
Abstract
Significance:
Oxidative stress refers to excessive intracellular levels of reactive oxygen species (ROS) due to an imbalance between ROS production and the antioxidant defense system. Under oxidative stress conditions, cells trigger various stress response pathways to protect themselves, among which repression of messenger RNA (mRNA) translation is one of the key hallmarks promoting cell survival. This regulation process minimizes cellular energy consumption, enabling cells to survive in adverse conditions and to promote recovery from stress-induced damage.
Recent Advances:
Recent studies suggest that transfer RNAs (tRNAs) play important roles in regulating translation as a part of stress response under adverse conditions. In particular, research relying on high-throughput techniques such as next-generation sequencing and mass spectrometry approaches has given us detailed information on mechanisms such as individual tRNA dynamics and crosstalk among post-transcriptional modifications.
Critical Issues:
Oxidative stress leads to dynamic tRNA changes, including their localization, cleavage, and alteration of expression profiles and modification patterns. Growing evidence suggests that these changes not only are tightly regulated by stress response mechanisms, but also can directly fine-tune the translation efficiency, which contributes to cell- or tissue-specific response to oxidative stress.
Future Directions:
In this review, we describe recent advances in the understanding of the dynamic changes of tRNAs caused by oxidative stress. We also highlight the emerging roles of tRNAs in translation regulation under the condition of oxidative stress. In addition, we discuss future perspectives in this research field. Antioxid. Redox Signal. 40, 715–735.
Introduction
Oxidative stress is defined as “an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control, and/or molecular damage” (Sies, 2020; Sies et al., 2017). This imbalance causes elevated intracellular levels of reactive oxygen species (ROS), resulting in damage to various biomolecules, including proteins, lipids, and nucleic acids.
As oxidative stress is linked to the pathophysiology of numerous diseases such as diabetes, cancers, and neurodegenerative disorders, it is important to better understand the mechanism of how cells cope with oxidative stress.
As protein synthesis is a complex, energy-consuming process that is essential to maintain cellular homeostasis, this process is strictly controlled to produce required proteins at optimal rates without wasting energy. When cells encounter a variety of intracellular and environmental stresses, including oxidative stress, they trigger stress response pathways to protect themselves until the stress is removed.
One of the major hallmarks of cellular stress response is repression of mRNA translation initiation. Protein synthesis is globally reduced in response to stress, whereas the translation of a subset of messenger RNAs (mRNAs) is maintained or paradoxically upregulated. Such mRNAs generally encode proteins responsible for cell survival, stress coping, and recovery.
By prioritizing the translation of stress-responsive genes, cells can not only survive in adverse conditions but also save energy and nutrients for recovery (Costa-Mattioli and Walter, 2020; Pakos-Zebrucka et al., 2016; Wek et al., 2023).
Recent studies have focused on non-canonical roles of transfer RNAs (tRNAs). Beyond their canonical role as adapter molecules delivering amino acids to the ribosome during protein synthesis, tRNAs are now known to play critical roles in cell signaling, survival, and apoptosis, especially under stress conditions (Su et al., 2020).
Growing evidence shows that oxidative stress induces dynamic and diverse changes in tRNAs, including nuclear accumulation, alteration of expression profiles, cleavage, modifications, misacylation, and inhibition of tRNA splicing. Interestingly, evidence is accumulating that these changes can, in turn, modulate translation rates, likely contributing to cell- or tissue-specific responses to oxidative stress.
Here, we outline the current understanding of a variety of changes in tRNAs in response to oxidative stress. We also describe the functional crosstalk among oxidative stress, tRNA metabolism, and translation.
Overview of ROS, and Experimental Models of Oxidative Stress
ROS are produced during normal aerobic metabolism in cells. The most common ROS include superoxide anion radicals (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH). Superoxide anions are produced by mitochondria and NADPH oxidases.
Once generated, superoxide is rapidly metabolized into H2O2 by superoxide dismutases. H2O2 can be further converted to •OH in the presence of ferrous ions (Fe2+), a process known as Fenton reaction. As hydroxyl radicals indiscriminately damage biomolecules due to their highly oxidizing activity, cells possess various enzymes that convert H2O2 to H2O such as glutathione peroxidase, peroxiredoxins, and catalase, preventing the accumulation of cytotoxic levels of H2O2 (Schieber and Chandel, 2014; Winterbourn, 2018). H2O2 also plays an important role as a regulatory molecule in redox signaling at physiological concentrations.
Increased levels of H2O2 modulate the activity of enzymes and transcription factors, resulting in the maintenance of redox homeostasis (Schieber and Chandel, 2014; Sies, 2017; Winterbourn, 2018). It should be noted that such physiological protein oxidation by H2O2 is reversible. However, an excess amount of H2O2 can cause cell death due to irreversible damage to biomolecules.
As H2O2 is chemically stable and commercially available at an affordable price, in addition to being a central mediator of redox signaling, exogenous H2O2 treatment has been widely used to mimic cellular oxidative stress. Because H2O2 is not only a major biological ROS but also permeable to the plasma membrane, intracellular oxidative stress can be induced by exogenous H2O2 treatment, which enables us to examine the direct effect of intracellular H2O2 on cellular stress responses.
However, the results obtained using exogenous H2O2 treatment should be carefully interpreted because the addition of exogeneous H2O2 to the cell does not necessarily reflect the cellular response to endogenously produced H2O2 (Forman, 2007). One reason for caution in H2O2 stress interpretation is the difficulty in setting optimal intracellular H2O2 levels.
Because the plasma membrane retards diffusion, there is a more than 100-fold concentration gradient between intracellular and extracellular H2O2 levels (Sies, 2017; Winterbourn, 2018). Also, intracellular H2O2 levels can be increased by uptake mediated by Aquaporins (Bienert and Chaumont, 2014). These factors make it difficult to estimate actual intracellular H2O2 levels. Another challenge in determining the effects of H2O2 stress is that spatial distribution of H2O2 is not uniform in the cell.
The H2O2 levels vary significantly depending on the localization of sources of ROS and antioxidant systems, compounding difficulties caused by the gradient between intracellular and extracellular levels. To both maximize the efficiency of redox signaling and minimize the adverse effects, H2O2-mediated oxidation of target proteins generally takes place close to the sources of H2O2.
Exogenous H2O2 treatment may increase the intracellular H2O2 in some locations to supraphysiological levels, and may not affect other locations at all. Finally, as H2O2 is generated from superoxide, exogenous H2O2 treatment cannot mimic actual intracellular superoxide generation and its impact on cellular stress response.
Arsenic compounds, especially sodium arsenite (SA), are also some of the most common chemicals used as an oxidative stress inducer. It is believed that arsenic compounds induce increased cellular production of various ROS such as peroxyl radicals (ROO•), superoxide anion radicals (O2 •−), singlet oxygen (1O2), hydroxyl radicals (•OH), and H2O2, although the exact mechanism has yet to be fully elucidated (Jomova et al., 2011).
Arsenic compounds also decrease the activity of antioxidant enzymes, such as glutathione reductase, glutathione peroxidase, and thioredoxin reductase (Shen et al., 2013). However, it is worth noting that arsenic compounds do not function just as an oxidative stress inducer. Arsenic compounds, especially trivalent inorganic arsenite, bind to sulfhydryl groups in various proteins with high affinity, resulting in the inhibition of a variety of enzymes (Shen et al., 2013).
One study identified 360 arsenic-binding proteins using human proteome microarray. Among them, the authors showed that hexokinase-2, a rate-limiting enzyme in the glycolytic pathway, is inhibited by arsenic in human cells (Zhang et al., 2015). Although oxidative stress induces cleavage of tRNAs (see Cleavage section), SA-induced tRNA cleavage can take place independently of oxidative stress (Akiyama et al., 2022b).
Therefore, when arsenic compounds are used as an oxidative stress inducer, it should be carefully considered whether the results are due to genuine oxidative stress or pleiotropic effects through binding to various proteins.
Two Major Stress-Responsive Pathways Involved in Translation Inhibition
When cells encounter various stress stimuli, including oxidative stress, they rapidly repress global protein synthesis. Mechanistically, most of such repression comes from inhibition of the initiation step of translation. Such inhibition saves energy used for protein synthesis toward future repair of cellular damage.
