
Abstract
Significance:
Chronic inflammation has emerged as a major underlying cause of many prevalent conditions in the Western world, including cardiovascular diseases. Although targeting inflammation has emerged as a promising avenue by which to treat cardiovascular disease, it is also associated with increased risk of infection.
Recent Advances:
Though previously assumed to be passive, resolution has now been identified as an active process, mediated by unique immunoresolving mediators and mechanisms designed to terminate acute inflammation and promote tissue repair. Recent work has determined that failures of resolution contribute to chronic inflammation and the progression of human disease. Specifically, failure to produce pro-resolving mediators and the impaired clearance of dead cells from inflamed tissue have been identified as major mechanisms by which resolution fails in disease.
Critical Issues:
Drawing from a rapidly expanding body of experimental and clinical studies, we review here what is known about the role of inflammation resolution in arterial hypertension, atherosclerosis, myocardial infarction, and ischemic heart disease. For each, we discuss the involvement of specialized pro-resolving mediators and pro-reparative cell types, including T regulatory cells, myeloid-derived suppressor cells, and macrophages.
Future Directions:
Pro-resolving therapies offer the promise of limiting chronic inflammation without impairing host defense. Therefore, it is imperative to better understand the mechanisms underlying resolution to identify therapeutic targets. Antioxid. Redox Signal. 40, 292–316.
Introduction
Cardiovascular disease is a broad classification of health problems encompassing heart attack, stroke, and heart failure among others. While the underlying etiology of each specific condition varies, one commonality among them is chronic inflammation. Significant progress has been made in understanding how excessive inflammation contributes to these diseases; however, only recently, have we begun to understand that failures of a counter-regulatory program known as “inflammation resolution” are equally contributory to disease. Here, we will provide an overview of the concept of inflammation resolution and review the evidence suggesting a role for impaired resolution in a variety of cardiovascular diseases and conditions.
1. General Overview of Inflammation and Resolution
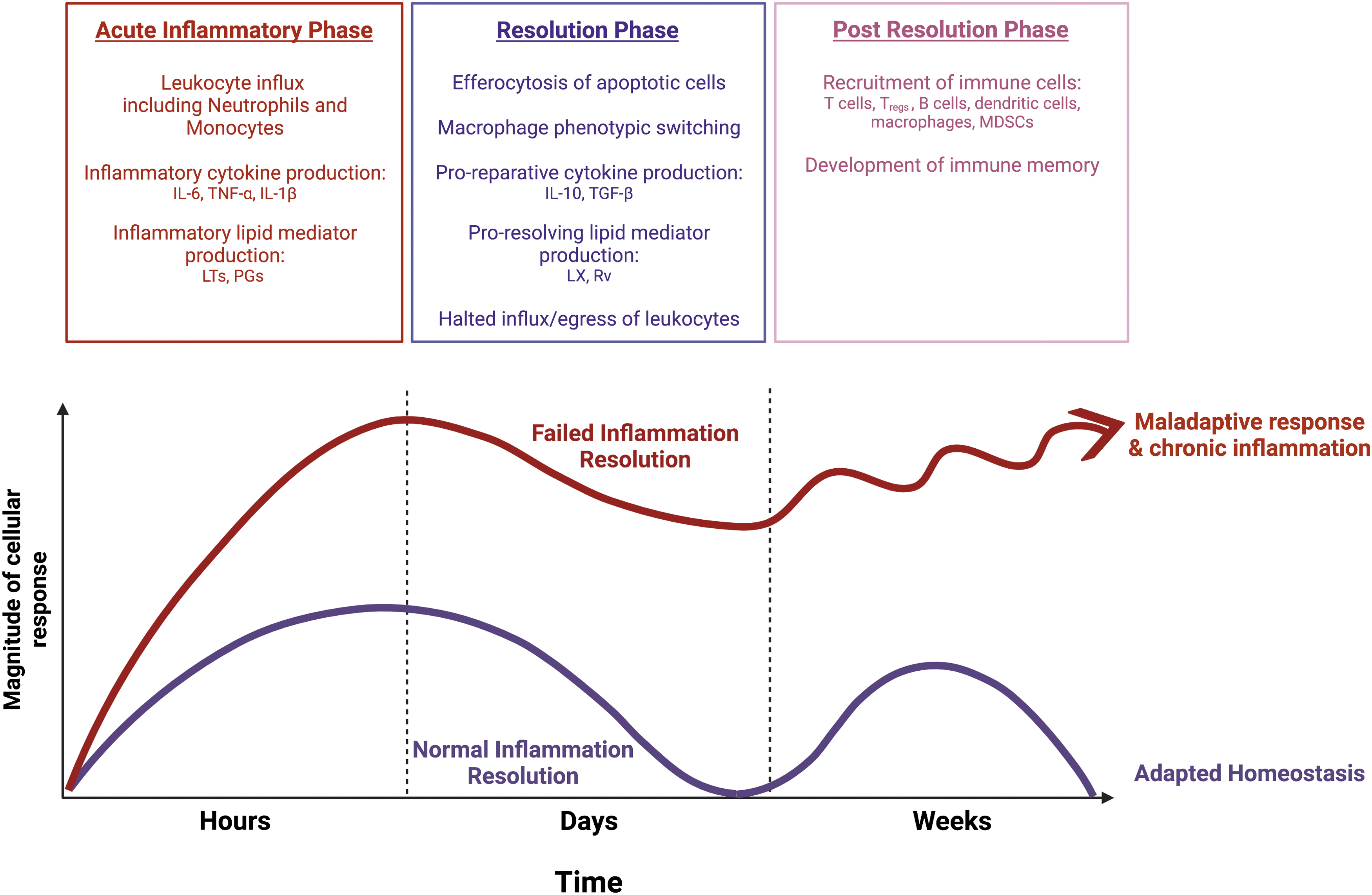
In response to infection or injury, the immune system initiates a coordinated program of acute inflammation to eradicate pathogens, limit tissue injury, and ultimately repair damage to return the host toward homeostasis (Fig. 1) (Serhan and Savill, 2005). Pathologically, this can be divided into an acute inflammatory phase, a resolution phase, and a post-resolution phase. Although it was once believed that resolution was a passive process by which inflammation was merely turned off, we now understand it to be its own active and delicately orchestrated program that may equally contribute to homeostasis and disease (Serhan et al., 2020; Serhan et al., 2000).
a. Inflammation
Acute inflammation is a protective response that occurs at the molecular, cellular, and tissue levels. Injury of tissue or invasion by pathogens generates damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) that are sensed by tissue-resident cells (Goulopoulou et al., 2016). These cells, which include dendritic cells, macrophages, endothelial cells, and fibroblasts, express pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) (Goulopoulou et al., 2016).
Signaling through these PRR pathways leads to expression of genes that initiate the inflammatory response, including chemokines, cytokines, and complement. Vasoactive amines such as bradykinin and histamine are released from cells, leading to increased vascular permeability (Di Rosa et al., 1971; Kareinen et al., 2015). Omega-6 polyunsaturated fatty acids, including arachidonic acid (AA), are released from phospholipids and catalyzed by cyclooxygenases (COX) and lipoxygenases (LO) to form prostaglandins (PGs) and leukotrienes (LTs), such as LTB4, respectively (Fig. 2).
