor types, missense mutations with loss of transcription factor activity dominate in GBM. These missense mutations often give rise to the oncogenic “gain-of-function” TP53 protein mutant (TP53mut) that can promote proliferation and resistance to apoptosis (Dittmer et al., 1993).
The six most frequent missense or “hotspot” mutations account for 25% of TP53 mutations across all tumor types (Zhang et al., 2018a). There is a dearth of studies on the role of TP53 pathway alterations on glucose metabolism in GBM, but studies on other tumor models provide some insight on the potential metabolic consequences of TP53 mutations.
Using lung and breast cancer cells, Zhang et al. (2013) observed that introduction of several “hotspot” mutations in the TP53 gene increases glucose uptake and lactate production, akin to a Warburg effect, whereas knocking down endogenous TP53mut reversed this phenotype. Mechanistically, TP53 hotspot mutations, through rho-associated protein kinase (ROCK) signaling, promote plasma membrane translocation of GLUT1 enhancing glucose uptake (Zhang et al., 2013). In contrast, in osteosarcoma cells, WT TP53 (TP53WT) downregulates GLUT1 expression by direct transcriptional repression, whereas mutations in the binding domain of TP53 prevents its binding to the SLC2A1 promoter abrogating transcriptional repression (Schwartzenberg-Bar-Yoseph et al., 2004). It seems, therefore, that alterations that suppress TP53WT transcriptional regulation will likely lead to a higher availability of GLUTs and an enhanced capacity for taking up glucose by tumor cells, especially in the context of hotspot TP53 mutations (Fig. 1).
Once glucose is transported into the cell, it is phosphorylated by HKs into glucose-6-phosphate (G6P), a modification that traps glucose inside the cell. HK2 is the predominant isoform of HK expressed in GBM relative to LGG and normal brain, which generally express HK1 (Wolf et al., 2011). The effect of TP53mut on HK levels in GBM remains unknown, but there is evidence in hepatoma cells that TP53mut binds to TP53 motifs in the HK2 promoter and increases its expression (Mathupala et al., 1997) (Fig. 1).
Following phosphorylation by HKs, G6P reversibly isomerizes into fructose-6-phosphate (F6P), which is then converted into fructose-1,6-bisphosphatase (1,6-FBP) by phosphofructokinase 1 (PFK1). Alternatively, PFK2 can convert F6P to 2,6-FBP, which is a potent allosteric activator of PFK1. Opposing the PFKs, fructose bisphosphatase (FBP1 and FBP2) catalyze the dephosphorylation of 1,6-FBP/2,6-FBP back to F6P, thus tightly regulating flux through glycolysis (Fig. 1). The TP53-induced glycolysis and apoptosis regulator (TIGAR), a TP53WT-induced protein (Bensaad et al., 2006), has bisphosphatase activity and can reduce glycolytic flux by degrading 2,6-FBP, the activator of PFK1. This results in the use of upstream glycolytic metabolites by the PPP to generate intermediates for nucleotide synthesis and the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) (Bensaad et al., 2006) (see more below).
It is no surprise, therefore, that TIGAR plays an important role in promoting cancer growth, including in GBM where its overexpression is associated with enhanced growth, lower oxidative stress, reduced overall survival (OS), and resistance to RT and TMZ (Tang and He, 2019). The effect of TP53mut on TIGAR is not clear in GBM although transcriptionally active TP53mut, such as R175P, has the potential to induce TIGAR expression (Bensaad et al., 2006) with metabolic responses that can affect therapeutic efficacy.
TP53 can also regulate PKM2 at the transcriptional and enzymatic activity levels. Although not shown in GBM, in pancreatic cancer, TP53mut (R273H) can stimulate mammalian target of rapamycin complex 1 (mTORC1) activity, most likely through growth factor receptors upstream of mTORC1 (Dando et al., 2016). This leads to mTORC1-induced phosphorylation of PKM2, a modification that prevents tetramer formation and reduces its pyruvate kinase activity (Hitosugi et al., 2009) (Fig. 1). mTOR can also increase HIF1-α expression, which can activate the transcription of PKM2 in different tumor models (Sun et al., 2011) (Fig. 1).
The above studies, largely performed in tumor types other than GBM, point to the potential for various TP53 mutations to directly impact GBM glucose metabolism (question marks on Fig. 1). In line with this, in a GBM-specific study, Mai et al. (2017) show that a fraction of GBM patient-derived lines respond to EFGR inhibition by decreasing glucose consumption, a response that primes them for apoptosis in a (cytoplasmic) TP53WT-dependent manner. The GBM cells with hotspot TP53 mutations that decrease their glucose consumption following EGFR inhibition seem to lack this “apoptotic priming” (Mai et al., 2017). Clearly, TP53 mutational status exerts a complex regulation on GBM metabolism that requires context-specific investigations.
Glycolytic Branches: A Reservoir of Building Blocks and Antioxidant Defenses That Can Promote Therapy Resistance
As the cell oxidizes glucose via glycolysis, the resulting intermediates are often diverted away from glycolysis to fulfill other metabolic needs. For rapidly proliferating cancer cells, a priority is generating enough energy and building blocks to support cell growth and division while keeping in check the oxidative stress that accompanies enhanced metabolic activity. Oxidative stress happens to also be one of the primary targets of RT. A collapse of the redox state would significantly disrupt biochemical reactions, a potentially lethal situation, making its stabilization a major requirement for cancer cells during RT. One central balancing mechanism is the production of reducing equivalents in the form of NADPH, a main cofactor of antioxidant systems. NADPH can neutralize oxidative stress either directly by buffering reactive oxygen species (ROS) or indirectly by recycling oxidized glutathione (GSSG) into its reduced form (GSH).
As demonstrated by recent studies, the ability of cancer cells to reconfigure metabolic fluxes to optimize the production of reduced redox cofactors seems to be critical in resisting the cytotoxic effects of RT (Lewis and Kemp, 2021; Lewis et al., 2021).
Three glycolytic intermediates feed into auxiliary pathways that ultimately provide the energy, building blocks and reducing equivalents needed for uninhibited proliferation and maintenance of redox homeostasis: G6P feeds the oxidative PPP; 3-phosphoglycerate (3PG) feeds into the de novo SBP; and pyruvate feeds into the TCA cycle (Fig. 2). How common oncogenes and tumor suppressor genes regulate these pathways in cancer cells in general has recently been reviewed (De Santis et al., 2018). In this section, we focus on the role of the PPP and the SBP in promoting tumor growth and therapy resistance, highlighting GBM-relevant studies whenever possible.

The PPP
The functions of PPP are twofold, to provide phosphopentoses for synthesis of nucleotides and to generate reducing equivalents in the form of NADPH via the oxidative arm of the PPP (Fig. 2). Nucleotides are necessary for DNA replication and repair, whereas NADPH contributes to reductive biosynthesis of lipids and is essential for maintaining redox homeostasis (Patra and Hay, 2014). A shift in primary carbon metabolism to enhance the PPP is the fastest way to produce NADPH, and therefore, activation of the PPP is a known metabolic response in protecting eukaryotic cells from oxidative stress (Filosa et al., 2003; Kuehne et al., 2015; Ralser et al., 2007). There is evidence, in normal cells and yeast systems, that shifting of glycolytic flux toward the PPP can occur within seconds of exposure to oxidative stress in a transcription-independent manner, followed by a gene expression-dependent antioxidant response (Kuehne et al., 2015; Ralser et al., 2009).
