
Abstract
Significance:
Autophagy is a self-degrading process that determines cell fate in response to various environmental stresses. In contrast to autophagy-mediated cell survival, the signals, mechanisms, and effects of autophagy-dependent cell death remain obscure. The discovery of autophagy-dependent ferroptosis provides a paradigm for understanding the relationship between aberrant degradation pathways and excessive lipid peroxidation in driving regulated cell death.
Recent Advances:
Ferroptosis was originally described as an autophagy-independent and iron-mediated nonapoptotic cell death. Current studies reveal that the level of intracellular autophagy is positively correlated with ferroptosis sensitivity. Selective autophagic degradation of proteins (e.g., ferritin, SLC40A1, ARNTL, GPX4, and CDH2) or organelles (e.g., lipid droplets or mitochondria) promotes ferroptosis by inducing iron overload and/or lipid peroxidation. Several upstream autophagosome regulators (e.g., TMEM164), downstream autophagy receptors (e.g., HPCAL1), or danger signals (e.g., DCN) are selectively required for ferroptosis-related autophagy, but not for starvation-induced autophagy. The induction of autophagy-dependent ferroptosis is an effective approach to eliminate drug-resistant cancer cells.
Critical Issues:
How different organelles selectively activate autophagy to modulate ferroptosis sensitivity is not fully understood. Identifying direct protein effectors of ferroptotic cell death remains a challenge.
Future Directions:
Further understanding of the molecular mechanics and immune consequences of autophagy-dependent ferroptosis is critical for the development of precision antitumor therapies. Antioxid. Redox Signal. 39, 79–101.
Introduction
Cellular homeostasis is an essential prerequisite for living cells to perform their biological functions, which requires autophagy to participate in the degradation of intracellular waste, including unused proteins or damaged organelles. In response to environmental stimuli, mammalian cells have evolved different types of autophagy, including microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy (hereafter referred to as autophagy). Autophagy can be further divided into bulk autophagy and selective autophagy according to the types and characteristics of the degraded substrates (Dikic and Elazar, 2018).
Substances degraded by autophagy can often be recycled for macromolecular synthesis and energy production (Kroemer et al, 2010). Thus, autophagy is generally a survival mechanism by which cells protect themselves from damage, such as starvation, hypoxia, and drug toxicity. However, when autophagy exceeds its physiological function, unlimited autophagy can lead to cell damage or death, known as autophagy-dependent cell death (Tang et al, 2019).
Ferroptosis is a nonapoptotic form of regulated cell death primarily driven by oxidative damage from iron overload, leading to lipid peroxidation (Tang and Kroemer, 2020). Since the autophagy inhibitor chloroquine cannot block the activity of ferroptosis activators in RAS-mutated cancer cells, ferroptosis has historically been described as a type of autophagy-independent cell death (Dixon et al, 2012). However, recent genetic evidence suggests that the onset and execution of ferroptosis may require autophagic machinery to trigger iron accumulation and/or lipid peroxidation.
Compared with previous reviews (Liu et al, 2020a; Zhou et al, 2020), we not only summarize the latest progress in the process and regulation of autophagy and ferroptosis in the last 3 years, but also discuss the dual role of autophagy-dependent ferroptosis in tumorigenesis and therapy. This knowledge may aid the design of future clinical trials to target autophagy-dependent cell death in patients with cancer.
The Dynamic Process and Regulation of Autophagy
This dynamic process of autophagy is complex and can be roughly divided into four stages: autophagy initiation and phagophore formation, autophagosome assembly and formation, the fusion of autophagosomes with lysosomes, and cargo degradation (Fig. 1). Two protein complexes are activated to participate in the induction of the formation of phagophores, namely the unc-51–like autophagy-activating kinase (ULK) complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (Kang et al, 2018b).

The ULK1 complex integrates signals from two master regulators or sensors of nutrient (mammalian target of rapamycin kinase [MTOR]) and energy stress (AMP-activated protein kinase [AMPK]) to initiate autophagy in response to changes in intracellular energy and nutrient status (Lahiri et al, 2021; Xie et al, 2015). Two ubiquitin-like conjugation systems are responsible for phagophore elongation to form autophagosomes. The first system involves the covalent conjugation of ATG12 and ATG5, assisted by the E1-like enzyme ATG7 and the E2-like enzyme ATG10.
The second system involves the processing and lipidation of ubiquitin-like microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), which is translated as pro-MAP1LC3, followed by the cleavage of the C-terminal polypeptide by ATG4 (a cysteine protease), exposing the glycine site and distributing it to the cytoplasm as MAP1LC3-I. MAP1LC3-I covalently associates with phosphatidylethanolamine (PE) in the presence of the E1-like enzyme ATG7 and the E2-like enzyme ATG3 to form the membrane-associated protein MAP1LC3-II, leading to subsequent autophagosome formation and cargo protein degradation (Nakatogawa, 2013; Scherz-Shouval et al, 2019).
Autophagosomes combine with lysosomes to produce autolysosomes. This process is regulated by a variety of non-ATG proteins, such as lysosome-associated membrane protein 2 (LAMP2), the GTPase-activating protein RAB7A, the homotypic fusion and protein sorting complex, and the soluble N-ethylmaleimide–sensitive fusion factor attachment protein receptor family (Bernard and Klionsky, 2015; Chi et al, 2019; Shen et al, 2021; Tian et al, 2021; Tian et al, 2020). The degradation of cargo in lysosomes is mediated by lysosomal acid enzymes, including cathepsins, acid phosphatases, and lipases (Saftig and Haas, 2016; Zhang et al, 2022a).
Notably, the inner membrane of autophagosomes, including MAP1LC3-II, is also degraded by lysosomal enzymes. Therefore, the reduction of MAP1LC3-II does not necessarily represent an inhibition of autophagic activity. The monitoring of autophagic flux used in conjunction with late-stage autophagy inhibitors may be one of the best ways to assess autophagic activity (Klionsky et al, 2021).