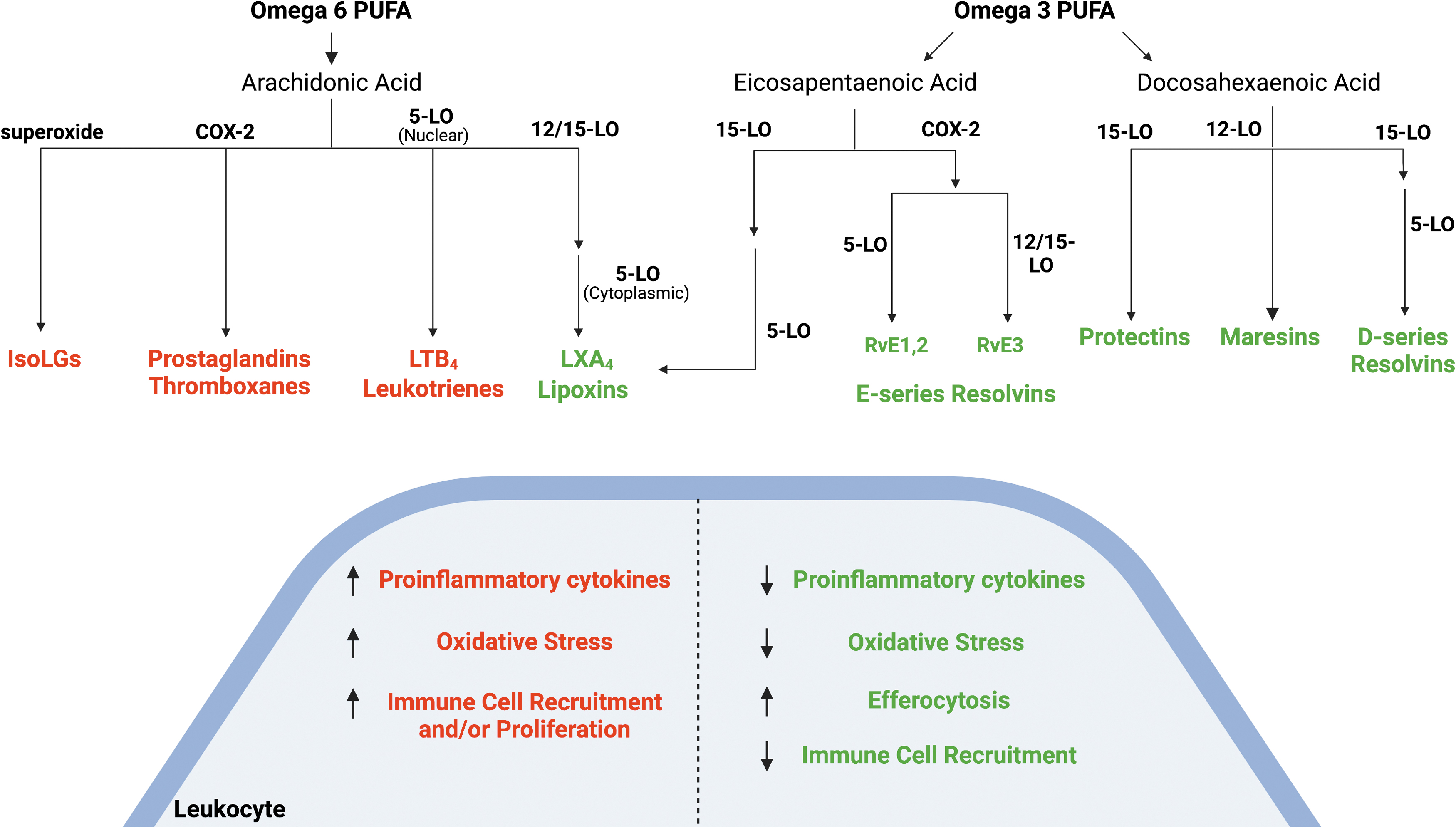
These bioactive lipids act through their cognate G-protein coupled receptors to increase expression of inflammatory cytokines and augment recruitment of immune cells. AA can also give rise to the isolevuglandins (IsoLGs) through peroxidation by free radicals. In response to these pro-inflammatory lipid mediators, endothelial cells and circulating leukocytes upregulate cellular adhesion molecules on their surface, which promotes recruitment of cells into the tissue at the site of injury (Watson et al., 1997; Zouki et al., 2000).
In the early phase of inflammation, these recruited cells are primarily neutrophils that neutralize pathogens through release of free radicals and production of myeloperoxide, lactoferrins, proteases, and neutrophil extracellular traps (NETs) (Segal, 2005; Serhan et al., 2010). The reactive oxygen species (ROS) produced by the action of phagocyte NADPH oxidase (Nox2) in neutrophils can serve as both signaling molecules and mediators of inflammation that cause collateral damage to surrounding tissue and vasculature, enhancing inflammation and endothelial permeability (Mittal et al., 2014).
Neutrophils also act as phagocytes to engulf and degrade pathogens or dead cells, after which they undergo apoptosis. Monocytes subsequently arrive to inflamed tissue and differentiate into macrophages. In the early stages of inflammation, macrophages have pro-inflammatory functions, including inflammatory cytokine production. The overall effect of these cascades at the tissue level is redness, heat, swelling, and pain that are recognized as the clinical syndrome of inflammation (Serhan and Savill, 2005; Serhan et al., 2010).
Although much of the early inflammatory response is governed by the innate immune system, the adaptive immune system also plays a role. T cell populations can be either CD4+ or CD8+ cells. Activated CD8+ T cells are generally pro-inflammatory cells that produce cytokines or cytotoxic molecules. CD4+ T cells, generally known as “helper T cells,” can be further divided into subsets that may be either pro-inflammatory, anti-inflammatory, or pro-resolving depending on the context.
For example, T helper (TH) 1 cells produce pro-inflammatory interferon gamma (IFN-γ), whereas TH2 cells produce interleukin (IL)-4, IL-5, and IL-13, which can be both pro- and anti-inflammatory. CD4+ cells also give rise to TH17 cells that produce pro-inflammatory IL-17. T regulatory cells (Tregs), which are predominantly derived from CD4+ T cells, are generally anti-inflammatory and pro-resolving based on their production of IL-10 and transforming growth factor β (TGF-β) (Mcmaster et al., 2015). LTB4 recruits CD4+ and CD8+ cells to sites of inflammation, and LTB44 and prostaglandin E2 (PGE2) can promote the differentiation of naive CD4+ T cells to TH1 and TH17 cells, while inhibiting polarization toward Tregs (Perez-Hernandez et al., 2021).
b. Resolution
Mounting an acute inflammatory response simultaneously triggers the initiation of the resolution phase. There are three components required to successfully resolve inflammation: termination of pro-inflammatory signals, switching to pro-resolving signals, and clearance of cells and debris from tissue (Serhan et al., 2020). Once the inciting stimulus has been cleared, neutrophil activation and recruitment are actively terminated through a variety of immunoinhibitory receptors (Azcutia et al., 2017). Dendritic cells and neutrophils express negative regulators of inflammation, including leucine-rich repeat-containing proteins, members of the suppressors of cytokine signaling (SOCS) family, and CD180 that inhibit TLR signaling and halt cytokine production.
Cytokine production is also dampened via post-translational mechanisms. For example, several pro-inflammatory messenger RNAs (mRNAs), including granulocyte-macrophage colony-stimulating factor (GM-CSF) (Carballo et al., 2000), IL-6 (Sauer et al., 2006), COX-2 (Phillips et al., 2004), IFN-γ (Ogilvie et al., 2009; Sauer et al., 2006), and tumor necrosis factor (TNF)-α (Carballo et al., 1998; Phillips et al., 2004), contain adenine- or uridine-rich elements (AREs) within their 3′ untranslated regions that act as binding sites for translational silencers and destabilizers.
MicroRNAs (miRs) also contribute to halting cytokine production in models of sterile inflammation. miRs targeting cytokines and TLRs are upregulated during inflammation and their expression is further enhanced by the actions of the specialized pro-resolving lipid mediators (SPMs) (Recchiuti et al., 2011). Cytokines and chemokines already present within the tissue undergo cleavage and sequestration, preventing them from binding and activating their cognate receptors (Charo and Ransohoff, 2006; Feehan and Gilroy, 2019).
