
Abstract
Significance:
Ferroptosis is featured by the accumulation of polyunsaturated-lipid peroxidation on cellular membranes in an iron-dependent manner. Ferroptosis has been implicated in various pathophysiological processes, including cancer, neurodegeneration, and ischemia-reperfusion tissue injury. However, our understanding about the dynamic and context-specific regulation of ferroptosis remains incomplete.
Recent Advances:
As the major substrate for peroxidation, the cellular lipidome regulates ferroptosis sensitivity and execution by controlling the abundance and availability of polyunsaturated-lipids for peroxidative modifications. In turn, the cellular lipidome is regulated by a complex network of enzymes and transporters, as well as upstream layers of receptors, kinases, and transcription factors. A number of research has shed light on the link between lipid metabolism and ferroptosis. Here, we summarize our current knowledge on the role of the lipidome and associated protein regulators in various stages of ferroptosis, ranging from initiation, execution to cell death evasion by cells experiencing ferroptotic stress.
Critical Issues:
This review provides an overview of the mechanisms underlying lipid peroxidation and ferroptosis by discussing the lipid species that directly contribute to lipid peroxidation and ferroptosis, how cells regulate the abundances of these pro-ferroptosis lipids, how lipid peroxidation causes cell death, and how cells prevent and repair membrane lipid damage under ferroptotic conditions.
Future Directions:
Cell fate regulation in vivo could be different from in vitro culture settings. We envision that a comprehensive and detailed understanding about these important questions in the dynamic regulation of ferroptosis in vivo will accelerate our development of ferroptosis-targeted therapies to improve human health.
Biological membranes drive the compartmentalization of cellular space for efficient and uninterrupted execution of biological processes. Physical and chemical insults to cellular membranes could compromise organelle integrity, block biological functions, and culminate in cell death. Ferroptosis is one of such cell death modalities, specifically driven by aberrant accumulation of lipid peroxidation in an iron-dependent manner (Dixon et al., 2012). In the past decade, numerous genetic and chemical tools that selectively modulate ferroptosis have emerged, enabling in-depth characterization of the biochemical basis of ferroptosis and its context-specific roles in biological systems (reviewed in Green, 2019; Hassannia et al., 2019; Jiang et al., 2021; Nehring et al., 2020; Stockwell, 2022; Stockwell et al., 2020).
Currently, ferroptosis is operationally defined by: (i) upregulation of lipid peroxidation reported by fluorogenic probes such as BODIPY-C11 and Liperfluo or by redox lipidomics (Drummen et al., 2002; Kagan et al., 2017), and (ii) functional rescue by lipophilic radical-trapping antioxidants (RTAs) including ferrostatin-1 and liproxstatin-1, and by iron chelators including deferoxamine (Dixon et al., 2012; Friedmann Angeli et al., 2014; Zilka et al., 2017). At the subcellular level, ferroptotic cell death is featured by distinct morphological changes, including mitochondria condensation and outer mitochondrial membrane rupture, though the trigger for these changes and its direct contribution to cell death remain to be determined (Dixon et al., 2012; Friedmann Angeli et al., 2014). Since the term was coined, ferroptosis has been implicated in the pathogenesis of a variety of human diseases (Stockwell, 2022), highlighting the potential therapeutic value of targeting ferroptosis.
Lipids are the major components of cellular membranes, and the cellular lipidome is composed of diverse lipid classes. In ferroptosis, lipids are indispensable substrates in the lipid peroxidation reaction, and their impact on cell death largely follows the rules governing the influence of substrates on the overall reaction outputs. On the other hand, lipids in cellular systems are extremely diverse and are extensively regulated by their import, synthesis, degradation, and distribution.
Emerging single-cell metabolomics and lipidomics technologies have shed some light on the metabolic heterogeneity of cells in situ (Capolupo et al., 2022; Tajik et al., 2022). The complexity of the cellular lipidome and associated metabolic network poses a challenge in drawing a unified conclusion about the regulatory role of lipids in ferroptosis. Illuminated by the recent reviews summarizing the interplays between lipid metabolism and ferroptosis in various contexts (Liang et al., 2022; Mishima and Conrad, 2022; Zheng and Conrad, 2020), here we discuss four questions centered on the direct link between lipids and ferroptosis.
First, what are the primary chemical basis of lipids that are subjected to lipid peroxidation and what are the cellular lipid classes that contribute to ferroptosis? Second, how are ferroptosis-related lipids regulated biochemically? Third, how does lipid peroxidation drive ferroptotic cell death? Does this event involve damage to specific cellular compartments or through downstream production of lipid-derived electrophiles? Fourth, how does the cell prevent and repair membrane damage induced by lipid peroxidation during ferroptosis? Finally, we briefly illustrate our perspective on the gaps in targeting ferroptosis for treating human diseases.
Question 1: What Is the Chemical Basis of Lipids That Contribute to Ferroptosis?
Primary chemical features enabling lipid peroxidation
Role of the pentadienyl structure
A characteristic event in ferroptosis is oxidative modification of polyunsaturated fatty acid (PUFA)-lipids on cellular membranes (Dixon et al., 2012; Yang et al., 2014). PUFA-phospholipids are important cellular membrane components. In animal cells, most PUFA species contain unconjugated double bonds, that is, every two double bonds are separated by a methylene bridge (R1-CH = CH-CH2-CH = CH-R2, Fig. 1a).

The bis-allylic H atoms render PUFA-phospholipids susceptible to peroxidation because the resulting radical is stabilized by resonance with the neighboring double bonds (Conrad and Pratt, 2019). In autoxidation, abstraction of a labile hydrogen atom from phospholipids by an initiating radical affords a carbon-centered radical (L•), which undergoes rapid rearrangement of the double bonds to form a conjugated diene (Fig. 1a) (Yin et al., 2011).
This reactive species then combines with molecular oxygen (O2) to generate a lipid peroxyl radical (LOO•), which can abstract an H atom from another PUFA-lipid moiety to form a new lipid radical and a lipid hydroperoxide (LOOH), further fueling the autocatalytic chain reaction. The radical chain reaction terminates when a reductant reduces the peroxyl radical (Fig. 1a). In this non-enzymatic process, the initiating radical can be lipid alkyl/hydroxyl radicals derived from a reduction of hydroperoxide (H2O2) in iron-mediated Fenton reaction whereas the reductant can be a one-electron donor, a low-valent iron, or another redox-active metal (Conrad and Pratt, 2019).
Replacing bis-allylic H with deuterium lowers PUFA peroxidation rate by ∼100-fold for enzymatic reactions and 6-fold for non-enzymatic reactions due to the kinetic isotope effects, underscoring the importance of bis-allylic H (Kitaguchi et al., 2006; Knapp et al., 2002; Muchalski et al., 2015). Deuterium-labeled PUFAs at the bis-allylic positions also suppress lipid peroxidation and ferroptosis in cellular contexts (Firsov et al., 2022; Firsov et al., 2019; Yang et al., 2016), offering an interventional strategy to prevent ferroptosis in disease settings.
Though uncommon in animal cells, FAs containing ≥2 double bonds in the conjugated form are present in certain plant seeds, microorganisms, and ruminant animal products (Gong et al., 2019). Conjugated linoleic acid and conjugated linolenic acid are the most widely reported conjugated fatty acids (Fig. 1b). Conjugated PUFAs containing three or more double bonds show a 2.5-fold faster rate of triene-adjacent H atom abstraction compared with that of non-conjugated PUFA due to the stronger electron attraction effect of the double bonds in an in vitro radical clock experiment (Do et al., 2021).
In line with this, a recent study showed that treating cancer cells with conjugated α-eleostearic acid (α-ESA) triggered ferroptosis with high potency in a subset of triple negative breast cancer cell lines, consistent with the high oxidizability of α-ESA (Fig. 1b) (Beatty et al., 2021). Hence, exploiting conjugated PUFAs as an anti-cancer treatment appears an attractive strategy (Beatty et al., 2021), though their in vivo efficacy in suppressing human cancers remains to be investigated.
One key physiological advantage of incorporating PUFA into membrane lipids is to increase membrane fluidity. This role can be partially fulfilled by monounsaturated fatty acids (MUFAs) containing a cis double bond in the acyl tail (Fig. 1b). However, without the pentadienyl structure and bis-allylic H atoms, MUFAs are not susceptible to lipid peroxidation.
Likely due to MUFAs competing with PUFAs for incorporation into lysophosphatidic acid (LPA) and lyso-phospholipids, excess MUFA treatment could reduce membrane PUFA-lipid contents and suppress cellular ferroptosis susceptibility (Magtanong et al., 2019; Yang et al., 2016). Hence, the cellular MUFA/PUFA-lipid ratio has a significant impact on ferroptosis sensitivity.
Proximity provided by membrane packing
Though the initial formation of the lipid radical initiator does not require a collective of PUFA molecules, the driving molecular event in the propagation of lipid peroxidation is a seeding lipid peroxyl radical abstracting a hydrogen atom from a neighboring PUFA-lipid to form another lipid radical. Therefore, PUFA-lipids packed in sufficient proximity with similar orientations in membrane lipid bilayers facilitate the diffusion of peroxyl radicals and promote ferroptosis more readily than free PUFA molecules.