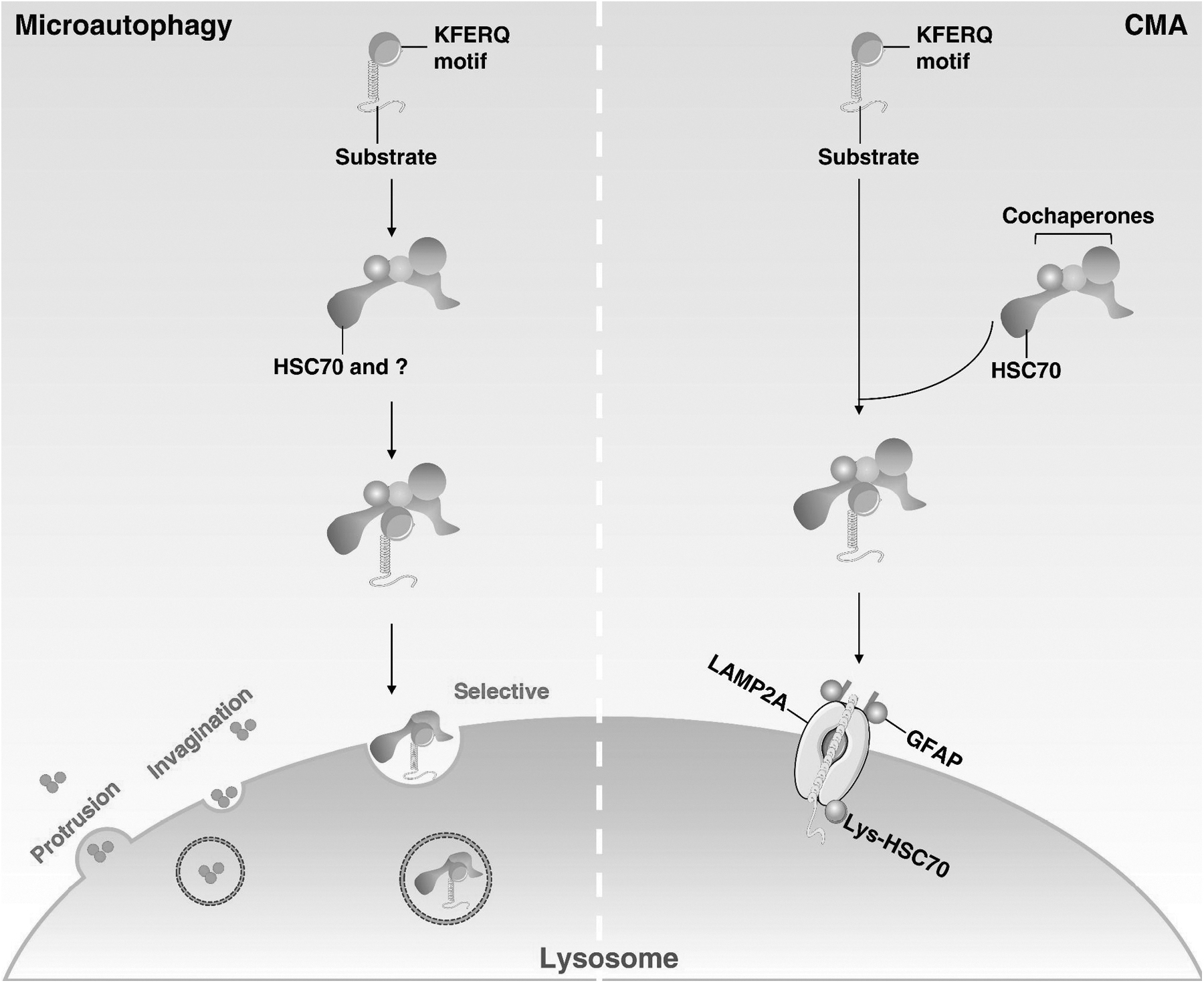
In contrast, CMA and microautophagy have different processes and mechanisms (Fig. 2). CMA only degrades proteins with a specific pentapeptide KFERQ motif, which is specifically recognized by the heat shock homologous 71 kDa protein (HSC70, also known as HSPA8) and further regulated by lysosome-associated membrane protein 2 (LAMP2A) in the lysosomes (Kaushik and Cuervo, 2018). Microautophagy is a process of direct phagocytosis of degraded cargo through lysosomes or late endosomes (Wang et al, 2022).
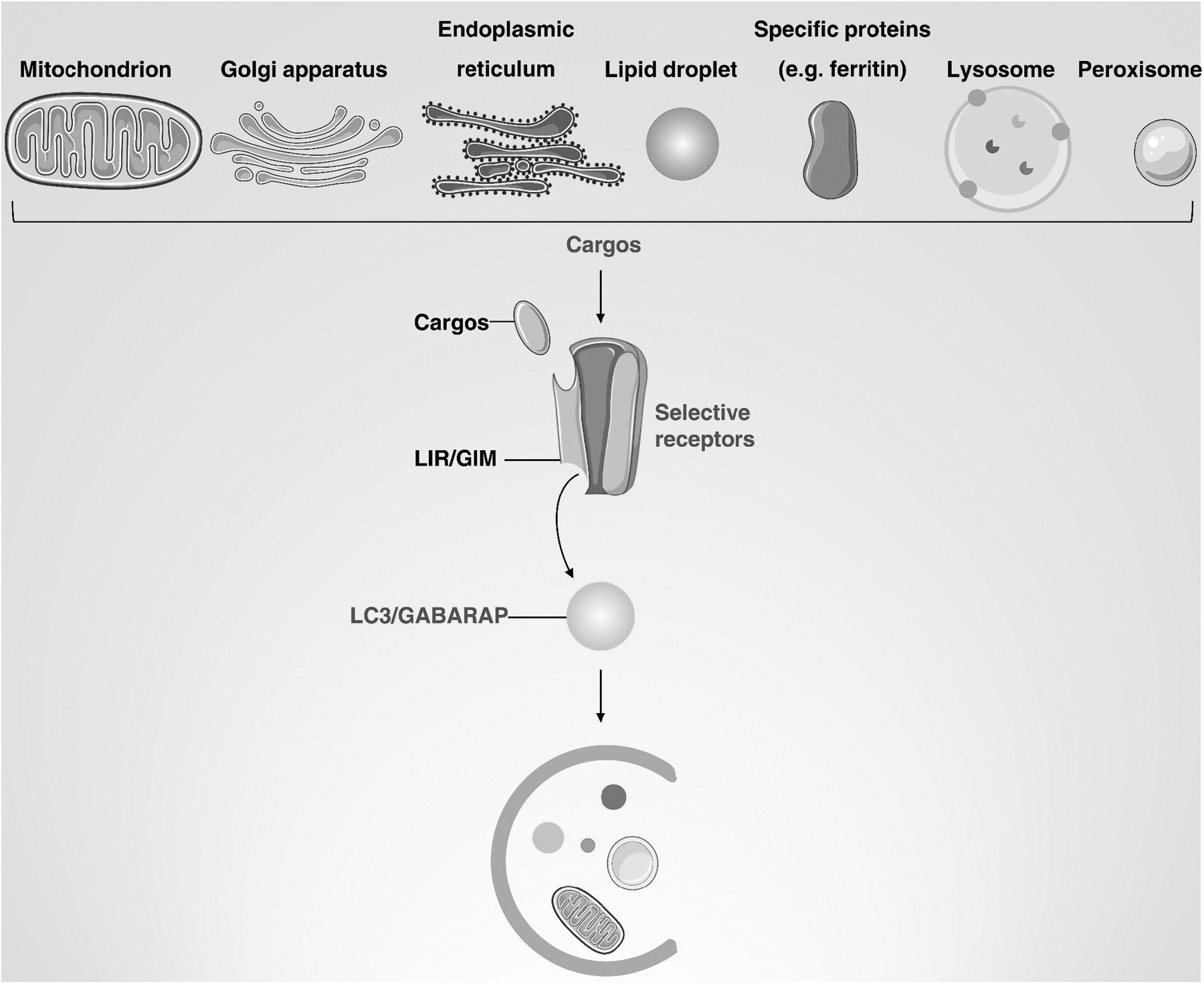
The Receptor and Cargo Recognition of Selective Autophagy
The binding of specific autophagy receptors to cargo is critical to ensure selectivity and response to various stresses. Autophagy receptors can be divided into two categories: ubiquitin-binding receptors, which recognize ubiquitinated cargoes, and cargo-localizing receptors, which localize directly to degraded cargoes. Selective autophagy receptors contain an Atg8-interacting motif/LC3-interacting region (AIM in yeast/LIR in mammalian cells) to bind the lipidated LC3/GABARAP/Atg8 protein family (Chen et al, 2022b). Below, we describe selective autophagy receptors and highlight the cargo-LIR-Atg8/LC3 interaction (Fig. 3).
Selective autophagy receptors
The first selective autophagy identified was the cytoplasm-to-vacuole transport of Saccharomyces cerevisiae, involving Atg11- and Atg19-dependent aminopeptidase 1, aspartate aminopeptidase, and α-mannosidase (Ape1, Ape4, and Ams1, respectively) processing (Baba et al, 1997). Other kinds of selective autophagy in budding yeast also rely on Atg receptors, which, in addition to Cue5, normally bind Atg8 and Atg11 (Table 1) (Lu et al, 2014).
Selective Autophagy Receptors in Mammals and Yeast
In mammalian cells, the receptors involved in the selective autophagy pathway are more complex and diverse, and >30 receptors have been studied. The selective removal of mitochondria by autophagy, that is, mitophagy, has the most studied type of autophagy receptor. Mitophagy can be induced by the recognition of ubiquitinated mitochondrial proteins and has a well-characterized mechanism involving PTEN-induced putative kinase 1 (PINK1) and Parkin RBR E3 ubiquitin protein ligase (Parkin) proteins.