The initial acute response is thought to be mediated by the inhibition of the enzymatic activity of downstream redox-sensitive glycolytic enzymes, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Ralser et al., 2009) and PKM2 (Anastasiou et al., 2011), which create a block in lower glycolysis making upstream metabolites available for the PPP. Others have reported, however, that this can also happen independently of GAPDH and PKM2 inhibition by oxidative stress (Kuehne et al., 2015). In the latter study, Kuehne et al. (2015) report that NADPH is a direct inhibitor of glucose-6-phosphate dehydrogenase (G6PD), the first rate-limiting enzyme in the PPP, and that acute depletion of NADPH by oxidative stress, in this case H2O2 or ultraviolet, removes the inhibitory effect on G6PD thus enhancing flux through the PPP.
HKs are redox-sensitive enzymes inhibited by oxidative stress (Heneberg, 2019) (Fig. 2) and there is some evidence that they regulate NADPH levels in ovarian cancer cells (Šimčíková et al., 2021), however, it remains unclear whether this is through the PPP. Although not performed in GBM, studies show that binding of HK2, the main isoform in GBM, to the mitochondria membrane protects cells from oxidative stress by reducing ROS generated in the mitochondria (da-Silva et al., 2004; Sun et al., 2008) (Fig. 2).
GAPDH is present in all normal cells, whereas PKM2 tends to be preferentially expressed in cancers and in GBM its expression increases with grade (Mukherjee et al., 2013). In a lung cancer model, oxidative stress induces oxidation of Cys358 residue on PKM2 and inhibition of its enzymatic activity (Anastasiou et al., 2011). Others have identified additional oxidation events that inhibit PKM2 activity (Irokawa et al., 2021). We have shown a similar inhibitory effect induced by oxidative stress generated during radiation of breast cancer cells (Zhang et al., 2019). In the lung cancer model (Anastasiou et al., 2011), oxidative stress-induced inhibition of PKM2 results in increased flux through the PPP and enhanced NADPH production that supports a robust antioxidant response, promoting survival under oxidative stress conditions (Anastasiou et al., 2011) (Fig. 2).
In parallel, the PPP generates precursors for nucleotides, which are essential for repairing DNA damage (Fig. 2). It seems, therefore, that rewiring glycolytic flux to enhance the PPP would be of particular importance in the context of RT in GBM where purine metabolism has been associated with radiation resistance (Zhou et al., 2020).
Two enzymes in the PPP directly contribute to the NADPH pool, the first enzyme, G6PD, and the third enzyme, 6-phosphogluconate dehydrogenase (6PGD) (Fig. 2). It is reported that 6PGD is upregulated in GBM compared with normal tissue, whereas G6PD is lower (Stanke et al., 2021). Pointing to a tumor promoting role for these enzymes, analysis of The Cancer Genome Atlas (TCGA) data sets (Goldman et al., 2020) on gliomas reveals that gene expression levels of both enzymes are highly correlated with OS of LGG (Fig. 3A, B), whereas in GBM the gene expression levels of only G6PD are correlative, trending toward significance for OS (sample numbers with gene expression data are much lower in the GBM cohort) (Fig. 3C), but significantly correlating with progression-free survival (Fig. 3D).

The SBP
The nonessential amino acid serine contributes to numerous biosynthetic pathways and its role in metabolic plasticity in cancer is increasingly being appreciated (Geeraerts et al., 2021). Cells can obtain serine via multiple ways, including de novo synthesis from glucose, conversion from glycine, catabolism from proteins and serine-containing phospholipids (PLs), and by transporter-mediated uptake from the extracellular environment (de Koning et al., 2003). However, some cancers, such as aggressive breast cancer, become addicted to de novo production of serine (Possemato et al., 2011). In the normal brain, serine is critical for neurotransmission and is newly synthesized by glial cells via the de novo SBP (Maugard et al., 2021). 3PG is converted into serine via three consecutive enzymatic reactions, catalyzed by phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme in the SBP, phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH).
In a final reversible step, serine is converted to glycine via serine hydroxymethyl transferases, SHMT1/2 (cytoplasmic/mitochondrial), concomitantly charging one-carbon metabolism (1CM) with one carbon units (Fig. 2). 1CM couples the folate and methionine cycles, which generate amino acids, nucleotides, lipids, and NADPH (Fig. 2). Flux into the SBP can be regulated by serine itself, which is an allosteric activator of PKM2, thus restricting 3PG routing into the SBP when serine levels are high (Fig. 2). Enolase (ENO) generates 3PG from 2PG. ENO1 seems to promote cell growth and invasion in GBM (Song et al., 2014) and a subset of gliomas harbor homozygous deletion of ENO1 that makes them vulnerable to the inhibition of its paralogue ENO2 (Lin et al., 2020).
Mechanistically, a recent study shows that ENO1 binds cellular messenger RNAs (mRNAs) resulting in inhibition of its enzymatic activity and rewiring of glycolysis. One of the consequences is increased synthesis of serine via the SBP (Huppertz et al., 2022) (Fig. 2).
While it is generally accepted that the largest contributor to cytosolic NADPH in proliferating cells is the PPP, a comprehensive study quantifying NADPH fluxes demonstrated that the serine-driven 1CM generates NADPH at levels comparable with those in the PPP (Fan et al., 2014). SBP also generates the precursors for glutathione, which comprised glycine, cysteine, and glutamate (derived from glutamine). GSH synthesis, therefore, is sustained through direct uptake and/or de novo synthesis of serine and glycine and the generation of cysteine through the methionine cycle of 1CM that directly consumes serine (Fig. 2). Analysis [via cBioPortal (Cerami et al., 2012; Gao et al., 2013)] of TCGA data sets on LGG and GBM revealed the presence of copy number gains and amplifications in all the SBP enzymes in both tumor types, but an increased frequency in GBM (Fig. 4).

Copy number gains and amplifications occur with the greatest frequency in the PSPH gene with a dramatic increase in GBM. ∼80% of GBM have copy number gains and ∼12% have copy number amplifications (Fig. 4). Copy number variations (CNVs, include deletions) in PHGDH correlate with OS in only LGG and not GBM (Fig. 5A, B), whereas CNVs in the PSPH gene significantly correlate with OS in both LGG and GBM (Fig. 5C, D). Interestingly, although CNVs are highly correlated with PSPH gene expression levels in both LGG and GBM (Fig. 5E, F), only copy numbers significantly correlate with OS (Fig. 5C, D), whereas gene expression levels do not (not shown).