Indeed, stress-induced attenuation of protein synthesis directly contributes to cell survival (Han et al., 2013). Here, we overview the molecular mechanism of translational regulation under both physiological and stress conditions.
In eukaryotes, canonical translation initiation starts with the formation of the ternary complex (TC) comprising the eukaryotic initiation factor 2 (eIF2), guanosine 5′-triphosphate (GTP), and charged initiator methionine tRNA (tRNAiMet). The TC next binds to the 40S ribosomal subunit associated with eIF1, eIF1A, eIF3, and eIF5, forming the 43S preinitiation complex (43S PIC).
In parallel with 43S PIC assembly, the trimeric eIF4F complex (consisting of a cap-binding eIF4E, a scaffolding protein eIF4G, and an RNA helicase eIF4A) binds to the 7-methylguanosine (m7G) cap structure at the 5′-end of mRNAs. The 43S PIC is then recruited to the 5′-end of mRNAs through the interaction of eIF3 with eIF4G. Once attached, the 43S PIC starts to scan down the mRNA until it finds the AUG start codon.
The interaction between the start codon and the anticodon of tRNAiMet in the favorable nucleotide context (“Kozak sequence”) terminates the scanning. It then triggers several processes such as the hydrolysis of eIF2-bound GTP and the release of GDP-bound eIF2, disassociation of eIF1, eIF1A, and eIF5, and the recruitment of 60S ribosomal subunit.
This results in the assembly of the 80S ribosome, which is competent for translation elongation (Jackson et al., 2010; Leppek et al., 2018). As the binding of eIF4F to the 5′-cap of mRNA is essential for this canonical translation initiation, this mechanism is commonly known as “cap-dependent translation” (Fig. 1). Although cells need to cope with a wide range of environmental and physiological changes, they generally modulate activities of two common adaptive pathways in response to diverse stress conditions, called the integrated stress response (ISR) and the mechanistic target of rapamycin complex 1 (mTORC1) pathway.
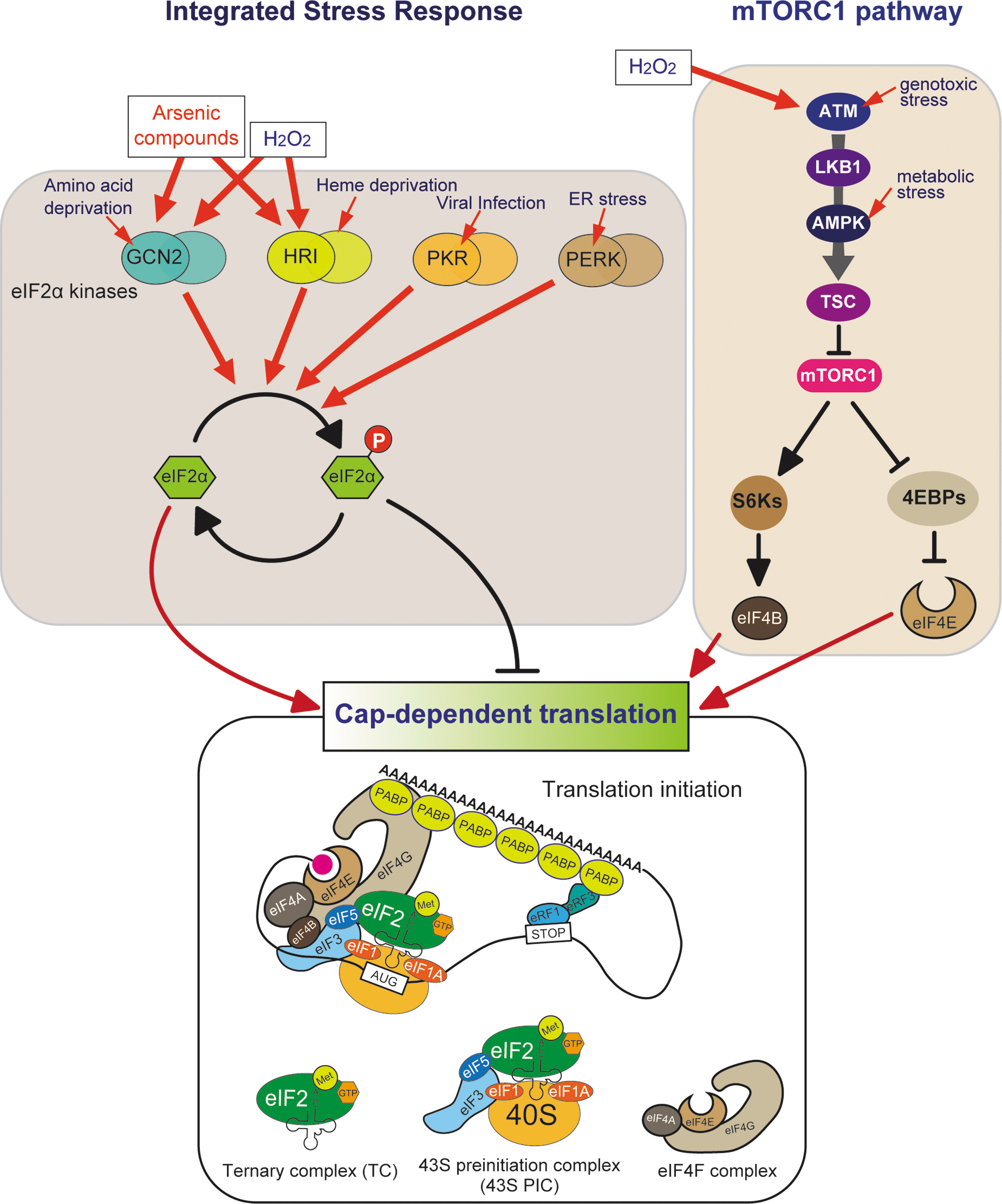
Both pathways target the initiation step of cap-dependent translation, implying the importance of strict regulation of translation initiation under stress conditions, although other less common protein synthesis pathways also exist (Bhatter et al., 2023).
The ISR is an evolutionarily conserved signaling pathway in eukaryotes. The ISR modulates activities of the key molecule of translation initiation, the initiator tRNAiMet (Fig. 1) (Costa-Mattioli and Walter, 2020; Pakos-Zebrucka et al., 2016), and this issue by (Wek et al., 2023). In mammals, the ISR is triggered by four different protein kinases: general control nonderepressible 2 (GCN2), heme-regulated inhibitor (HRI), protein kinase R (PKR), and PKR-like endoplasmic reticulum kinase (PERK).
All four kinases sense distinct environmental and physiological stresses. GCN2 is activated in response to amino acid deprivation and ultraviolet (UV) irradiation. Although HRI is typically activated by heme deprivation in erythroid cells, it is also activated by other stress stimuli such as oxidative stress, osmotic stress, and heat shock. PKR is activated by the presence of double-stranded RNAs during viral infection. PERK is activated in response to endoplasmic reticulum (ER) stress, which is caused by the accumulation of abnormal unfolded proteins in the ER. Both SA and H2O2 trigger the ISR via the activation of GCN2 and HRI (Fig. 1) (Taniuchi et al., 2016; Zhan et al., 2004).
Although these kinases are activated by distinct stress stimuli, once activated, they all specifically phosphorylate the serine 51 residue of the α-subunit of eIF2α. Phosphorylated eIF2α (p-eIF2α) inhibits the nucleotide exchange from GDP to GTP mediated by guanine nucleotide exchange factor eIF2B. Since the exchange of eIF2-bound GDP for GTP is the rate-limiting step for assembly of active TC, eIF2α phosphorylation efficiently causes global repression of cap-dependent translation initiation.
Paradoxically, the translation of a subset of mRNAs is however upregulated, causing preferential expression of stress-responsive genes necessary for cell survival. The mechanisms of changes of gene expression profiles under stress conditions are comprehensively reviewed in this issue by (Wek et al., 2023).