PINK1 is a mitochondrial serine/threonine kinase with a mitochondrial targeting sequence, while Parkin is an E3 ubiquitin ligase present in the cytosol (Lazarou et al, 2015). Under normal nonstress conditions, PINK1 protein is rapidly degraded by its translocation from mitochondria to cytosol. Conversely, when mitochondria are damaged, the depolarization of the membrane potential promotes the accumulation and activation of PINK1 in the mitochondrial outer membrane, leading to the subsequent recruitment and activation of Parkin.
Activated Parkin can polyubiquitinate multiple mitochondrial protein substrates, causing autophagosomes to target mitochondria in the presence of other autophagy receptors, including sequestosome 1 (SQSTM1, best known as p62) (Geisler et al, 2010), optineurin (OPTN) (Heo et al, 2015), nuclear dot protein 52 (NDP52) (Di Rita et al, 2021), TAX1-binding protein 1 (TAX1BP1) (Fan et al, 2022), or neighbors of BRCA1 gene protein (NBR1) (Turco et al, 2021). PINK1 also induces mitophagy in a Parkin-independent manner, highlighting the complexity of the mitophagy machinery (Xie et al, 2021; Xie et al, 2020).
LC3 proteins or LC3 receptors can induce autophagy through a ubiquitin-independent mechanism, which involves a direct association of LC3 proteins with mitochondrial proteins that include BCL2-interacting protein 3-like (BNIP3L, also known as NIX), FUN14 domain containing 1 (FUNDC1), BCL2-interacting protein 3 (BNIP3), nucleotide-binding domain and leucine-rich repeat containing protein X1 (NLRX1), Bcl-2–like protein 13 (BCL2L13), FK506-binding protein 8 (FKBP8), prohibitin-2 (PHB2), and cardiolipin (Chu et al, 2013). In other types of selective autophagy, different receptors mediate different sorts of cargo degradation, and thus participate in the regulation of cellular homeostasis, as summarized in Table 1.
Atg8/LC3 family proteins and LIR interaction
The binding of cargo-containing autophagy receptors to the Atg8/LC3 family of proteins ensures autophagy selectivity. In yeast, there is only one Atg8 protein, whereas mammalian cells contain two Atg8 protein families, MAP1LC3 and GABA type A receptor-associated protein (GABARAP), which are further subdivided into seven isoforms: MAP1LC3A, MAP1LC3B1, MAP1LC3B2, MAP1LC3C, GABARAPL, GABARAPL1, and GABARAPL2. Mammalian selective autophagy receptors bind LC3/GABARAP proteins through conserved LIR and GABARAP-interacting motif (Wirth et al, 2019).
The core motif of LIRs and GABARAPs is [W/F/Y]-XX-[L/V/I], where X represents any amino acid, and [W/F/Y] and [L/V/I] interact with the gap between the N-terminal α-helix and the ubiquitin fold of the Atg8/LC3 family and within the ubiquitin fold (Rogov et al, 2017). In addition, LIRs contain residues that can be phosphorylated to increase interaction with the Atg8/LC3 family (Chino et al, 2022).
The lipidated Atg8/LC3 family binds to autophagy receptors via LIR docking site-LIR, thereby promoting the recruitment of core autophagic components and effectively expanding autophagosome formation. Several mammalian autophagy proteins also have LIR motifs, such as ULK1 and ATG13. Similarly, ATG14, BECN1, and VPS34 of the PtdIns3K complex also bind to the Atg8/LC3 family through LIR motifs (Grunwald et al, 2020).
ATG4B interacts with the Atg8/LC3 family through catalytic binding sites and LIR motifs. LIR-containing proteins preferentially bind GABARAP and GABARAPL1 (Birgisdottir et al, 2019). In mammalian cells, autophagosome formation and lysosome fusion are impaired by the lack of MAP1LC3 proteins, and GABARAP family proteins act as the corresponding compensatory mechanisms (Nguyen et al, 2016). Thus, targeting a single member of the Atg8/LC3 family may be not sufficient to shut down bulk autophagy, although it is possible to inhibit specific selective autophagy in the short term.
Autophagy in Cell Death
Historically, autophagic cell death has been considered a type 2 cell death defined based on ultrastructural morphology, accompanied by abundant autophagic vacuoles (e.g., autophagosome and autolysosome) in the cytoplasm (Kroemer and Levine, 2008; Zhang et al, 2013). The current view is that modest induction of autophagy generally promotes survival, whereas unrestricted autophagy leads to cell death.
The concept of “autophagy-dependent cell death” is defined in terms of the consequences of activation of the autophagy pathway to promote cell death (Kriel and Loos, 2019). Mechanistically, autophagy-dependent cell death is mediated by the components of autophagy machinery, including ATG genes and autophagic protein complexes (Galluzzi et al, 2018). This regulated cell death is associated with human diseases, including cancer.
The induction of autophagy-dependent cell death provides a therapeutic option for cancers that evade apoptosis, and the mechanisms for inducing autophagy-dependent cell death involve multiple layers: (1) lysosomal overload-mediated cytotoxicity (Kessel et al, 2012), (2) energy deficiency due to active mitophagy (Xie et al, 2021), and (3) the degradation of negative regulators of cell death (Hou et al, 2010).
Although both ferroptosis and autophagy have their own unique features (Table 2), accumulating evidence indicates that ferroptosis is a type of autophagy-dependent cell death (Liu et al, 2020a; Zhou et al, 2020). First, ferroptosis activators lead to autophagosome formation and increase autophagic flux (Gao et al, 2016; Hou et al, 2016). Second, increased autophagosome formation positively correlates with susceptibility to ferroptosis induction (Li et al, 2021d).
The Difference and Relationship Between Autophagy and Ferroptosis
Third, genetic deletion of core regulators of autophagy, such as ATG5, ATG7, and BECN1, suppresses ferroptosis induction (Gao et al, 2016; Hou et al, 2016). Fourth, pharmacological inhibition of autophagy prevents ferroptotic injury or death in various disease models such as liver injury, tumorigenesis, pancreatitis, and neurodegradative diseases (Chen et al, 2022d; Liu et al, 2021c, Liu et al, 2021d; Yang et al, 2022a; Zhang et al, 2020c). Further elucidating the molecular and cellular links between degradation systems and ferroptosis may provide new targets for the treatment of diseases (Chen et al, 2021f).
The Defense and Induction of Ferroptosis
Ferroptosis is an oxidative cell death that is primarily regulated by antioxidant defenses (Fig. 4), although other signals or molecules are also involved in the control of ferroptosis sensitivity (Fig. 5). Below, we summarize the major antioxidant systems and mechanisms that control ferroptosis.