ROS production is halted, partly through the upregulation of the Negative Regulator of ROS (NRROS), which facilitates the degradation of Nox2 (Noubade et al., 2014). Nox2 itself also plays a role in the resolution phase; genetic deletion of Nox2 leads to enhanced production of pro-inflammatory cytokines and continued recruitment of neutrophils (Zhang et al., 2009). Pro-inflammatory lipid mediators are catabolized by sequential enzymatic changes that lead to the formation of short chain metabolites that can be excreted (Fullerton and Gilroy, 2016; Hahn et al., 1998). Simultaneously, production of lipid mediators shifts to favor pro-resolving mediators.
This occurs through the release of omega-3 fatty acid precursors, including eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), which give rise to the resolvins (Rvs), protectins, and maresins. Signaling by PGE2 and PGD2 in the acute inflammatory phase primes the production of SPMs during resolution by increasing production of 12/15-LO, one of the major enzymes required for the synthesis of SPMs derived from both AA and DHA (Levy et al., 2001). Acting on AA, 12/15-LO initiates production of the pro-resolving mediator, lipoxin (LX) A4. Because LTB4 and LXA4 are both derived from AA, the net effect of this is increased production of LXA4 at the expense of the proinflammatory mediator (LTB4) (Levy et al., 2001; Serhan and Savill, 2005).
LXs, Rvs, and resolving cytokines such as IL-10 are able to reduce vascular permeability, slow the influx of inflammatory neutrophils to tissue, and increase recruitment of non-inflammatory monocytes (Buckley et al., 2014). In addition, LXs and Rvs promote differentiation of CD4+ cells to Tregs while inhibiting differentiation of TH1 and TH17 cells (Perez-Hernandez et al., 2021). The resolving mediators also promote a phenotypic shift in tissue macrophages toward a more pro-resolving phenotype, including decreased inflammatory cytokine secretion, decreased oxidative stress, and increased efferocytosis (Fig. 2).
Biologically active gases, including nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), also contribute to resolution by promoting apoptosis and the clearance of bacteria, debris, and cells (Wallace et al., 2015). In addition, they limit leukocyte trafficking to sites of inflammation by promoting the downregulation of adhesion molecules on the endothelium (Li et al., 2009; Wallace et al., 2015). All three gaseous mediators blunt pro-inflammatory cytokine production in macrophages, contributing to their phenotypic switch (Dufton et al., 2012; Hernandez-Cuellar et al., 2012).
H2S has been shown to reduce COX-2-dependent pro-inflammatory lipid mediator production and to enhance the activity of Annexin-A1, a pro-resolving peptide, which blocks leukocyte infiltration and enhances IL-10 production (Wallace et al., 2015). Similarly, CO leads to a reduction in LTB4 and a concomitant increase in RvD1, maresin 1, and IL-10 production in neutrophils and macrophages (Chiang et al., 2013; Lee and Chau, 2002).
Once leukocyte influx is halted, the remaining cells within the tissue either rejoin the circulation, exit through the lymphatics, or undergo cell death. Cell death occurs through a variety of mechanisms, including apoptosis, pyroptosis, necroptosis, or necrosis (Qian et al., 2021; Rayner, 2017). The signaling pathways by which these types of death occur have been reviewed extensively elsewhere (Fullerton and Gilroy, 2016; Green et al., 2009; Perez-Figueroa et al., 2021). Dead cells are cleared from inflamed tissue through phagocytosis by macrophages and dendritic cells. Of particular importance is the clearance of apoptotic cells through a specific type of phagocytosis known as efferocytosis (Doran et al., 2020).
Efferocytosis is the receptor-mediated process by which phagocytic cells recognize, bind to, internalize, and degrade apoptotic cells (Doran et al., 2020) (Fig. 3). Professional efferocytes include macrophages, as well as neutrophils and subsets of dendritic cells (Lam and Heit, 2021). When cells undergo apoptosis, they release “find me” signals, including chemokines such as C-X3-C motif chemokine ligand 1 (CX3CL1), lipids such as sphingosine 1-phosphate or lysophosphatidylcholine, and the nucleotides adenosine triphosphate (ATP) and uridine triphosphate (UTP) (Elliott et al., 2009; Gude et al., 2008; Mueller et al., 2007).

These signals induce rapid recruitment of efferocytes, such as macrophages, to the site of inflammation. These macrophages then interact with apoptotic cells through cell surface receptors either directly or indirectly through bridging molecules. Receptors including the stabilins, brain-specific angiogenesis inhibitor 1 (BAI1), the T cell immunoglobulin and mucin domain-containing receptors (TIMs), and low-density lipoprotein receptor-related protein 1 (LRP1) all bind directly to apoptotic cells.
The TAM receptors, including Tyro3, Axl, and Mer proto-oncogene tyrosine kinase (MerTK), CD36, and the integrins, all require bridging molecules to bind to Apoptotic cells. Bridging molecules include Gas6, protein S, and milk fat globule-EGF factor 8 (MFG-E8). These interact with a variety of signals on the surface of the apoptotic cells, including phosphatidylserine and calreticulin. Once bound, apoptotic cells are internalized and then must be degraded quickly by the macrophage.
This requires activation of Rac1 to promote cytoskeletal rearrangement and formation of the phagocytic cup around the apoptotic cell (Castellano et al., 2000). The clearance of dead cells prevents their degradation within tissue that would otherwise act as an inflammatory stimulus (Poon et al., 2014). In addition, efferocytosis promotes a phenotypic shift in macrophages and upregulates anti-inflammatory, tolerogenic, and pro-resolving pathways.
Macrophages exposed to apoptotic cells downregulate production and release of pro-inflammatory mediators, including thromboxane A2, IL-1β, IL-12, TNF-α, and LTs (Meagher et al., 1992; Voll et al., 1997). At the same time, they upregulate expression of pro-resolving mediators, including LXA4, and RvD1, which contribute to decreased recruitment of leukocytes (Voll et al., 1997). Efferocytosis also leads to increased production of IL-10 and TGF-β, which further blunt inflammatory processes and promote the induction of Tregs both in vitro and in vivo (Kleinclauss et al., 2006; Maeda et al., 2005).
c. Post-resolution
In the post-resolution phase, additional immune cells, including dendritic cells, myeloid-derived suppressor cells (MDSCs), memory T cells, and memory B cells, accumulate. Resident macrophages repopulate the tissue as well as proinflammatory Ly6chigh monocyte-derived macrophages that persist for up to months (Fullerton and Gilroy, 2016). These macrophages are tolerized compared with their pro-inflammatory counterparts, preventing further activation of inflammatory responses.
This occurs through inhibition of TLR signaling by the macrophages as well as through negative regulation by signals such as CD200, which prevents activation of both monocytes and surrounding vascular cells (Didierlaurent et al., 2008; Kassiteridi et al., 2021; Snelgrove et al., 2008). Interestingly, PGE2, which has long been appreciated for its pro-inflammatory role, also appears to play an important role in the post-resolution phase. In a mouse model of peritonitis as well as in a human model of skin inflammation, PGE2 levels rise during the acute inflammatory phase, fall with resolution, and then rise again during the post-resolution phase to levels three to five times higher than in the acute phase (Motwani et al., 2017; Newson et al., 2017).
In this setting, PGE2 was found to inhibit T cell proliferation, limit pro-inflammatory cytokine production by macrophages, and promote MDSC formation (Feehan and Gilroy, 2019). These studies emphasize that despite resolution of the initial inflammation, the tissue remains in a state of “adaptive homeostasis” that differs from the naive state in which it began (Fullerton and Gilroy, 2016).
