
Abstract
Significance:
Reactive oxygen species (ROS) are essential in maintaining normal intestinal physiology. Inflammatory bowel disease (IBD) is a relapsing chronic inflammatory disease of the intestine that is a major risk factor for colorectal cancer (CRC). Excess ROS are widely implicated in intestinal inflammation and cancer.
Recent Advances:
Clinical data have shown that targeting ROS broadly does not yield improved outcomes in IBD and CRC. However, selectively limiting oxidative damage may improve the efficacy of ROS targeting. An accumulation of lipid ROS induces a novel oxidative cell death pathway known as ferroptosis. A growing body of evidence suggests that ferroptosis is relevant to both IBD and CRC.
Critical Issues:
We propose that inhibition of ferroptosis will improve disease severity in IBD, whereas activating ferroptosis will limit CRC progression. Data from preclinical models suggest that methods of modulating ferroptosis have been successful in attenuating IBD and CRC.
Future Directions:
The etiology of IBD and progression of IBD to CRC are still unclear. Further understanding of ferroptosis in intestinal diseases will provide novel therapies. Ferroptosis is highly linked to inflammation, cell metabolism, and is cell-type dependent. Further research in assessing the inflammatory and tumor microenvironment in the intestine may provide novel vulnerabilities that can be targeted. Antioxid. Redox Signal. 39, 551–568.
Introduction
Reactive oxygen species (ROS) are a group of highly reactive molecules formed through the reduction of molecular oxygen (Sies and Jones, 2020). They include free radicals, superoxide anions (O2 •−), hydroxyl (HO•), peroxyl (RO2 •−), hydroperoxyl (HO2 •), and alkoxyl radicals (RO•), and molecules that produce free radicals such as hydrogen peroxide (H2O2), hydroxide ion (OH−), and organic peroxides (ROOH) (Shields et al., 2021). Free radicals are short-lived and react rapidly in subcellular locations where they are produced, while nonfree radical ROS pass through membranes due to decreased reactivity (Shields et al., 2021). Excess transition metals such as cuprous copper (Cu+) or ferrous iron (Fe2+) can also react with H2O2 to generate hydroxyl radicals through the Fenton reaction (Shields et al., 2021). While many studies have focused on the detrimental effects of ROS (Alfadda and Sallam, 2012; Liu et al., 2018), it is important to understand that ROS are essential for normal physiological function and health.
ROS are generated in subcellular compartments such as the cytoplasm, endoplasmic reticulum (ER), cell membrane, and peroxisome. The main source of ROS is the mitochondria (mtROS) by the electron transport chain (ETC) during oxidative phosphorylation (OXPHOS) (Fig. 1) (Tirichen et al., 2021). mtROS account for ∼90% of cellular ROS (Tirichen et al., 2021). During OXPHOS, O2 •− is converted into H2O2 and OH− (Tirichen et al., 2021). O2 •− is generated primarily in complex I at the flavin mononucleotide (FMN) site and in complex III during the Q cycle (Tirichen et al., 2021).

In complex I, two electrons are transferred from nicotine adenine dinucleotide (NADH) generated in the tricarboxylic acid cycle to ubiquinone (Q). These electrons can be passed on to form FMNH2 at FMN sites or circulated through a series of iron–sulfur clusters to reduce Q to uniquinol (QH2) (Tirichen et al., 2021). In complex III, O2 •− is generated as a result of autooxidation of ubisemiquinone, the primary electron donor that reduces O2 to O2 •− (Tirichen et al., 2021). In comparison with complex I and III, complex II generates small amounts of O2 •− at the FAD site (Tirichen et al., 2021).
Mitochondria are the main source of ROS in cells; however, an important source of ROS is within the plasma membrane generated by NADPH oxidase (NOX/DUOX) enzymes (Aviello and Knaus, 2017). ROS production by innate immune cells is often a first-line defense toward pathogens (Arnold and Heimall, 2017). Innate immune cells utilize oxidative bursts to clear microbial pathogens, which is generated by the reduction of molecular oxygen to superoxide anions by NOX (Lambeth and Neish, 2014). Mutations in NOX genes and the subsequent decrease of oxidative burst capacity in innate immune cells lead to a primary immune deficiency known as chronic granulomatous disease (CGD) (Arnold and Heimall, 2017). These patients are at increased risk for infection and experience significant morbidity (Arnold and Heimall, 2017).
In addition, as part of the acquired immune response, ROS are continuously secreted by activated phagocytes to reinforce intracellular T cell signaling present at the sites of infection (Dröge, 2002). Moreover, NOX enzymes in other cell types generate ROS for the regulation of growth, survival, and apoptosis (Lambeth and Neish, 2014).
Under normal physiological conditions, ROS play an important role in regulating homeostasis. However, under conditions of decreased cellular oxygen, or hypoxia, the production of mtROS increases due to dysregulation of the ETC. The enhanced ROS in hypoxia are an essential mechanism of inducing the transcription factor hypoxia-induced factor (HIF)-1α (Guzy and Schumacker, 2006; Wang et al., 2007). HIF-1α, in turn, induces metabolic responses that normalize ROS levels and mediate redox homeostasis (Guzy and Schumacker, 2006). Physiological processes such as programmed cell death, metabolism, cell proliferation, and autophagy are regulated at the transcriptional level by ROS through modulation of chromatin dynamics and transcriptional activity (Rajendran et al., 2011).
Excess cellular ROS production has many detrimental effects, such as damaging DNA, proteins, and lipids (Pham-Huy et al., 2008). One example of this is the accumulation of phospholipid hydroperoxide (PLOOH) lipid ROS in cells, which leads to a form of iron-dependent programmed cell death called ferroptosis (Dixon et al., 2012). Ferroptosis is a nonapoptotic form of cell death that is characterized by an accumulation of lipid peroxides and reactive iron. Ferroptotic cells display morphological changes such as shrunken mitochondria with degenerated cristae and membrane rupture.