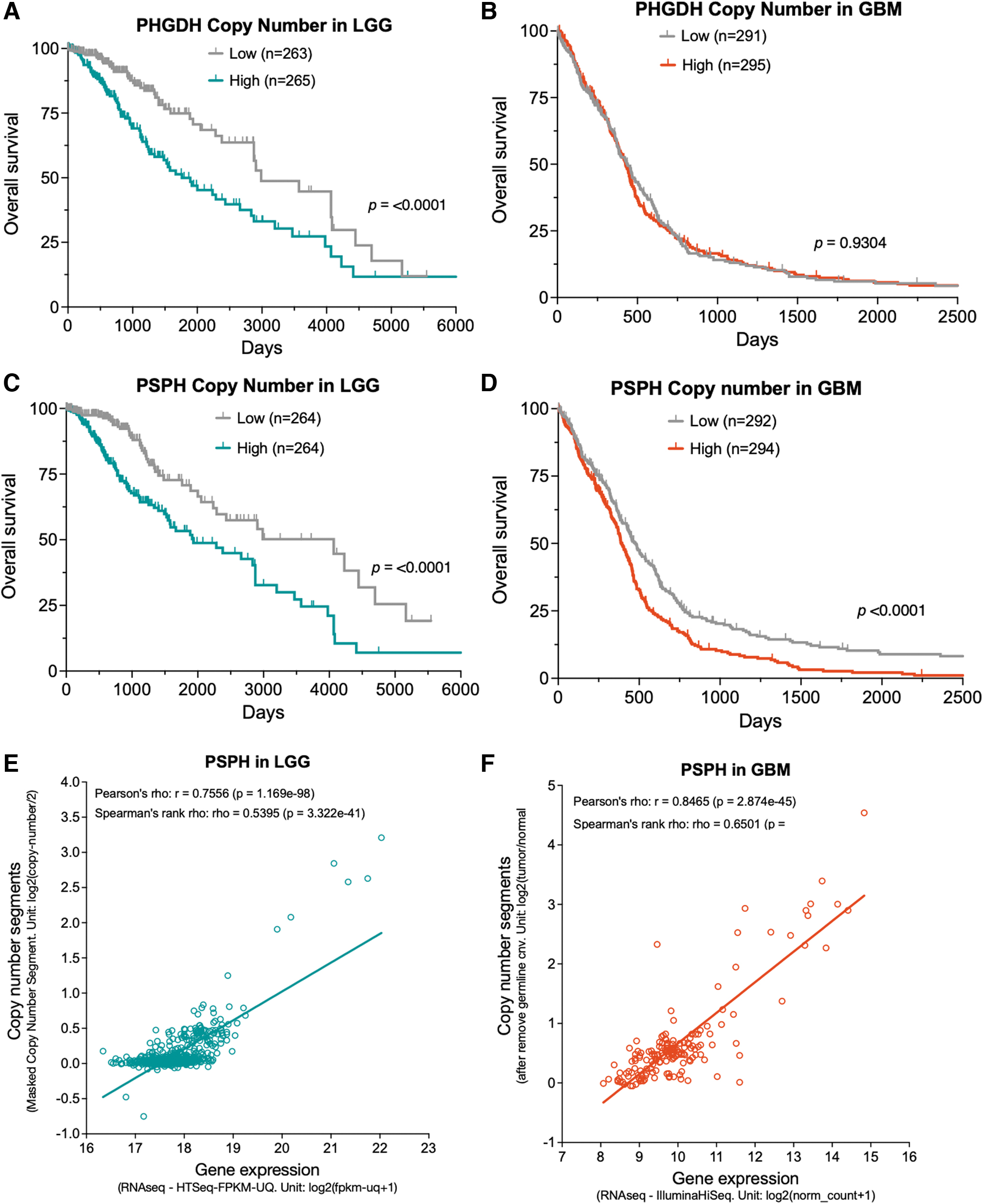
A recent study offers some insight into the curiously low copy number gains and lack of amplifications in the rate-limiting enzyme PHGDH, relative to the other SBP enzymes (Fig. 4). It seems that higher PHGDH levels, at either the gene expression or protein level, correlate with a favorable prognosis in GBM. This suggests a functional role of PHGDH in limiting tumor aggressiveness and thus selection for PHGDH-low cells during tumor evolution (Oh et al., 2020).
Although low in frequency, copy number amplifications are only found in the SHMT2 isoform (mitochondrial) and not the SHMT1 (cytoplasmic) (Fig. 4), pointing to a potentially greater importance of SHMT2 over SHMT1 in GBM. In line with this, recently, Engel et al. (2020) showed that under hypoxic conditions, GBM cells upregulate SHMT2 but not SHMT1, indicating that SHMT2 plays a crucial role in the survival of hypoxic GBM cells, such as in areas of pseudopalisading, which is common in GBM (Evans et al., 2004). Adding mechanistic insight, another study connected high levels of SHMT2 with lower PKM2 activity and hypothesized that lower PKM2 activity limits flux into the TCA cycle resulting in decreased oxygen consumption and enhanced GBM cell survival under hypoxia (Kim et al., 2015).
Mitochondrial folate metabolism has been linked to cancer development (Yang and Vousden, 2016). Under starvation conditions, as in the core of GBM tumors, for example, PSAT1, the mitochondrial methylenetetrahydrofolate dehydrogenase (MTHFD2) and serine-dependent 1CM are upregulated to counteract the effects of nutrient deprivation and allow GBM cells to survive harsh microenvironmental conditions. In tumors with TP53 mutations or loss, including GBM, MTHFD2 is often upregulated and required for cancer cell proliferation and DNA repair (Li et al., 2021), pointing to a potential mechanism for radiation resistance in TP53mut GBMs. Surprisingly little is known about the effect of localized RT-induced oxidative stress on serine metabolism in GBM. This warrants further investigation, as funneling glucose carbons into the de novo SBP might be an alternative, or even complementary, pathway via which irradiated GBM cells support a robust antioxidant response (Engel et al., 2020), DNA repair (Sánchez-Castillo et al., 2021), and resistance to RT.
Glutamine Metabolism Facilitates Therapy Resistance by Replenishing Antioxidant and Nucleotide Pools
Another important source of energy and antioxidants for cancer cells is the catabolism of glutamine. Glutamine, a nonessential amino acid, is the most abundant free amino acid in human blood (Brosnan, 2003). Despite this, astrocytes in the normal brain have high expression of glutamine synthetase (GS) (Tardito et al., 2015) and can synthesize glutamine de novo from glutamate (Bagga et al., 2014), which is derived from extracellular sources or formed from α-ketoglutarate (α-KG) in the TCA cycle. GBM cells mainly obtain glutamine from in situ sources, taking advantage of astrocytic synthesis (Tardito et al., 2015), with only 20% maximum originating from the circulation in the normal mouse brain (Bagga et al., 2014). Glutamine enters the cell via the amino acid antiporter ASCT2 (Alanine, Serine, Cysteine Transporter 2) (Scalise et al., 2018) and is converted to glutamate by glutaminase (GLS) (Fig. 2).
Glutamate can then participate in numerous cellular processes, including glutathione synthesis, SBP, and anaplerosis, defined as the replenishment of metabolites in the TCA cycle (Owen et al., 2002) (Fig. 2). Glutamine metabolism is therefore intimately interconnected with the cell's antioxidant systems. GBM glutamine metabolism and targeting opportunities have been recently reviewed (Obara-Michlewska and Szeliga, 2020).