The other major stress-responsive pathway is the mTORC1 pathway. The mTOR is a serine/threonine kinase belonging to PI3K-related protein kinases (PIKK) family that constitutes two distinct complexes, mTORC1 and mTORC2. mTORC2 promotes cell proliferation and migration mainly through phosphorylation of Akt, a key effector of the insulin/PI3K (phosphatidylinositol 3-kinases) signaling pathway. On the other hand, mTORC1 plays a central role in regulating cellular metabolism such as the synthesis of proteins, lipids, and nucleotides.
To maintain balance between anabolism and catabolism, mTORC1 senses cellular nutrients such as ATP, glucose, and amino acids (Liu and Sabatini, 2020; Saxton and Sabatini, 2017).
mTORC1-mediated translational regulation largely depends on its two key targets, eIF4E-binding proteins (4EBPs) and S6 kinases (S6Ks). The unphosphorylated form of 4EBPs suppresses translation by preventing the assembly of eIF4F complex through the binding to eIF4E, whereas mTORC1-mediated 4EBP phosphorylation causes its dissociation from eIF4E, thus enhancing cap-dependent translation initiation.
The phosphorylated form of S6Ks activates eIF4B, a positive regulator of the interaction between eIF4F complex and the 5′-cap of mRNAs. Therefore, under nutrient-rich conditions, mTORC1 promotes cap-dependent translation through phosphorylation-mediated 4EBP inactivation and S6K activation. Under various stress conditions, mTORC1 is inactivated mainly by tuberous sclerosis complex (TSC), a key negative regulator of mTORC1 signaling. Various metabolic and environmental stresses converge on TSC activation.
For example, metabolic stress stimuli, such as glucose deprivation and low ATP levels, are sensed by AMP-activated protein kinase (AMPK), which phosphorylates and activates TSC, resulting in mTORC1 inactivation. The elevated levels of ROS are sensed by the protein encoded by ataxia telangiectasia mutated (ATM) gene (Alexander et al., 2010; Guo et al., 2010), an upstream component of the mTORC1 pathway that senses genotoxic stresses (Shiloh and Ziv, 2013).
Although ATM exists as an inactive homodimer under normal conditions, oxidative stress activates ATM homodimers through intermolecular disulfide bond formation (Guo et al., 2010). Active ATM homodimers phosphorylate downstream targets, liver kinase B1 (LKB1) and AMPK, resulting in the inhibition of mTORC1 via the LKB1-AMPK-TSC signaling cascade (Alexander et al., 2010) (Fig. 1).
tRNAs: Basic Characteristics and Transcriptional Repression Under Oxidative Stress
tRNAs are small (ranging from 74 to 90 nucleotides) non-coding RNAs whose main function is to deliver amino acids to the ribosomes and aid in the decoding of mRNA codons (Berg and Brandl, 2021). tRNAs are the second most abundant RNA species following ribosomal RNAs (rRNAs) in the cell, representing 10–15% of total RNA (by mass). tRNAs are also characterized by high diversity and multiplicity.
In the human genome, there are 429 tRNA genes (with ∼half of them functional) according to the genomic tRNA database (Chan and Lowe, 2016), which are classified into subgroups, including isoacceptors and isodecoders. Isoacceptors are defined as tRNAs that are charged by the same amino acid. There are 21 types of isoacceptors because human cells use 21 kinds of amino acids (standard 20 amino acids and selenocysteine [SeC]) to synthesize proteins. Isodecoders are defined as tRNAs that have the same anticodon.
For example, glycine-decoding isoacceptors consist of 28 genes that are classified into three kinds of isoacceptors, tRNAGly-CCC (5 genes), tRNAGly-GCC (14 genes), and tRNAGly-TCC (9 genes) (Chan and Lowe, 2016). The tRNAGly-CCC isodecoders are further classified into three types (tRNAGly-CCC-1, tRNAGly-CCC-2, and tRNAGly-CCC-3) depending on their sequences.
Further, tRNAGly-CCC-1 consists of two identical genes (tRNAGly-CCC1-1 and tRNAGly-CCC-1-2), and similarly, tRNAGly-CCC-2 has two identical genes. In the possible 64 (43) codons, three codons (UAA, UAG, and UGA) are stop codons, thus the other 61 codons should code amino acids. Interestingly, not all the 61 codons have corresponding tRNAs with a complementary anticodon (Rak et al., 2018), although there are enough tRNA genes to fully cover all the codons.
There are only 46 anticodons (except SeC TCA anticodon) in human tRNA genes, which means that the other 15 codons do not have fully complementary tRNAs. Decoding of such codons relies on wobble base-pairing, non-Watson-Crick base-pairing between the third base of the codon in mRNAs, and the first base of the anticodon (position 34) in tRNAs (Quax et al., 2015). In tRNAs, the position 34 (so-called the wobble position) is frequently modified, which further modulates the specificity of the base-pairing between codons and anticodons.
tRNAs are transcribed by RNA polymerase III (Pol III), an RNA polymerase complex comprising 17 subunits (Wang et al., 2022). Pol III is specialized to transcribe short, non-coding RNAs such as 5S rRNA, U6 small nuclear RNA and Y RNAs, as well as tRNAs. These genes are differentially transcribed by three distinct Pol III promoter types (Fig. 2A) (Arimbasseri and Maraia, 2016; Kessler and Maraia, 2021; Schramm and Hernandez, 2002; Wang et al., 2022).

First, the type 1 promoter is used exclusively for the transcription of 5S rRNA genes. 5S rRNA genes have an internal promoter element called internal control region (ICR), which is recognized by a type 1 promoter-specific transcription factor TFIIIA. The interaction between TFIIIA and ICR is an essential step in 5S rRNA transcription. In addition, two transcription factor complexes, TFIIIC and TFIIIBβ, are required to recruit Pol III to transcription start sites (TSS).
Second, all the tRNA genes except the SeC tRNA (tRNASeC) gene are transcribed through the type 2 promoter. tRNA genes have highly conserved internal promoter elements called A box and B box, which are recognized by TFIIIC. While TFIIIA is not required for the type 2 promoter, TFIIIBβ is necessary for the recruitment of Pol III to TSS.
Finally, the type 3 promoter, which is used for the transcription of tRNASeC and other non-coding RNAs such as U6 snRNA and Y RNAs, exists only in higher eukaryotes. In contrast to type 1 and type 2 promoters, the type 3 promoter consists of elements that locate the upstream of TSS, called proximal sequence element (PSE) and TATA box. TATA box is recognized by a type 3 promoter-specific TFIIIB, called TFIIIBα, whereas PSE is recognized by a five-subunit complex called snRNA-activating protein complex (SNAPc) (Fig. 2A).
When transcribed by Pol III, precursor tRNAs (pre-tRNAs) have immature sequences at both 5′- and 3′-end, called 5′-leader and 3′-trailer, respectively. In addition, some pre-tRNAs, such as human pre-tRNATyr and pre-tRNALeu, possess introns (total of 28 in Homo sapiens) (Chan and Lowe, 2016). These redundant sequences must be removed during the maturation process.
Endoribonucleases RNase P and RNase Z are responsible for the cleavage of 5′-leader and 3′-trailer, respectively (Esakova and Krasilnikov, 2010; Rammelt and Rossmanith, 2016). pre-tRNA introns are excised by the tRNA-splicing endonuclease (TSEN) complex, and the resulting exons are ligated by the RTCB complex, an RNA ligase complex whose catalytic subunit is RTCB, the human homolog of bacterial RtcB (Popow et al., 2012).
It is worth noting that loss or decrease of splicing activity may cause the accumulation of pre-tRNA-derived fragments (pre-tRFs) (described later in detail). After splicing as well as 5′- and 3′-end processing, a CCA trinucleotide is post-transcriptionally added by the CCA-adding enzyme tRNA nucleotidyl transferase 1 as the final step of maturation (Rammelt and Rossmanith, 2016).
During the maturation process described earlier, various kinds of post-transcriptional modifications are induced at several positions of tRNAs (Boccaletto et al., 2022).
In addition to the protein synthesis levels, tRNA expression levels are tightly regulated to maintain optimal levels of translation. As expected, Pol III-mediated transcription of tRNA genes is also stress-sensitive, and repressed in response to oxidative stress. Pol III activity is strictly regulated by MAF1, a conserved negative regulator of Pol III (Johnson et al., 2007; Pluta et al., 2001; Upadhya et al., 2002), and MAF1 activity is negatively regulated by mTORC1 (Zhang et al., 2018). Therefore, mTORC1 is an essential positive regulator of Pol III-mediated tRNA transcription.