The intestine is exposed to a large number of microbes, dietary antigens, and the hypoxic nature of the gut lumen, making it necessary to have a robust mechanism to maintain physiological ROS levels. In this review, we examine the role of ROS in the intestine and in two intestinal diseases: inflammatory bowel disease (IBD) and colorectal cancer (CRC). We summarize findings that suggest ferroptosis is associated with these two disease states. Lastly, we review potential mechanisms for leveraging the role of ferroptosis in IBD and CRC as therapeutic treatments.
Intestinal Physiology
The gastrointestinal (GI) epithelium plays a crucial role in maintaining nutrient absorption, secreting entero-hormones, and serving as a physical barrier to the microbiota. In a state of homeostasis, the cellular interactions between immune cells and the GI epithelium mediate mucosal tolerance toward the microbiota (Chieppa et al., 2006; Hepworth et al., 2015; Nanno et al., 2008; Peterson and Artis, 2014). Dysregulation or damage to the intestinal barrier initiates bacteria translocation across the lamina propria eliciting antimicrobial defenses. ROS production by immune cells functions to both promote pathogen clearance and regulate intracellular signals for barrier healing and repair (Neurath, 2014). Redox imbalance promotes a sequalae of events that lead to a myriad of complications such as cancer, fibrosis, and systemic symptoms (Neurath, 2014).
In the GI tract, the primary sources of ROS are from NOX/DUOX enzymes and ETC (Aviello and Knaus, 2017). Superoxides from ETC reacts with superoxide dismutase (SOD)-2 within the mitochondrial matrix to form hydrogen peroxide, which modulates cell activity through altering levels of transcription factors, kinases, and phosphatases (Mittal et al., 2014; Tanner et al., 2011). To maintain the redox balance, both endogenous and exogenous antioxidants are necessary. Thioredoxin (Trx) and glutathione (GSH) are two major antioxidant mechanisms used by cells to protect against ROS. Trx and GSH can act as electron acceptors to limit ROS through nonenzymatic mechanisms.
In addition, Trx and GSH are essential factors in enzymatic-mediated detoxification of ROS. Glutathione peroxidase (GPX) converts GSH into glutathione disulfide (GSSG), which reduces H2O2 into water and lipid peroxides into alcohols (Ursini et al., 2022). Trx also serves as an electron donor to peroxidase to reduce ROS. Oxidized Trx and GSSG are reduced in an NADPH manner to maintain proper redox balance (Ursini et al., 2022). Other endogenous enzymatic antioxidants include SOD and catalase. SOD reacts with O2 •− to form O2 and H2O2, and catalase converts H2O2 into H2O and O2 (Ighodaro and Akinloye, 2018).
Nuclear factor erythroid 2-related factor 2 (NRF2) is a transcription factor that is activated by ROS and plays a crucial role in the adaptive response to limit ROS toxicity (Mazur-Bialy and Pocheć, 2021). Under basal conditions, NRF2 binds to Kelch-like epichlorohydrin-related protein (KEAP1), resulting in the ubiquitination and subsequent degradation of NRF2. Under conditions of oxidative stress, NRF2 is stabilized and translocates to the nucleus to activate a battery of antioxidant genes (Cuadrado et al., 2019). In addition, exogenous antioxidants are another source of ROS detoxification. This category includes vitamins such as B12 and E, amino acids, peptides, fatty acids, minerals, and other bioactive natural products (Wang et al., 2020c).
ROS in Inflammation and Colon Cancer
Redox imbalance leads to insufficient microbial clearance, compromised wound healing, prolonged inflammation, and altered immune responses (Neurath, 2014). One such consequence of altered GI ROS imbalance is IBD. IBD is a group of chronic idiopathic GI inflammatory conditions that primarily include ulcerative colitis (UC) and Crohn's disease (CD). IBD is clinically characterized by symptoms such as diarrhea, nausea, vomiting, abdominal pain, bloody stool, weight loss, as well as extraintestinal symptoms such as fever, fatigue, skin rashes, and ulcers (Abraham and Cho, 2009). UC is characterized by inflammation that affects the mucosa and/or submucosa of the rectum, and in some cases may extend proximally to involve the entire colon in a continuous pattern (Abraham and Cho, 2009; Gajendran et al., 2018; Khor et al., 2011; Monteleone et al., 2006).
In contrast, CD can affect any part of the GI tract in a patchy distribution, and is often characterized by transmural inflammation, granulomas, fissuring ulceration, and a thickened submucosa (Abraham and Cho, 2009; Gajendran et al., 2018; Khor et al., 2011; Monteleone et al., 2006).
The exact cause of IBD is not fully understood, however, several ROS-related factors have been found to contribute to its pathogenesis. There is a strong genetic component to IBD. Nucleotide-binding oligomerization domain 2 (NOD2) gene, a member of the Nod-like receptor family responsible for microbial detection, was the first gene linked to IBD (Yamamoto and Ma, 2009). Alterations in mucosal bacterial recognition can lead to increased nuclear factor kappa-light-chain-enhancer of activated B cell signaling and the subsequent inflammation (Ogura et al., 2001). Host-microbiota dysregulation also contributes to the pathogenesis of IBD. While many commensal microorganisms are necessary for healthy gut homeostasis, pathogenic microbes can disrupt this balance leading to an inflammatory environment (Guan, 2019).
Environmental factors, such as diet, specifically Western diets high in polyunsaturated fatty acids (PUFAs), have been shown to contribute to the development of IBD through metabolic inflammation (Guan, 2019). Interestingly, while the rate of IBD incidence is highest among developed Western nations, the fastest acceleration of IBD incidence is among developing nations undergoing westernization (Kaplan and Windsor, 2021). In addition, abnormal immune response has been linked to the development of IBD, with CD associated with an overreactive T helper 1 (Th1) immune response leading to excessive interleukin-23/T helper 17 (IL-23/Th17) activation in response to bacterial antigens in genetically susceptible individuals (Monteleone et al., 2006; Souza and Fiocchi, 2016; Tavakoli et al., 2021). UC displays a more characteristic overreactive T helper 2 (Th2) immune response with increased IL-5 and IL-13 (Strober and Fuss, 2011). Overall, IBD is a complex, multifactorial disease with many contributing factors.