The post-resolution phase is context dependent and appears to be programmed by the initial acute inflammatory response. For example, in a model of acute sterile peritonitis, there is a predictable accumulation of adaptive immune cells that peaks around day 10 after injection of a low dose (0.1 mg/mouse) of zymosan, which is a glucan derived from the cell wall of yeast. When mice are instead injected with high-dose (10 mg/mouse) zymosan, minimal post-resolution adaptive immune cell accumulation is observed (Newson et al., 2014). Similarly, when mice are chronically exposed to inflammatory cytokines, such as TNF-α, they develop blunted adaptive immune responses (Cope et al., 1997; Feehan and Gilroy, 2019; Iwasaki and Medzhitov, 2015). Although the mechanisms behind these findings are not yet fully elucidated, it underscores the importance of the resolution in bridging the innate and adaptive immune systems.
If the resolution program fails because the inflammatory stimulus persists or a pathological process lowers the levels of resolving mediator or their effectiveness, the inflammatory response is sustained, and the organism may fail to develop an appropriate adaptive immune response. There is mounting evidence that failures in resolution and post-resolution lead to chronic inflammatory conditions, including chronic obstructive pulmonary disease, inflammatory bowel disease, cancers, rheumatoid arthritis, asthma, and Alzheimer's disease (Nathan and Ding, 2010; Tabas and Glass, 2013). Here, we will review the role of inflammation resolution in cardiovascular diseases.
2. Failures of Resolution in the Cardiovascular System
Common to many forms of cardiovascular diseases is vascular dysfunction, whether it is from a hemodynamic, metabolic, or traumatic insult, that then triggers an inflammatory response. We now know that, in addition to excess or chronic inflammation, multiple failures of resolution also contribute to progression of disease and adverse clinical outcomes. Although atherosclerosis is one of the best explored examples of failed resolution in cardiovascular disease, there is mounting evidence of a role for resolution in a variety of cardiovascular diseases and conditions.
a. Hypertension
Hypertension is defined as a sustained systolic blood pressure greater than or equal to 130 mmHg or diastolic blood pressure greater than or equal to 80 mmHg (Whelton et al., 2018). The underlying cause is multifactorial, with disturbances of the central nervous system, kidneys, and vasculature all implicated in the pathogenesis (reviewed in Harrison et al., 2021; Norlander et al., 2018). At the molecular level, there is increased production of vasoconstrictors, including endothelin-1, PGs, and angiotensin II (Ang II), and loss of vasodilators such as NO (Norlander et al., 2018).
In addition, the immune system plays a critical role in the development of hypertension and hypertensive target organ damage with almost every cell type involved in both innate and adaptive immunity shown to play a role (Madhur et al., 2021). Importantly, patients who achieve reasonable blood pressure control still have an increased risk of cardiovascular events compared with their untreated counterparts with similar levels of blood pressure (Blacher et al., 2010; Lieb et al., 2015).
This suggests that there is a component of residual risk not addressed by currently available anti-hypertensive strategies (Madhur et al., 2021). A potential explanation for this residual risk is untreated or undertreated inflammation. The pro-inflammatory mechanisms in the pathogenesis of hypertension have been well reviewed elsewhere, therefore we will briefly summarize next and focus on the emerging mechanisms of resolution (Madhur et al., 2021; Norlander et al., 2018) (Fig. 4).
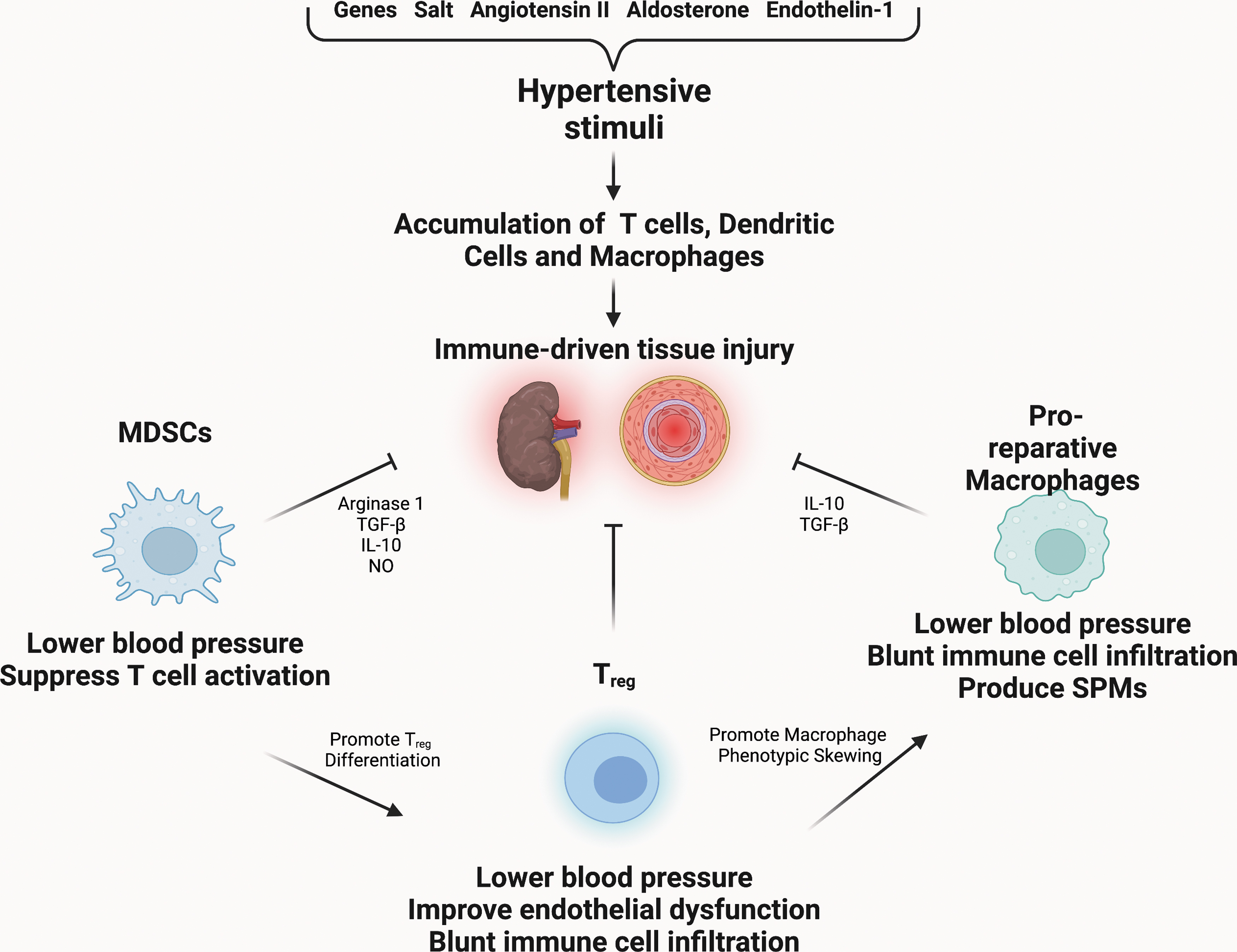
i. Inflammation
A causal role for T cells (Guzik et al., 2007; Saleh et al., 2015), dendritic cells (Kirabo et al., 2014), and macrophages (Wenzel et al., 2011) that accumulate in hypertensive organs has been demonstrated. During the development of hypertension, monocytes, macrophages, and dendritic cells are a key source of pro-inflammatory cytokines and chemokines, including IL-1β, IL-6, IL-12, IL-18, monocyte chemoattractant protein-1 (MCP-1), and TNF-α (Harwani, 2018; Norlander et al., 2018; Wenzel, 2019).