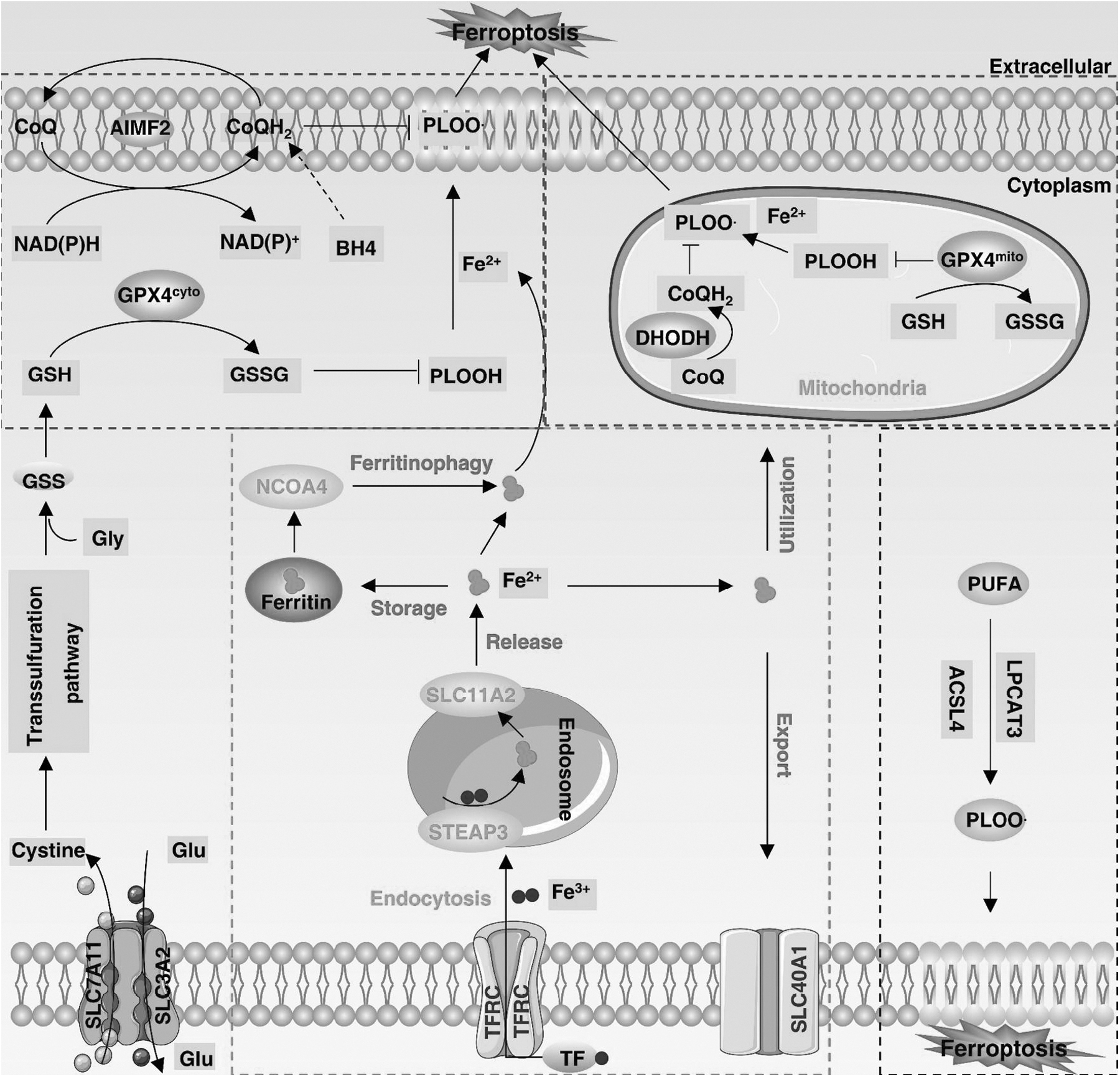
The GPX4-GSH antioxidant system
Glutathione peroxidase 4 (GPX4) is the only GPX member in mammalian cells that converts phospholipid (PL) hydroperoxides to alcohols (Yang et al, 2014). Despite differences in distribution (cytosolic, mitochondrial, and nuclear GPX4), cytosolic GPX4 plays a major role in the inhibition of ferroptosis, based on the finding that the re-expression of cytosolic GPX4 rescues Gpx4 deletion-induced ferroptosis in mouse embryonic fibroblasts (Yant et al, 2003). In some cases, mitochondrial GPX4 plays a role in blocking mitochondrial oxidative damage-induced ferroptosis in cancer cells (Mao et al, 2021a).
The antiferroptosis activity of GPX4 requires a cofactor of reduced glutathione (GSH), a tripeptide derived from glycine, glutamate, and cysteine, of which cysteine is the rate-limiting precursor (Aquilano et al, 2014). Most cells acquire cysteine through system xc−-mediated uptake and subsequent transformation of cystine, a complex consisting of solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2).
Nutritional deprivation of cysteine or the pharmacological inhibition of the SLC7A11-GPX4 pathway (using erastin or RSL3) can trigger ferroptosis in various cells (Tang et al, 2021). Although GPX4 is considered a central repressor of ferroptosis, the conditional inactivation of GPX4 does not always induce ferroptosis, suggesting the existence of a GPX4-independent pathway (Kang et al, 2018a; Viswanathan et al, 2017).
The AIFM2-CoQH2 antioxidant system
CRISPR-Cas9 screening has revealed apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as FSP1) as a ferroptosis defense protein parallel to the GPX4 system (Doll et al, 2019). Mechanistically, N-myristoylation is required for AIFM2 translocation from mitochondria to the plasma membrane, which inhibits lipid peroxidation and ferroptosis by reducing ubiquinone (also known as coenzyme Q or CoQ) to ubiquinol (CoQH2) via its NADH:ubiquinone oxidoreductase activity (Doll et al, 2019).
CoQH2 can trap lipid peroxyl radicals to arrest lipid autoxidation or indirectly oxidized α-tocopheryl radicals (vitamin E, a lipid antioxidant), thus functioning as a nonmitochondrial electron transport chain (Bersuker et al, 2019). AIFM2 also acts as a vitamin K reductase, reducing vitamin K to hydroquinone (VKH2) and protecting GPX4-depleted cells from harmful lipid peroxidation and ferroptosis (Mishima et al, 2022).
In addition to these reported enzymatic functions, AIFM2 can activate endosomal sorting complexes required for transport-III–dependent membrane repair mechanisms to inhibit ferroptosis (Dai et al, 2020c). Taken together, these results highlight three distinct molecular and cellular mechanisms of AIFM2 against ferroptosis.
The DHODH-CoQH2 antioxidant system
Mitochondria are the regulatory centers of cellular metabolism and death. During oxidative phosphorylation, a large amount of reactive oxygen species (ROS) is generated in the electron transport chain located in the inner membrane. Lipid peroxidation occurs when the mitochondrial antioxidant system is out of balance. Dihydroorotate dehydrogenase (DHODH) is an enzyme that controls pyrimidine synthesis in the inner mitochondrial membrane and can transfer electrons to CoQ in the inner mitochondrial membrane for reduction to CoQH2.
DHODH-mediated CoQH2 production is enhanced to compensate for lipid peroxidation induced by GPX4 inactivation, an effect rescued only by mitochondrial GPX4 or DHODH, but not cytosolic GPX4 (Mao et al, 2021b). Furthermore, mitochondrial StAR-related lipid transfer domain containing 7 (STARD7) can mediate the translocation of CoQ to the plasma membrane to activate subsequent CoQH2 production during ferroptosis (Deshwal et al, 2023).
This finding establishes a mitochondrial antioxidant defense mechanism that prevents ferroptosis. In addition, the GTP cyclohydrolase 1 (GCH1)–tetrahydrobiopterin (BH4) and the Kelch-like ECH-associated protein 1 (KEAP1)–NFE2-like BZIP transcription factor 2 (NFE2L2) pathways play a content-dependent antioxidant role in blocking ferroptosis (Kraft et al, 2020; Sun et al, 2016b).
Iron toxicity in ferroptosis
Iron is an indispensable trace element for living organisms and exists in two redox states: ferrous (Fe2+) or ferric (Fe3+). The maintenance of iron homeostasis is important for oxygen transport, electron transfer, and DNA synthesis. This homeostasis process involves the uptake, utilization, storage, and export of iron (Crielaard et al, 2017; Torti et al, 2018). Cells acquire transferrin (TF)-bound Fe3+ through transferrin receptor (TFRC)-mediated endocytosis, and Fe3+ is reduced to Fe2+ by STEAP3 metalloreductase in the endosome, and then released into the cytosol via solute carrier family 11 member 2 (SLC11A2).