Chronic inflammation has been linked to the development of cancer, particularly CRC, which is a malignancy of the colon or rectum and one of the most common and deadly cancers in the United States (Siegel et al., 2022). IBD is major risk factor for CRC (Porter et al., 2021). Other risk factors include diets high in PUFAs, smoking, obesity, red meat, and alcohol (Hossain et al., 2022). Genetic causes of CRC include familial adenomatous polyposis and hereditary nonpolyposis colon cancer (Lynch Syndrome) (Hossain et al., 2022). The development of CRC is characterized by a well-established mutational pathway, with the earliest and most prevalent mutation involving the tumor suppressor gene APC. Subsequent mutations in KRAS and p53 lead to the development of carcinoma and metastatic cancer (Fig. 2) (Fearon and Vogelstein, 1990; Muzny et al., 2012).
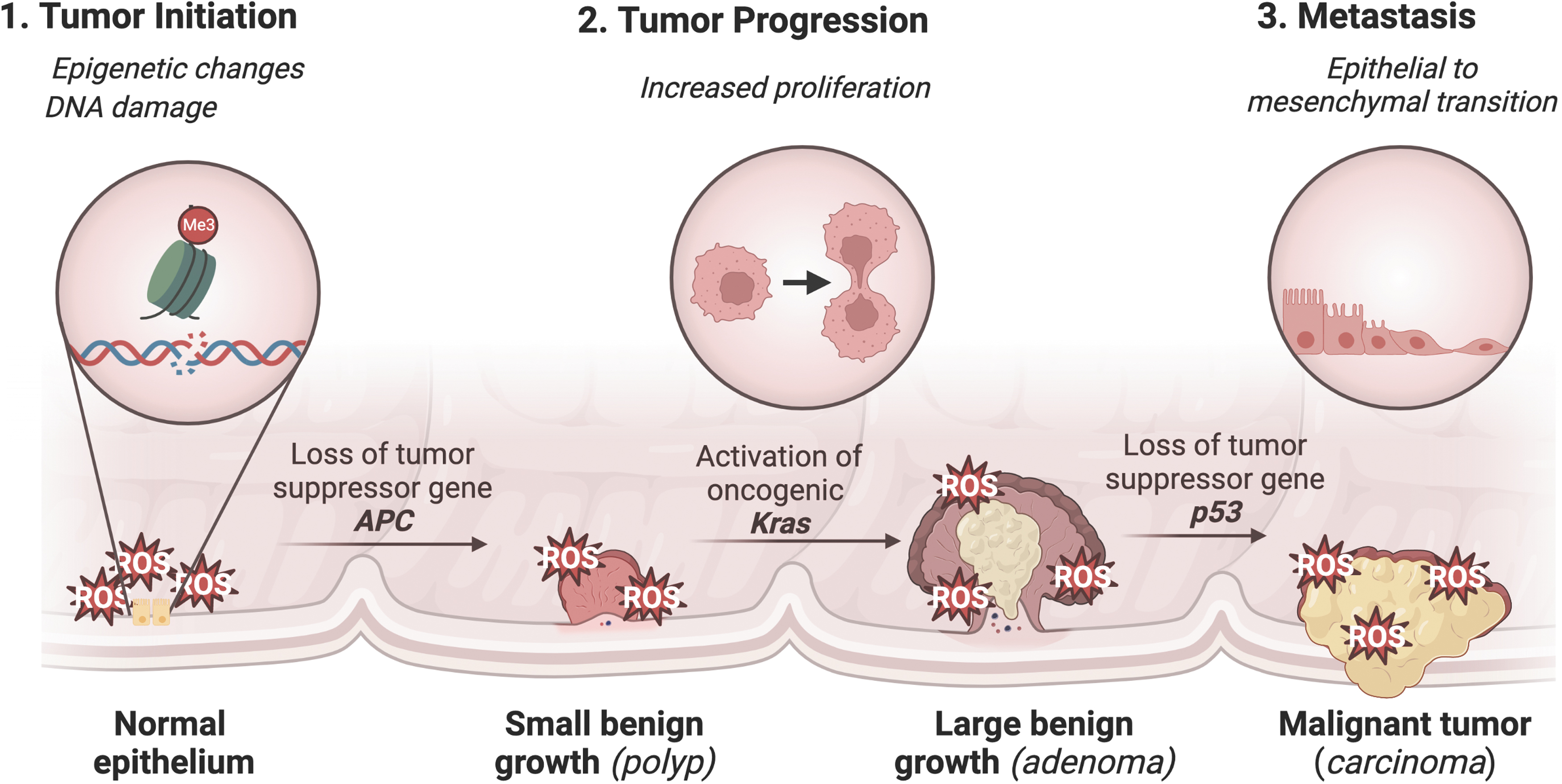
In IBD and CRC, ROS play an important role in the disease pathogenesis, which has led to several human clinical trials assessing the efficacy of antioxidant therapy (Moura et al., 2015). A systematic meta-analysis review of the use of antioxidant therapy in the treatment of IBD concluded that there was not enough evidence to suggest that antioxidant therapy is effective in treating IBD. Therefore, further research is needed in this field (Table 1) (Moura et al., 2015).
Antioxidant Therapies Used in Preclinical Models and Clinical Trials of Inflammatory Bowel Disease
AA, arachidonic acid; ACE, angiotensin-converting-enzyme; ARB, angiotensin receptor blocker; CD, Crohn's disease; CRC, colorectal cancer; DSS, dextran sulfate sodium; IBD, inflammatory bowel disease; IL, interleukin; KO, knockout; MEL, melatonin; NAC, N-acetylcysteine; PLC, propionyl-
The discrepancy between promising preclinical models and disappointing clinical trials in IBD suggests that the models currently used do not fully replicate all aspects of this complex disease. In addition, since ROS play both positive physiological roles and detrimental effects, targeting all ROS may not yield positive results. ROS can damage DNA, proteins, and lipids, and so, selectively targeting a specific aspect of ROS damage may be more effective in treating IBD. The role of antioxidants in CRC therapy is even more uncertain. Given the role of ROS in various stages of CRC progression, antioxidant agents could potentially inhibit aggressive cancer phenotypes. However, several effective chemotherapy, radiotherapy, and immunotherapy treatments rely on inducing cell death through oxidative stress (Table 2).