Glutamine's contribution not only to the carbon pool but also as a nitrogen source makes it indispensable for many tumor types, including triple-negative breast cancer (Quek et al., 2022), lung cancer (Hassanein et al., 2015), pancreatic cancer (Son et al., 2013), and as discussed below, in GBM.
The dependence on glutamine seems to be heterogeneous across GBM subtypes, as different cell lines have differing sensitivities to glutamine starvation (Tardito et al., 2015). Mesenchymal GBMs, for example, increase glutamine uptake and utilization (Oizel et al., 2020; Oizel et al., 2017), whereas GS expression seems to be restricted to nonmesenchymal GBM (Oizel et al., 2020). GBM tumor tissue accumulates a large pool of intracellular glutamine in orthotopic GBM models due to higher synthesis and import (Marin-Valencia et al., 2012). However, it remains unclear if glutamine is used by GBM tumors as a mean to support anaplerosis. Some studies have shown an enrichment of glutamine-derived carbons in the TCA cycle of GBM cells compared with normal brain (Oizel et al., 2020) associated with increased NADPH production most likely via the malic enzyme (DeBerardinis et al., 2007) (Fig. 2).
DeBerardinis et al. (2007) have demonstrated that GBM cells use glutamine for the synthesis of fatty acids and aspartate, a precursor for nucleotide synthesis and asparagine/arginine synthesis. However, others suggest that GBM tumors mainly use glucose to supply the TCA cycle (Marin-Valencia et al., 2012) and that glutamine-induced anaplerosis is not essential for GBM cell proliferation (Tardito et al., 2015).
A recent study showed that the EGFR pathway promotes glutamate dehydrogenase-1 (GDH1) transcription in GBM (Yang et al., 2020), thereby stimulating glutamine metabolism. GDH1 catalyzes the formation of α-KG from glutamate (Fig. 2), suggesting that GDH1 upregulation promotes anaplerosis. Introduction of EGFR-vIII into GBM cells increases the expression of other key enzymes in glutaminolysis, such as GLS and GS (Tanaka et al., 2015). GBMs often resist EGFR inhibitors, which is mediated, in part, by the mTOR pathway (Mellinghoff et al., 2005). However, mTOR inhibition fails to sensitize EGFR-vIII GBM tumors to EGFR inhibition, but rather increases glutaminolysis (Tanaka et al., 2015), pointing to glutamine metabolism as a player in therapy resistance.
Glutamate is exported via the cystine/glutamate antiporter (SLC7A11/xCT), in exchange for cystine (Jyotsana et al., 2022) (Fig. 2). SLC7A11/xCT is overexpressed in GBM cells relative to normal human astrocytes (Polewski et al., 2016) and glutamate export supports tumor progression in GBM (Takano et al., 2001), most likely because of the neurotoxicity of high levels of glutamate. Under glutamine starvation, GBM cells still export glutamate, but prioritize glutamine synthesis from glutamate (Tardito et al., 2015), thereby promoting cataplerosis [reactions involved in the removal of TCA cycle intermediates (Owen et al., 2002)], to fuel de novo purine synthesis (Tardito et al., 2015).
Effects of anticancer therapies on glutamine metabolism
Very few studies have addressed the impact of anticancer therapies on glutamine metabolism in GBM. At the transport level, xCT is associated with chemo- and radioresistance in GBM. Knocking down xCT leads to increased oxidative stress, depletion of GSH, and enhanced sensitivity to TMZ (Polewski et al., 2016). Other studies have connected glutamine metabolism to radiation resistance although without directly addressing the impact of RT on how tumors use glutamine. xCT targeting sensitizes breast cancer cells to RT due to depletion of GSH pools (Cobler et al., 2018). There is also evidence that radioresistant cancer cells, including GBM, enhance glutamine anabolism via upregulation of GS expression and suppress glycolysis, TCA cycle, and mitochondrial respiration (Fu et al., 2019).
The interpretation is that the boosted glutamine levels provide the necessary nitrogen for the increased need of pyrimidine and purine synthesis that ultimately facilitates DNA repair (Fu et al., 2019). Indeed, nucleotide metabolism, which largely depends on glutamine metabolism, is recognized as an actionable pathway for sensitizing cancer cells, including GBM, to RT (Fu et al., 2019; Zhou et al., 2020).
It Is Not All About Glucose and Glutamine in GBM: Lipid Metabolism Has Emerged as a Key Player
Although the normal brain relies mainly on glucose as its primary energy source, it is now widely accepted that the brain is capable of oxidizing fatty acids that cross the blood–brain barrier (BBB) (Ebert et al., 2003; Panov et al., 2014). Fatty acids are key players in energy storage and production and a major component of cellular lipids. Lipids exist in three main classes of esters—triacylglycerol (TAG), PLs, and cholesteryl esters—and can be obtained extracellularly from the diet or synthesized from glucose and glutamine carbons. Catabolism of glucose and glutamine via the TCA cycle provides citrate, which can be converted to acetyl-CoA, a precursor for lipogenesis (synthesis of PL and TAG), as well as cholesterol synthesis via the mevalonate pathway (Beloribi-Djefaflia et al., 2016) (Fig. 6). Acetyl-CoA can also be generated from the breakdown of TAGs in the mitochondria, a process known as β-oxidation.