Indeed, treating cells with oxygenated arachidonyl-(C20:4) and adrenyl (C22:4)-phosphatidylethanolamine (PE)-OOH, but not free arachidonyl-OOH or adrenyl-OOH, leads to ferroptosis (Kagan et al., 2017). Further supporting this distinction, most cells responding to genetic or pharmacological induction of ferroptosis require esterification of PUFA catalyzed by long-chain (LC) fatty acyl–CoA ligases, including acyl-CoA synthetase long chain family member 4 (ACSL4) and downstream acyltransferases (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017; Zou et al., 2019).
These observations strengthen the notion that lipid peroxidation requires free PUFAs to be enzymatically incorporated into phospholipids to cause sufficient membrane damage and ferroptosis. Meanwhile, the quantitative measurements in phospholipid proximity and packing densities required to support efficient lipid peroxidation spreading remain to be further defined. Of note, peroxidation of free PUFA can also trigger ferroptosis in some contexts.
In a recent study, Cui et al. (2022) reported that Fas-associated factor 1 (FAF1) could directly bind free PUFA and form a globular structure, protecting PUFA from labile iron-mediated peroxidation, which may explain the limited contribution of free PUFA to ferroptosis under physiological conditions. However, with FAF1 knockdown in SV589 cells or mouse liver, PUFA stimulation can cause severe cell death and liver injury respectively (Cui et al., 2022).
Cellular lipid classes contributing to ferroptosis
Though the primary chemical features required for lipid peroxidation are relatively well-defined, the specific lipid classes fueling the burst of cell-killing peroxidative damage in various cellular contexts are rather poorly characterized. Cellular membrane lipids are diverse in structure and distribution, with the most abundant lipid classes being glycerophospholipids, sphingolipids, and cholesterols (Harayama and Riezman, 2018).
We envision that three lines of evidence may support the involvement of specific lipid classes in ferroptosis: (i) detection of the lipid peroxides or lipid radicals in ferroptotic cells and tissues using redox lipidomics or other analytical approaches; (ii) genetic perturbation of lipid substrate-specific enzymes causing a shift in ferroptosis sensitivity or cell death kinetics; and (iii) treatment with synthetic oxygenated lipid species leads to ferroptosis induction or sensitization. Thus far, the involvement of phospholipids and sterols, at least, in lipid peroxidation and ferroptosis, have been substantiated by orthogonal approaches.
Free radical oxidation of phospholipids
Phospholipids share the common structures of a glycerol backbone, a head group, and one or two fatty acid (FA) chain(s) (Fig. 2a). Glycerophospholipids can be subgrouped by their distinct head groups, FA tails, and chemical bonds connecting FA chains and glycerol moiety. Grouped by the head structure, phospholipids include PE, phosphatidylcholine (PC), phosphatidylinositol, phosphatidylserine (PS), phosphatidylglycerol, etc. By varying the unsaturation level of the FA chains, phospholipids contain combinations of saturated fatty acid (SFA), MUFA, PUFA, respectively (Fig. 1b). The linkage chemical bond includes, at least, acyl- (ester linked), alkyl- (ether-linked), or alkenyl- (vinyl ether-linked) groups.
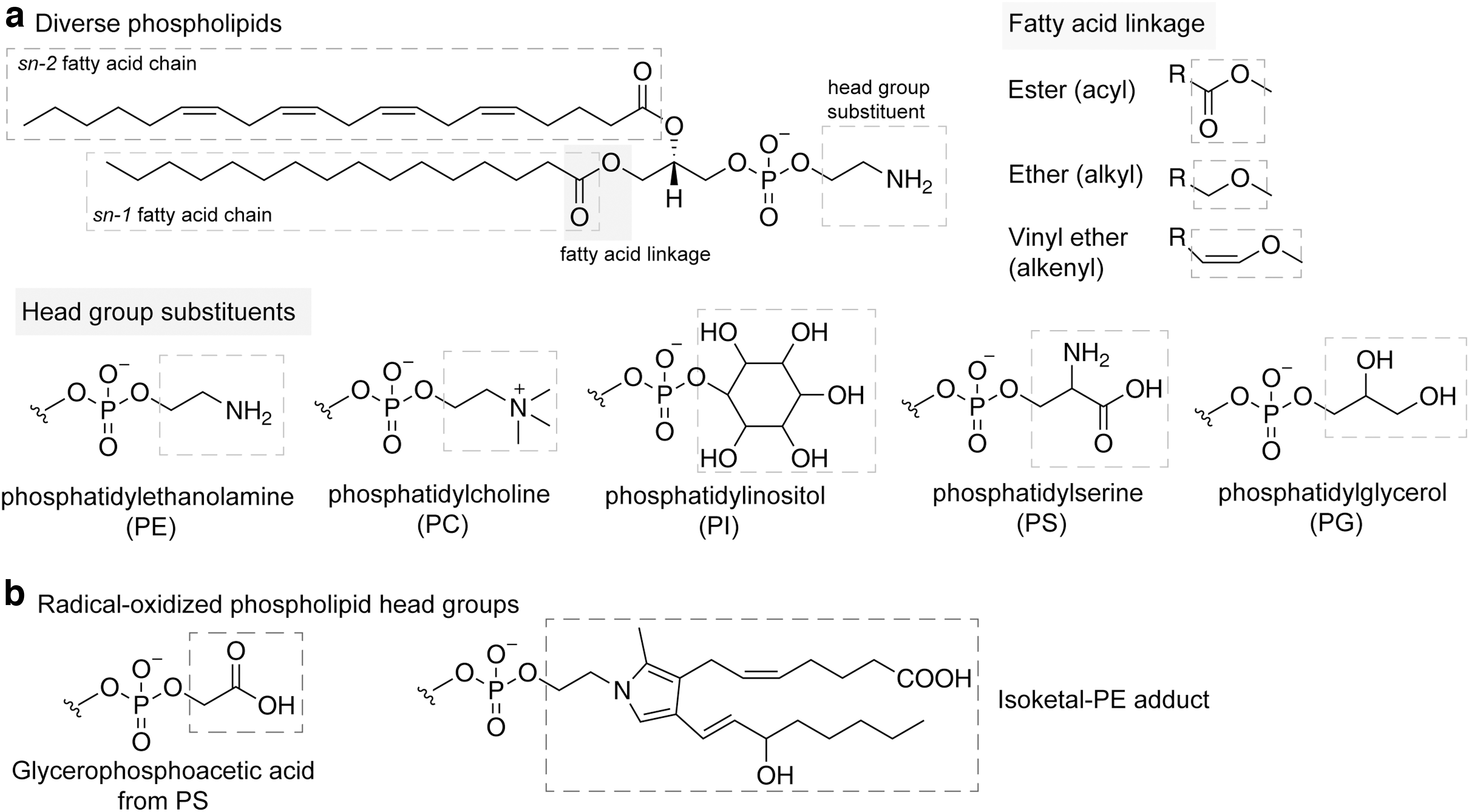
Though the structure of the phospholipid head group is often the first parameter used to categorize a phospholipid, its contribution to ferroptosis remains rather enigmatic. On one hand, the head groups play an important role in determining the subcellular localization of phospholipids across various membrane-bound compartments (Harayama and Riezman, 2018; Yang et al., 2018), though technologies with higher spatiotemporal resolutions are in demand to empower more systematic characterizations of membrane-specific lipid distribution patterns.
On the other hand, the chemical structures in phospholipid head groups are also targets of radical modification. Amine-containing phospholipid head groups such as PE and PS are susceptible to oxidation by radical or electrophile (Spickett, 2020). Radical oxidation of PS results in a glycerophosphoacetic acid moiety, a serine head group containing acetic acid linked to the phosphate group, as the most abundant derivative (Fig. 2b) (Maciel et al., 2014; Spickett, 2020).
In addition, the ethanolamine moiety of PE can be modified by the arachidonic acid-derived reactive isoketals (Fig. 2b) (Bernoud-Hubac et al., 2004; Sullivan et al., 2010). Of note, the glycation of both PS and PE enhances the propensity of oxidation of these head groups due to the potential oxidation sites in the glucose moiety (Maciel et al., 2013; Melo et al., 2013). These modifications underscore the key roles of the head group in membrane structure and function, as well as cell signaling. The contributions of other head groups of phospholipids in lipid peroxidation during ferroptosis require further investigations.
The presence of bis-allylic sites in the side chain renders PUFA-phospholipids susceptible to oxidation; however, the extent of oxidizability within the membrane varies depending on the degree of unsaturation of the FA chains and the diffusibility (Yin et al., 2011). The general consensus is that phospholipids containing more bis-allylic sites tend to exert stronger toxicity in cells potentially due to elevated susceptibility to peroxidation. Arachidonic acid (C20:4n6), for instance, is more prone to peroxidation than linoleic acid (C18:2n6) due to the three possible bis-allylic positions for radical initiation.
The oxidizability of membrane lipids is also limited by the diffusibility within the membrane and the exposure of polar peroxyl radical to the surface (Yin et al., 2011). Redox lipidomic analysis in RSL3-treated Pfa1 cells support this supposition, revealing abundant levels of oxidized PEs containing arachidonic acid or adrenic acid at the sn-2 position (Kagan et al., 2017), which might be partially contributed by their higher oxidizability.