Anticancer Therapies Used for Colorectal Cancer That Rely on Reactive Oxygen Species Generating Effects
5-Fu, 5-fluorouracil; CAPOX, capecitabine; EGFR, epidermal growth factor receptor; FOLFOX, folinic acid and oxaliplatin; GSH, glutathione; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; RSL3, RAS-selective lethal 3; VEGF-A, vascular endothelial growth factor A.
Chemotherapeutic drugs, such as anthracyclines, platinum complexes, arsenic agents, and alkylating agents, have anticancer effects by generating high levels of ROS and/or inhibiting cellular antioxidant systems to mediate cytotoxicity in cancer cells (Loenhout et al., 2020). Radiotherapy uses ionizing radiation to induce DNA damage in cancerous tissue thus inducing cell death (Baskar et al., 2012). Immunotherapy leverages the immune system to accurately identify cancer cells and mediate cell death, with ROS playing a necessary role in coordinating the cross talk between various immune cells and creating a toxic environment for cancer cells (Wang et al., 2021). It is believed that cells with a normal level of oxidative stress would not be sufficiently affected, thus conveying a selective advantage for normal cells during oxidative treatment.
It is generally recommended that supplemental antioxidants should be avoided in cancer patients undergoing pro-oxidant therapies (Loenhout et al., 2020). A better understanding of ROS and selectively targeting specific ROS may serve to improve anticancer therapies.
Ferroptosis
There is increasing evidence that lipid ROS and ferroptosis are implicated in the pathophysiology of IBD and CRC. The primary regulator and inhibitor of ferroptosis is the selenoprotein GPX4 (Fig. 3) (Jiang et al., 2021). GPX4 inhibits ferroptosis by reducing lipid ROS through the conversion of GSH to GSSG to maintain redox balance (Ighodaro and Akinloye, 2018). Another important inhibitor of ferroptosis is the cystine/glutamate antiporter system composed of SLC7A11 and SLC3A2. Cystine is converted to cysteine, an essential amino acid for GSH. Inhibiting GPX4 or the cystine/glutamate antiporter system can lead to an accumulation of lipid ROS and an imbalance in redox balance. Certain drugs and compounds, such as RAS-selective lethal 3 (RSL3) and erastin, can induce ferroptosis by inhibiting GPX4 or the cystine/glutamate antiporter system. Antioxidants such as ferrostatin, liproxstatin-1, vitamin E, and CoQ10 can act as inhibitors of ferroptosis (Jiang et al., 2021).
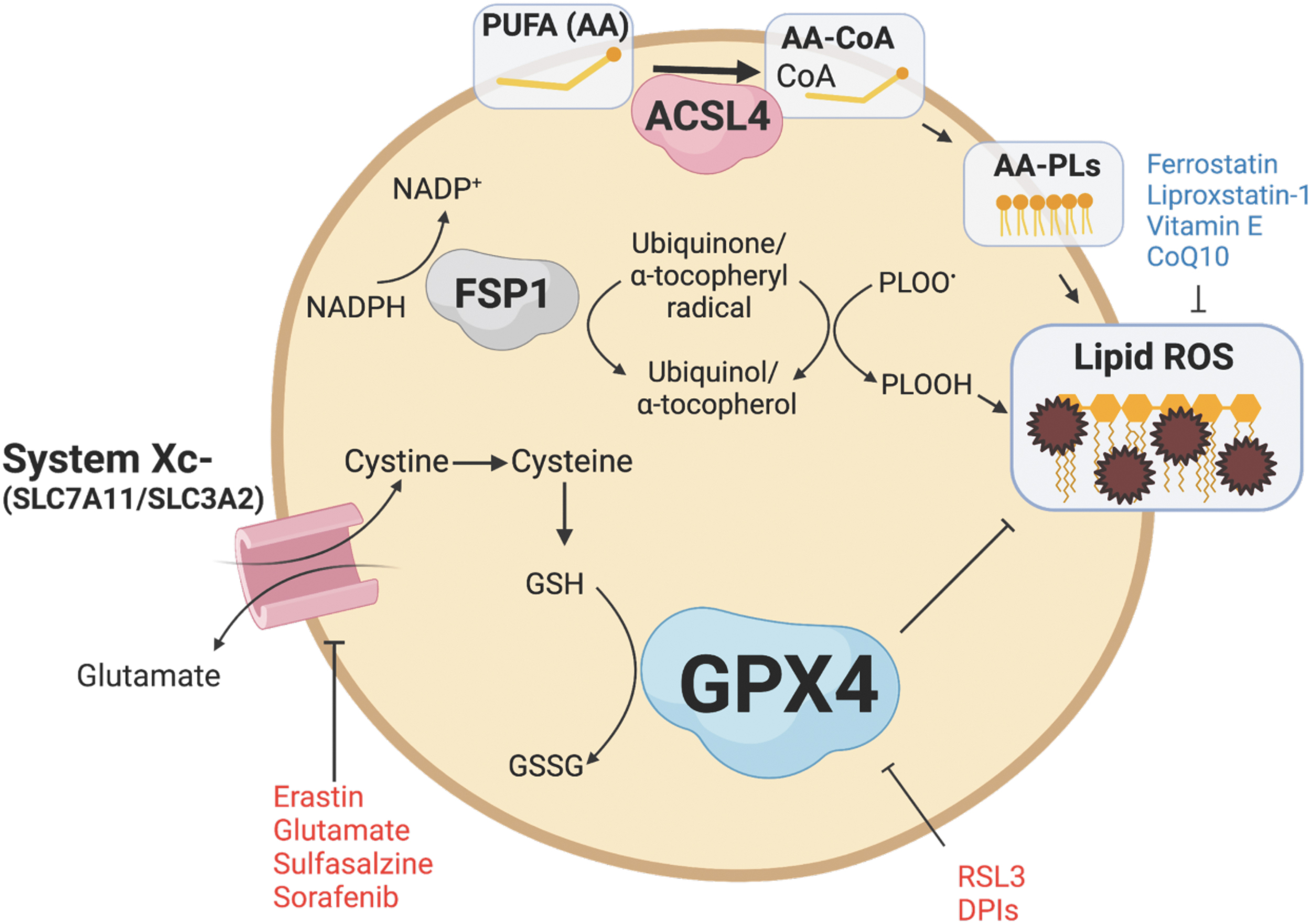
Although GPX4 is the primary inhibitor of ferroptosis, other pathways contribute to regulating ferroptosis. Ferroptosis suppressor protein 1 (FSP1; also known as AIFM2) reduces lipid peroxides and prevents ferroptosis through its NADH:ubiquinone oxioreductase function (Elguindy and Nakamaru-Ogiso, 2015). FSP1 reduces ubiquione to ubiquinol, which directly reduces lipid peroxides and indirectly recovers oxidized vitamin E (Bersuker et al., 2019; Doll et al., 2019). GCH1 inhibits ferroptosis through the production of tetrahydrobiopterin (BH4) and dihydrobiopterin (BH2). BH4 traps ROS and plays a role in ubiquinone synthesis (Kraft et al., 2020). In addition, acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are also mediators of ferroptosis.