Under basal conditions, mTORC1 predominantly phosphorylates serine residues in human MAF1 (Ser60, Ser68 and Ser75) (Michels et al., 2010), preventing the interaction between MAF1 and Pol III (Michels et al., 2010; Shor et al., 2010), enabling Pol III to transcribe tRNA genes at optimal levels to maintain cellular homeostasis. Under oxidative stress conditions, MAF1 phosphorylation is inhibited due to mTORC1 inactivation, which leads to nuclear translocation of MAF1, resulting in transcription repression by the direct binding of MAF1 to Pol III (Fig. 2A).
Recent cryo-EM studies have revealed the detailed mechanism of MAF1-mediated Pol III inhibition (Girbig et al., 2021; Vorlander et al., 2020). The yeast Maf1 binds between the clamp, wall, and protrusion domains of Pol III, which blocks the interaction between Pol III and the transcription factor TFIIIB, resulting in the repression of transcription initiation (Vorlander et al., 2020).
It is worth noting that in humans, there are two isoforms of RPC7, one of the Pol III subunits, in contrast to only one in yeast. Girbig et al. showed that RPC7α-containing Pol III is resistant to MAF1-mediated inhibition, whereas Pol III containing RPC7β, which is more closely related to the yeast RPC7/C31, is susceptible to MAF1-mediated repression (Girbig et al., 2021).
As RPC7α-containing Pol III is enriched in embryonic stem cells and cancer cells, and related to cancer cell growth (Haurie et al., 2010), cancer cells may escape MAF1-mediated repression by preferentially selecting RPC7α.
In addition to MAF1, there are additional mechanisms to regulate Pol III-mediated transcription in response to oxidative stress. In the TFIIIB complex for the type 3 promoter (TFIIIBα), Brf1, one of the subunits, is replaced by Brf2 (Ramsay and Vannini, 2018; Willis and Moir, 2018). It has been reported that Brf2 acts as a redox sensor through the conserved cysteine 361 residue (Gouge et al., 2015).
Mechanistically, under oxidative stress conditions, the assembly of TFIIIBα complex is impaired likely through disulfide bond formation between Cys361 and Cys370 residues of Brf2, resulting in the repression of selenocysteine tRNA (tRNASeC) transcription (Fig. 2A). If tRNASeC downregulation is prolonged by severe oxidative stress, cells undergo apoptosis due to the decreased synthesis of selenoproteins, which function as antioxidant factors.
Pol III activity is also repressed by tumor suppressors RB (retinoblastoma) and p53 under oxidative stress conditions (Fig. 2B). Although RB is inactivated by phosphorylation under non-stress conditions, once exposed to oxidative stress, it is activated through dephosphorylation by protein phosphatase 2A (PP2A) (Macleod, 2008). p53 is also inactivated under basal conditions by its negative regulators, MDM2 and MDMX.
MDM2 has an intrinsic E3 ubiquitin ligase activity, leading to ubiquitination-mediated proteosomal degradation of p53. Although MDMX does not have similar intrinsic ubiquitin ligase activity, the hetero-oligomerization of MDM2 and MDMX is important for the effective inhibition of p53 function (Wade et al., 2013). In response to oxidative stress, p53 is post-translationally modified by phosphorylation, methylation, acetylation, and sumoylation.
These modifications lead to the dissociation of p53 from its negative regulators, resulting in the activation of p53 (Liu and Xu, 2011). Activated p53 and RB interact with TFIIIB, repressing Pol III-mediated transcription by sequestering TFIIIB (White, 2004). A recent study showed that Pol III activity can interfere with RNA polymerase II-mediated transcription (Gerber et al., 2020). Such crosstalk is essential for MAF1-mediated transcription repression of tRNAs under serum starvation.
In summary, Pol III activity is constantly monitored and tightly regulated simultaneously by many factors. A variety of transcription regulation mechanisms described here contribute to fine-tuning tRNA expression. Such modulation results in differential transcription regulation of specific tRNA genes in response to various stimuli. Alteration of tRNA gene expression under oxidative stress conditions is further described next.
tRNA Metabolism Under Oxidative Stress
RNAs can be oxidized by ROS, especially through the Fenton reaction. Oxidized RNAs commonly bear 8-hydroxyguanosine as a result of guanosine oxidation. However, tRNAs are not as easily oxidized by ROS compared with rRNAs or mRNAs because tRNAs rarely interact with ferrous iron (Honda et al., 2005). Instead, oxidative stress indirectly induces diverse changes in tRNAs through triggering cellular stress response pathways.
Intriguingly, these changes affect translation, implying that tRNA metabolism contributes to stress-induced translation regulation. Here, we describe a variety of changes in tRNAs induced by oxidative stress and the impact of stress-induced tRNA metabolism on translation regulation. We also summarize representative protein molecules that regulate tRNA metabolism in response to oxidative stress in Table 1.
Representative Molecules That Regulate tRNA Metabolism in Response to Oxidative Stress
AMPK, AMP-activated protein kinase; ANG, angiogenin; ATM, ataxia telangiectasia mutated; mTORC1, LKB, liver kinase B; mTORC, mechanistic (or mammalian) target of rapamycin complex; tDR, tRNA-derived RNA; TFIIIB, transcription factor IIIB complex; TSC, tuberous sclerosis complex.
Expression profiles
The profile of cellular tRNA expression differs significantly between tissues and cells (Dittmar et al., 2006), suggesting that tissue-specific expression patterns may have a role in optimizing translation. Accumulating evidence proposes that such tissue-specific tRNA expression is associated with various diseases. For example, one of five tRNAArg-TCT genes is predominantly expressed in the central nervous system in mice, whereas the other four genes are ubiquitously expressed in all tissues.
This tRNA is proposed to contribute to the maintenance of neural homeostasis and preventing neurodegeneration (Ishimura et al., 2014). Because oxidative stress represses Pol III activity as described earlier, the profile of tRNA expression can be altered by oxidative stress stimuli (Fig. 3A). In Saccharomyces cerevisiae, H2O2 exposure significantly changed the expression levels of many tRNA genes with some tRNAs being significantly upregulated (Pang et al., 2014).

A recent study showed that the alteration of tRNA expression patterns regulates protein synthesis in response to stress in S. cerevisiae (Torrent et al., 2018). For example, if mRNAs encoding stress-responsive genes are enriched for specific codons, stress-induced upregulation of tRNAs decoding these codons facilitates the mRNA translation of these stress-responsive genes, thus contributing to the maintenance of cellular homeostasis.
The comprehensive profiling of cellular tRNA expression is still challenging despite recent advances in high-throughput quantification methods such as small RNA sequencing for tRNAs (tRNA-seq) and tRNA microarrays. For example, tRNA-seq requires reverse transcription (RT) step to prepare cDNA libraries.
Because of the high number of nucleotide modifications and the rigid secondary structure of tRNAs, RT can result in the arrest or nucleotide misincorporation at specific modification sites, which subsequently causes a bias in tRNA expression profile. Various approaches are used to overcome such bias, such as the removal of methylation modifications using a bacterial demethylase AlkB (Cozen et al., 2015; Zheng et al., 2015). Further considerations before (Karaca et al., 2014) and during library preparation (Shigematsu et al., 2017), and a comprehensive computational analysis considering modification status (Behrens et al., 2021), also reduced the bias.
Complementary methods have been developed to quantify tRNAs based on the hybridization approaches, such as classic tRNA microarray (Dittmar et al., 2006), quantification of oligo DNA probes hybridized to tRNAs (Goodarzi et al., 2016), quantification of tRNA expression based on the thermal motion of tRNA-probe hybrids (Jacob et al., 2019), and fluorescence labeling at the 3′-terminus of tRNAs (Nagai et al., 2021). However, hybridization-based methods also have their limitations.
Hybridization efficiency of probes can significantly differ depending on the presence of modifications within target sites, which makes difficult the estimation of the relative abundance of tRNAs. Although there has not been a golden standard method due to limitations across all methodologies, the advances described earlier and comparative analysis of the results across various methods have already led us to less biased profiling of tRNA expression patterns and relative tRNA levels.