With regard to the linkage bond, recent genetic studies revealed that PUFA-ether-phospholipids (PUFA-ePLs) are critical contributors to ferroptosis sensitivity in some cellular contexts, including renal and ovarian cancer cells (Cui et al., 2021; Zou et al., 2020a), but not others, including HT-1080N fibrosarcoma cells (Magtanong et al., 2022). The alkene group in plasmalogens, a subclass of ePL containing the -O-CH = CH- linkage at sn-1, was previously suggested to act as an antioxidant reservoir on membranes (Engelmann, 2004; Reiss et al., 1997; Zommara et al., 1995).
The presence of an sn-2 PUFA, however, appears to dominate the oxidative potential of PUFA-plasmalogens. On the other hand, PUFA-ePLs are not intrinsically more sensitive to peroxidation than other PUFA-phospholipids, given that PUFA-ePLs or their non-ether-linked phospholipid counterparts have comparable sensitizing effects on ferroptosis (Zou et al., 2020a). The relative contribution of PUFA-ePLs to ferroptosis is likely dictated by both their high abundance and their likelihood to locate on oxidation-susceptible sites such as the plasma membrane inner leaflet that is more accessible to cytosolic reactive oxygen species (ROS) and labile iron.
Notably, compared with di-ester phospholipids, PUFA-ePLs play a unique role in shaping the changes in ferroptosis sensitivity by exhibiting significant plasticity when cells undergo cell-fate and -state transitions. For instance, clear-cell renal cell carcinoma (ccRCC) cells that were initially susceptible to ferroptosis induction became glutathione peroxidase 4 (GPX4)-independent after in vivo adaptation and selection, and a key feature of cancer cells evading from ferroptosis is the downregulated PUFA-ePLs (Zou et al., 2020a).
Hence, plasticity of the cellular lipidome may undermine the durability of ferroptosis-targeted anti-cancer therapies. On the other hand, neurons and cardiomyocytes gain ferroptosis sensitivity via active upregulation of PUFA-ePLs during differentiation (Zou et al., 2020a). In a Parkinson's disease model, ɑ-synuclein induces dopaminergic neuron's vulnerability to lipid peroxidation and ferroptosis, and resistance to ferroptosis in ɑ-synuclein-depleted neurons is associated with reduced PUFA-ePLs (Mahoney-Sanchez et al., 2022). The molecular mechanism underlying the adaptability in the ether-linked fraction of cellular phospholipids remains unclear.
Free radical oxidation of sterols
In addition to PUFA-containing lipids, cholesterol and other sterols with double bond on the ring are also potential targets for radical oxidation (Fig. 3a) (Zielinski and Pratt, 2016). Sterols are susceptible to free radical oxidation due to the presence of allylic H atoms. Sterol peroxidation is generally slower than PUFA peroxidation, as the propagation rate constant of cholesterol peroxidation is 1/23 of that of arachidonic acid (Xu et al., 2009).

However, the precursor of cholesterol synthesis, 7-dehydrocholesterol (7-DHC), and its isomer 8-dehydrocholesterol (8-DHC) are highly sensitive to peroxidation since the conjugated double bonds on their rings further weaken the hydrocarbon bonds at position 9 and 14, and 7, respectively (Fig. 3a, b) (Porter et al., 2020). 7-DHC and 8-DHC have 5- and 11-fold higher peroxidation propagation rate constant than that of arachidonic acid, respectively (Xu et al., 2009; Yin et al., 2011), making them the most autoxidizable lipids in cells.
Indeed, treating neuroblastoma cells with the oxysterol mixture from 7-DHC peroxidation induces strong cytotoxicity (Xu et al., 2010). Notably, the high abundance of cholesterol in cellular membranes, up to 50% of lipid content in certain cell types (van Meer et al., 2008), makes them prominent substrates for peroxidation. The collective contribution of sterols to ferroptosis in specific cell types remains poorly defined.
Given its high oxidizability, 7-DHC was speculated to promote lipid peroxidation, thereby sensitizing cells to ferroptosis. However, the accumulation of 7-DHC protects Pfa1, HT-1080, and MDA-MB-435 cells from ferroptosis by potentially suppressing the formation of (phospho)lipid-truncated species, proposed downstream executors of ferroptosis (Angeli et al., 2021). The context-specific roles of 7-DHC in radical chain oxidation reactions may be attributed by factors, including subcellular localization, overall cellular lipidomes, and the surrounding antioxidants. Further studies are required to understand the precise role of 7-DHC in lipid peroxidation and ferroptosis in various biological contexts.
Question 2: How Are Ferroptosis-Related Lipids Regulated?
Protein regulators of lipids that contribute to ferroptosis
The cellular lipidome is regulated by a complex molecular network (Abumrad et al., 2021; Ko et al., 2020; Peck and Schulze, 2019). Given the indispensable role of PUFA–phospholipids in ferroptosis, the uptake, biosynthesis, and degradation pathways of PUFAs and their incorporation into phospholipids are critical in dictating ferroptosis sensitivity in cells (Fig. 4).
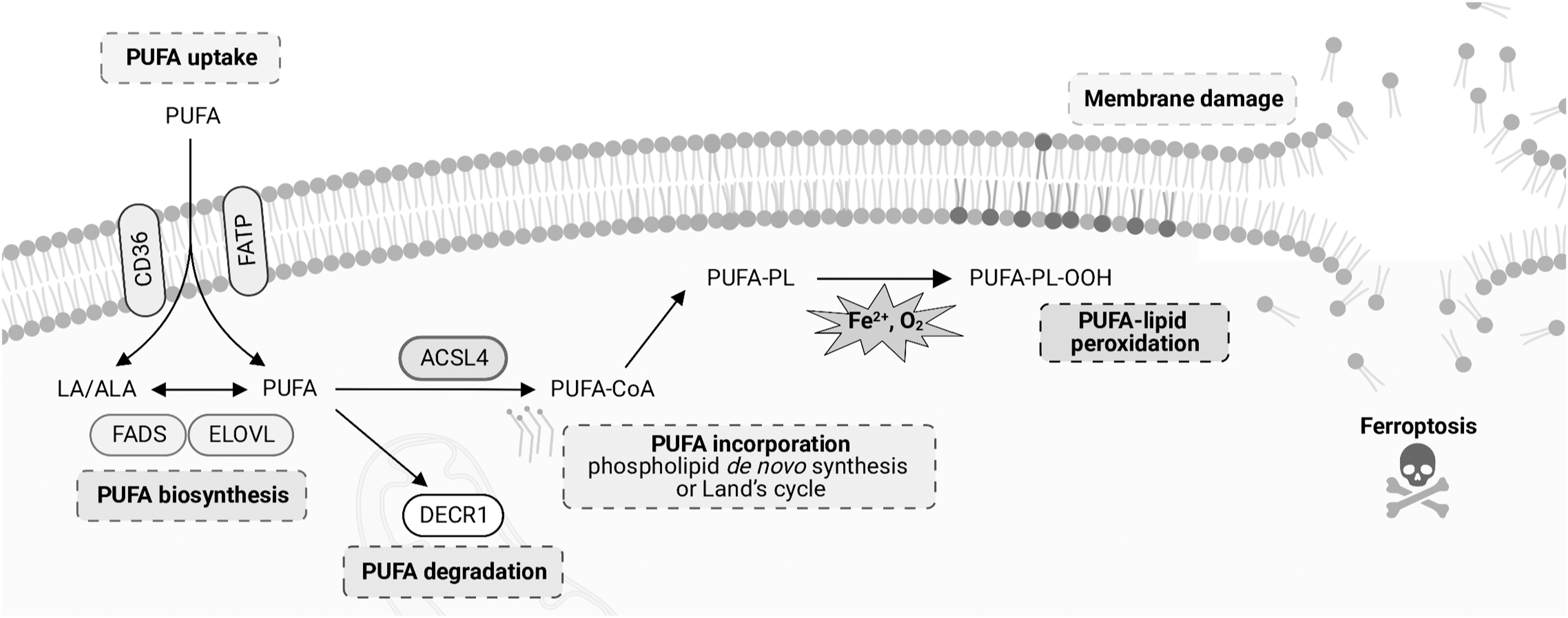
PUFA biosynthesis
As the key building blocks of phospholipids, PUFAs cannot be de novo synthesized by mammalian cells through acetyl-CoA, with mead acid (C20:3n9) being an exception (Chilton et al., 2014). Instead, mammals ingest essential FAs, including linoleic acid and α-linoleic acid (C18:3n3) from diets to synthesize n-6 or n-3 LC (long-chain, 20–24 carbons) PUFAs, including arachidonic acid and eicosapentaenoic acid (EPA, C20:5n3).
Elongation of very long chain FA (ELOVL) proteins and fatty acid desaturases (FADSs) on the endoplasmic reticulum (ER) membrane involved in LC-PUFA biosynthesis, with FADS1/2 touted to be rate-limiting factors (Chilton et al., 2014). Recent studies have revealed that ELOVL5 and FADS1 are pro-ferroptosis factors in gastric cancer and that FADS2 mediates ferroptosis sensitivity in hepatocytes on hepatitis C virus infection (Lee et al., 2020b; Yamane et al., 2022).