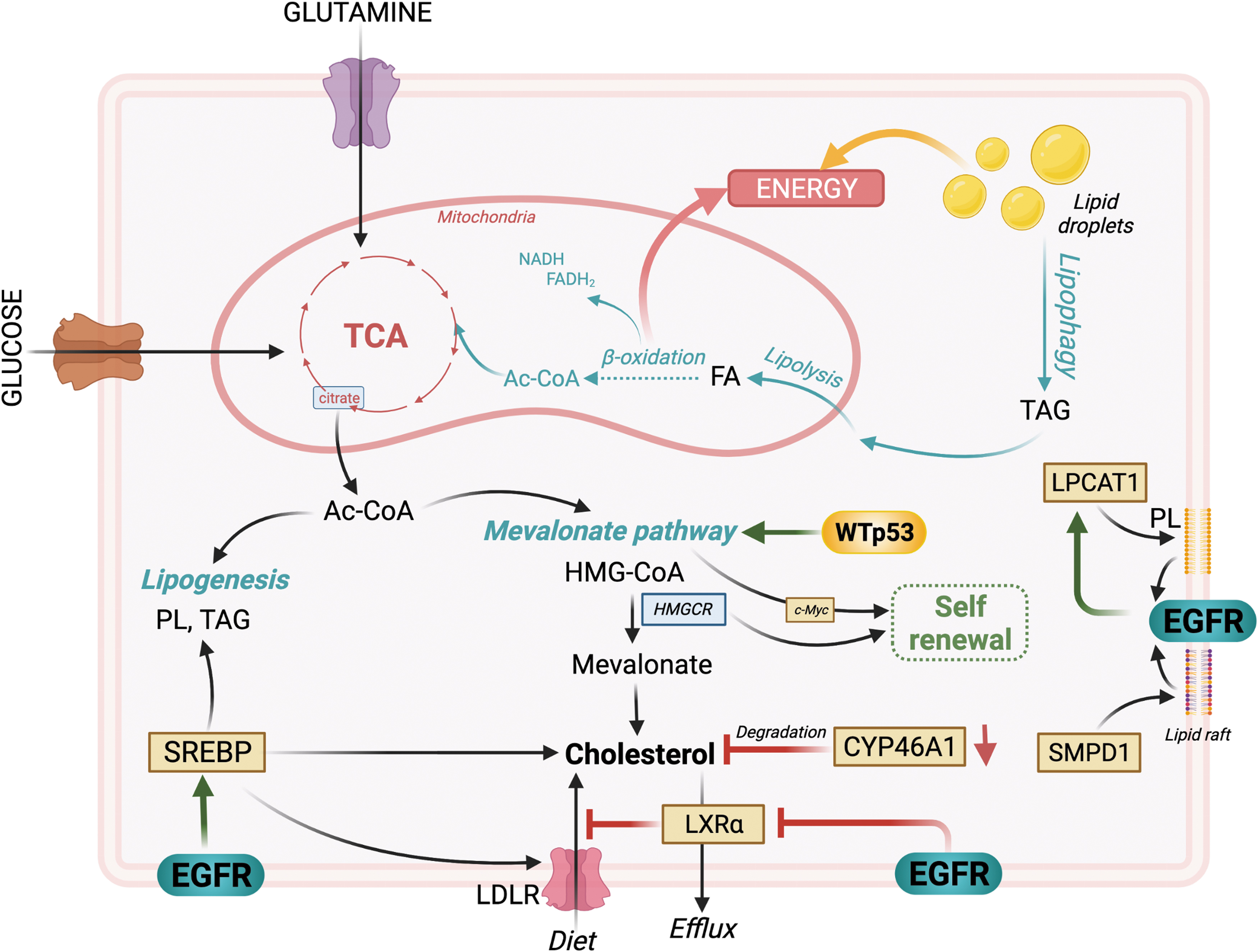
Within the cell, TAGs can also be stored inside lipid droplets, highly dynamic organelles that regulate the storage and hydrolysis of lipids. Lipid droplets are generally composed of a neutral lipid core encapsulated in a monolayer of PLs. They are fairly ubiquitous in cells and often reflect their metabolic state (Olzmann and Carvalho, 2019).
Lipid metabolism, the synthesis and degradation of lipids, has emerged as a critical contributor to tumor progression and therapy response, including in GBM (Guo et al., 2013; Shakya et al., 2021; Sperry et al., 2020; Taib et al., 2019). Rapidly proliferating cancer cells often have increased levels of lipids stored in droplets. GBM tumors display an overall increase in lipid levels compared with normal tissue (Guo et al., 2013) and further enhance lipid metabolism in a nutrient-poor environment (Shakya et al., 2021; Sperry et al., 2020). A study integrating metabolomics, lipidomics, and proteogenomics in GBM tumors revealed differences in lipid composition between GBM subtypes. The mesenchymal subtype stood out with a lipid metabolism profile distinct from other subtypes, characterized by an increased abundance of TAGs accompanied by enhanced β-oxidation (Kant et al., 2020; Wang et al., 2021).
Others show that human GBM tissues express fatty acid oxidation enzymes, suggesting that they can perform β-oxidation (Lin et al., 2017). The expression of these enzymes seems essential for aerobic respiration (Lin et al., 2017), perhaps pointing to a more important contribution of fatty acids to the TCA cycle than glucose (Sperry et al., 2020). High levels of unsaturated fatty acids in GBM, such as oleic acid, induce the accumulation of lipid droplets, which promote cell proliferation upon their hydrolysis (Taib et al., 2019) by acting as an energy reservoir (Wu et al., 2020) (Fig. 6). Although lipid droplets can be broken down via enzymatic hydrolysis mediated by lipases (lipolysis), lipophagy (lipid-selective autophagy) seems to be the main contributor to lipid droplet hydrolysis under glucose deprivation conditions in GBM (Wu et al., 2020) (Fig. 6).
Cholesterol, originating from acetyl-CoA via the mevalonate pathway, is essential for normal brain function with the brain containing almost a quarter of the body's cholesterol (Martin et al., 2014). Since the BBB prevents import of cholesterol, the brain cholesterol is synthesized locally in astrocytes and oligodendrocytes (Martin et al., 2014). Cholesterol plays an important role in tumor growth (Pirmoradi et al., 2019; Villa et al., 2016). Surprisingly, however, GBM cells lack the ability to synthesize cholesterol and must import it (Pirmoradi et al., 2019). One potential reason is that the mevalonate pathway is driven by TP53WT, and one study shows that GBM cells with a TP53mut (R273H), which lacks DNA-binding activity, downregulate several mevalonate pathway-related genes (Laezza et al., 2015) (Fig. 6).
The high prevalence of TP53 pathway alterations in GBM tumors (∼80%) potentially explains their inability to synthesize cholesterol, although this might be tissue- and cell type-specific. For example, the same mutation in the TP53 gene, in breast cancer cells, leads to the activation of the mevalonate pathway, whereas its knockdown results in decreased expression of several mevalonate pathway-related genes (Freed-Pastor et al., 2012). GBM cells may obtain cholesterol through its import from tumor-associated astrocytes (TAAs) (Perelroizen et al., 2022). TAAs accumulate at the tumor margins, produce cholesterol, and export it through the sterol transporter ATP-binding cassette transporter A1 (ABCA1). Astrocyte-derived cholesterol then promotes GBM cell survival and growth, while targeting of ABCA1 leads to tumor regression (Perelroizen et al., 2022).
There is some evidence that the mevalonate pathway maintains GBM stem cells (GSCs), which overexpress mevalonate pathway-related genes compared with differentiated GBM cells (Wang et al., 2017), suggesting a role in GBM maintenance (Fig. 6), although the role of TP53 mutations is unclear in this context. The rate-limiting enzyme in mevalonate synthesis, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), which converts HMG-CoA to mevalonate, is highly expressed in GSCs, in an Myc-dependent manner, and its inhibition hampers GSC self-renewal capacity and tumorigenicity in vivo (Wang et al., 2017), suggesting a targetable vulnerability to specifically eradicate therapy-resistant GSCs (Fig. 6).
Amplified EGFR signaling also leads to a remodeling of cholesterol metabolism in GBM (Fang et al., 2021; Gabitova et al., 2015; Guo et al., 2011; Guo et al., 2009; Villa et al., 2016). GBM cells profit from cholesterol biosynthesis in the brain to sustain their rapid growth, by maintaining high intracellular levels of cholesterol. This represents a growth advantage for GBM as cholesterol synthesis uses many nicotinamide adenine dinucleotide (NADH) reducing equivalents, which deprive other cellular processes (Lunt and Vander Heiden, 2011). GBM cells maintain their high cholesterol levels by preventing activation of liver X receptor (LXR). LXRs are nuclear receptors that regulate intracellular cholesterol levels by controlling the expression of genes related to cholesterol export. Constitutively activated EGFR signaling downregulates the expression of LXRα mRNA (Fang et al., 2021) (Fig. 6).