The labile Fe2+ can be stored by ferritin (including ferritin heavy polypeptide 1 [FTH1] and ferritin light polypeptide 1 [FTL1] subunits). Fe2+ is also involved in oxygen transport and the production of mitochondrial iron–sulfur clusters. Excess Fe2+ is exported out of the cell via solute carrier family 40 member 1 (SLC40A1), producing Fe3+. Thus, the dysregulation of any steps in iron metabolism may affect ferroptosis sensitivity (Chen et al, 2021e).
Mechanistically, iron contributes to ferroptosis not only by producing ROS through a nonenzymatic Fenton reaction, but also by being a cofactor of metabolic enzymes (such as arachidonate lipoxygenase [ALOX] and cytochrome P450 oxidoreductase [POR]) to induce lipid peroxidation (Ajoolabady et al, 2021). Thus, increased intracellular iron overload can trigger ferroptosis.
For example, nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy degrades ferritin, resulting in iron release into the labile pool and the promotion of ferroptosis in pancreatic cancer cells (Gao et al, 2016; Hou et al, 2016). In contrast, glutamate oxaloacetate transaminase 1 (GOT1) inhibits ferritinophagy-mediated ferroptosis in pancreatic cancer cells (Kremer et al, 2021).
Lipid toxicity in ferroptosis
Unrestricted lipid peroxidation and the production of toxic lipid metabolites are hallmarks of ferroptosis. Arachidonic acid (AA) and adrenic acid (AdA) are the lipids most susceptible to peroxidation, thus causing cell membrane rupture. The metabolism of polyunsaturated fatty acid (PUFA)-PL synthesis involves two key mediators, namely acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). ACSL4 catalyzes the linkage of PUFAs (AA or AdA) with CoA to produce PUFA-CoAs.
Subsequent esterification of PUFA-CoAs with PL via LPCAT3 results in PUFA-PLs (Dixon et al, 2015; Yuan et al, 2016). In addition, acetyl CoA carboxylase (ACC)-catalyzed carboxylation of acetyl CoA to produce malonyl-CoA is required for the synthesis of some PUFAs (Lee et al, 2020). Thus, the inactivation of ACSL4 or LPCAT3 can prevent or reduce ferroptosis sensitivity, although there is also an LPCAT3- or ACSL4-independent pathway (Lin et al, 2022; Magtanong et al, 2022).
Lipoxygenases (ALOXs), a nonheme iron-dependent dioxygenase that can directly oxidize PUFA-containing lipids in biological membranes, play a content-dependent role in ferroptosis. For example, ALOX12 and ALOX15 are dispensable for ferroptosis in mice (Friedmann Angeli et al, 2014). These data suggest that other regulators (e.g., POR) contribute to lipid peroxidation during ferroptosis.
Monounsaturated fatty acids produced by stearoyl-CoA desaturase 1 can completely inhibit PUFA-mediated ferroptosis (Magtanong et al, 2019). In addition, 4-hydroxy-2-nonenal, a well-known by-product of lipid peroxidation, has recently been implicated as a direct mediator of ferroptosis, underscoring that the effector of ferroptosis may not be a protein (Chen et al, 2022c).
The Mechanism of Autophagy-Dependent Ferroptosis
Depending on the context, autophagy plays a dual role in inhibiting or promoting cell death. Here, we outline the known mechanisms by which autophagy regulates ferroptosis in cancer cells (Fig. 6).

NCOA4-mediated ferritinophagy
Ferritinophagy is a selective autophagy that degrades ferritin, a complex composed of FTH1 and FTL1 (Fig. 6A). NCOA4 is a cytosolic autophagy receptor that binds to key surface arginine in FTH1 to mediate ferritinophagy-dependent ferritin degradation, thereby promoting ferroptosis (Hou et al, 2016). The knockdown of NCOA4 rescues erastin-induced ferritin degradation, and subsequent iron accumulation and ferroptosis in multiple cancer cell lines (e.g., HT1080 and PANC1). The inhibition of ATG genes (e.g., ATG3, ATG5, ATG7, and ATG13) had similar effects on iron-dependent ferroptosis (Xie et al, 2016). As a feedback mechanism, NCOA4 can be degraded by the ubiquitin–proteasome pathway in response to high levels of intracellular iron (Mancias et al, 2015).
Different signal pathways can promote or inhibit ferritinophagy-dependent ferroptosis. For example, O-GlcNAcylation of serine179 residues of FTH1 inhibits FTH1 binding to NCOA4 during RSL3-induced ferroptosis, whereas pharmacological or genetic inhibition of O-GlcNAcylation enhances ferritinophagy as well as mitophagy-dependent ferroptosis (Yu et al, 2022).
Similarly, poly(rC)-binding protein 1 (PCBP1) inhibits ferritinophagy-dependent ferroptosis from the translational level by directly binding to CU-rich elements on the 3′ untranslated region (3′-UTR) of BECN1 mRNA, thus suppressing the activation of autophagy (Lee et al, 2022). Hypoxia downregulates NCOA4 and increases FTH/FTL protein levels, thus inhibiting ferritinophagy (Fuhrmann et al, 2020). In contrast, ferritinophagy-mediated ferroptosis can be regulated by ELAV-like RNA-binding protein 1 (ELAVL1/HuR), which is required for sorafenib-induced human hepatic stellate cell fibrosis (Zhang et al, 2018).
Unlike starvation-induced autophagosome formation, which requires ATG9A, transmembrane protein 164 (TMEM164) selectively mediates autophagosome formation to degrade ferritin, GPX4, and lipid droplets (LDs) during ferroptosis in cancer cells (Liu et al, 2022a). In addition, SQSTM1-dependent autophagic degradation of SLC40A1 overcomes ferroptosis resistance in human pancreatic cancer cells by increasing intracellular iron overload (Li et al, 2021d).
Bafilomycin A1, a lysosomal V-ATPase inhibitor, inhibits ferritin degradation and ferroptosis (Gao et al, 2018), whereas dihydroartemisinin (DHA) induces ferritinophagy-dependent ferroptosis (Lin et al, 2016). Together, these data suggest that ferritinophagy plays a major role in promoting ferroptosis by increasing the accumulation of toxic iron. Because NCOA4 is widely expressed in cancer and noncancer cells, targeting NCOA4-dependent ferritinophagy or ferroptosis in cancer therapy may require careful monitoring of toxicity (Santana-Codina et al, 2022).
RAB7A-mediated lipophagy
Fatty acids are esterified intracellularly to triglycerides and cholesterol esters, which undergo a complex process in the endoplasmic reticulum (ER) and are stored in a unique spherical organelle, the LD. In response to stress (e.g., starvation or RSL3), LDs can be engulfed by autophagosomes for lysosomal breakdown, thereby releasing free fatty acids. Lipophagy-mediated degradation of LDs promotes RSL3-induced ferroptosis in primary mouse hepatocytes and human hepatoma cell line HepG2, which is regulated by the LD cargo receptor RAB7A (Fig. 6B) (Bai et al, 2019; Schroeder et al, 2015).
Mechanistically, RAB7A selectively recognizes LDs and mediates ATG5-dependent autophagosome formation. Thus, the knockdown of ATG5 or RAB7A prevents RSL3-induced ferroptosis in vitro and in vivo.
In addition, the knockdown of tumor protein D52 (TPD52), a regulator of LD formation, enhances RSL3-induced ferroptosis, whereas the overexpression of TPD52 limits ferroptosis (Kamili et al, 2015). These results reveal that increasing LD storage can prevent lipid toxicity, whereas lipophagy increases ferroptosis sensitivity by releasing more lipids for subsequent lipid peroxidation. Further identification of the signals and mediators of lipophagy is important for the development of ferroptosis-related therapies in LD-rich cancers, such as liver cancer.
SQSTM1-mediated clockophagy
Circadian rhythm is an endogenous mechanism regulated by circadian clock proteins, that is, aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL) and clock circadian regulator (CLOCK), which control various cellular processes, including iron and lipid metabolism (Fig. 6C).