In contrast, stearoyl-CoA desaturase 1 (SCD1), the rate-limiting enzyme in synthesizing MUFA from SFA, largely inhibits ferroptosis sensitivity (Luis et al., 2021; Tesfay et al., 2019; Wang et al., 2020; Yi et al., 2020). Therefore, factors that induce SCD1 expression, such as SREBP1, could contribute to acquired ferroptosis resistance in cancer cells (Tesfay et al., 2019; Yi et al., 2020). Notably, PUFA biosynthesis is also regulated by other metabolic modules, including the energy stress sensor AMP-activated protein kinase (Lee et al., 2020a).
PUFA uptake
Cellular PUFA-lipid levels are regulated not only by processing shorter PUFA precursors, but also by directly importing from the environment as free FAs, carrier protein such as albumin-bound FAs or lipoproteins. FA transporters include CD36, plasma membrane-associated fatty acid binding proteins, and fatty acid transport proteins (FATPs) with yet-to-be completely defined selectivities (Doege and Stahl, 2006; Glatz et al., 2010; Ménégaut et al., 2019; Nagarajan et al., 2021). CD36 internalizes FAs into cells through endocytosis (Hao et al., 2020). Specifically in the tumor microenvironment, CD36-mediated arachidonic acid uptake induces ferroptosis in the tumor-infiltrating CD8+ T cells and high CD36-expression impairs anti-tumor immunity (Ma et al., 2021); whereas in the germinal center, B1 and marginal zone B cells express higher levels of CD36 than follicular B2 cells and exhibit higher sensitivity to Gpx4 depletion-induced lipid peroxidation and ferroptosis, presumably due to elevated FA uptake (Muri et al., 2019).
FATPs directly promote FA translocation across the plasma membrane. Alternatively, the acyl-CoA synthetase (ACS) activity of FATPs allow them to trap FAs as acyl-CoA derivatives to promote diffusion processes (Ménégaut et al., 2019). A recent study showed that pathologically activated neutrophils, or myeloid-derived suppressor cells selectively upregulate FATP2 (Slc27a2) to uptake arachidonic acid from the extracellular environment during differentiation (Veglia et al., 2019), leading to spontaneous accumulation of oxidized PE containing arachidonic acid and ferroptosis (Kim et al., 2022), stressing that the dynamic regulation of fatty acid transporter expression could have a significant impact on immune cell fate.
Lipid uptake is also limited by its availability in tissue environments. For metastatic cancer cells, the lymphatic circulation contains higher levels of ferroptosis-protective MUFAs than that of blood circulation, a difference that plays a critical role in supporting the higher metastatic potential of melanoma cancer cells migrating through the lymphatic system than the hematogenous route (Ubellacker et al., 2020). It would be intriguing to dissect the influence of environmental FA availability and composition on cell-fate and -state regulation.
PUFA degradation
A key component of cellular PUFA homeostasis is its degradation. A recent study showed that 2,4 dienoyl-CoA reductase (DECR1), an effector in mediating PUFA β-oxidation, is significantly upregulated in castration-resistant prostate cancer tissues (Blomme et al., 2020; Nassar et al., 2020). DECR1 depletion causes PUFA accumulation and enhanced sensitivity to ferroptosis (Nassar et al., 2020), highlighting that blocking PUFA degradation might be another strategy to elevate cellular PUFA-lipid content and induce ferroptosis susceptibility.
In contrast, the degradation of SFA/MUFA may increase cellular sensitivity to ferroptosis by providing more ROS via β-oxidation and increasing the relative density of PUFA-lipids. In ccRCC, chemerin, an adipokine, inhibits FA β-oxidation and prevents ferroptosis, and a chemerin blocking antibody suppresses tumor growth (Tan et al., 2021). Whether elevated susceptibility to ferroptosis is a generalizable consequence to SFA/MUFA β-oxidation warrants further study.
Incorporation into phospholipids
Imported or synthesized PUFAs are incorporated into phospholipids through two pathways: phospholipid de novo synthesis and the Land's cycle, that is, the phospholipid remodeling pathway. In both pathways, free PUFAs are first activated to PUFA-CoA through the ACSL enzyme family. Among these, ACSL4 selectively catalyzes thioesterification of PUFA to PUFA-CoA, especially arachidonic acid and EPA (Kang et al., 1997).
Therefore, ACSL4 directly regulates the PUFA-lipid content of the cellular membranes to influence ferroptosis sensitivity in a broad array of contexts (Dixon et al., 2015; Doll et al., 2017; Kagan et al., 2017; Zou et al., 2019). Chemical inhibition and downregulation of ACSL4 in vivo can, in part, rescue ischemia/reperfusion-induced kidney or intestine injury partially induced by ferroptosis (Li et al., 2019; Tao et al., 2022; Wang et al., 2022b). In contrast to ACSL4, ACSL3 preferentially catalyzes MUFA activation and protects cells from ferroptosis.
ACSL3-depletion increases ferroptosis sensitivity in cultured cells and metastasizing melanoma cells (Magtanong et al., 2019; Ubellacker et al., 2020). In addition to ACSL3/4, ACSL1 was reported to incorporate conjugated PUFAs to triglyceride (TG) and further transfer them to lipid droplets (LDs) (Beatty et al., 2021). Unlike normal PUFAs, conjugated PUFAs on LDs can cause lipid peroxidation and induce ferroptosis (Beatty et al., 2021). Of note, ACSL4-depletion partially rescued the α-ESA-induced cell death to a lesser extent than that of ACSL1 knockout. Hence, the ACSL family collectively plays an important role in shaping the cellular lipidome and regulating ferroptosis sensitivity.
Following activation, PUFA-CoA is incorporated into complex lipids, including phospholipids and TGs. Glycerophosphate acyltransferases (GPATs) catalyze the first step of phospholipid de novo synthesis by transferring SFA from acyl-CoA to sn-1 site of G3P to afford LPA, 1-acyl-G3P. Subsequently, acylglycerophosphate acyltransferases (AGPATs) adds another fatty acid moiety from fatty acyl-CoA to the sn-2 site of LPA, resulting in phosphatidic acid. Among this protein family, 1-acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3) exhibits selectivity on incorporating PUFA-CoA into lysophosphatidylcholine (LPC), indicating its role in regulating ferroptosis sensitivity (Lu et al., 2005; Yuki et al., 2009).
Genetic screens in human haploid cell KBM7, ccRCC cell line 786-O, and ovarian cancer cell line OVCAR-8 further confirmed AGPAT3 as a key acyltransferase important for ferroptosis sensitivity (Dixon et al., 2015; Zou et al., 2020a). The generality of AGPAT3's pro-ferroptosis role in other cellular contexts awaits to be tested.
In phospholipid remodeling, the FA moieties in the glycerol backbone are replaced by other FAs to form diverse phospholipids. Two steps of the reaction, deacylation and reacylation of phospholipids, are involved in the remodeling processes. First, in the deacylation step, the FAs on the sn-2 site of phospholipid is cleaved by phospholipase A2 (PLA2). The resultant lysophospholipids are acylated by acyl-CoA:lysophospholipid acyltransferases (LPCATs).
Among these, LPCAT3 was suggested to preferentially transfer PUFA-CoA, especially arachidonoyl-CoA, to LPC, lysophosphatidylethanolamine, and lysophosphatidylserine, and this selectivity was supported by the atomic structure of LPCAT3 (Hishikawa et al., 2008; Lagrost and Masson, 2022; Rong et al., 2015; Zhang et al., 2021). LPCAT3-mediated PUFA-CoA transfer results in PUFA-phospholipid enrichment in the ER membrane (Hishikawa et al., 2008; Rong et al., 2017; Yamashita et al., 2017; Yamashita et al., 2014).
Genetic or pharmacological inhibition of LPCAT3 in HAP1 and THP-1 protected cells from ferroptosis, stressing an important role of LPCAT3 in modulating ferroptosis sensitivity (Dixon et al., 2015; Reed et al., 2022a). Notably, a recent study in KRAS-mutant lung cancer (KMLC) cells showed that LPCAT3 uses palmitoyl(C16:0)-CoA as an acyl donor, and it prefers 16:0-CoA over 20:4-CoA provided that both substrates are available at the same molar concentration (Bartolacci et al., 2022). In the context of KMLC, LPCAT3 played a ferroptosis-suppressive role instead (Bartolacci et al., 2022). Further research is warranted to reconcile the substrate selectivity of LPCAT3 and its context-specific contributions to the cellular lipidome composition and ferroptosis sensitivity. In addition, AGPAT3 also contributes to the remodeling of phospholipids via the acyltransferase activity toward lysophospholipids (Yamashita et al., 2017; Yamashita et al., 2014). Collectively, perturbing the PUFA-lipid biosynthesis regulators will likely have a significant impact on membrane PUFA-lipid content and cellular ferroptosis sensitivity.
Incorporation into ePLs
Through CRISPR-Cas9 screens, genes involved in ether-lipid biosynthesis have been identified as top pro-ferroptosis regulators, including glyceronephosphate O-acyltransferase, fatty acyl-CoA reductase 1 (FAR1), alkylglycerone phosphate synthase, AGPAT3, and transmembrane protein 164 (TMEM164) (Zou et al., 2020a). On the other hand, peroxisome-localized FAR1, which reduces C16/C18 SFA to fatty alcohol, can promote ferroptosis by supplying SFA alcohol for the synthesis of alkyl-ether lipids and plasmalogens (Cui et al., 2021).