Localization
Although tRNAs play an essential role in protein translation in the cytoplasm, it is well known that tRNAs can shuttle between nucleus and cytoplasm. This regulated tRNA transport provides the balance between the import into the nucleus and the export to the cytoplasm, termed “tRNA retrograde nuclear import” and “tRNA re-export,” respectively.
A variety of stress conditions are known to induce the accumulation of mature tRNAs in the nucleus (Fig. 3B), probably because tRNA retrograde nuclear import becomes relatively dominant compared with tRNA re-export under stress conditions (Chatterjee et al., 2018). Although first identified in yeast, nuclear tRNA accumulation also has been observed in mammalian cells under stress conditions, such as amino acid deprivation (Shaheen et al., 2007), nutrition starvation (Dhakal et al., 2019; Huynh et al., 2010), treatment with translation elongation inhibitor puromycin (Barhoom et al., 2011), and heat stress (Watanabe et al., 2013), suggesting that this stress-responsive process is evolutionarily conserved.
In addition, it has been recently reported that oxidative stress (H2O2 treatment) induces the accumulation of selective tRNAs in the nucleus in human cells via ER stress-induced ISR (Schwenzer et al., 2019). Yeast do not have the PERK-eIF2α phosphorylation pathway (Masson, 2019), which might hint at the existence of stress-responsive tRNA transport pathways that are specific to higher eukaryotes.
Inhibition of pre-tRNA splicing
As intron-containing tRNAs must be spliced during their maturation, abnormalities of pre-tRNA splicing machineries can cause the generation of pre-tRFs. It has been reported that mutation of Clp1, one of the components of TSEN complex, causes the accumulation of pre-tRNATyr-derived fragments and a progressive loss of spinal motor neurons in mice (Hanada et al., 2013).
This pre-tRNATyr-derived fragments, comprising 5′-leader followed by 5′-exon sequences of pre-tRNAsTyr, sensitized cells to oxidative stress-induced p53 activation, and hence p53-mediated cell death. Although the mechanism of pre-tRF generation had been unknown, it was recently reported that H2O2 inactivates the enzymatic activity of RTCB ligase complex, inhibiting exon-exon ligation (Fig. 3C) (Asanovic et al., 2021). As H2O2 treatment induced the accumulation of the same pre-tRNATyr-derived fragments as those accumulated in Clp1 mutant mice (Hanada et al., 2013), it is possible that the loss of Clp1 activity causes oxidative stress-mediated inhibition of Rtcb ligase complex.
A recent study showed that H2O2 treatment induces the production of pre-tRFs, including pre-tRNATyr fragments as well as the depletion of some mature tRNAs, especially tRNATyr (Huh et al., 2021). H2O2-induced tRNATyr depletion impaired translation of mRNAs enriched in cognate tyrosine codons. It is likely that mature tRNATyr depletion and pre-tRNA fragments observed in this study are at least partly due to impaired exon ligation caused by H2O2-mediated RTCB inhibition.
Cleavage
tRNAs are rich sources for various tRNA-derived RNAs (tDRs) (Holmes et al., 2023). These tDRs are produced by tRNA cleavage, especially cleavage of mature tRNAs in response to various stress stimuli, including oxidative stress (Fig. 3D) (Akiyama and Ivanov, 2023).
In vertebrates, this stress-induced tRNA cleavage is mediated predominantly by an endoribonuclease angiogenin (ANG), a secreted ribonuclease that is a member of RNase A superfamily (Lyons et al., 2017a; Sheng and Xu, 2016). Two groups independently reported that human ANG cleaves tRNAs within anticodon loops in response to stress, including SA treatment, generating two types of tDRs called tRNA-derived stress-induced RNAs (5′-tiRNAs and 3′-tiRNAs) (Fu et al., 2009; Yamasaki et al., 2009).
Although the nucleolytic activity of ANG toward purified RNAs is very weak (∼105–6-fold lower than that of RNase A), under the condition where tRNAs are physiologically folded, ANG preferentially and efficiently cleaves anticodon loops of tRNAs (Akiyama et al., 2021). It is worth noting that ANG is a vertebrate-specific RNase; however, oxidative stress-induced tRNA cleavage around anticodon loops is an evolutionarily conserved phenomenon (Thompson et al., 2008).
In S. cerevisiae, Rny1, an RNase T2 family member enzyme, is responsible for the cleavage (Thompson and Parker, 2009). Recently, it has been shown that tiRNAs can be generated in an ANG-independent manner (Akiyama et al., 2022a; Su et al., 2019). We showed that SA-induced, ANG-independent tiRNA production largely depends on RNase A superfamily enzymes other than ANG (Akiyama et al., 2022a). Importantly, some tiRNAs directly regulate translation, contributing to the fine-tuning of translational regulation under stress conditions. The precise mechanisms of ANG-mediated tiRNA generation and the roles of tiRNAs in stress response are further described next.
tDRs are also generated even under non-stress conditions. These constitutively produced fragments, often called tRFs, are generally shorter (15–26 nt) than tiRNAs (30–50 nt). Although how these tRFs are generated is yet to be clarified, some of them are suggested to be produced from tiRNAs, implying the possibility that these tRFs may also function in response to stress (Su et al., 2020).
Recent studies showed that tRFs also impact translation by different mechanisms from that of tiRNAs. Because of the similarity to miRNAs in length, tRFs can function in a miRNA-like manner, causing degradation of specific mRNAs (Guan et al., 2020; Kumar et al., 2014). In addition, 5′-end derived tRFs can also repress translation independently of miRNA-like mechanism (Sobala and Hutvagner, 2013).
A recent study showed that a 3′-end-containing tRF derived from tRNALeu binds mRNAs encoding ribosomal proteins and enhances production of these proteins, resulting in the promotion of ribosome biogenesis (Kim et al., 2017).
Modifications
tRNAs are known to be the most heavily modified RNA species (Boccaletto et al., 2022). These modifications are thought to have at least two critical roles: (1) to stabilize tertiary structure, and (2) to modulate translation accuracy by contributing to codon-anticodon interactions (Lorenz et al., 2017). Therefore, modification patterns of tRNAs can affect both tRNA stability and mRNA translation. Generally, modifications within the anticodon can make a significant impact on translation (Chou et al., 2017).
For example, modifications of uridines at the wobble position (U34) are essential to optimize translation rates (Nedialkova and Leidel, 2015; Rapino et al., 2018). Interestingly, modifications located apart from the anticodon can also affect translation. ALKBH1-mediated demethylation of 1-methyladenosine at position 58 (m1A58) significantly attenuated translation (Liu et al., 2016). Under glucose deprivation, m1A modifications were decreased due to upregulation of ALKBH1, contributing to stress-responsive translation control.
tRNA modifications induced by oxidative stress
Several lines of evidence have shown that oxidative stress alters tRNA modification profiles (Fig. 3E). In S. cerevisiae, H2O2 exposure increased the levels of 2′-O-methylcytidine (Cm), 5-methylcytidine (m5C), and N2,N2-dimethylguanosine (m2 2G). Strains that lack the enzymes responsible for these modifications showed hypersensitivity to H2O2, suggesting that such modifications have a role in protecting cells from oxidative stress (Chan et al., 2010).
H2O2 exposure induced Trm4 methyltransferase-mediated m5C modification at the wobble position in tRNALeu-CAA, which specifically facilitates the translation of genes enriched with TTG codon (Leucine-coding codon decoded by tRNALeu-CAA), such as a ribosomal protein PRL22A, thus contributing to cellular protection from oxidative stress (Chan et al., 2012). It was also shown that SA treatment repressed the expression of Nsun2, a homolog of yeast Trm4, resulting in the decreased levels of m5C modification at the wobble position in tRNALeu-CAA in mouse cells (Gkatza et al., 2019).
tRNA levels can also affect selenoprotein synthesis in response to ROS. Selenoproteins refer to proteins that contain SeC, a selenium-containing amino acid, including ROS detoxification enzymes such as glutathione peroxidases (Labunskyy et al., 2014). SeC is encoded by a UGA stop codon in the selenoprotein mRNAs. In mammals, the wobble position (position 34) of uridine in tRNASeC is modified to either 5-methoxycarbonylmethyluridine (mcm5U) or 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um).