In addition, macrophages and dendritic cells, along with endothelial cells and smooth muscle cells (SMCs), generate ROS (Li et al., 2014; Wenzel, 2019). Deletion of myelomonocytic cells from mice leads to blunted hypertensive responses to Ang II, accompanied by decreased aortic inflammation, improved endothelial cell and SMC function, and less oxidative stress (Wenzel et al., 2011). Similarly, macrophage-colony stimulating factor (M-CSF)-deficient mice (Op/Op mice), which have markedly reduced numbers of circulating monocytes and fewer peritoneal macrophages, are protected from Ang II-induced hypertension (De Ciuceis et al., 2005; Ko et al., 2007).
In response to oxidative stress induced by hypertensive stimuli, dendritic cells present a class of potential neoantigens composed of isoLG-modified peptides that ultimately drive T cell proliferation and pro-inflammatory cytokine production (Kirabo et al., 2014). Mice or rats that lack recombinase activating gene 1 (Rag1 −/−) fail to develop mature B and T lymphocytes, and these mice are protected from the full development of hypertension and end organ damage (Crowley et al., 2010; Guzik et al., 2007). The adoptive transfer of T lymphocytes into Rag1 −/− mice restored their hypertensive response (Crowley et al., 2010; Guzik et al., 2007). Loss of CD8+ T cells alone confers protection from hypertension, likely due to the ability of CD8+ T cells to generate multiple inflammatory mediators, including IFN-γ, TNF-α, and granzyme B (Norlander et al., 2018; Trott et al., 2014).
CD4+ TH17 cells, as well as gamma delta T cells, are a major source of IL-17A, which plays a critical role in hypertension (Davis et al., 2021; Madhur et al., 2010; Saleh et al., 2016). In response to Ang II infusion, Il17a −/− mice develop a blunted hypertensive response compared with wild type mice (Madhur et al., 2010). Importantly, the renal and vascular end-organ damage induced by Ang II infusion is virtually abolished in mice lacking IL-17A (Madhur et al., 2010; Norlander et al., 2016). Treatment of mice with IL-17A alone is sufficient to increase blood pressure, at least partly through its ability to impair NO production through inhibition of endothelial nitric oxide synthase (eNOS) activity (Nguyen et al., 2013). Human hypertension is also associated with increased IL-17A levels (Davis et al., 2021).
ii. Resolution
Although the inflammatory basis of hypertension is well understood, less is known about the possible role of inflammation resolution. Tregs appear to serve an anti-inflammatory and pro-resolving role in hypertension. In response to Ang II-infusion, the number of Tregs decreases. Conversely, infusion of Tregs to hypertensive mice lowered blood pressure, improved endothelial function, and led to less macrophage and T cell infiltration within the aorta (Barhoumi et al., 2011; Matrougui et al., 2011). Treatment of mice with IL-2/anti-IL-2 immune complex, which leads to the expansion of Tregs in vivo, did not change overall blood pressure but did lead to less aortic stiffening, less infiltration of the aorta by TH17 cells and macrophages, as well as lower expression of IL-17A (Majeed et al., 2014).
Hypertension appears to shift the balance of pro-inflammatory and pro-resolving macrophages in mice and rats toward a pro-inflammatory state in the aorta, liver, and kidney (Harwani, 2018; Harwani et al., 2016; Harwani et al., 2012; Lin et al., 2016; Ndisang and Mishra, 2013). Monocytes isolated from patients with hypertension also skew toward pro-inflammatory phenotypes (CD14+/CD16−) and express high levels of IL-1β and angiotensin-converting enzyme (ACE) (Dorffel et al., 1999). Interventions that decrease pro-inflammatory macrophages or increase pro-resolving macrophages lead to reductions in blood pressure and decreased inflammatory infiltration of renal and aortic tissues (Gomolak and Didion, 2014; Ndisang and Mishra, 2013).
In addition, inhibition of a variety of functions typically attributed to pro-inflammatory macrophages, including secretion of IL-1β, IL-6, or ROS production leads to improvements in blood pressure (Harwani, 2018). Further, although not yet specifically examined in hypertension, it is worth noting that in models of atherosclerosis, Ang II leads to the cleavage of the efferocytosis receptor, MerTK, thereby impairing efferocytosis and resolution (Zhang et al., 2019b).
The MDSCs provide another example of pro-resolving cells that are important in hypertension. The MDSCs are a heterogenous group of immature myeloid cells that possess both anti-inflammatory and pro-resolving properties. These cells produce Arginase-1, TGF-β, and IL-10 that promote Tregs. In addition, they produce NO and hydrogen peroxide, which inhibit T cell activation and proliferation (Gabrilovich, 2017). In response to Ang II infusion, MDSCs increase in number within the circulation and the spleen.
When isolated from hypertensive mice, these cells are able to suppress T cell activation in vitro in an ROS-dependent manner. Depletion of MDSCs from hypertensive mice raises blood pressure and renal inflammation and adoptive transfer of MDSCs from normotensive mice to hypertensive mice led to a reduction in blood pressure (Shah et al., 2015).
Drugs used to treat hypertension have been shown to reduce inflammation, but potential effects on inflammation resolution are less well understood (Silva et al., 2019). For example, there is evidence that drugs that interfere with the renin-angiotensin-aldosterone system (RAAS) lower inflammation in rodents (Amador et al., 2014; Platten et al., 2009) and humans (Bendtzen et al., 2003; Gamboa et al., 2012; Yang et al., 2020). In addition, calcium channel blockers have been shown to have anti-inflammatory effects on macrophages (Das et al., 2009; Martin-Ventura et al., 2008).
Interestingly, the calcium channel blocker, amlodipine, induces regression of atherosclerosis in mice via anti-inflammatory and anti-oxidative stress pathways independent of blood pressure, suggesting that amlodipine may induce inflammation resolution (Yoshii et al., 2006). However, more research is needed to determine the precise mechanisms by which amlodipine or other anti-hypertensive drugs potentially modulate inflammation resolution pathways.
1. Pro-resolving mediators
a. Lipid mediators
The SPMs are not yet well studied in hypertension; however, there are several suggestions that they may play a role. Patients with hypertension have elevated levels of AA, the precursor to LX and LT formation (Tsukamoto and Sugawara, 2018; Xu et al., 2020). Deletion of 12/15-LO, the enzyme responsible for the first step in conversion of AA to other eicosanoids, led to protection from hypertension in two different mouse models (Kriska et al., 2012). LXA4 and its analogues have been shown to be potent inducers of NO through effects on both eNOS and inducible nitric oxide synthase (iNOS), suggesting that AA, LXA4, and its analogs may contribute to vascular tone (Paul-Clark et al., 2004).
In support of this, administration of intravenous LXA4 to hypertensive rats led to a transient but significant decrease in blood pressure, consistent with its short half life. Ex vivo, treatment of pre-contracted mesenteric arteries or aortic rings with LXA4 led to relaxation. Therefore, it is possible that LXA4 produced by a resolving response plays a role in local blood flow and possibly systemic blood pressure (Von Der Weid et al., 2004). Rv, which are derived from EPA and DHA, can also modulate contractility of vascular smooth muscle.
Treatment of pre-contracted aortic rings from rats or intact segments of human pulmonary arteries in vitro with RvD1, RvD2, or RvE1 led to vascular relaxation (Jannaway et al., 2018). In addition, Ang II infusion in mice elevated levels of RvD2-producing enzymes as well as the RvD2 receptor. Preventive RvD2 treatment during Ang II infusion lowered systolic blood pressure, improved vascular function, and increased pro-resolving macrophages, but RvD2 treatment after hypertension was already established did not lower blood pressure and had more limited effects (Diaz Del Campo et al., 2023).