Notably, a recent study uncovered TMEM164 as an ether-lipid acyltransferase, piecing together the full biochemical pathway mediating ether-lipid biosynthesis (Reed et al., 2022b). TMEM164 selectively transfers C20:4 acyl chains from diester PC to lyso-ether-phospholipids, forming PUFA-ePLs. In addition, TMEM164 may regulate ferroptosis by promoting ATG5-dependent autophagosome formation and one consequence is to increase the labile iron available for lipid peroxidation (Liu et al., 2022b).
Among the phospholipid biosynthesis enzymes involved in PUFA-lipid formation, ACSL4 is not only a generally necessary and non-redundant factor mediating ferroptosis sensitivity, but its expression and activity is also tightly modulated under various pathological contexts. ACSL4 expression is upregulated in cells or tissues undergoing ferroptosis stress, including kidneys experiencing ischemia/reperfusion-induced injury (Li et al., 2019; Müller et al., 2017) and cancer cells undergoing ionizing radiation (Lei et al., 2020); moreover, ACSL4 expression positively correlates with ferroptosis sensitivity in cancer cells and its genetic depletion dramatically abolishes cellular ferroptosis sensitivity (Doll et al., 2017; Yuan et al., 2016).
In addition, in anti-tumor immune response, T cell-derived IFN-γ sensitizes cancer cells to ferroptosis through stimulating ACSL4 expression in cancer cells (Liao et al., 2022). In certain cell types, such as glioma cells, ACSL4 overexpression alone was sufficient to trigger cell death (Cheng et al., 2020). Importantly, the activation of protein kinase C βII, a proposed sensor of lipid peroxides, promotes ferroptosis sensitivity by phosphorylation of ACSL4 at Thr328 and dimerization-mediated activation in MDA-MB-231 breast cancer and HT-1080 fibrosarcoma cells (Zhang et al., 2022).
Non-phosphorylatable ACSL4 mutant attenuated ferroptosis induced by erastin or RSL3-treatment (Zhang et al., 2022). Although the precise mechanism underlying the stress-responsive ACSL4 expression and activation remains to be fully elucidated, these observations highlight ACSL4 as an attractive point of intervention for pharmacological control of ferroptosis in various contexts.
Despite a wealth of information on their biosynthesis and regulation, much less is known whether membrane phospholipid transporters such as scramblases, flippases, and floppases contribute to ferroptosis. In a recent study, Lin et al. (2022) uncovered lipid flippase solute carrier family 47 member 1 (SLC47A1) as an anti-ferroptosis regulator in pancreatic cancer cells and tumor xenograft models. SLC47A1 protects cells from pharmacological ferroptosis induction via lipid remodeling, specifically, by inhibiting the production of PUFA-cholesterol esters for lipid peroxidation. This work underscores the critical roles of PUFA-related metabolic regulators ranging from biosynthesis, membrane transportation and incorporation, to degradation in ferroptosis.
Organelle hosts of pro-ferroptosis lipids and catalysts
Endoplasmic reticulum
The ER membrane contains the highest level of PUFA-lipids (Harayama and Riezman, 2018), and is touted as the source where lipid peroxidation initiates. By monitoring lipid peroxidation in mouse embryonic fibroblasts undergoing ferroptosis, Kagan et al. (2017) reported that the ER, but not the mitochondria, show an increased signal, suggesting that lipid peroxidation enrichment occurs initially in the ER. In line with this, the accumulation of ferroptosis inhibitor analogs, ferrostatins, in ER, but not lysosome and mitochondria, protects the cells from erastin-induced ferroptosis (Gaschler et al., 2018). Future research is required to determine whether there are membrane microdomains on the ER that are particularly susceptible or resistant to lipid peroxidation.
In addition to harboring high levels of PUFA-lipids, the ER is also the host for oxidoreductases involved in lipid peroxidation. By two independent genome-wide CRISPR-Cas9 screens in ccRCC 786-O and UACC-257 melanoma cells, cytochrome P450 oxidoreductase (POR) was found to be involved in ferroptosis (Yan et al., 2021; Zou et al., 2020b). POR is an ER-resident oxidoreductase that forms a complex with an electron acceptor partner such as cytochrome P450 (CYP) isoenzyme to detoxify xenobiotic chemicals (Foti and Dalvie, 2016; Pandey and Flück, 2013).
By redox lipidomic analysis in 786-O, 769-P, and Hela cells, POR knockdown significantly reduced intracellular levels of peroxidized PUFA-PEs, suggesting that POR induces ferroptosis in cells by promoting lipid peroxidation (Yan et al., 2021; Zou et al., 2020b). Importantly, POR alone is sufficient to trigger peroxidative damage in PUFA-phospholipid-containing membranes (Yan et al., 2021; Zou et al., 2020b).
Downstream of POR, cytochrome P450s were recognized as monooxygenases or mixed-function oxidases directly oxidizing lipids contributing to phospholipid peroxidation (Dey et al., 2002; Ghosh et al., 1997; Guengerich, 2018; Minoda and Kharasch, 2001; Sevanian et al., 1990). However, disrupting the interaction between POR and its electron acceptor by truncating POR's ER-resident motif does not affect its ability to promote ferroptosis. Instead, H2O2 that is produced due to electron leakage has been shown to increase the cellular susceptibility to ferroptosis (Yan et al., 2021; Zou et al., 2020b).
By further ER-residing oxidoreductase focused CRISPR screen in POR-depleted Hela cells, CYB5R1 was also identified to increase cellular ferroptosis sensitivity. Mechanistically, CYB5R1 also promotes H2O2 formation to drive lipid peroxidation (Yan et al., 2021). These findings confirm the role of ER in initiating lipid peroxidation under cellular contexts and highlight H2O2 as a potential intermediate reactive oxidant in this process.
Mitochondria
As the main intracellular site of oxidative metabolism, mitochondria produce large amounts of ROS during respiration, and they have evolved an antioxidant system to counteract ROS-induced membrane damage. Nevertheless, the collective contribution of mitochondria on lipid peroxidation and ferroptosis remains unclear and rather context-specific. In RSL3-induced HT-1080N cell ferroptosis, an antioxidant localized in mitochondria shows a stronger protective effect compared with its analog, which is uniformly distributed in cells (Krainz et al., 2016).
However, the effect of the radical chelator ferrostatin-1 is not dependent on mitochondria. Ferrostatin-1 retains its ferroptosis inhibiting activity even in mitochondria-depleted cells (Gaschler et al., 2018).
Mitochondria use membrane integrative, endogenous antioxidants to alleviate potential oxidative damage of the inner mitochondrial membrane. In a metabolomic analysis of GPX4 inhibitor-treated HT-1080N cells, the uridine metabolic pathway showed a significant increase. Further, the mitochondrial dihydroorotate dehydrogenase (DHODH) involved in this pathway showed significantly higher activity during ferroptosis (Mao et al., 2021).
DHODH converts dihydroorotic acid to orotate by a redox reaction of ubiquinone to generate ubiquinol, which, in turn, repairs lipid peroxidation damage in mitochondria. Inhibition of DHODH in GPX4 low-expressing NCI-H226 cells or in HT-1080 cells treated with GPX4 inhibitors greatly increased intracellular lipid peroxidation levels (Mao et al., 2021).
These findings suggest that mitochondrial repair of lipid peroxidation is an important cellular defense mechanism against ferroptosis. However, a recent preprint from Mishima et al. argued that the ferroptosis-sensitizing effect of high concentrations of brequinar, the main DHODH inhibitor used in the Mao et al. (2021) study, is mediated via inhibition of Ferroptosis suppressor protein 1 (FSP1) but not DHODH (Mishima et al., 2022b), suggesting that the contribution of DHODH in mitochondrial lipid peroxidation and ferroptosis might be highly context-specific.
On the other hand, a recent study showed that RSL3 treatment in U2OS cells induces a short phase of O-GlcNAcylation accumulation, followed by its decrease (Yu et al., 2022). Blocking the upregulation of O-GlcNAcylation promotes ferritinophagy and mitophagy, leading to more release of labile iron and enhanced sensitivity to ferroptosis (Yu et al., 2022).
This study highlights yet another mechanism by which mitochondria and the autophagy process may contribute to ferroptosis sensitivity regulation. A recent review provided an overview on the multifaceted role of mitochondria in ferroptosis (Guo et al., 2022).
Lysosomes
Lysosomes may affect the cellular susceptibility to ferroptosis by both regulating the intracellular iron content via autophagy and recycling the lipid substrates required for phospholipid biosynthesis, and amino acids required for glutathione (GSH) regeneration (Chen et al., 2021b; Torii et al., 2016). In a recent study, impaired lysosome function leads to lipofuscin formation and has a significant impact on aging. Lipofuscin traps iron and generates ROS, thus increasing the sensitivity to ferroptosis in neurons (Tian et al., 2021b).