ACSL4 incorporates long-chain PUFAs, namely arachidonic acid (20:4) and adrenic acid (22:4), with coenzyme A to form PUFA-CoA molecules, which can then be converted to phospholipids via LPCAT enzymes and thereby increasing phospholipid-PUFAs into cellular membranes (Dixon et al., 2015; Doll et al., 2017; Zou et al., 2019). Inhibiting ACSL4 through pharmacological or genetic means can alter the composition of phospholipids toward the incorporation of monounsaturated fatty acyl tails instead of long-chain PUFA tails, which can protect against ferroptosis (Doll et al., 2017).
As the name implies, iron is essential in ferroptosis (Fig. 4). Iron is necessary for a multitude of redox reactions that generate cellular ROS (Jiang et al., 2021). Through the Fenton reaction, iron amplifies PLOOHs and generates additional free radicals such as PLO• and PLOO• to further oxidize lipids (Jiang et al., 2021). Iron importers, divalent metal transporter-1 (DMT1), transferrin (Tf), and transferrin receptor (TfR) promote ferroptosis, while iron storage (ferritin) and export proteins (ferroportin) inhibit ferroptosis (Jiang et al., 2021; Liang et al., 2022). Nuclear receptor coactivator 4 (NCOA4) is responsible for the degradation of ferritin through a process called ferritinophagy, which releases free iron and promotes ferroptosis (Bogdan et al., 2016; Hou et al., 2016; Mancias et al., 2014).

In addition, HIF-2α, the master transcription factor critical for iron import, sensitizes CRC cells to ferroptosis by increasing cellular iron and altering cellular lipid composition (Singhal et al., 2021; Zou et al., 2019). HIF-2α has also been found to increase systemic iron via NCOA4-mediated intestinal ferritinophagy (Das et al., 2020). The association between HIF-2α and ferroptosis suggests a possible link between hypoxia, iron, and ferroptosis.
Identifying cellular death as ferroptosis poses many challenges. Unlike other markers of regulated cell death pathways such as capase-3 for apoptosis or RIP1/3 for necroptosis, there are no widely accepted markers of ferroptosis (Chen et al., 2016, 2021a; Ward et al., 2008). Recently, TfR1 has been proposed as a potential marker of ferroptosis, but more work is needed to understand its utility (Feng et al., 2020). Ferroptosis is typically identified by several biochemical and morphological hallmarks, including the ability to be rescued by ferroptosis inhibitors and the inability to respond to other cell death inhibitors (Chen et al., 2021a). Biochemical hallmarks include accumulation of cellular iron, induction of lipid peroxidation, and loss of antioxidant defense (Chen et al., 2021a).
Assays used to detect lipid peroxidation include the use of lipid-soluble fluorescent probes, antibodies directed against the final products of ferroptosis such as malonyl dialdehyde and 4-hydroxynonenal, and antibodies directed against oxidative DNA damage such as the phosphorylated H2A.X variant histone (γH2AX) and 8-hydroxydeoxyguanosine. Morphological features include cell enlargement, plasma membrane rupture, reduction in mitochondrial volume, disappearance of mitochondrial cristae, and increase in mitochondrial membrane density (Chen et al., 2021a).
Physiological Roles of Ferroptosis
Ferroptosis may play a physiological role in immune surveillance. It is known that other forms of regulated cell death such as apoptosis, necroptosis, and pyroptosis play a role in modulating immune responses (Green, 2019). Interferon γ (IFNγ) has been shown to downregulate the expression of system Xc − in macrophages (Sato et al., 1995). IL-4 and IL-13 suppress the expression of GPX4 in the kidney, lung, spleen, and cardiomyocytes. These same cells also exhibited upregulation of arachidonate lipoxygenase 15 (ALOX15), allowing for increased production of arachidonic acid metabolites, which serve as important inflammatory signaling molecules (Schnurr et al., 1999). GPX4 suppresses the function of LOXs and cyclooxygenases by reducing lipid peroxidation. In the context of reduced GPX4 expression, it is possible that secretion of lipid signaling molecules is altered and in turn alerts immune cells about ferroptosis-sensitive cells.
In plants, induction of ferroptosis prevents fungal Magnaporthe oryzae infection in rice through the removal of afflicted cells, thus preventing the spread of pathogen (Shen et al., 2020). It was also discovered that ferroptotic cell death in M. oryzae is necessary for the development of infection in its host (Shen et al., 2020).
Inhibition of ferroptosis plays a role in the development and homeostasis of adult tissues. GPX4 knockout embryos do not survive past midgestation (E7.5), indicating that inhibition of ferroptosis is necessary for embryonic development (Yant et al., 2003). When GPX4 is disrupted globally in adult mice via a whole-body tamoxifen-inducible Cre, mice die within 2 weeks and exhibit significant tissue injury (Angeli et al., 2014; Yoo et al., 2012). Interestingly, when both copies of GPX4 are deleted under an intestinal epithelial cell (IEC)-specific constitutive Cre driver, the mice are embryonic lethal, but ∼11% of mice were able to be rescued with the use of in utero supplementation of α-tocopherol (Mayr et al., 2020).