Lastly, PGD2 signaling has been implicated in age-related hypertension as deletion of the PGD2 in CD4+ T cells leads to an exaggerated rise in blood pressure with aging and increased pro-inflammatory cytokine production (Kong et al., 2020). Our current understanding of SPMs in hypertension remains limited; however, based on their beneficial immunomodulatory effects in models of atherosclerosis, it is interesting to hypothesize that they may play a role in hypertension as well (Diaz Del Campo et al., 2023).
b. Gaseous transmitters
NO, CO, and H2S have all been shown to promote vasodilation and reduce blood pressure; however, less is known about the effect of these gaseous mediators on the inflammation associated with hypertension. The best studied transmitter in hypertension in NO, which is produced by one of three isoforms of nitric oxide synthase (NOS), including the constitutively active neuronal NOS and eNOS as well as iNOS. The physiological levels of NO produced by eNOS are associated with anti-inflammatory properties of endothelial cells, including reduced endothelial permeability, reduced apoptosis, and downregulation of cellular adhesion molecules that promote leukocyte trafficking (Forstermann et al., 2017).
In macrophages, NO can nitrosylate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to blunt pro-inflammatory gene expression as well as NLRP3 to blunt inflammasome activation and downstream IL-1β production (Fernando et al., 2019; Hernandez-Cuellar et al., 2012). Genetic deletion of eNOS (Nos3−/− ) in mice promotes hypertension and end-organ damage, including renal injury (Huang et al., 1995). These mice have higher numbers of renal macrophages and increased expression of inflammasome-associated genes (Sogawa et al., 2018).
Deletion of the inflammasome-associated gene, Asc, in Nos3−/− mice leads to improvement in renal function, further linking eNOS and inflammation (Sogawa et al., 2018). In contrast to the role of eNOS, deletion or inhibition of iNOS protects against the development of hypertension, blunts renal interstitial inflammation, and limits cardiac fibrosis (Hong et al., 2000; Sun et al., 2009a). Although the mechanisms are not fully elucidated, this may be because iNOS is capable of producing excessive amounts of NO.
As the name implies, iNOS is absent from healthy vasculature but is upregulated in response to inflammation. High iNOS activity sequesters cofactors necessary for eNOS activity, leading to uncoupling of eNOS that favors superoxide generation over NO (Forstermann et al., 2017). Therefore, although typically NO would be considered a pro-resolving mediator, it appears to fail under chronic inflammatory conditions. Further research is necessary to understand these mechanisms.
Low levels of H2S are correlated with human hypertension (Sun et al., 2007) and deletion of cystathionine γ-lyase (CSE), the enzyme that produces H2S, leads to hypertension in mice (Yang et al., 2008). Treatment of spontaneously hypertensive rats or Ang II-infused mice with sodium hydrosulfide or thiosulfate, donors that increases levels of H2S, led not only to improvements in blood pressure but also to a reduction in fibrosis, the CD4:CD8 T cell ratio, and circulating pro-inflammatory cytokine levels with a concomitant increase in circulating IL-10 (Ni et al., 2018; Snijder et al., 2014; Van Goor et al., 2016).
CO also attenuates hypertension in rodent models and increasing its levels in vivo has been shown to reduce expression of COX2, IL-1, and IL-6 in tissue (Zhang et al., 2019a).
b. Atherosclerosis
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of death and disability worldwide. Further, its prevalence is rising as rates of the major risk factors for ASCVD, including hypertension, obesity and diabetes, rapidly increase (Roth et al., 2020). Conventional therapy for ASCVD is aimed at lowering cholesterol levels; however, even with optimal treatment many patients still have progression of disease and recurrent clinical cardiac events (Aday and Ridker, 2019). A series of recent landmark clinical trials have demonstrated that blocking inflammation in addition to lipid lowering leads to lower rates of clinical cardiovascular disease (Nidorf et al., 2020; Ridker et al., 2017; Tardif et al., 2019).
Despite these successes, these anti-inflammatory trials were hindered by infectious complications that resulted from compromised host defense (Nidorf et al., 2020; Ridker et al., 2017; Tardif et al., 2019). Therefore, understanding the mechanisms that promote resolution is imperative to develop innovative solutions.
i. Inflammation
Atherosclerosis results from the accumulation of apolipoprotein B100-containing proteins within vulnerable portions of the artery where the endothelial barrier layer has been compromised. Multiple factors, including hypertension, smoking, and age, impair the vasodilatory capacity of vessels and enhance ROS production, leading to increased endothelial permeability and the initiation of lesion development (Libby et al., 2019; Warnholtz et al., 1999) (Fig. 5). Lipoproteins then infiltrate the vessel wall and become immobilized via interactions with matrix proteoglycans. Once trapped, low-density lipoprotein (LDL) can undergo oxidation (oxLDL), aggregation, and cleavage, which generates epitopes that are capable of acting as DAMPs recognized by TLRs (Miller et al., 2011).
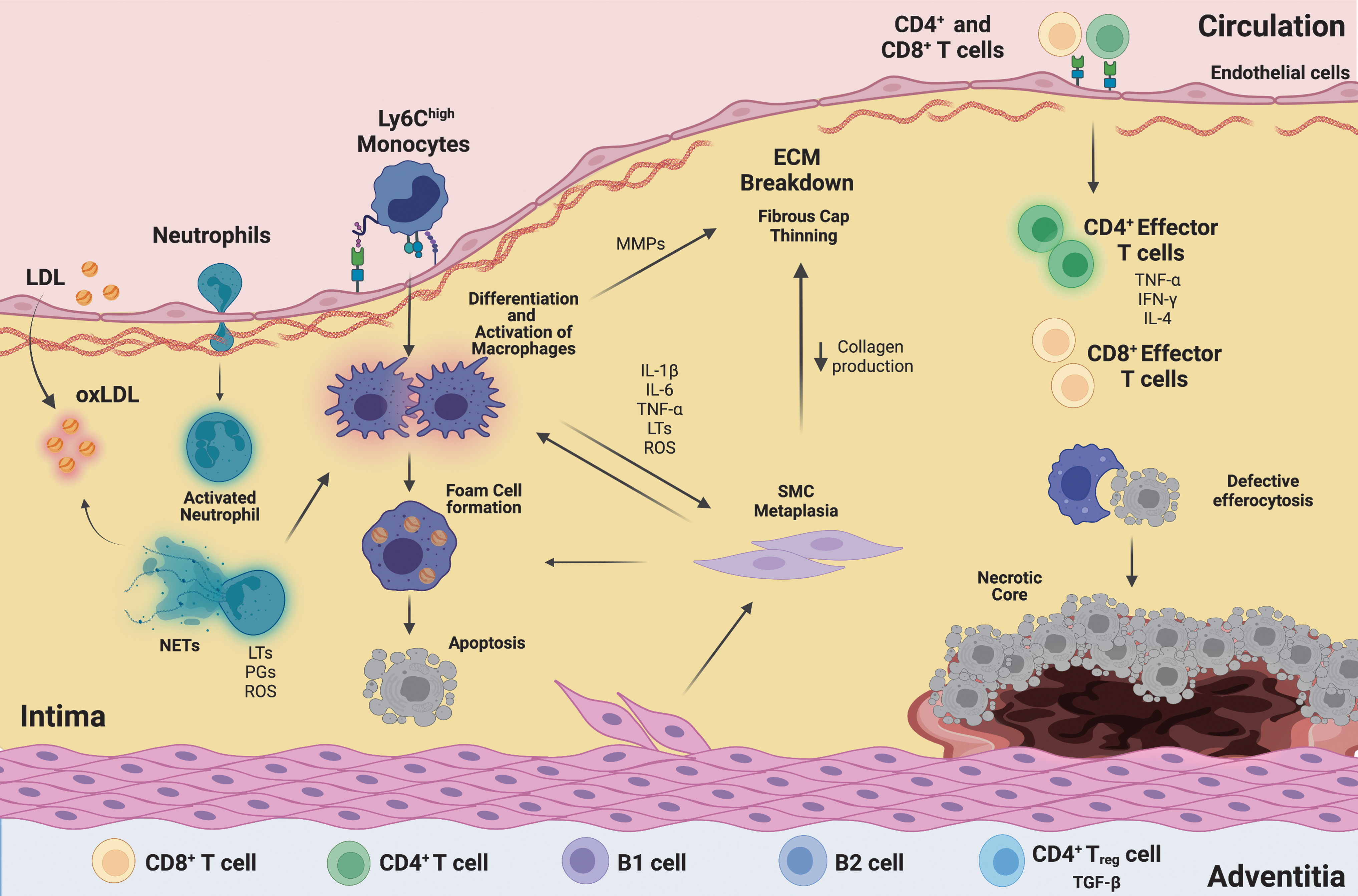
This, in turn, stimulates the endothelial layer to produce cytokines, growth factors, and adhesion molecules that recruit circulating leukocytes, particularly Ly6chigh monocytes (Hansson and Hermansson, 2011; Libby et al., 2019). In response to systemic hypercholesterolemia, there is heightened myelopoiesis in the spleen and bone marrow, leading to increased numbers of circulating monocytes as well as neutrophils (Rahman et al., 2017). Neutrophils can deposit cathepsin G on arterial endothelial cells, which promotes neutrophil and monocyte recruitment (Ortega-Gomez et al., 2016).