Lysosomes could also contribute to ferroptosis inhibition by degrading albumin to supply cysteine for GSH biosynthesis (Armenta et al., 2022). On the other hand, supported by the observation that a lysosome-targeting analog of ferrostatin-1 failed to inhibit ferroptosis (Gaschler et al., 2018), the lysosome membrane does not appear to directly contribute to lipid peroxidation.
Peroxisomes
The peroxisome contains catalase and superoxide dismutase 1/2, both of which are involved in the metabolism of H2O2 (Lodhi and Semenkovich, 2014). Although H2O2 is believed to trigger lipid peroxidation, it lacks direct evidence that these peroxisomal redox enzymes are involved in the regulation of cellular ferroptosis susceptibility. There is also no evidence that peroxisomal membrane directly contributes to lipid peroxidation initiation or propagation. Instead, the peroxisome is the host site for enzymes catalyzing the initial steps of ether-lipid biosynthesis, hence it modulates the abundances of PUFA-phospholipids available for lipid peroxidation (Zou et al., 2020a). The contribution of peroxisome in ferroptosis in specific cellular contexts warrants further investigations.
Lipid droplets
LDs are organelles where cells store lipids in neutral forms such as TG and cholesterol esters. Compared with other organelles, LDs have less oxidizing environment; thus, PUFA-TGs and cholesterol esters stored in LDs are more difficult to be peroxidized than PUFA-phospholipids. This has led LDs to be considered as a sanctuary place for lipid peroxidation propagation (Bailey et al., 2015). Diglyceride acyltransferases 1/2 (DGAT1/2) acylate fatty acyl-CoA to diglycerides, thus playing a critical role in TG biosynthesis.
Inhibition of DGAT1/2 promotes cellular ferroptosis susceptibility, possibly because inhibition of TG synthesis leads to a greater tendency for PUFA to be incorporated into phospholipids, hence promoting the occurrence of lipid peroxidation (Dierge et al., 2021). The specific mechanism by which TGs containing conjugated PUFAs in LDs employ to promote ferroptosis sensitivity remains elusive (Beatty et al., 2021). It is possible that the contribution of LDs to ferroptosis is dependent on where and how lipid peroxidation is initiated.
For instance, in the context of GSH depletion or GPX4 inhibition-induced ferroptosis, lipid peroxidation was largely initiated in the ER and the LDs are relatively insulated from the penetration and spreading of polarized lipid radicals; whereas elevated conjugated PUFA-containing TGs may trigger LD-originated peroxidation reactions directly. Further research is warranted to assess the impact of lipid storage in ferroptosis susceptibility in specific tissue contexts.
Question 3: How Does Lipid Peroxidation Drive Ferroptotic Cell Death?
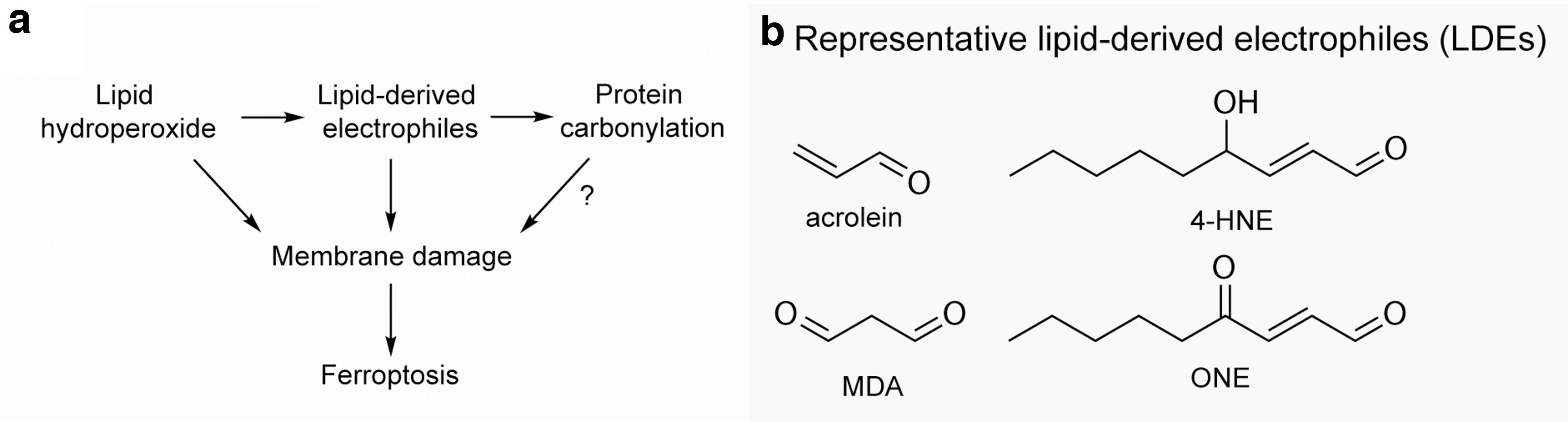
A poorly resolved question in our mechanistic understanding about ferroptosis is what specifically during lipid peroxidation drives ferroptotic cell death. Two non-mutually exclusive hypotheses prevail: lipid hydroperoxides can directly destabilize membrane integrity, and lipid peroxidation by-products modify other macromolecules including proteins to disable essential cellular physiology (Fig. 5a).
In the direct membrane disruption model, lipid peroxidation initiated by an enzymatic or non-enzymatic process results in accumulation of polar lipid radicals and lipid hydroperoxides, which destabilize membrane integrity, disrupt ion gradients, and increase the membrane permeability. This leads to membrane damage and, passing a threshold, culminates in cell death (Borst et al., 2000; Yin et al., 2011).
In a molecular dynamics study, oxidized PUFA-phospholipids accumulated during ferroptosis induction led to membrane thinning and increased curvature, driving a vicious cycle of increasing accessibility to oxidants and micelle formation, which exacerbates membrane destruction and cell death (Agmon et al., 2018).
In addition to compromising the integrity of the phospholipid bilayer directly, lipid peroxidation generates secondary by-products that can induce oxidation of other macromolecules, including proteins. Lipid hydroperoxides are intrinsically unstable and could undergo non-enzymatic degradation to yield a variety of diffusible reactive lipid-derived electrophiles (LDEs, Fig. 5b).
These reactive species consisted of two important classes of compounds: (i) α,β-unsaturated aldehydes such as acrolein and 4-hydroxynonenal (4-HNE); (ii) lipid dicarbonyls such as isoketals, malondialdehyde (MDA), and 4-oxo-nonenal (Yin et al., 2011). These LDEs are highly reactive toward proteins, DNA, and phospholipids to generate a variety of intra- and inter-molecular covalent adducts. Whether diffusible LDEs contribute to intracellular spreading of lipid peroxidation during ferroptosis remains to be investigated.
For protein targets, LDEs are highly reactive toward nucleophilic residues, including lysine, cysteine, and histidine in a process termed protein carbonylation. Proteomic profiling of protein carbonylation in RSL3-induced ferroptotic fibrosarcoma HT-1080N cells revealed >400 carbonylated proteins (Chen et al., 2018), including ferroptosis-relevant factors transferrin receptor (Feng et al., 2020; Gao et al., 2015; Park and Chung, 2019), VDAC2/3 (Yagoda et al., 2007; Yang et al., 2020), and ACSL4. The specific contributions of carbonylated proteins in ferroptosis remain unclear.
Under physiological conditions, cells have evolved mechanisms to detoxify LDEs, including aldehyde dehydrogenases (ALDHs) that can oxidize LDEs to their corresponding acids for further detoxification (Ayala et al., 2014). A recent study reveals an impaired 4-HNE detoxification mediated by ALDH1 family member B1 promotes ferroptosis in HT-1080 cells and tumor xenograft model. This study highlights the contribution of lipid peroxidation-derived aldehyde byproducts in ferroptosis.
In addition to ALDH-mediated detoxification, LDE can react with GSH via conjugation reaction followed by GSH-LDE adduct exportation. In a recent study, Van Kessel et al. (2022) report an impaired LDE detoxification during
This study proposes that downstream of lipid peroxidation, the accumulation of LDE species, and on reaching a certain threshold, could be considered as the point of “no return” in ferroptosis. However, the accumulation of intracellular LDE-adducts is due to not only the GSH-LDE, but also LDE-modified proteins (Lincoln et al., 2017). Thus, in addition to failure in LDE detoxification, the potential contribution of LDE-modified proteins cannot be ruled out. This observation suggests the potential involvement of repair mechanisms for protein carbonylation involved in ferroptosis induction.
It remains unknown, however, whether specific pore-forming proteins are actively driving cytoplasm leakage and executing ferroptotic cell death. Considering that in liposome studies lipid peroxidation is sufficient to trigger disruption of liposome topology (Yan et al., 2021), it seems unlikely that a protein-mediated channel would be an additional requirement for sustained opening of the plasma membrane; hence, ferroptosis could be solely driven by changes in polarity of the oxidized lipids, altered membrane packing, and consequent osmotic imbalance.
The counter argument is that membranes in liposomes lack the extensive mechanical support of intracellular cytoskeleton and extracellular matrix structures, hence damages that were sufficient to disrupt liposomes may be readily repairable in cellular settings and stronger insults are required for cell death to occur. This topic remains ambiguous at the moment and further research is required to determine the precise mechanisms driving cell death.