These modifications are mediated by a tRNA methyltransferase named mammalian alkylation repair homolog 8 (ALKBH8). Importantly, these wobble modifications are necessary for decoding of the UGA codon as SeC incorporation. In mouse embryonic fibroblasts (MEFs), 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um) modification at the wobble position in tRNASeC was upregulated by H2O2 treatment through Alkbh8 upregulation, facilitating the translation of selenoproteins (Endres et al., 2015).
Conversely, Alkbh8 knockout MEFs showed elevated ROS levels, suggesting that the Alkbh8-mediated translation regulation of selenoproteins is critical for cellular redox homeostasis. Similar to ALKBH8-mediated uridine modification, some modifications are suggested to play a role in redox homeostasis. For example, TRMT1 is a methyltransferase that is responsible for catalyzing N2,N2-dimethylguanosine (m2 2G) modification at position 26.
The loss of m2 2G modification by TRMT1 knockout showed translation repression and increased levels of ROS in HEK293T cells (Dewe et al., 2017). Similarly, 1-methylguanosine (m1G) modification at position 9 catalyzed by TRMT10A was also reported to be involved in redox homeostasis (Cosentino et al., 2018). Knockdown of TRMT10A induced oxidative stress and apoptosis in both human and rat pancreatic β cells. Collectively, these data suggest that the modification status of tRNAs is closely related to oxidative stress.
tRNA modifications can affect tRNA cleavage
Several tRNA modifications have been reported to affect the efficiency of tRNA cleavage, especially ANG-mediated tiRNA production (Fig. 4). These modifications generally inhibit cleavage by ANG. The location of such modifications is not limited to the anticodon loop where ANG directly targets for cleavage.

As one of the major roles of tRNA modifications is to stabilize the tertiary structure of tRNAs (Lorenz et al., 2017), it can be speculated that modifications located apart from the anticodon can inhibit ANG-mediated cleavage by stabilizing or modifying the tertiary structure of tRNAs. As ANG-induced tiRNAs can regulate translation rates, tRNA modification status is an important factor in cellular translational regulation.
Five-methylcytosine (m5C) is the first identified modification that inhibits ANG-mediated tRNA cleavage. Two methylases, NSUN2 and DNMT2, are responsible for m5C modification in tRNAs. In higher eukaryotes, DNMT2 methylates cytosines in the anticodon loop at position 38 (Schaefer et al., 2010; Tuorto et al., 2012), whereas NSUN2 is responsible for m5C modification at position 34 in the anticodon as well as at position 48–50 in the variable loop (Blanco et al., 2014).
Loss of m5C modifications via deletion of DNMT2 or NSUN2 promotes ANG-mediated tiRNA production (Blanco et al., 2014; Schaefer et al., 2010; Tuorto et al., 2012). Deletion of both Dnmt2 and Nsun2 in mice showed a complete lack of m5C modifications in tRNAs, and showed a decreased translation rate, suggesting that m5C modifications are crucial for translational regulation.
ALKBH3 is an RNA demethylase that catalyzes the demethylation of 1-methyladenosine (m1A) and 3-methylcytosine (m3C). ALKBH3 overexpression increased ANG-mediated tRNA cleavage, whereas ALKBH3 deletion decreased cleavage, suggesting that m1A and m3C modifications negatively regulate tiRNA production (Chen et al., 2019).
The inhibitory effect of m1A at position 58 on stress-induced tiRNA production was also shown by overexpression/knockdown of ALKBH1 (Rashad et al., 2020), a demethylase targeting m1A at position 58 in tRNAs.
Queuosine modification is a modified 7-azaguanosine-derivative at the wobble position at 34 mediated by queuine tRNA-ribosyltransferase heterodimeric complex (QTRT1/QTRT2). It is found in the wobble anticodon position of tRNAs for amino acids Asn, Asp, His, and Tyr (Fergus et al., 2015). Queuosine (Q) was also reported to protect Q-modified tRNAs from ANG-mediated cleavage (Wang et al., 2018).
Similarly, 2′-O-methylcytidine modification at the wobble position (Cm34) of human elongator tRNAMet was also reported to prevent ANG-mediated cleavage (Vitali and Kiss, 2019). Interestingly, this modification is mediated by small nucleolar and small Cajal body-specific ribonucleoprotein (RNP) complexes, carrying SNORD97 and SCARNA97 as guide RNAs, respectively (Vitali and Kiss, 2019).
It has also been recently reported that TRMT2A-mediated 5-methyluridine (m5U) modification at position 54 is related to the efficiency of ANG-induced tiRNA production (Pereira et al., 2021). Inhibition of TRMT2A induced hypomodification of m5U54, resulting in the increased production of ANG-mediated tiRNAs and subsequent translation inhibition.
Intriguingly, exposure of cells to SA induced downregulation of TRMT2A (Pereira et al., 2021), suggesting that oxidative stress conditions may facilitate tiRNA production by the inhibition of TRMT2A-mediated m5U54 modification. In addition, it has been reported that SA decreases m5C modifications through downregulation of NSUN2, resulting in translation repression due to enhancement of ANG-mediated tRNA cleavage (Gkatza et al., 2019).
Altogether, these data suggest that tRNA modifications play a role in translation regulation under oxidative stress conditions.
Misacylation
The accuracy of aminoacylation by aminoacyl-tRNA synthetases is critical for maintaining translational fidelity (Steiner and Ibba, 2019). Oxidative stress increases the frequency of methionine misacylation, that is, methionine is erroneously conjugated to non-methionine tRNAs (Fig. 3F), resulting in the increased levels of methionine incorporation into newly synthesized proteins. Surprisingly, these methionine-incorporated proteins play a protective role in cell survival against oxidative stress (Netzer et al., 2009).
Mechanistically, oxidative stress activates extracellular signal-related kinases (ERK1/2), causing phosphorylation of the methionyl-tRNA synthetase (MARS). Phosphorylated MARS shows increased and decreased affinities for non-methionine tRNAs and tRNAMet, respectively (Lee et al., 2014). Because the sulfur group of methionine is highly reactive with ROS, methionine residues in proteins have the potential to act as an endogenous antioxidant (Luo and Levine, 2009).
It is interesting that oxidative stress-induced methionine misacylation seems to serve as an antioxidant defense system at the expense of translational fidelity. Such stress-induced methionine misacylation is conserved in all domains of life (Jones et al., 2011; Schwartz and Pan, 2016; Wiltrout et al., 2012), implying a pivotal role in adaptation to stress conditions.
Molecular Mechanisms of Stress Response Mediated by tiRNAs
While the phenomena of tRNA cleavage have been recognized for a long time, the cleavage products of tRNAs, tDRs (tRNA-derived small RNAs), had been considered simply as degradation products until recent years. Accumulating evidence has recently revealed essential roles of tDRs in regulating proliferation, differentiation, and translation, especially under stress conditions (Anderson and Ivanov, 2014; Keam and Hutvagner, 2015; Su et al., 2020).
One of the key mechanisms of stress response mediated by tRNA cleavage is the repression of translation initiation in parallel to the ISR and mTORC1 pathways, further indicating the importance of regulating translation initiation to adapt to stress.
Under normal conditions, ANG is mainly localized in the nucleus to promote the synthesis of rRNAs (Moroianu and Riordan, 1994). Although a small proportion of ANG resides in the cytoplasm, it is inactivated by RNH1, an endogenous inhibitor of RNase A superfamily enzymes, including ANG (Dickson et al., 2005; Sarangdhar and Allam, 2021). As shown in Figure 5, once cells are exposed to stress stimuli, ANG becomes activated through dissociation from RNH1.

In addition, nuclear ANG translocates into the cytoplasm (Pizzo et al., 2013). Although the efficiency varies depending on tRNA species, activated ANG can cleave all the mature tRNAs (Akiyama et al., 2022a; Saikia et al., 2012), generating two subclasses of tiRNAs, 5′-tiRNAs (30–35 nt) and 3′-tiRNAs (40–50 nt). Among them, specific 5′-tiRNAs derived from tRNAAla and tRNACys (5′-tiRNAAla and 5′-tiRNACys) have the ability to repress translation by targeting translation initiation (Ivanov et al., 2011).