Although ARNTL degradation is largely dependent on the ubiquitin–proteasome pathway, an autophagy-dependent degradation pathway, called clockophagy, was found to specifically degrade ARNTL1 and promote ferroptosis in cancer cells in response to GPX4 inhibitors (e.g., RSL3 and FIN56), but not SLC7A11 inhibitors (e.g., erastin, sulfasalazine, and sorafenib) (Yang et al, 2019).
Mass spectrometry analysis revealed that SQSTM1 acts as an autophagy receptor that binds ARNTL to ATG5- and ATG7-dependent autophagosomes for lysosomal degradation. In contrast, ATG9A is not required for autophagosome formation during this process. Mechanistically, the degradation of ARNTL represses target gene egl-9 family hypoxia-inducible factor 2 (EGLN2, also known as PHD1) expression, which controls hypoxia-inducible factor 1 subunit alpha (HIF1A)-mediated lipid metabolism (Yang et al, 2019).
In addition, the knockdown of the circadian protein PER1 inhibits hippocampal neuronal autophagy, which may contribute to ferroptosis vulnerability during cerebral ischemia (Rami et al, 2017). The ferroptosis inhibitor liproxstatin-1 blocks L-arginine–induced acute pancreatitis in pancreatic Arntl-specific knockout mice (Liu et al, 2020b). These results demonstrate a new link between circadian rhythms, autophagy, and ferroptosis.
Additional work is required to assess whether regulators of ferroptosis are controlled by circadian rhythms. Furthermore, extracellular SQSTM1 induces ACSL4 expression to enhance autophagy-dependent ferroptosis and subsequent pancreatitis (Yang et al, 2022a), providing alternative strategies to mediate ferroptosis-induced sterile inflammation.
CMA- and autophagy-mediated GPX4 degradation
Both CMA and autophagy mediate GPX4 degradation to enhance ferroptosis in cancer cells (Fig. 6D). The autophagy inducer rapamycin and the ferroptosis inducer RSL3 can inactivate MTOR and degrade GPX4, suggesting that ferroptosis may be regulated by autophagy-mediated GPX4 degradation (Liu et al, 2021d). Heat shock protein 90 (HSP90) was identified as a molecular chaperone that binds to the CMA cargo receptor LAMP2A and thus mediates GPX4 degradation during ferroptosis, whereas the inhibition of HSP90 blocks CMA-mediated ferroptosis in mouse neuronal cell line HT-22 (Wu et al, 2019).
In addition, HSP90 stabilizes erastin-induced ACSL4 expression by dephosphorylating Ser637 at dynamin 1 like (DNM1L, a member of the dynamin superfamily of GTPases), thereby inducing ferroptosis in glioma (Miao et al, 2022). In contrast, HSPA5 (an ER-associated molecular chaperone) prevents erastin-induced ferroptosis in human pancreatic cancer cell lines by blocking the degradation of GPX4 protein (Zhu et al, 2017). These findings suggest that different HSP family proteins exert different molecular chaperone functions to enhance or inhibit GPX4 protein degradation.
HPCAL1-mediated CDH2 degradation
There are many forms of autophagy, and a key unresolved question is whether there are specific autophagy receptors that only target ferroptosis. A recent membrane protein screening combined with functional studies showed that hippocampal calcin-like protein 1 (HPCAL1), as a specific autophagy receptor, promotes ferroptosis in multiple cancer cells by directly mediating the degradation of cadherin 2 (CDH2) protein, but not the degradation of ferritin, SLC40A1, or GPX4. HPCAL1 is also not required for starvation-induced autophagy and cancer cell apoptosis (Fig. 6E).
Notably, the known function of HPCAL1 as a Ca2+-binding protein is not essential for autophagy-dependent ferroptosis. Subsequent analysis of kinase activity revealed that binding to LC3 requires protein kinase C theta (PRKCQ)-mediated phosphorylation of Thr149 on HPCAL1 for further recognition and degradation of CDH2 (Chen et al, 2022d). The finding of HPCAL1-mediated degradation of CDH2 provides new insight for understanding the specific role of autophagy-dependent ferroptosis in cancer therapy.
STAT3-mediated lysosomal membrane permeabilization
A lysosomal luminal iron-mediated Fenton response leads to increased lysosomal membrane permeability (LMP) and subsequent cell death (Gao et al, 2018). In contrast, lysosomal ferritin can reduce oxidative stress. LMP leads to the leakage of lysosomal contents, such as cathepsin B (CTSB), cathepsin D (CTSD), and iron, thereby promoting ROS production and subsequent ferroptosis (Fig. 6F), which may involve V-ATPase–mediated selective autophagy (Chen et al, 2022a). Ferroptosis can also be regulated by erastin-induced lysosomal cell death.
Mechanistically, signal transducer and activator of transcription 3 (STAT3) induces the expression of CTSB, which is required for lysosomal cell death, whereas pharmacological suppression (using CA-074Me, a cathepsin inhibitor) or genetic suppression of STAT3 limits ferroptosis in cancer cells (Gao et al, 2018).
In 5-FU–resistant gastric cancer, STAT3 negatively regulates ferroptosis by binding to consensus DNA response elements in the promoters of genes (e.g., GPX4, SLC7A11, and FTH1) involved in the negative regulation of ferroptosis (Ouyang et al, 2022). These results suggest that STAT3 plays a content-dependent role in ferroptosis, and the cargo receptors and mechanisms mediating STAT3 degradation require further exploration.
Mitophagy-dependent ferroptosis
Mitophagy is one of the quality control mechanisms of mitochondria, thereby regulating mitochondrial ROS production and cell death sensitivity (Jiao et al, 2021). PINK1-Parkin–dependent or –independent mitophagy is the central mechanism of mitochondrial quality control. However, the relationship between mitochondria and ferroptosis is uncertain and even paradoxical. For example, BAY 87-2243 (a mitochondrial respiratory chain inhibitor) mediates mitochondrial ROS production and subsequent ferroptosis by inducing excessive mitophagy in BRAFV600E melanoma cell lines, whereas the overexpression of GPX4 or the administration of ferrostatin-1 reverses BAY 87-2243–induced ferroptosis (Basit et al, 2017).
Conversely, a loss of function of fumarate hydratase, a mitochondrial tricarboxylic acid cycle component, confers resistance to ferroptosis induced by cysteine deprivation (Gao et al, 2019). WJ460, a compound that interacts with myoferlin protein, induces mitophagy and mitochondrial fission, leading to an increased sensitivity in pancreatic cancer cells to ferroptosis (Rademaker et al, 2022).
Moreover, zalcitabine-induced mitochondrial DNA stress triggers autophagy-dependent ferroptosis through the stimulator of interferon response cGAMP interactor 1 (STING1) pathway (Li et al, 2021b), which plays an important role in infection and immunity (Chen et al, 2022b; Zeng et al, 2017; Zhang et al, 2022b; Zhang et al, 2020b). STING1 also binds with MFN1/2 to enhance mitochondrial fusion during ferroptosis (Li et al, 2021a) (Fig. 6G). These studies increase our understanding of the interplay between mitophagy, mitochondrial dynamics, DNA sensor, and ferroptosis (Zhang et al, 2021). However, whether specific autophagy receptor-mediated mitophagy is involved in ferroptosis remains to be further explored.
BECN1-mediated system xc− inhibition
BECN1 is a multifaceted protein that, in addition to inducing autophagy, plays an important role in ferroptosis regulation (Kang et al, 2018b). In response to SLC7A11 inhibitors (e.g., erastin, sulfasalazine, and sorafenib), but not GPX4 inhibitors (e.g., RSL3 and FIN56), BECN1 binds directly to SLC7A11, resulting in cysteine deprivation-dependent ferroptosis (Kang et al, 2018b; Song et al, 2018) (Fig. 6H). Furthermore, the phosphorylation of BECN1 at S90 and S93 induced by AMPK promotes the formation of the BECN1-SLC7A11 complex, and mutations at these phosphorylation sites prevent ferroptosis.