Q4: How Does the Cell Defend Against Lipid Peroxidation and Repair Membrane Damage?
Lipid peroxidation occurs at a basal level during normal cellular physiology, and oxidative modifications of PUFA are prominent ways to generate signaling molecules including eicosanoids and leukotrienes (Wymann and Schneiter, 2008). Cells have evolved mechanisms to keep the lipid radical levels below the detrimental threshold, including using (Fig. 6): (i) specific enzymes to reduce non-radical lipid hydroperoxides, featured by the GPX4-GSH enzymatic axis; (ii) radical scavengers to quench the lipid radicals; and (iii) enzymes to cleave and release the oxidized fatty acyl tail from packed phospholipid bilayers.

Reducing and detoxifying oxidized fatty acyl chains
GPX4, a seleno-containing oxidoreductase, is the sole enzyme that reduces LOOH to non-toxic lipid alcohol (L-OH) at the expense of reductants such as GSH or thiol-containing proteins (Ursini et al., 1982; Yang et al., 2014). Given its indispensable activity, homozygous Gpx4 knockout in the mice is embryonic lethal, and conditional Gpx4-depletion in adult mice leads to acute kidney failure (Friedmann Angeli et al., 2014).
Numerous studies also support the evolutionarily conserved role of the GPX4-GSH axis in defending against accumulation of lipid hydroperoxides (Carlson et al., 2016; Conrad et al., 2018; Hangauer et al., 2017; Ingold et al., 2018; Viswanathan et al., 2017; Yang et al., 2016; Zou et al., 2019). Molecular simulations predicted a model in which cationic residues of GPX4 are responsible for the interaction with lipid substrate (Cozza et al., 2017), which was later confirmed by structural analyses of GPX4 protein (Furuita et al., 2022; Janowski et al., 2016; Labrecque et al., 2022; Moosmayer et al., 2021).
The highly cationic patch for membrane lipid interaction is located adjacent to the GPX4 active site, positioning the peroxidized lipid tail for Sec46 reduction (Labrecque and Fuglestad, 2021). Of note, mitochondrial cardiolipin exhibits the strongest interaction with the cationic patch compared with that of the arachidonic acid, whereas neutral cholesterol reveals no observable interaction.
This result highlights GPX4's preference in interacting with anionic lipids when bound to a membrane, and this observation corresponds to GPX4's lower catalytic activity for cholesterol reduction (Thomas et al., 1990). The biochemical basis of GPX4-mediated lipid hydroperoxide detoxification and -associated regulations is recently reviewed (Forcina and Dixon, 2019; Ursini and Maiorino, 2020).
Shedding light on the cellular compartment-specific accumulation of and then defense against lipid peroxidation, alternative transcription initiation results in three isoforms of GPX4: cytosolic, mitochondrial, and nuclear GPX4 (c/m/nGPX4, respectively) (Maiorino et al., 2003). Both mGPX4 and nGPX4 isoforms perform moonlighting functions that are independent of the lipid hydroperoxidase activity. mGPX4 polymerizes through surface-exposed cysteines, resulting in a structural feature of the mitochondrial capsule in mature spermatozoa (Scheerer et al., 2007).
nGPX4, on the other hand, regulates the compactness of chromatin partly by involving in DNA cross-linking during sperm development (Conrad et al., 2005). Structural insights shed some light on how GPX4 achieves diverse biological functions. Crystal structure of full-length GPX4 in complex with a covalent inhibitor, ML162, reveals a shallow active site of GPX4 where the catalytic Sec46 interacts with three other residues (Gln81, Trp136, and Asn137), forming a catalytic tetrad (Moosmayer et al., 2021).
In this GPX4•ML162 complex, the chloroacetamide and methoxychlorophenyl moiety of ML162 interacts with the catalytic tetrad blocking the active site. These insights, together with a recent discovery on the second pocket (Liu et al., 2022a), point toward the avenues for targeting GPX4.
As the reducing agent used by GPX4, the availability of GSH also affects the sensitivity of cells to ferroptosis. GSH synthesis in cells is mainly limited by the rate of cystine uptake, which is primarily achieved by cystine/glutamate exchange mediated by the protein complex system Xc −. Inhibition of system Xc − and its component SLC7A11 leads to increased cellular sensitivity to ferroptosis (Dixon et al., 2012; Jiang et al., 2015; Yang et al., 2014).
Surveillance by antioxidants
In addition to the GPX4-GSH axis, cells also deploy various endogenous antioxidative small molecules to defend against oxidative lipid radicals.
FSP1-CoQ10/Vitamin K
Using genetic screens, two independent studies identified the FSP1–CoQ10–NAD(P)H pathway that acts in parallel to the canonical GSH-GPX4 axis to scavenge harmful lipid peroxides and inhibit ferroptosis (Bersuker et al., 2019; Doll et al., 2019). FSP1, previously known as apoptosis-inducing factor mitochondrial 2, belongs to the type II NADH: quinone oxidoreductase NDH-2 family, which is mainly localized in LDs and plasma membranes.
Ubiquinone, also known as coenzyme Q (CoQ or CoQ10), is a class of lipid-soluble quinones found in the inner mitochondrial membrane or cell membrane. Ubiquinone not only assists in electron transport and energy production in mitochondria, but also has significant anti-lipid peroxidation activity. Mechanistically, FSP1 reduces ubiquinone (CoQ) to ubiquinol (CoQH2) using NAD(P)H at the cell membrane. And the reduced form ubiquinol (CoQH2) acts as a membrane-resident RTA to halt the propagation of lipid peroxides and inhibit ferroptosis.
A new path for FSP1 to suppress lipid peroxidation was recently uncovered (Mishima et al., 2022a). Mishima et al. (2022a) systematically studied naturally available vitamin compounds and found that the vitamin K family, including phylloquinone, menaquinone-4 and menadione, act as potent anti-ferroptotic agents in multiple cell lines and inducible Gpx4 liver knockout model. Because of the structural similarities between vitamin K and FSP1 substrate CoQ10, FSP1 was tested to be the warfarin-resistant vitamin K reductase.
This non-canonical FSP1-vitamin K cycle suppresses ferroptosis by supplying an RTA, vitamin K-hydroquinone (VKH2), at the expense of NAD(P)H to suppress lipid peroxidation.
Vitamin E
Another natural lipophilic antioxidant, vitamin E, also plays an important role in suppressing lipid peroxidation. Fat-soluble vitamin E, including both tocopherol (Toc) and triene-tocopherols, is an endogenous antioxidant in the body (Munné-Bosch et al., 2022). The ɑ-isomer of vitamin E has the highest potency in suppressing lipid peroxidation, followed by the β- and γ-isomers, which are equipotent, whereas the δ- isomer is the least potent (Miyazawa et al., 2019). The antioxidant effect of vitamin E is mainly to inhibit lipid peroxidation due to its presence in cellular membranes.
In the process of lipid peroxidation, α-tocopherol, the main component of vitamin E, is prone to deliver H atoms to peroxyl radicals or other radicals due to its relatively weak bonding energy of the O-H bond. For example, α-tocopherol (α-TocH) donates an H atom to lipid peroxyl radical, yielding an α-tocopheroxyl radical (α-Toc•) and a lipid hydroperoxide.
The α-Toc• can then recombine with another LOO• to generate inert adducts, which results in the termination of the lipid peroxidation chain reactions (Miyazawa et al., 2019; Traber and Head, 2021). Thus, one molecule of vitamin E can trap two molecules of peroxyl radicals. In addition, vitamin E can also inhibit lipoxygenase (LOX) activity, and block LOX-catalyzed peroxidation of PUFA-phospholipids (Jiang et al., 2011).
In several models, vitamin E has been shown to scavenge lipid peroxides and inhibit ferroptosis. Pretreatment of animals with vitamin E could reduce liver necrosis and cirrhosis induced by CCl4 (Parola et al., 1992). Moreover, pre-feeding mice with a vitamin E-deficient diet before conditional Gpx4 knockout in the endothelial cells causes systemic thrombus formation and early death (Wortmann et al., 2013).
Squalene
Squalene is a precursor in cholesterol de novo synthesis and was previously implicated in preventing the diffusion of free radical reactions (Kohno et al., 1995) and influencing the spatial assembly of membrane lipid bilayers (Gilmore et al., 2013). Using barcoded cell line screens, Garcia-Bermudez et al. (2019) revealed that squalene accumulates in cholesterol-auxotrophic, ALK+ anaplastic large cell lymphoma cells and confers resistance to lipid peroxidation and ferroptosis induced by GPX4 inhibitors (Garcia-Bermudez et al., 2019). Due to the low abundance of squalene in most human cell lines, the relative contribution of squalene to ferroptosis protection in other cellular contexts awaits to be further characterized.
Tetrahydrobiopterin
Two recent genetic screens identified the GTP cyclohydroxylase-1 (GCH1)-tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) axis as an essential protective pathway in Jurkat cells or immortalized murine fibroblasts experiencing ferroptosis induction (Kraft et al., 2020; Soula et al., 2020). GCH1 catalyzes the first and rate-limiting step converting GTP to 7,8-dihydroneopterin triphosphate, which is later converted to BH4.