In addition, the intestinal GPX4 knockout pups that were rescued by α-tocopherol were born with a lower weight but regained weight by 7 weeks of age, suggesting that GPX4 is not necessary to maintain adult gut homeostasis and that there may be compensatory mechanisms in the intestines to reduce lipid ROS (Mayr et al., 2020).
Although the role of ferroptosis in normal physiology is not well understood, its implications in pathophysiological conditions are broadly studied. Ferroptosis has been linked to obesity, atherosclerosis, ischemia/reperfusion (I/R) injury, IBD, and cancer. In this section, we focus on the role of ferroptosis in IBD and CRC and how ferroptosis can be leveraged to develop therapeutics for these diseases.
Ferroptosis in IBD
There is evidence to suggest that ferroptosis may play a role in IBD; however, the specific relationship remains unclear. It is well-established that the levels of iron in the diet can have an impact on colitis in several mouse models. Studies have shown that high levels of iron supplementation induce gut dysbiosis and promote intestinal injury (Uritski et al., 2004; Werner et al., 2011; Zhang et al., 2022). In humans, high levels of dietary iron are associated with elevated risk of IBD (Kobayashi et al., 2019). In addition, levels of mucosal ROS production are correlated with disease severity, and iron chelators reduce ROS production and improve symptoms in IBD (Millar et al., 2000; Minaiyan et al., 2011).
These findings suggest that one possible mechanism by which excess iron in the intestines can lead to injury is by promoting the production of ROS through the Fenton reaction. This in turn leads to lipid peroxidation and the death of cells through a process called ferroptosis.
Ferroptosis-associated genes are elevated in IBD tissue (Xu et al., 2020). Furthermore, inhibition of ferroptosis by ferrostatin-1, liproxstatin-1, and desferoxamine reduced intestinal injury in dextran sulfate sodium (DSS)-induced murine models of colitis (Chen et al., 2020b; Wang et al., 2020b; Xu et al., 2020). Curculigoside (CUR), a botanical antioxidant, was also found to protect against ferroptosis through promoting GPX4 activity in a DSS model of colitis (Wang et al., 2020b). In an intestinal rat epithelial cell line (IEC6), CUR was shown to sensitize cells to selenium and upregulate GPX4 expression (Wang et al., 2020b). GPX4 knockdown by siRNA attenuated the protective effects of CUR against ferroptosis (Wang et al., 2020b). In IBD patients, impaired GPX4 activity and elevated markers of lipid peroxidation were present in small IECs (Mayr et al., 2020).
Similar to other mechanisms of ferroptosis, PUFAs induced inflammatory cytokine release in GPX4 knockdown IECs in response to high levels of iron, lipid peroxidation, and increased activity of ACSL4 (Mayr et al., 2020). Intestinal-specific GPX4 knockout mice fed on a PUFA-enriched Western diet displayed increased intestinal damage and symptoms similar to IBD. Together, these findings indicate that ferroptosis plays a crucial role in IBD and targeting this pathway may be a promising therapeutic approach for treating IBD (Mayr et al., 2020).
Ferroptosis in CRC
Targeting ferroptosis is a promising approach in many cancers, including CRC (Jiang et al., 2021). The first evidence that targeting ferroptosis as a therapeutic intervention in vivo was shown in mouse models of pancreatic ductal adenocarcinoma (PDAC) (Badgley et al., 2020). Cyst(e)ine is a central dependency for PDAC. Depletion of cyst(e)ine through genetic deletion of SLC7A11 or pharmacologically through cyst(e)inase induced tumor-selective ferroptosis and inhibited PDAC growth. These findings demonstrate that activating ferroptosis is a promising approach for cancer therapy (Badgley et al., 2020).
CRC accumulates large amounts of iron to meet the metabolic requirement for growth. Elevated HIF-2α levels in CRC mediate colon carcinogenesis through increased expression of the iron importer DMT1 (Xue et al., 2012). In addition, CRC activates ectopic hepcidin in the tumor microenvironment to sequester iron (Schwartz et al., 2018). Mining The Cancer Gene Atlas, GPX4 and System Xc − are highly induced in CRC (Muzny et al., 2012), potentially to inhibit iron toxicity and ferroptosis. Studies have shown that GPX4 overexpression in CRC cell lines can prevent cell death following RSL3 treatment, suggesting that ferroptosis may be a potential mechanism of cell death in CRC (Sui et al., 2018).
In vitro experiments have also demonstrated that RSL3 is able to induce ferroptosis and increase levels of ROS and cellular labile iron pools in a time- and dose-dependent manner in three CRC cell lines (Sui et al., 2018). Moreover, cisplatin induces ferroptosis, and a combination of cisplatin and erastin is synergistic in suppressing the growth of CRC cell lines suggesting that ferroptosis provides alternative mechanisms of cell death in CRC (Guo et al., 2018).
The physiological roles of ferroptosis have not been fully understood, but there is a growing body of evidence to suggest that ferroptosis plays a role in tumor suppression. Several tumor suppressors such as p53 and BAP1 can sensitize tumor cells to ferroptosis. It was discovered that acetylation of p53 lysine sites potentiates ferroptosis through suppression of System Xc − transcription (Jiang et al., 2015; Wang et al., 2016). However, the role of p53 in affecting cancer is context dependent. In CRC, p53 has also been shown to inhibit ferroptosis by blocking dipeptidyl-peptidase 4 (DPP4) activity and preventing localization to the nucleus to form the DPP4-NOX1 complex that promotes lipid peroxidation (Xie et al., 2017). Furthermore, loss of p53 in tumor-bearing mice synergistically increased the efficacy of erastin (Xie et al., 2017). Disassembly of the DPP4-NOX1 complex in the context of p53 loss restored the erastin-induced sensitivity of CRC (Xie et al., 2017).