The BECN1 activator Tat-beclin 1 protein peptide enhances autophagy and ferroptosis-induced tumor suppression in mice (Song et al, 2018). However, the mechanism by which AMPK selectively activates BECN1 remains unclear. In addition, the RNA-binding protein ELAVL1/HuR promotes autophagy activation by binding AU-rich elements within the 3′-UTR F3 of BECN1 mRNA, thus inducing autophagy-dependent ferroptosis in human hepatic stellate cells, which may be enhanced by exosome-mediated delivery of BECN1 secreted by human umbilical cord mesenchymal stem cells (Tan et al, 2022; Zhang et al, 2018).
BECN1-mediated autophagy-dependent ferroptosis is further demonstrated in spinal cord ischemia–reperfusion injury, which binds ubiquitin-specific protease 11 (USP11) to prevent its degradation (Rong et al, 2022). Thus, BECN1 can mediate ferroptosis by directly limiting SLC7A11 activity or by activating an autophagy-dependent pathway.
Reticulophagy-related ferroptosis
The ER is an organelle composed of membranes and tubules responsible for the synthesis of proteins and lipids (such as triglycerides), and is dynamically modulated to meet different cellular requirements. Unfolded proteins that accumulate during protein synthesis induce the unfolded protein response (UPR) to mitigate cellular damage. Conversely, excess UPR induces cell death (Oakes and Papa, 2015). Selective removal of ER by autophagy, namely reticulophagy, occurs during ER stress. Reticulophagy removes either unnecessary parts of the ER or proteins that the UPR cannot process (Cinque et al, 2020; Zielke et al, 2021).
A series of autophagy receptors mediate reticulophagy, including cell-cycle progression 1 (CCPG1) (Smith et al, 2018), reticulophagy regulator 1 (RETREG1, also known as FAM134B) (Khaminets et al, 2015), atlastin GTPase 3 (ATL3) (Chen et al, 2019), and testis-expressed 264 ER-phagy receptor (TEX264) (Chino et al, 2019). Interestingly, sorafenib induces FAM134B-mediated reticulophagy, thereby limiting ferroptosis in hepatocellular carcinoma cells.
Although the effector mechanism remains unclear, FAM134B may be transcriptionally activated by poly(A)-binding protein cytoplasmic 1 (PABPC1) through its mRNA binding during ferroptosis (Liu et al, 2022d) (Fig. 6I). These findings also suggest that different types of selective autophagy may play dual roles in the control of ferroptosis sensitivity.
Autophagy-Dependent Ferroptosis in Cancer
Ferroptosis offers a new therapeutic option to clear drug-resistant cancer cells, especially those that evade apoptosis. However, tumor heterogeneity presents challenges for ferroptosis therapy (Li et al, 2021d). Autophagy-dependent ferroptosis may not only provide more effective treatment designs for clinical trials, but also reduce or delay the emergence of drug resistance or side effects. In addition, autophagy-dependent ferroptosis plays a content-dependent role in regulating tumor immunity and macrophage polarization (Fig. 7).

Drug resistance
Most chemotherapeutic anticancer drugs exert their anticancer effects by inducing apoptosis. However, cancer cells are prone to apoptotic evasion through different mechanisms, such as autophagy-mediated degradation of proapoptotic proteins (Liu et al, 2020a). Although combining the chemotherapy drugs with some autophagy inhibitors (e.g., hydroxychloroquine) can enhance the drugs' anticancer effect, the therapeutic process can be hindered by side effects and off-target effects caused by autophagy inhibitor-mediated inactivation of lysosomal acidification (Fernandez, 2017).
Several stress proteins may limit the therapeutic effect of ferroptosis induction in animals or patients. For example, sorafenib-induced upregulation of metallothionein-1G (MT-1G), RSL3, or erastin-induced nuclear protein 1, transcriptional regulator (NUPR1), and pirin expression, and high expression of the TYRO3 protein tyrosine kinase drive ferroptosis resistance in mice or cancer patients (Hu et al, 2021; Huang et al, 2021; Jiang et al, 2021; Liu et al, 2021b; Sun et al, 2016a).
A complex tumor microenvironment, such as increased formation of cancer-associated fibroblasts, can create favorable conditions for evading ferroptosis induction (Zhang et al, 2020a). Different oncogenes or tumor suppressor genes can selectively control ferroptosis sensitivity by integrating the autophagy response (Li et al, 2021b, 2021c; Liu et al, 2021a).
The upregulation of components of the autophagy-dependent ferroptosis pathway in tumors, such as SLC40A1, NCOA4, TFRC, and BECN1, may provide unique targets for overcoming ferroptosis resistance (Chen et al, 2021b). For example, human retinoblastoma cells can develop multidrug resistance through autophagy. In contrast, 4-octyl itaconate induces ferritinophagy-dependent ferritin degradation to promote ferroptosis, thereby eliminating multidrug-resistant human retinoblastoma cells (Liu et al, 2022c).
Although it is unclear whether this model is applicable to other drug-resistant cancer cells, the induction of autophagy-dependent ferroptosis provides a new strategy for overcoming cancer drug resistance. Furthermore, DHA induces AMPK-mediated autophagic ferritin degradation, leading to ferroptosis in acute myeloid leukemia cells (Du et al, 2019).
In contrast, cell division cycle 25A (CDC25A) blocks autophagy-dependent ferroptosis in cervical cancer cells by upregulating Erb-B2 receptor tyrosine kinase 2 (ERBB2) expression through the dephosphorylation of pyruvate kinase M2 (PKM2) (Wang et al, 2021) (Fig. 7A). These findings highlight the signaling and regulatory networks of autophagy-dependent ferroptosis in drug-resistant cells involving stress responses and metabolic reprogramming.
Tumor immune microenvironment
Unlike robust cell death induction, which is an established method of removing cancer cells, chronic cell death-mediated inflammation results in an immunosuppressive tumor microenvironment. Several damage-associated molecular patterns (DAMPs) released by ferroptotic cells play a dual role in tumor immunity, leading not only to inflammation-related immunosuppression but also to immunogenic cell death to suppress tumor growth. Elevated autophagy of tumor cells plays a protumor role in TME.
The antitumor effect of autophagy deficiency on pancreatic tumor growth is seen in immunoreactive mice but not in immunodeficient mice (Eng et al, 2016; Yamamoto et al, 2020). This suggests a potential influence of the immune microenvironment on the role of autophagy in tumorigenesis. Targeting autophagy-dependent ferroptosis also requires consideration of the tumor immune microenvironment (Chen et al, 2021a).
Macrophages are characterized by their diversity and plasticity, and can rapidly transform in response to specific environmental stimuli, a process called polarization. They can be simply classified as M1 (proinflammatory macrophages) and M2 (anti-inflammatory macrophages) based on their genetic profiles and metabolic adaptations. M2 macrophages can be further divided into four types, M2a, M2b, M2c, and M2d, based on surface markers, functions, and secreted mediators.
Specifically, M2d macrophages, also known as tumor-associated macrophages (TAMs), are present in the tumor microenvironment, and lead to tumor angiogenesis and tumor growth (Yang et al, 2022b). A direct link between ferroptosis and TAMs has been established in pancreatic cancer. Indeed, in response to oxidative damage, ferroptotic pancreatic ductal adenocarcinoma (PDAC) cancer cells release KRASG12D protein in an autophagy-dependent manner, which is then taken up by surrounding macrophages in an advanced glycosylation end product-specific receptor (AGER)-dependent manner (Dai et al, 2020b).