Forced activation of LXR via agonists negatively affects GBM growth (Guo et al., 2011; Villa et al., 2016), likely by forcing cholesterol export from the cell. In addition, EGFR induces the cleavage and nuclear translocation of sterol regulatory element-binding protein (SREBP) (Guo et al., 2009), another important transcriptional regulator of fatty acid synthesis, cholesterol synthesis, and uptake by inducing the expression of low-density lipoprotein receptor (LDLR), again leading to an increased intracellular availability of fatty acids in GBM cells (Fig. 6). Further enhancing intracellular cholesterol levels in GBM is the downregulation of cholesterol 24-hydroxylase (CYP46A1) gene expression. CYP46A1 converts cholesterol into 24(S)-hydroxycholesterol, thereby eliminating cholesterol (Moutinho et al., 2016). Its overexpression or forced activation reduces cholesterol levels and decreases GBM tumor growth in vivo (Han et al., 2020) (Fig. 6).
Recent evidence suggests that the composition of cholesterol and PLs may be radically different in the plasma membrane of GBM cells and that this profoundly impacts EGFR signaling (Bi et al., 2021; Bi et al., 2019). Lysophosphatidylcholine acyltransferase 1 (LPCAT1) (Bi et al., 2019) and sphingomyelin phosphodiesterase 1 (SMPD1) (Bi et al., 2021) were identified as two important enzymatic regulators of plasma membrane reorganization in GBM. LPCAT1 seems to control membrane PL saturation (Bi et al., 2019), whereas SMPD1 converts sphingomyelin to ceramide and is crucial for lipid raft composition and clustering of membrane signaling molecules, such as EGFR (Bi et al., 2021). Targeting of both LPCAT1 and SMPD1 has proven beneficial for controlling GBM growth (Bi et al., 2021; Bi et al., 2019), especially in combination with TMZ (Bi et al., 2021).
Taken together, the current body of work on GBM lipid metabolism reveals a beneficial interplay between lipid, cholesterol, and glucose metabolism that seems to support prosurvival metabolic plasticity in GBM. However, many questions remain regarding the molecular drivers of such plasticity and the impact of microenvironmental conditions and therapies. For example, it seems that in the presence of exogenous fatty acids, GBM cells upregulate their glucose consumption (Taib et al., 2019), but the mechanisms remain unclear.
Lipid metabolism modifies the response to anticancer therapies
There is evidence that lipid metabolism in GBM may modify the response to anticancer therapies (Caragher et al., 2020; Gupta et al., 2020). Studies performed in GBM, and other tumor models, point to a role for lipid droplets in protecting cancer cells from oxidative stress-induced lipid peroxidation (Jarc and Petan, 2019; Shyu et al., 2018) and nutrient/hypoxic stress (Bensaad et al., 2014; Jarc and Petan, 2019; Jarc et al., 2018) by buffering and delaying the release of lipids. ROS can cause lipid peroxidation of polyunsaturated fatty acids, which will become unstable and break down into oxidative stress second messengers (Barrera, 2012; Dalleau et al., 2013). Ultimately, lipid peroxidation negatively affects the integrity of cellular membranes (Barrera, 2012).
Under hypoxia-induced oxidative stress conditions, inhibition of lipid droplet synthesis increases ROS levels, whereas addition of free fatty acids decreases ROS levels (Bensaad et al., 2014), suggesting that lipid droplets may maintain cellular redox homeostasis under stress conditions. It remains unclear if therapy-induced oxidative stress increases the number of lipid droplets in GBM cells. However, one study shows that RT increases lipid droplet levels, and a higher number is associated with increased radiation resistance in glioma (Tirinato et al., 2021). Hydrolysis of lipid droplets can also provide the energy needed to survive therapy. Therapy-induced increase in autophagy is recognized in most cancers, including in GBM (Tsai et al., 2021; Zois and Koukourakis, 2009) and lipophagy can hydrolyze lipid droplets (Wu et al., 2020).
A hypothesis emerging from these observations is that therapy-induced increase in autophagy/lipophagy may enhance lipid droplet hydrolysis, providing the tumor cells with an alternative way to maintain redox homeostasis and with the energy needed to survive therapy.
GSCs are often associated with increased therapy-resistance and invasion (Bao et al., 2006; Beier et al., 2011) and are characterized by a distinct metabolic phenotype and enhanced metabolic plasticity (Caragher et al., 2020; Hoang-Minh et al., 2018; Vlashi et al., 2011). There is evidence that slow cycling GSCs have higher lipid levels and are enriched in lipid droplets, compared with fast cycling cells, thanks to a basal increase in fatty acid uptake, and this is accompanied by increased resistance to TMZ (Hoang-Minh et al., 2018). Under stress conditions such as low glucose, the slow cycling GSCs break down (by autophagy) their lipid droplets as a source of energy. Targeting fatty acid uptake via inhibition of fatty acid binding protein 7 (FABP7) sensitizes slow cycling GSCs to glucose deprivation or glycolysis inhibition (Hoang-Minh et al., 2018).
It is unclear, however, if TMZ treatment may induce such a dependency on lipid droplets and lipid catabolism, although there is evidence that TMZ treatment increases fatty acid uptake in GBM, with a concomitant decrease in glucose uptake in vitro and in vivo (Caragher et al., 2020). This effect is enhanced in the GSC population and is accompanied by a higher oxygen consumption rate and respiratory capacity that seems to be supported by endogenous fatty acids (Caragher et al., 2020). The TMZ-treated cells also increase the expression of genes involved in fatty acid uptake and β-oxidation (Caragher et al., 2020).
While fatty acid metabolism plays a role in TMZ resistance in GBM, it seems that cholesterol metabolism is preferentially involved in the response to RT. The expression of the master regulator of cholesterol metabolism, SREBP2, and its target genes are upregulated in irradiated GBM cells, and this response is further enhanced when RT is combined with a dopamine receptor (DR) antagonist (Bhat et al., 2021). Although the mechanisms remain unknown, treating GBM with a combination of RT and DR-antagonists induces specific metabolic rewiring in the form of enhanced cholesterol metabolism, likely aimed at promoting survival under these stressors. Inhibiting cholesterol metabolism, via atorvastatin, further reduces GBM survival, pointing to an acquired dependence on cholesterol metabolism after RT/DR-antagonist treatment (Bhat et al., 2021).
This study illustrates that targeting metabolic plasticity has the potential to enhance the therapeutic efficacy, although further investigations are needed to fully appreciate the importance of lipid metabolism in GBM and the contribution to therapy resistance.
Combating Therapy Resistance by Dismantling GBM's Metabolic Back-Up Plans
Given that glucose is also an essential nutrient for normal cells, especially the brain (Mergenthaler et al., 2013), denying glucose to tumors does not provide an optimal therapeutic window, although attempts have been made in GBM using the glucose competitor, 2-deoxyglucose (2DG), in combination with RT (Singh et al., 2005). While the cancer's greed for glucose may not be the ideal therapeutic target, it has proven quite useful for imaging purposes in the form of FDG-positron emission tomography (PET), widely used for cancer detection, diagnosis, and treatment responses (Kostakoglu et al., 2003), including in GBM (Verger and Langen, 2017). Targeting the flexibility with which GBM metabolizes glucose, glutamine, or lipids, rather than specific metabolic nodes, by targeting molecular regulators of metabolic plasticity, will likely prove a more effective way to slow GBM growth and/or render GBM more responsive to current therapies.