In contrast to Jiang et al. (2015), it was found that p53 upregulates SLC7A11 expression in CRC and thus protects CRC cells from ferroptosis (Xie et al., 2017). It has also been reported that BAP1 promotes ferroptosis through downregulation of SLC7A11, one of the two subunits in system Xc − (Zhang et al., 2018). However, the exact role of tumor suppressors in modulating ferroptosis is not yet fully delineated. In addition, unlike the sufficient ferroptosis-mediated tumor suppressing activity of p53 alone, it is unclear to what extent BAP1 contributes to tumor suppression through ferroptosis (Zhang et al., 2018). The Ras mutation, one of the most common genetic mutations in CRC, is widely linked to ferroptosis (Bartolacci et al., 2022; Chen et al., 2020a; Park et al., 2018).
In Kras-mutant lung cancer, it has been reported that fatty acid synthase protects cancer cells against ferroptosis through the generation of oxidation-resistant fatty acids, while it sensitizes the tumor microenvironment to ferroptosis through producing more PUFAs (Chen et al., 2020a). Moreover, GPX4 and ACSL3 appear to be central dependencies in Kras-mutant lung cancer, highlighting the sensitivity of ferroptosis in Ras-mutant cancers (Andreani et al., 2022).
It has also been shown that during immunotherapy, IFNγ secreted from CD8+ T cells sensitizes tumor cells to ferroptosis through the downregulation of SLC3A2 and SLC7A11, resulting in reduced cystine uptake by tumor cells and the subsequent tumor cell lipid peroxidation and ferroptosis (Wang et al., 2019). Moreover, CD8+ T cell-derived IFNγ increased tumor ACSL4 expression to promote ferroptosis (Liao et al., 2022).
Several anticancer drugs have been found to inhibit CRC through a ferroptotic-dependent manner (Table 3). Apatinib induces ferroptosis in CRC cell lines through activation of the ELOVL6/ACSL4 pathway (Tian et al., 2021). A benzopyran derivative, 2-imino-6-methoxy-2H-chromene-3-carbothioamide, induces ferroptosis in CRC through downregulation of SLC7A11 (Zhang et al., 2020b). Talaroconvolutin A induces ferroptosis in CRC cells in a time- and dose-dependent manner (Xia et al., 2020). Honokiol induces ferroptosis in CRC cells through inhibiting the activity of GPX4 (Guo et al., 2020). Camellia nitidissima Chi induces ferroptosis in CRC cells through downregulation of GPX4 (Chen et al., 2021b).
Drugs That Have Been Found to Induce Ferroptosis in Colorectal Cancer
ACSL4, acyl-CoA synthetase long-chain family member 4; BSO, buthionine sulfoximine; CNC, Camellia nitidissima Chi; DCA, dichloracetate; GPX4, glutathione peroxidase 4; IMCA, 2-imino-6-methoxy-2H-chromene-3-carbothioamide; TalaA, talaroconvolutin A.
The role of ferroptosis in CRC is not yet fully elucidated. However, the results demonstrate that ferroptosis serves as a targetable pathway and that regulating this form of cell death may provide a new avenue of anticancer therapy.
Conclusions and Future Perspectives
ROS play a vital role in maintaining homeostasis in the body by mediating immune function, metabolism, and cellular signaling. However, an imbalance in the levels of ROS can contribute to disease states such as sepsis, atherosclerosis, and CGD. Moreover, there is growing evidence to suggest that lipid ROS specifically are implicated in IBD and CRC. Accumulation of lipid ROS results in an iron-dependent programmed cell death pathway termed ferroptosis. Although evidence of modulating ferroptosis, either induction or inhibition, is beneficial in treating preclinical models of IBD and CRC, there are still no viable pharmacological agents available to treat patients in clinical trials. The development of safe and efficacious ferroptotic modulators could serve as important means of treating IBD and CRC.
To effectively target ferroptosis in IBD and CRC, it is important to identify the specific cell types and temporal dynamics involved in the process. While inhibition of ROS in general does not provide benefits in IBD, systemic inhibition of ferroptosis could lead to similar results. Systemic cyst(e)ine depletion selectively decreased PDAC growth, and there is a growing body of evidence to suggest that other specific cell types in the tumor microenvironment affect ferroptosis sensitivity in cancer cells. It was reported that exosome-miR-522 from cancer-associated fibroblasts inhibit ferroptosis in cancer cells through inhibition of ALOX15 and lipid ROS accumulation (Zhang et al., 2020a). Likewise, the role of ferroptosis in immune cells also mediates cancer growth. Ferroptosis in immune cells has been found to be both protumorigenic and antitumorigenic (Fig. 5).

Liao et al. (2022) demonstrated that IFNγ secretion from CD8+ T cells mediated ferroptosis-induced tumor killing through upregulation of ACSL4. In contrast, a recent report demonstrated that ferroptosis in neutrophils results in immune suppression and promotion of tumor growth (Kim et al., 2022). The context-dependent nature of ferroptosis in cancer highlights the need to accurately identify and target specific cell types to ensure proper antitumor activity.
Currently, there are no pharmacological agents that target ferroptosis in humans. However, research has indicated that the gut microbiome may play a role in ferroptosis (Fig. 6). Intestinal I/R injury alters the gut microbiota and results in elevated levels of capsiate that serves to protect against ferroptosis-dependent intestinal I/R injury (Deng et al., 2021). Mice treated with antibiotics exhibited worse intestinal injuries (Deng et al., 2021). In addition, it has been demonstrated that bromoacetic acid-induced hepatic injury alters the gut microbiome and results in elevated levels of the gut microbiota metabolite glycochenodeoxycholate (GCDCA). GCDCA activates TfR-ACSL4-mediated ferroptosis to promote fatty liver disease (Liu et al., 2022). Butyrate, a short-chain fatty acid produced by gut microbiota, promotes ferroptosis through inducing NCOA4-mediated ferritinophagy (Zhao et al., 2020). A more nuanced understanding of the context-dependent nature of ferroptosis in these settings will help direct better targeted therapies in the intestine and colon.