The uptake of extracellular KRASG12D protein promotes protumor-like polarization of macrophages through STAT3-dependent fatty acid oxidation (Dai et al, 2020b). In a KRASG12D-driven mouse model of spontaneous pancreatic tumors, pancreatic ferroptosis injury caused by the conditional knockout of GPX4 results in the release of the DNA oxidation product 8-hydroxy-2'-deoxyguanosine (8-OHdG), thereby activating STING1-dependent macrophage polarization to produce an inflammation-related immunosuppression microenvironment (Dai et al, 2020b) (Fig. 7B).
In the tumor microenvironment, immunogenic cell death plays a major role in restoring a dysfunctional antitumor immune system. The release or exposure of DAMPs by dead or dying cells promotes the maturation and activation of antigen-presenting cells (e.g., dendritic cells and macrophages), which stimulates specific cytotoxic T cell responses to clear cancer cells.
Ferroptotic cells can release the nuclear protein high-mobility group box 1 (HMGB1) and the membrane proteoglycan decorin (DCN) in an autophagy-dependent manner. Extracellular HMGB1 or DCN, acting as DAMPs, activate AGER-dependent inflammation or antitumor immunity (Liu et al, 2022b; Wen et al, 2019) (Fig. 7C). Ferroptotic cells also have the ability to suppress dendritic cell antigen presentation to activate cytotoxic T cell (CTL) (Wiernicki et al, 2022), indicating that early or late immune responses to ferroptosis may differ.
Furthermore, HMGB1 is a positive regulator of autophagy under various conditions, including oxidative stress (Kang et al, 2011; Livesey et al, 2012; Tang et al, 2010a, 2010b). These findings also reinforce the notion that DAMPs are important mediators of cell death-related autophagy and immune responses, although their activity depends on the receptor and the tumor microenvironment (Zhang et al, 2013). It is worth noting that a recent study concluded that ferroptotic cancer cell does not have an immunogenicity (Wiernicki et al, 2022), although ferroptotic injury can induce sterile inflammation (Chen et al, 2021c).
Metabolic reprogramming
The ferroptosis mechanism is a complex network regulated by different metabolic pathways, including the levels of amino acids, glucose, NADPH, fatty acids, and GSH. Functionally, these metabolic pathways regulate the levels of intracellular redox species to counteract or induce ferroptosis. Current therapeutic approaches aim to target vulnerability to ferroptosis by inhibiting metabolic pathways or increasing selected dietary nutrients.
For example, tumor cells in an acidic environment preferentially take up more n-3 and n-6 PUFAs, and incorporate them into LDs to counteract ferroptosis, and ferroptosis inducers significantly increase the cytotoxicity of PUFAs (Dierge et al, 2021). Similarly, interferon gamma (IFNγ) from CD8+ T cells binds AA from the microenvironment to induce immunogenic tumor ferroptosis via ACSL4 (Liao et al, 2022). These findings establish a new link between tumor immunometabolism and cell death.
As a feedback mechanism, cancer cells can activate autophagy to shape the metabolic demands of ferroptosis. For example, the RNA-binding protein partner of NOB1 (PNO1) inhibits autophagy-mediated ferroptosis in hepatocellular carcinoma cells through GSH metabolic reprogramming (Hu et al, 2022) (Fig. 7D).
In addition, glucose increases ferroptosis sensitivity in PDAC cells by blocking pyruvate dehydrogenase kinase 4 (PDK4)-mediated inhibition of pyruvate oxidative (Song et al, 2021). The lipid flippase solute carrier family 47 member 1 (SLC47A1) can inhibit ferroptosis in cancer cells by rewiring of PUFA metabolism (Lin et al, 2022). Further understanding of the crosstalk between autophagy and metabolic reprogramming is important to enhance ferroptosis-related therapies.
Nanomedicine therapy
Rapid advances in nanotechnology offer new perspectives for ferroptosis drugs in cancer (Fig. 8). Nanomedicine is a therapeutic approach based on engineered control of drug delivery and release to improve pharmacokinetics. Multifunctional iron-containing nanomaterials have been shown to scavenge GSH, generate hydrogen peroxide, catalyze the Fenton reaction, and synergize with ferroptosis inducers to exert antitumor effects. For example, liposomal nanosystems encapsulating copper peroxide nanodots and artemisinin induce ferritinophagy-dependent ferroptosis (Li et al, 2022b) (Fig. 8A).

Furthermore, induction of autophagy-dependent ferroptosis offers a therapeutic approach for tumors that lack targeting endogenous receptors. For example, ferroptosis inducer RSL3 and ferritinophagy initiator DHA are loaded in nanocomponents. When drug is released, DHA-induced ferritinophagy degrades intracellular ferritin to release stored iron and synergizes with RSL3-mediated GPX4 inhibition to enhance ferroptosis therapy (Li et al, 2022a) (Fig. 8B).
Conversely, copper nanodots based on sonodynamics and catalysis can inhibit autophagy and induce GPX4/SLC7A11 inhibition-dependent ferroptosis (Feng et al, 2022) (Fig. 8C). The heterogeneity of genetic background determines the inconsistency of tumor susceptibility to ferroptosis. Treatment strategies also need to be designed according to the characteristics of the tumor microenvironment. Nevertheless, the biosafety and off-target effects of nanomedicines are still issues that cannot be ignored.
Conclusion and Perspectives
In the past 5 years, we have witnessed the rapid development of ferroptosis research around the world. The mechanism of ferroptosis is context dependent. Our concepts about ferroptosis and its regulation may differ from the original findings about ferroptosis in 2012. For example, we now know that the modulation of ferroptosis can be independent of GPX4, ACSL4, or ALOX. We also know that ferroptosis is closely related to autophagy machinery in some conditions.
Elevated autophagic activity can promote ferroptosis by selectively degrading antioxidant proteins or organelles. These findings raise questions about the molecular or metabolic checkpoints of autophagy in promoting survival or cell death. In fact, both ferroptosis and autophagy are dynamic processes involving oxidative stress and changes in membrane structure.
Multiple mechanism-mediated oxidative and antioxidant systems are involved in the regulation of ferroptosis and autophagy (Chen et al, 2021e). There is currently an incomplete understanding of how different organelles selectively activate autophagy to regulate ferroptosis sensitivity (Chen et al, 2021d). Identifying different selective autophagy receptors as well as specific degradation substrates remains the key to understanding the mechanism of autophagy-dependent ferroptosis.
Both autophagy and ferroptosis can occur in normal cells or tissues, so developing autophagy- and/or ferroptosis-related targeted therapies for cancer cells presents challenges in assessing risks and benefits (Kang and Tang, 2017). In addition, the role of ferroptosis in noncancerous diseases, such as neurodegenerative diseases and ischemia–reperfusion injury, has been confirmed (Tang et al, 2021).
Brain tissue is highly susceptible to oxidative stress due to its high metabolic activity. Some natural antioxidants or ferroptosis inhibitors protect against neurodegenerative diseases by activating antioxidant pathways, such as the nuclear factor erythroid 2-related factor 2 (NFE2L2, also known as NRF2) pathway (Dai et al, 2020a; Sun et al, 2016b), which is consistent with hormesis-mediated protection against neuroinflammatory injury (Calabrese et al, 2012; Calabrese et al, 2010; Calabrese et al, 2007).
The possible implications of this redox property of ferroptosis signaling in health and disease may be twofold. Furthermore, although DCN is identified as a relatively specific DAMP for ferroptosis injury, the identification of additional biomarkers of autophagy-dependent ferroptosis (including DAMPs) is important for future clinical translational studies. Regardless, understanding the immune properties of autophagy-dependent ferroptotic cell death is required for the development of novel immunochemical antitumor therapies (Chen et al, 2021c).
Footnotes
Acknowledgment
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the article.
Authors' Contribution
J.L. and D.T. developed conceptualization; F.C. and D.T. assisted with writing; X.C., J.L., and R.K. performed review and editing. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
J.L. was supported by grants from the National Natural Sciences Foundation of China (82070613).