Targeting glycolytic and redox plasticity
Although hyperactive EFGR signaling instructs GBM cells to grow, this would be futile without sufficient metabolite pools to support growth. Therefore, a more coordinated effort to increase metabolite supply must also take place and some of these efforts are directly regulated by growth signaling. Enhanced levels of metabolite supply can generally be achieved in four ways: increase of exogenous uptake (i.e., glucose), increase of de novo synthesis (i.e., SBP), salvage from other macromolecules (i.e., autophagy), or creation of bottlenecks in metabolic fluxes to boost a specific metabolite pool. One example of the latter is the suppression of the enzymatic activity of PKM2, which can be achieved by growth factor signaling (Hitosugi et al., 2009; Li et al., 2016a; Yang et al., 2011) and various posttranslational modifications (Zahra et al., 2020).
This bottleneck in glycolytic flux results in upstream metabolites becoming available for use in glycolytic branches, that is, PPP and SBP, both of which generate macromolecule precursors as well as reducing equivalents in the form of NADPH and GSH (Fig. 2). “Turning off” PKM2 enzymatic activity, therefore, is one flexible way to couple oncogenic growth signaling with metabolic resources necessary to carry out instructions for rapid division and growth or ramp up antioxidant production and precursors for DNA repair under oxidative stress conditions, such as during RT (Fig. 2). In contrast, forcing PKM2 in the “on” position would deplete the upstream metabolites and prevent the increase in PPP and SBP flux resulting in insufficient amounts of nucleotides and NADPH and, consequently, insufficient GSH.
This would hamper the cell's ability to fight oxidative stress and repair DNA damage, offering a potential cancer-specific target, especially in the context of RT. This has motivated the development of small-molecule activators for PKM2 that lock the enzyme in a high-activity conformation that has shown preclinical therapeutic promise in different tumor types (Anastasiou et al., 2012; Li et al., 2018; Parnell et al., 2013). This approach seems to also slow down GBM growth, however, it has not yet been tested in orthotopic models of GBM (Ding et al., 2020; Guo et al., 2019). Newer generations of PKM2 activators also prevent PKM2 from entering the nucleus, thus further interfering with its moonlighting functions that could intensify therapy resistance (Ding et al., 2020).
Although there is evidence that some PKM2 activators cross the BBB as shown with PET radiotracer analogs of PKM2 activators (Witney et al., 2015), the successful use of such activators for treating GBM would require brain accumulation at therapeutic levels and this remains to be demonstrated. However, even if a brain penetrant PKM2 activator were to be developed, it seems unlikely that it will suppress GBM growth as a single agent, as the metabolically plastic GBM tumors would likely rewire metabolic fluxes to circumvent the blockage in the “PKM2 switch.” Combination with other therapies that exploit the metabolic vulnerabilities induced by PKM2 activation would be expected to enhance the effectiveness of this approach. For example, we have shown that combining PKM2 activators with RT can radiosensitize human breast cancers (Zhang et al., 2019), suggesting that this might be a feasible approach in treating GBM if PKM2 activators can effectively accumulate in intracranial GBM.
PGK1 represents another attractive metabolic target in GBM (Qian et al., 2019) for interfering with prosurvival metabolic rewiring. Radioresistant GBM cells display an increased expression of PGK1 (Ding et al., 2014), pointing to a radioprotective role. It is reasonable to postulate that increasing the precursors for the SBP protects GBM from RT-induced cytotoxicity, likely by generating NADPH, and precursors for GSH, as well as providing precursors for nucleotide biosynthesis promoting DNA repair (Fig. 2). A few mechanisms of PGK1 regulation have been identified, including posttranslational modifications [acetylation (Hu et al., 2017), O-GlcNAcylation (Nie et al., 2020), phosphorylation (Qian et al., 2019)], HIF1 regulation (Kress et al., 1998), and EGFR signaling (Li et al., 2016b). The common occurrence of hypoxia and aberrant EGFR signaling in GBM tumors argue in favor of a therapeutic potential for PGK1 targeting.
In addition, evidence that PTEN levels inversely correlate with PGK1 activation (Qian et al., 2019) suggest that targeting PGK1 may be more relevant in the context of a PTEN deletion. Studies have shown that loss of PTEN is associated with increased radioresistance (Pappas et al., 2007; Zhang et al., 2011), including in GBM (Zhang et al., 2011), suggesting a potential therapeutic opportunity in targeting PGK1 in combination with RT in PTEN-negative GBM tumors. It is worth mentioning that while PGK1 targeting would deplete the SBP from its precursor 3PG, GBM cells may still import extracellular serine and glycine (Fig. 2), which might contribute to resistance to this approach. In fact, although not performed in GBM, forced activation of PKM2, which would also divert 3PG away from the SBP has been shown to render cancer cells serine auxotroph (Kung et al., 2012).
Such a possible outcome could be counteracted by implementing a serine/glycine-depleted diet. Serine depletion has been tested in vivo, in a colorectal cancer model, without apparent systemic toxicity (Maddocks et al., 2017; Maddocks et al., 2013), but this remains to be tested in combination with RT in GBM.
Another node in the SBP with targeting potential is PHGDH, the enzyme that initiates serine biosynthesis. Engel et al. (2020) have tested the effects of PHGDH inhibition in GBM cells in vitro and found that it reduced proliferation and resistance to hypoxia. In other tumor types, PHGDH targeting has been suggested as a way to sensitize cancer cells to chemotherapy (Rathore et al., 2020). The amino acid transporter SLC7A11/xCT constitutes another promising target as its inhibition will prevent import of cysteine, thereby affecting GSH synthesis in cells (Chung et al., 2005; Cobler et al., 2018; Muir et al., 2017). In GBM, inhibition of xCT prevents tumor growth (Chung et al., 2005) and increases sensitivity to TMZ and oxidative stress (Polewski et al., 2016).
However, oxidative stress was induced by hydrogen peroxide treatment rather than radiation, and radiation-specific studies are needed, although there is evidence that in breast cancer, xCT inhibition has a radiosensitizing effect (Cobler et al., 2018). In an interesting observation, Polewski et al. (2016) show that GBM cells increase GSH levels in response to oxidative stress, but this increase is greater when xCT is overexpressed, suggesting a potential resistance mechanism to oxidative stress. Inhibition of xCT as a therapeutic approach would prevent the export of glutamate in the extracellular compartment, leading to a buildup of glutamate inside the cell. As high levels of extracellular glutamate are neurotoxic, inhibition of xCT could also help alleviate the neurologic symptoms of GBM (de Groot and Sontheimer, 2011).