Production of ROS and proteases by neutrophils further activates endothelial cells, extracellular matrix production, and continued LDL modification (Silvestre-Roig et al., 2020). Superoxide can peroxidate AA, producing pro-inflammatory IsoLGs that contribute to modification of LDL, priming it for uptake by macrophages. In addition, IsoLG can modify high-density lipoprotein (HDL), impairing HDL's ability to remove cholesterol from macrophages and the vessel wall (Davies and May-Zhang, 2018; Tao et al., 2020).
Within the arterial wall, monocytes differentiate into macrophages (Marchini et al., 2021). Macrophages ingest lipid through a variety of receptors, including scavenger receptor A (SR-A), SR-B1, MARCO, and CD36, among others. This leads to the conversion of macrophages to “foam cells” that form the basis of early atherosclerotic lesions. Ester hydrolases within macrophages convert cholesterol to its free form, which can also lead to cholesterol crystal formation (Moore and Tabas, 2011). These cholesterol crystals can activate the NLRP3 inflammasome, contributing to the production of pro-inflammatory IL-1β and augmenting plaque inflammation (Duewell et al., 2010).
Engagement of scavenger receptors, particularly SR-A, also promotes local macrophage proliferation. Macrophages within plaques take on a pro-inflammatory phenotype and produce pro-inflammatory IL-1β, IL-6, TNF-α, PGs, LTs, and thromboxane. In addition, SMCs can undergo a metaplastic process by which they take on a macrophage-like phenotype, contributing to inflammatory cytokine production (Miano et al., 2021). Further, evidence suggests that SMCs can also take up lipid, contributing to lesional foam cells (Bennett et al., 2016; Doran et al., 2008). Neutrophils also produce PGs and LTs and further contribute to the polarization of macrophages through their release of NETs, which are composed of webs of strand-like DNA and a variety of pro-inflammatory proteins (Serhan, 2014; Warnatsch et al., 2015).
Once the innate immune response is established, an inflammatory adaptive immune response is also initiated, and lymphocytes infiltrate the plaque. Naive CD4+ T cells are recruited to atherosclerotic lesions through their expression of L-selectin, C-C chemokine receptor (CCR)1, CCR5, and CXC chemokine receptor (CXCR)3 (Saigusa et al., 2020). These differentiate into a variety of T cell subsets, predominantly TH1 cells that elaborate the proinflammatory cytokines IFN-γ and TNF-α, but also small populations of TH2 and TH17 cells (Wolf and Ley, 2019).
The generation of these pro-inflammatory T cell subsets appears to come at the expense of Tregs, which decrease in number as atherosclerosis progresses (Witztum and Lichtman, 2014). B cells are also associated with the plaque, primarily on the adventitial side of the vessel. B-1 cells produce a natural IgM that recognizes oxidized epitopes of LDL and may serve to prevent oxLDL uptake and promote efferocytosis by macrophages, thereby slowing the progression of atherosclerosis (Sage et al., 2019). B-2 cells, however, promote the development of atherosclerosis through their ability to promote and sustain effector CD4+ T cells, including TH1 and TH17 subsets (Sage et al., 2019).
As the lesion grows, SMCs proliferate and migrate from the medial to the intimal side of the artery where they produce an extracellular matrix and form a fibrous cap to stabilize the plaques. In response to continued inflammation, the overlying fibrous cap of the lesion may begin to erode or rupture. Exposure of this highly inflammatory necrotic material to the bloodstream promotes thrombosis, which leads to acute clinical events such as heart attack and stroke.
ii. Resolution
In a healthy inflammatory process, the peak of inflammation would be followed by the initiation of a resolution program, whereas in atherosclerosis inflammation resolution fails as the lesion becomes more advanced (Back et al., 2019; Kasikara et al., 2018; Moore et al., 2018). Although lipid-lowering therapy is the mainstay of treatment for clinical atherosclerotic disease, LDL cannot be “removed” as an inciting stimulus, creating a cycle of chronic immune stimulation. This leads to ongoing recruitment of monocytes and neutrophils, accompanied by continued pro-inflammatory cytokine production and ROS generation (Kasikara et al., 2018).
In addition, macrophages are unable to egress from the plaque, both because of interactions with endothelial cells as well as upregulation of chemostatic factors by macrophages themselves, including netrin-1 and semaphorin 3E (Van Gils et al., 2012; Wanschel et al., 2013). These plaque macrophages retain a pro-inflammatory phenotype rather than transitioning toward a more pro-reparative phenotype. Recently, it was discovered that CD200 signaling through its cognate receptor (CD200R) restrains activation of lesional macrophages, leading to downregulation of CCL2 and reduced accumulation of Ly6chigh monocytes within the atherosclerotic aorta.
Further, the authors found that CD200R expression was significantly decreased on classical monocytes (CD14high CD16low) in patients with high atherosclerotic burden and plaque necrosis as compared with those with lower atheroma burden and percent necrosis, suggesting that CD200R expression is associated with more favorable plaque phenotypes (Kassiteridi et al., 2021). These findings suggest that impaired expression of CD200 may contribute to continued recruitment of monocytes to the developing lesion.
Tregs are present within human and murine atherosclerotic lesions and have been demonstrated to have a protective, pro-resolving role in atherosclerosis (De Boer et al., 2007; Saigusa et al., 2020; Tabas and Lichtman, 2017). Depletion of Tregs from hypercholesterolemic mice leads to increased lesion development, whereas transfer of Tregs slows plaque development (Ait-Oufella et al., 2006). This is believed to be at least in part due to the ability of Tregs to secrete TGF-β and IL-10, which suppresses the pro-inflammatory effects of Teffector (Teff) cells and macrophage activation, respectively (Witztum and Lichtman, 2014).