5′-tiRNAAla and 5′-tiRNACys possess five consecutive guanosines at their 5′-end, termed a terminal oligoguanine (TOG) motif. These TOG motif-containing 5′-tiRNAs form G-quadruplex (G4) structures (Akiyama et al., 2020; Ivanov et al., 2014; Lyons et al., 2017b), stable non-canonical RNA structures formed in guanine (G)-rich sequences (Kharel et al., 2020).
G4 assembly through the TOG motif is essential for translation inhibition activity of these 5′-tiRNAs. Mechanistically, TOG-containing 5′-tiRNAs (G4-tiRNAs) directly bind to eIF4G, one of the subunits of eIF4F complex, displacing the eIF4F complex from the cap structure of mRNAs (Fig. 5) (Lyons et al., 2020). TOG-containing 5′-tiRNAs also induce the assembly of stress granules (SGs) (Emara et al., 2010; Lyons et al., 2016), membraneless cytoplasmic RNA granules comprising translationally stalled mRNAs as well as associated translation initiation factors, and various RNA-binding proteins (Hofmann et al., 2021; Riggs et al., 2020). The SGs can further modulate translation status (Baymiller and Moon, 2023).
In addition to cleavage in the anticodons, ANG is suggested to cleave the 3′-CCA end of tRNAs in vitro (Czech et al., 2013). If the 3′-CCA end of tRNAs were globally trimmed in response to stress, translation would be significantly repressed because tRNAs would lose the ability to charge amino acids.
However, most of the intracellular 3′-tiRNAs that are generated by endonucleolytic cleavage by ANG or other RNases had intact CCA ends in cellulo (Akiyama et al., 2022a; Li et al., 2022; Su et al., 2019), suggesting that the 3′-CCA end is not preferentially targeted by stress-responsive RNases in the cell. Therefore, the impact of stress-induced CCA-trimming on translation regulation is estimated to be small.
Growing evidence has suggested that various molecules, in addition to eIF4G (as in the case of G4-tiRNAs), can also be targets of tDR-mediated stress response. We overview several examples here. First, tiRNAs, as well as full-length tRNAs, interact with cytochrome c released from mitochondria under stress conditions, preventing apoptosis by blocking the interaction between cytochrome c and Apaf-1, an essential step to trigger apoptosis (Mei et al., 2010; Saikia et al., 2014).
Second, tiRNAs directly bind to YB-1, an RNA-binding protein with a variety of binding partners (Lyabin et al., 2014). As YB-1 plays a role in stabilizing mRNAs, the interaction between tDRs and YB-1 causes destabilization of specific (especially pro-metastatic) mRNAs through displacement of YB-1 from mRNAs (Goodarzi et al., 2015), which alters the translational profile. YB-1 is also required for the SG assembly induced by TOG-containing 5′-tiRNAs (Lyons et al., 2016).
Third, specific tDRs similar to tiRNAs can target ribosomes. In S. cerevisiae, both 5′- and 3′- halves derived from tRNAHis interact with ribosome under stress conditions, resulting in translation repression (Bakowska-Zywicka et al., 2016). A recent study showed that 5′-tDR derived from tRNAPro (5′-tiRNAPro) causes global translation inhibition through interaction with ribosomes in mammalian cell lines (Gonskikh et al., 2020).
Finally, although the mechanism has yet to be clarified, 5′-tiRNAGly also inhibits translation initiation (Ivanov et al., 2011). Interestingly, the endogenous 5′-tiRNAGly showed stronger translation inhibitory effect than its synthetic counterpart (Akiyama et al., 2020), suggesting that the modification status is involved in the fine-tuning of tiRNA-mediated translational regulation. Recent studies have also revealed the impact of modifications, such as pseudouridine (Guzzi et al., 2018) and m1A (Su et al., 2022), on the biological activity of tDRs.
tiRNA Production Rates Determine Cell Fate
Accumulating evidence suggests that intracellular tDR production rates, especially ANG-mediated tiRNA production rates, are involved in a variety of physiological and pathophysiological processes in the cell. These include cell survival, stem cell differentiation (Blanco et al., 2016; Flores et al., 2017; Goncalves et al., 2016; Kfoury et al., 2021), chemoresistance in cancers (Blanco et al., 2016), neurodegenerative disorders (Ivanov et al., 2014; Su et al., 2020), tumorigenesis (Yu et al., 2020; Zhu et al., 2019), and T cell activation (Yue et al., 2021).
Some of them occur through the regulation of translation (Akiyama and Ivanov, 2023). A variety of factors are suggested to modulate tiRNA production rates, likely affecting cell fate (Fig. 6).
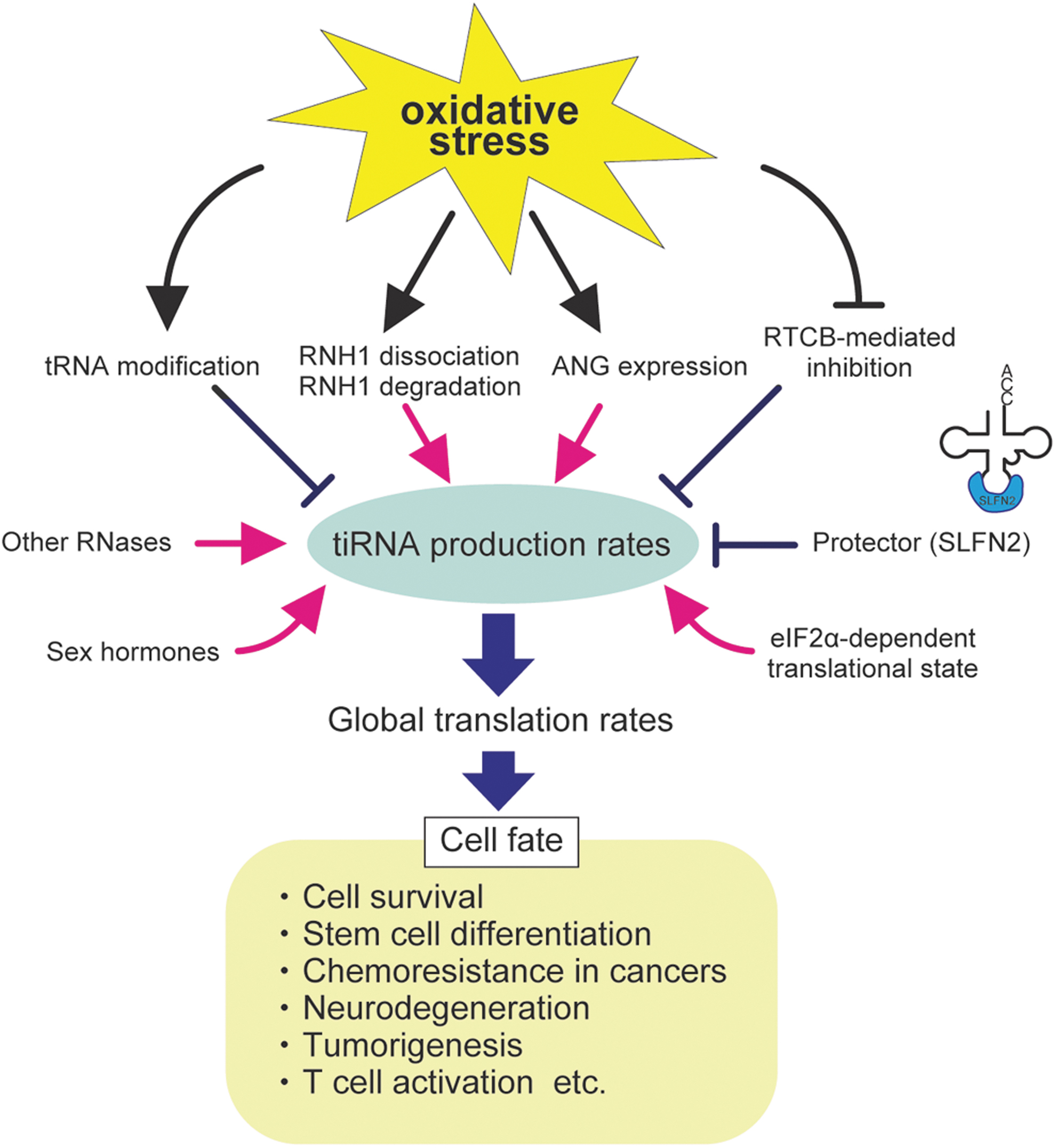
Factors regulating tDR production
First, intracellular ANG levels are the most direct determinant of tiRNA production. Other RNases capable of tRNA cleavage are also suggested to affect tiRNA production. Although such RNases theoretically include all the RNase A superfamily enzymes (Akiyama et al., 2022a), a recent study suggests that RNase 1 is mainly responsible for ANG-independent tRNA cleavage in the cell (Li et al., 2022).