Besides acting as a redox-active cofactor for fueling ubiquinone synthesis (Kraft et al., 2020), BH4 can act as an endogenous RTA to directly mitigate lipid peroxidation, alone and in synergy with α-tocopherol (Soula et al., 2020). Moreover, dihydrofolate reductase (DHFR) can regenerate BH4 from BH2 to suppress ferroptosis, and DHFR inhibition by methotrexate synergizes with GPX4 blockade (Soula et al., 2020). Blocking GCH1/BH4 resensitized colorectal cancer cells to erastin-induced ferroptosis, highlighting a potential strategy to increase ferroptosis induction efficiency in cancer (Hu et al., 2022; Kraft et al., 2020).
Hydropersulfides
Recently, two independent studies reveal the anti-ferroptosis role of hydropersulfides by reducing lipid-derived peroxyl radicals, and terminating radical chain reactions (Barayeu et al., 2022; Wu et al., 2022). Hydropersulfide is a potent RTA via one-electron reaction; however, its role in scavenging lipid radical remains largely unexamined. Using the fluorescence-enabled inhibited autoxidation approach, Wu et al. (2022) demonstrates that hydropersulfides (cumyl-SSH, benzyl-SSH, and t-dodecyl-SSH) are highly reactive toward phospholipid-derived peroxyl radicals whereas other sulfane sulfur species, such as H2S, disulfide, and trisulfide, are relatively poor lipid radical scavengers in vitro.
Exogenous hydropersulfide treatment protects mouse embryonic fibroblasts from GPX4 inhibition-induced ferroptosis, although with lower potency than ferrostatin-1. In a separate study, Barayeu et al. (2022) show that upregulating intracellular persulfide levels via cystathionine γ-lyase protects cells from lipid peroxidation-induced membrane damage. In contrast, downregulating persulfides via persulfide dioxygenase (ETHE1) overexpression results in membrane leakage and ferroptotic death (Barayeu et al., 2022).
Nevertheless, it remains unclear how these hydrophilic species inhibit lipid peroxidation within the hydrophobic lipid bilayer environment. Hydropersulfides may directly reduce the FA chain of lipid peroxyl radical, which swings out to the membrane surface due to increased polarity, terminating the radical chain reaction (Laguerre et al., 2017). Alternatively, hydropersulfides may indirectly suppress lipid peroxidation via reduction of lipophilic RTAs such as α-tocopherol and/or ubiquinone.
The coupling of hydrophilic and lipophilic RTAs is a recognized mechanism in redox homeostasis. For instance, α-tocopherol reduces lipid radicals, and the resulting α-tocopherol radicals are scavenged by water-soluble antioxidant, ascorbate (Kalyanaraman, 2013). In addition, in cells, both cysteine and GSH serve as the sulfur source for persulfide and polysulfide biosynthesis. These studies establish a link between the GPX4-GSH enzymatic axis and the radical scavenger axis in ferroptosis defense.
In addition to the aforementioned antioxidants, recent research revealed that intermediates of tryptophan metabolism, including serotonin and 3-hydroxyanthranilic acid, act as potent RTAs to attenuate lipid peroxidation and suppress GPX4 inhibition-induced ferroptosis in cancer cells (Liu et al., 2023). This finding reveals an important antioxidant mechanism that is independent of cysteine.
Cleavage of oxidized phospholipids and membrane repair
In addition to GPX4-mediated reduction of lipid hydroperoxides, oxidized FA chains can be released from the glycerol backbone of phospholipids by calcium-independent phospholipases, including phospholipase A2 group VI (iPLA2β), thus alleviating the cells from further spreading of lipid peroxidation and ferroptosis (Beharier et al., 2020; Chen et al., 2021a; Sun et al., 2021). The remaining lysophospholipids on the membrane likely will be recycled for generating new phospholipid moieties.
Expression of iPLA2β can be induced by activated p53, contributing to the tumor-suppressive role of p53. How the anti-ferroptosis roles of iPLA2β and GPX4 are coordinated under different contexts, and the extent of their anti-ferroptosis contributions remain to be characterized.
Membrane repair by endosomal sorting complexes required for transport-III
The endosomal sorting complexes required for transport (ESCRT) is among the key machineries involved in membrane repair in many cellular processes. In particular, the ESCRT-III machinery functions as an important membrane repair mechanism in necroptosis and pyroptosis (de Vasconcelos, 2019; Ros et al., 2017). In these scenarios, Ca2+ fluxes have been associated with the activation of membrane repair mechanisms.
A recent study by Dai et al. (2020a) posits the ESCRT-III machinery as a potential negative regulator in ferroptosis. In this work, the authors report the accumulation of CHMP5 and CHMP6, two subunits of ESCRT-III machinery, in the plasma membrane on erastin- and RSL3-induced ferroptosis in PANC1 and HepG2 cancer cells.
Silencing of CHMP5 or CHMP6 expression sensitized cells to erastin- or RSL3-induced ferroptosis in both human cancer cell lines and tumor xenograft models. Dai et al. (2020b) also suggested that FSP1 can abrogate ferroptosis through a CoQ10-independent mechanism by activating the ESCRT-III repair mechanism, though the direct downstream target of FSP1 mediating ESCRT-III activation remains unclear.
Morphologically, ferroptotic cells exhibit shrunken mitochondria, plasma membrane blebbing, and rather intact nuclei (Dixon et al., 2012); whereas necrosis involves cell swelling and fragmentation of plasma membranes exposing PS due to ESCRT-III activation (Gong et al., 2017). The exposure of PS due to ESCRT-III activation has not been observed in ferroptotic cells.
Thus, additional lines of evidence are required to firmly establish the link between ferroptosis and ESCRT-III-mediated plasma membrane repair. Of note, the potential involvement of ESCRT-III in ferroptosis might suggest that cells undergoing ferroptosis may express chemokines and other regulatory molecules resulting from the ESCRT-III activation to influence neighboring cells. Further research is warranted to broadly characterize the role of organelle-specific membrane repair pathways in ferroptosis.
Perspective
While we are rapidly advancing toward deeper understanding about the biochemical basis of ferroptosis and its regulation, the translational value of ferroptosis research also welcomes its boom. Two central ideas are actively pursued in modulating ferroptosis for disease treatment, including using ferroptosis blockers to alleviate diseases caused by excessive ferroptosis such as ischemia/reperfusion-induced organ injury, neurodegeneration, etc., and using ferroptosis inducers to control cancer and potentially autoimmune diseases (Hadian and Stockwell, 2021). The former concept is encouraged by the strong ferroptosis-suppressive effects of naturally available vitamins, including vitamin E and VKH2 (Mishima et al., 2022a; Miyazawa et al., 2019; Wortmann et al., 2013), as well as potent tool compounds, including liproxstatin-1 and ferrostatin-1 (Dixon et al., 2012; Friedmann Angeli et al., 2014; Zilka et al., 2017); and deeper and wider pharmacological studies are required to evaluate the role of ferroptosis inhibition in disease settings.
Meanwhile, the prospect of ferroptosis-targeted therapies in cancer faces complications from double-edged effects on tumor immunity (Kim et al., 2022; Wiernicki et al., 2022), and potential resistance (Zou et al., 2020a), and the final outcome of ferroptosis induction in the tumor microenvironment awaits to be dissected using specific, potent, and bioavailable ferroptosis inducers. We are optimistic, as important strides are made toward this goal (Doll et al., 2019; Eaton et al., 2020; Liu et al., 2022a; Yoshioka et al., 2022).
Along with the therapeutic development, another gap is the lack of markers and tools to both predict a biological sample's sensitivity to ferroptosis, as well as report an early, on-target response to suggest whether certain treatment has been effective. This need is further highlighted by the heterogeneity in cancer metabolism and ferroptosis sensitivity across subtypes of cancers, individual patients, and different tumor lesions from the same patient (Snaebjornsson et al., 2020; Yang et al., 2023; Zou et al., 2019).
Recently, methods including photochemical activation of membrane lipid peroxidation (Wang et al., 2022a) and machine learning-based analyses of TfR1 immunostaining (Feng et al., 2020; Jin et al., 2022) have been developed to tackle these challenges. Mass spectrometry imaging techniques enabling micrometer-resolution spatial multi-omics in tissues in situ are maturing (Capolupo et al., 2022; Tian et al., 2022; Tian et al., 2021a), and they have already provided important insights on lipid peroxidation and ferroptosis detection in tissues (Sparvero et al., 2021).
Collectively, as the field continues to develop a full assembly of arsenals, we envision that targeting the ferroptosis modality will have a significant impact in improving human health.
Footnotes
Acknowledgments
The authors thank members of the Zou lab for insightful discussions. They apologize to the colleagues whose relevant work cannot be cited here due to space limitations.
Authors' Contributions
N.N., T.W.W., and Y.Z. drafted the manuscript with contributions from Z.P., M.L., and J.R.H. All authors discussed and revised the manuscript.
Author Disclosure Statement
Y.Z. is a consultant for Keen Therapeutics. The remaining authors declare no competing interests.
Funding Information
This work was supported by the Westlake Laboratory of Life Sciences and Biomedicine (to Yilong Zou), Westlake Education Foundation (to Yilong Zou) and by the National Natural Science Foundation of China (Project No. 82273257, awarded to Yilong Zou).