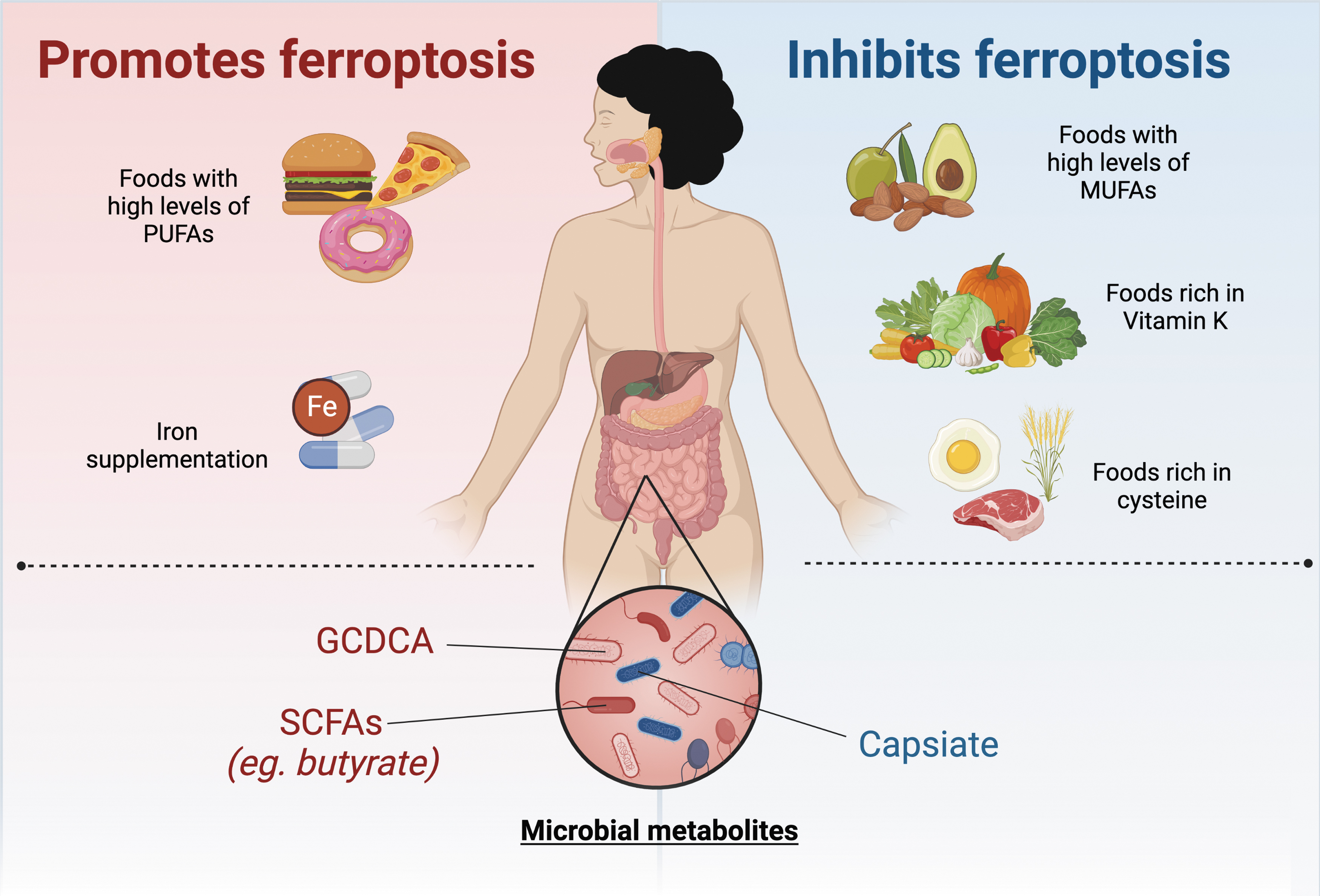
Lastly, modulation of diet through amino acids, vitamins, iron, and lipids also serve as a method of targeting ferroptosis (Fig. 6). In a model of cytosolic aspartate aminotransaminase (GOT1)-deficient PDAC, a diet lacking cysteine resulted in increased survival due to PDAC cell death by ferroptosis (Kremer et al., 2021). A recent study demonstrated that FSP1-dependent noncanonical vitamin K cycle protects against ferroptosis in human cancer cell lines (Mishima et al., 2022). In murine models of hemochromatosis, mice fed a high-iron diet exacerbated hepatic injury through ferroptosis (Wang et al., 2017). In addition, a high-fat diet promotes colitis-associated carcinogenesis through ferroptosis evasion in the ER stress-mediated pathway (Zhang et al., 2021).
However, it is important to note that the effect of dietary fat on ferroptosis may be specific to the type of fat consumed, as PUFAs have been shown to promote ferroptosis, while monounsaturated fatty acids have been shown to protect against ferroptosis (Magtanong et al., 2019; Yang et al., 2016).
Understanding the role of lipid ROS and ferroptosis in IBD and CRC is of central importance. As research continues to uncover the role of ferroptosis in CRC and IBD, this will open up new possibilities for treatments based on the modulation of ferroptosis.
Summary Points
ROS have physiological functions, and redox imbalance leads to pathophysiological states.
The roles of lipid ROS-induced ferroptosis in normal physiology are still relatively unclear; however, its roles in disease have been widely reported.
There is mounting evidence that ferroptosis contributes to the pathogenesis of IBD and CRC.
Inhibition of ferroptosis has been shown to be beneficial in preclinical models of IBD and CRC.
The role of ferroptosis in IBD and CRC may be leveraged for the development of new therapeutics toward these diseases.
Footnotes
Authors' Contributions
W.H. wrote the article and tables. W.H. and N.A. designed the figures. W.H. and Y.M.S. conceived the review and revised the article. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by NIH grants: R01CA148828, R01CA245546, and R01DK095201 (Yatrik M. Shah), and UMCCC Core Grant P30CA046592 (Yatrik M. Shah). Wesley Huang was supported by NIH grant: F30DK131851.