RNase L, an RNase activated by double stranded RNAs (dsRNAs), also cleaves specific tRNAs around the anticodon loops, generating tiRNA-like tDRs (Donovan et al., 2017). dsRNAs are hallmarks of viral infection, indicating further function of tiRNA-like tDRs in immune response. As described earlier, some tRNA modifications can affect the efficiency of tRNA cleavage by such RNases.
As stress-induced dissociation of RNH1 activates RNases, the efficiency of RNH1 dissociation in response to stress is an important factor. RNH1 is one of the most highly expressed cytosolic proteins. The intriguing feature of RNH1 is the abundance of cysteine residues. For example, human RNH1 has 32 cysteine residues (Papageorgiou et al., 1997), all of which must remain reduced to inhibit RNase A superfamily enzymes (Fominaya and Hofsteenge, 1992).
Knockdown of RNH1 decreased intracellular glutathione levels and caused more severe DNA damage under diethylmaleate-induced oxidative stress condition compared with the control, suggesting that RNH1 can contribute to redox homeostasis by acting as an intracellular oxidation sensor (Monti et al., 2007). Although oxidative stress induces the dissociation of RNH1 from RNases by oxidizing thiol residues in RNH1 (Ferreras et al., 1995), it is still unclear whether there exist mechanisms of RNH1 dissociation that are independent of RNH1 oxidation.
ANG-mediated tiRNA production is also promoted by sex hormones. In estrogen receptor-positive breast cancer cells and androgen receptor-positive prostate cancer cells, ANG-mediated tRNA cleavage is promoted in a sex hormone-dependent manner. This results in the accumulation of tRNA halves termed
ANG-mediated tiRNA production is also associated with T cell-mediated immunity. Schlafen 2 (SLFN2) acts as a protector to prevent ANG-mediated tRNA cleavage through direct binding to tRNAs (Yue et al., 2021). In T cell-specific Slfn2 knockout mice, T cell receptor (TCR) stimulation failed to induce T cell proliferation due to translation inhibition by ANG-induced tiRNAs. This suggests that SLFN2 is critical for keeping high enough translation rates to activate T cells (Yue et al., 2021).
eIF2α-dependent translational states also determine the efficiency of tiRNA production. ANG-induced tiRNA production is increased under the condition where translation is not inhibited in response to stress, such as eIF2α phosphorylation-dead mutation (Saikia et al., 2012) or HRI knockdown (Yamasaki et al., 2009). On the other hand, inhibitors of translation initiation, such as hippuristanol, repress ANG-mediated tiRNA production (Saikia et al., 2012). These data suggest that active translation is required for the efficient ANG-mediated tRNA cleavage.
The impact of oxidative stress on tDR production
Oxidative stress is suggested to further modify tDR production rates by affecting factors involved with tDR synthesis (Fig. 6). For example, some tRNA modifications induced by oxidative stress can promote ANG-induced tiRNA production (Gkatza et al., 2019; Pereira et al., 2021). When oxidized under oxidative stress conditions, RNH1 is degraded as well as loses the capability to bind RNases (Blazquez et al., 1996; Moenner et al., 1998), suggesting that prolonged oxidative stress further enhances tiRNA production through the decrease in functional RNH1 proteins in the cell.
Oxidative stress conditions have also been suggested to upregulate ANG expression (Yue et al., 2021). However, it is worth noting that upregulation of ANG expression does not necessarily reflect intracellular ANG levels because ANG is a secreted RNase. ANG is abundant in body fluids such as the plasma, amniotic and cerebrospinal fluids (Sheng and Xu, 2016).
The newly synthesized ANG must be secreted and taken up before becoming functional in the cytosol. Thus, expression levels of cell surface receptors responsible for ANG incorporation such as Plexin-B2 (Yu et al., 2017) and Syndecan-4 (Skorupa et al., 2012) are also important determinants of intracellular ANG levels. Finally, we have recently reported that the RTCB ligase complex represses tiRNA production by repairing nicks in the anticodon loops generated by ANG (Akiyama et al., 2022b).
As the RTCB complex is inhibited by oxidative stress (Asanovic et al., 2021), tiRNA production is boosted under oxidative stress conditions (Akiyama et al., 2022b), which enables cells to rapidly produce tiRNAs without transcriptional or translational regulation of RNases.
Concluding Remarks
Response to oxidative stress is an integral part of normal cellular physiology. Multiple non-overlapping pathways sensing oxidative stress converge on the regulation of protein synthesis, the most energy-consuming cellular process. It is not surprising that tRNA as an integral and fundamental component of protein synthesis also contributes to stress responses. It is, however, quite unexpected that the contribution of tRNAs to cellular pathways involving stress adaptation relies also on its non-ribosomal functions.
Advanced proteomics approaches (e.g., “redox proteome”) were instrumental for the investigations of cellular redox metabolism, and its connection with biological structures and functions (Go and Jones, 2013). Likewise, recent advances in tRNA sequencing and mass spectrometry in conjunction with other -omics techniques such as ribosomal profiling, chip-seq, and microarrays are paving the way to deepen the understanding of the complex tRNA world.
Besides these high-throughput methods, routine biochemical, cellular, and genetic approaches will be required to decipher the role of translational control and tRNA metabolism in response to stress.
One of the challenging tasks in tRNA biology that still remains is to decipher the function of isodecoders. It is quite likely that the expression of specific isodecoders is cell- and tissue-specific. It is also possible that their expression is interconnected with stress response pathways via both mRNA-translation-dependent and -independent (non-canonical) manners.
In addition, it will be important to understand how tRNA modifications contribute to stress adaptation in different cells, tissues, or organs. The answer to these questions will require a major technological breakthrough such as single cell tRNA-seq. Such an approach would uncover molecular fingerprints within specific cells based on the tRNA abundance, expression, and modification profiles.
Another emerging theme connecting tRNA and oxidative stress is tRNA release into extracellular space. It is thought that extracellular tRNAs, “nicked” tRNAs, and tDRs comprise a significant part of extracellular RNAome (Costa et al., 2023; Dhahbi et al., 2014; Gambaro et al., 2020; Tosar et al., 2020).
They can be found in different extracellular fractions (e.g., in the form of ribonucleoprotein complexes or within extracellular vesicles) and play roles in intercellular or even inter-species communication (Tosar and Cayota, 2020). Profiling of tRNA and tDRs in human biofluids is a promising area in the biomarker field.
As oxidative stress is a contributing factor in a number of human pathologies, it is important to understand the molecular mechanisms that govern physiological regulatory mechanisms and pathways that detect and deal with stress. Such understanding will allow manipulation of stress-induced genetic and biochemical programs, which can be therapeutically beneficial.
Importantly, with the discovery of oxidative stress-induced molecular mechanisms, we will greatly expand our knowledge about tRNA cell- and tissue-specific expression patterns and the role of inter-cellular communications in coping with stress. In conclusion, we have reviewed here multiple connections between oxidative stress, protein synthesis, and tRNA metabolism. We expect that new oxidative stress-related paradigms related to mRNA translation will emerge in future, which will further illuminate roles of these processes in normal cell physiology and human disease.
Footnotes
Acknowledgment
The authors thank Dr. Allison Williams for discussion and help with article proofreading.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Japan Society for the Promotion of Science, Grants-in-Aid for Scientific Research (PS KAKENHI) (21K06865 to Y.A.), and the National Institutes of Health (R01 GM126150 and R01 GM146997 to P.I.).