Targeting lipid metabolism
Evidence of lipid metabolism reprogramming in GBM basally and following therapies suggests that targeted interventions would benefit GBM patients. Preclinical investigations (Jiang et al., 2014; Zhu et al., 2019) and clinical trials (Altwairgi et al., 2021; Xie et al., 2020) have explored the therapeutic potential of targeting the mevalonate/cholesterol pathway. Statins, which are cholesterol-lowering drugs, block the synthesis of mevalonate by inhibiting HMGCR, the rate-limiting enzyme in the pathway, and are being investigated for their anticancer activity (Di Bello et al., 2020). All nine FDA-approved statins tested in vitro with GBM cells inhibited cell proliferation (Jiang et al., 2014; Zhu et al., 2019), with pitavastatin, the most effective, also inhibiting GBM growth in vivo, as a single agent (Jiang et al., 2014). Lovastatin and simvastatin increase TMZ-induced apoptosis and impair autophagic flux in GBM cells, although in vitro (Shojaei et al., 2020; Zhu et al., 2019) with additional preclinical investigations required to further assess the therapeutic potential.
To the best of our knowledge, one clinical trial has been conducted in GBM to assess the therapeutic potential of statins in combination with RT/chemotherapy. Atorvastatin showed no improvement of progression-free survival or OS at 6 months in GBM, in combination with standard therapy (RT and TMZ), compared with standard therapy alone (Altwairgi et al., 2021), however, newer evidence suggests that it might be beneficial in combination with other novel therapies (Bhat et al., 2021).
Therapeutic targeting of cholesterol metabolism may also be achieved by interfering with its regulation. An interesting target is LXR, a regulator of cholesterol transport. Under normal conditions, LXRs are activated by oxidized forms of cholesterols, named oxysterols. LXR activation upregulates the E3 ligase IDOL, which triggers the ubiquitination and degradation of LDLR. The LDLR family is essential for importing extracellular cholesterol into the cell. Therefore, LXR activation ultimately decreases cholesterol import and intracellular levels. GBM cells do not produce enough oxysterols. This results in inactive LXR and increased import of cholesterol. It follows therefore that forced activation of LXR even in the absence of oxysterols, via LXR agonists, may be beneficial in depleting cholesterol from GBM cells and inhibits growth (Guo et al., 2011; Mita et al., 2018; Nguyen et al., 2019; Villa et al., 2016).
The effect of such agonists in preclinical models of GBM seems to vary, with one study using orthotopic GBM tumors and the brain-penetrant LXR agonist, LXR-623, enhancing GBM cell death and significantly increasing survival when used on its own (Villa et al., 2016). In a different GBM model, LXR-623 alone did not significantly affect tumor growth (Nguyen et al., 2019). However, in a subcutaneous GBM model, it did significantly inhibit tumor growth when combined with B cell lymphoma-extra large (BCL-xL) inhibition (Nguyen et al., 2019), suggesting that combination therapies may be required. Another more potent LXR agonist, RGX-104, currently in a phase 1 clinical trial in patients with lymphoma or advanced solid tumors (Clinicaltrials.gov, NCT02922764), has shown promise in various preclinical tumor models, including GBM (Tavazoie et al., 2018).
Given the known dependency of GBM cells to glucose metabolism, there are many ongoing investigations on the therapeutic potential of a ketogenic diet for GBM patients. Ketogenic diets are based on low carbohydrates and high-fat uptake and have been used as a therapy for epilepsy (Simeone et al., 2017). The rationale behind its use in GBM is that while neurons and glial cells use glucose as a primary energy source they can also switch to fatty acid use under glucose deprivation (Panov et al., 2014). Since GBM cells are glucose-addicted, they would suffer from a glucose-restricted diet. However, a multitude of studies now indicate that GBM cells can use fatty acids as an energy source (Guo et al., 2013; Shakya et al., 2021), and that under nutrient stress, they may hydrolyze lipid droplets and use their fatty acid content (Jarc and Petan, 2019).
A recent study demonstrated that the ketogenic diet, on its own, did not slow GBM growth, whereas the inhibition of the rate-limiting enzyme in fatty acid oxidation, carnitine palmitoyltransferase 1A (CPT1A), did (Sperry et al., 2020). This study suggests that it is very likely that GBM cells will be able to switch to fatty acid oxidation as their energy source in replacement of glucose. Another study demonstrated that lipid loading in GBM leads to decreased survival, by potentiating the paracrine activation of stromal cells, such as macrophages, and angiogenesis (Offer et al., 2019). Clearly, more investigations are needed on the therapeutic potential of the ketogenic diet in combination with standard therapies, or fatty acid oxidation inhibition (Sperry et al., 2020).
Summary
The current knowledge from published studies strongly supports the notion that robust metabolic plasticity can act as a (flexible) shield against the cytotoxic effects of standard GBM therapies, thus driving therapy resistance. Relevant to RT, two recent studies using personalized genome-scale metabolic flux models to determine the role that tumor metabolism plays in RT resistance across different tumor types, including GBM, identified tumor redox metabolism as a major predictor for RT resistance (Lewis and Kemp, 2021; Lewis et al., 2021). It was demonstrated that radioresistant tumors reroute metabolic fluxes to boost the levels of reducing factors of the cell, most notably NADPH and GSH, thus enhancing clearance of ROS that are generated during RT (Lewis et al., 2021).
Conceptually, these studies posit that the demand for antioxidants to neutralize radiation-induced ROS could outstrip the existing cellular supplies of tumors and that radioresistant tumors compensate by rerouting metabolic fluxes to efficiently fuel antioxidant pathways. Such appreciation also reveals an Achilles' heel, opening up the possibility that interventions designed to target regulators of “metabolic plasticity,” rather than specific metabolic pathways, have the potential to improve the therapeutic outcomes in GBM (van Gisbergen et al., 2021). Since metabolic plasticity is made possible by the redundancy in metabolic pathways, such an approach might be even more effective in the context of loss of isozyme diversity (LID).
An exciting recent study by Marczyk et al. (2022) systematically analyzed isozymes affected by cancer-specific LID (relative to a corresponding normal tissue) across a broad range of tumor types and demonstrated that LID may have unsalvageable effects on the redundancy of cancer cell metabolism. Although GBM was not included in the study, LID seems to affect ∼5%–30% of tumor isozymes (Marczyk et al., 2022). If LID exists in GBM metabolic enzymes and their intersection with therapy-induced metabolic rewiring is understood, targeting pathways regulated by enzymes with LID would present a promising strategy for creating lethal bottlenecks in metabolic plasticity.
We pose therefore that identifying and appropriately targeting the molecular regulators of metabolic plasticity in GBM to force “metabolic rigidity,” especially as it applies to radiation response with regard to maintaining redox homeostasis and properly and swiftly repairing potentially lethal DNA damage, hold promise for further improving this already effective form of therapy in GBM and by extension, patients' outcomes.
Footnotes
Acknowledgment
All schematics were created with BioRender.com
Authors' Contributions
Conceptualization, writing, editing, figure design, and preparation: J.B. and E.V. Funding acquisition: E.V.
Author Disclosure Statement
The authors have no conflict of interest.
Funding Information
E.V. and J.B. were supported by the National Cancer Institute CA251872 (E.V.).