In addition, Tregs promote macrophage efferocytosis through the secretion of IL-13, which enhances macrophage production of IL-10, activates Rac1 to promote apoptotic cell engulfment, and limits expansion of necrotic cores (Proto et al., 2018). In addition, the presence of Tregs leads to lower expression of matrix metalloproteinase (MMP)-2 and MMP-9, which slows collagen remodeling and enhances fibrous cap stability (Meng et al., 2013). In humans, Treg numbers are lower in “vulnerable” plaques than in stable plaques (Dietel et al., 2013). In a large prospective cohort study, low levels of circulating Tregs were associated with an increased risk of myocardial infarction (MI) (Wigren et al., 2012); although this was not replicated in a more recent population-based cohort study (Olson et al., 2020).
In mice, circulating Tregs initially increase in number during high-fat diet feeding, but unlike Teff cells, their numbers decline in the setting of sustained hypercholesterolemia (Maganto-Garcia et al., 2011). More specifically, the Treg:Teff ratio declines with more advanced disease (Tabas and Lichtman, 2017). This appears to be, at least in part, due to a phenotypic shift in Tregs in which they take on more Teff-like functions.
Multiple studies using lineage tracing have shown that, under pressure from an atherogenic milieu, classical Tregs (CD4+ CD25+ FoxP3+) can convert to TH1-Treg cells that produce IFN-γ and TNF-α or to TH17-Treg cells that elaborate IL-17A, IL-22, and IFN-γ although the direct consequences of Treg conversion on atherosclerotic plaque development are not yet fully known (Tabas and Lichtman, 2017). Further, whether this occurs in humans is not yet understood. Interestingly, it has been shown in vitro that RvD1, RvD2, and maresin 1 are able to promote Treg differentiation while preventing TH1 and TH17 differentiation and blocking Teff production of pro-inflammatory cytokines, further linking these resolution processes (Chiurchiu et al., 2016).
1. Efferocytosis
Sustained inflammation promotes cell death and apoptotic cells begin to accumulate within the artery wall. Early in lesion development, efferocytosis is efficient, however, as lesions progress the process begins to fail. Interaction of apoptotic cells with macrophages leads to downregulation of inflammatory cytokine expression, upregulation of pro-resolving cytokines IL-10 and TGF-β, and increased SPM production. Therefore, when efferocytosis is compromised, these key components of the resolution response are also dysregulated.
Further, apoptotic cells that fail to be cleared through efferocytosis become secondarily necrotic and serve as a nidus for the formation of a necrotic core along with extracellular lipid that accumulates within advanced plaques.
Efferocytosis fails in advanced atherosclerosis by various mechanisms, and we have previously reviewed these in detail (Doran et al., 2020; Yurdagul et al., 2017). Several efferocytosis receptors have been shown to play a causal role in plaque progression. Genetic deletion of MerTK from western diet-fed atheroprone mice (Apoe−/− or Ldlr−/− ) led to increased lesion size, reduced lesional efferocytosis, and large necrotic cores (Ait-Oufella et al., 2008; Thorp et al., 2008). Similarly, loss of LRP1 from macrophages of Ldlr−/− or Apoe−/− mice leads to increased lesion area and larger necrotic cores (Overton et al., 2007; Yancey et al., 2011).
In an atherogenic milieu, these receptors can be downregulated or inactivated. In response to pro-inflammatory signals, the protease a disintegrin and metalloproteinase domain-containing protein 17 (ADAM17) is activated to cleave MerTK. This renders the membrane-bound receptor fragment inactive and releases a soluble fragment (soluble MerTK [sol-Mer]) (Sather et al., 2007; Thorp et al., 2011). Although a disease-specific role of sol-Mer is not yet definitively proven, sol-Mer has been shown to accumulate in human atherosclerotic lesions (Cai et al., 2017; Garbin et al., 2013).
In vitro, sol-Mer scavenges the bridging molecule, Gas6, and impairs efferocytosis, suggesting that this mechanism may also play a role in vivo (Sather et al., 2007; Thorp et al., 2011). In support of this, western diet-fed Ldlr−/− mice that were genetically engineered to express a cleavage-resistant MerTK had enhanced lesional efferocytosis, smaller necrotic cores, and higher levels of SPMs as compared with control mice (Cai et al., 2017; Cai et al., 2016). Like MerTK, LRP1 can also be inactivated by ADAM17, although it is not yet certain whether this is causal in atheroprogression (Liu et al., 2009).
LRP1 can be downregulated via epsin-mediated, ubiquitin-dependent internalization and degradation (Brophy et al., 2019). Deletion of epsin 1 and 2 from myeloid cells enhances cell surface expression of LRP1, decreases overall lesion size, increases efferocytosis, and reduces necrotic cores (Brophy et al., 2019). Inflammation also alters the levels of pro-efferocytic bridging molecules, including MFG-E8, transglutaminase 2 (TG2), and complement component 1q (C1q) (Yurdagul et al., 2017).
Various studies have demonstrated that their expression declines in atherosclerosis and that genetic disruption of these genes leads to an increase in lesion size and necrotic core area (Yurdagul, 2021). Cells that become apoptotic in an inflammatory milieu also undergo changes that make them poor substrates for efferocytosis. In response to TNF pathway signaling, the “don't-eat-me” signal CD47 is upregulated on dying cells, rendering them resistant to uptake by lesional efferocytes (Kojima et al., 2016).
Treatment of Ldlr−/−- mice with an anti-CD47 antibody leads to improved lesional efferocytosis and smaller necrotic cores (Kojima et al., 2016). Further, consistent with the knowledge that efferocytosis enhances SPM production, mice treated with an anti-CD47 antibody also had higher levels of lesional SPMs, particularly RvD1 (Gerlach et al., 2020). This mechanism is of particular interest as it has recently been shown in a retrospective pre-clinical trial of patients with known cardiovascular risk factors that administration of a humanized anti-CD47 antibody led to a significant reduction in carotid artery inflammation as measured non-invasively by 18F—fluorodeoxyglucose positron-emission tomography and computer tomography (18F-FDG PET/CT) (Jarr et al., 2021).
2. Pro-resolving mediators
a. Lipid mediators
Advanced atherosclerosis is characterized by an imbalance of pro-resolving and pro-inflammatory lipid mediators. Regions of advanced human plaque characterized by large necrotic cores and thin collagen caps have low levels of RvD1 and high levels of LTB4, whereas those with less advanced features had higher RvD1:LTB4 ratios (Fredman et al., 2016). Similarly, Ldlr−/− mice with advanced lesions (after 17 weeks of high-fat diet) had a lower RvD1:LTB4 ratio than mice with early lesions (after 8 weeks of high-fat diet) (Fredman et al., 2016).
In vitro, the balance of SPMs versus pro-inflammatory mediators produced by macrophages shifts depending on the environment. In response to pro-inflammatory stimuli, one of the major biosynthetic enzymes involved in SPM generation, 5-LO, localizes to the nuclear membrane where it favors synthesis of LTB4. Under pro-resolving conditions and in response to efferocytosis, 5-LO instead localizes to the plasma membrane where it favors the synthesis of LXA4 (Fig. 2).
Enhanced nuclear localization of 5-LO also occurs in atherosclerosis, favoring LTB4 over LXA4 production (Fredman et al., 2014). This inflammatory pathway is promoted by calcium/calmodulin-dependent protein kinase II (CaMKII) and dampened by LXA4, RvD1, and signaling through the MerTK efferocytosis receptor (Cai et al., 2016; Fredman et al., 2014). Lower levels of the SPMs within atherosclerotic plaques have several implications.
In general, treatment of cells with the SPMs leads to decreased pro-inflammatory cytokine production (Kasikara et al., 2018). RvD2 has been shown to inhibit the priming and activation of the NLRP3 inflammasome, leading to decreased expression of IL-1β (Lopategi et al., 2019). This is of particular interest because of recent clinical trials demonstrating that targeting IL-1β using a monoclonal antibody, canakinumab, led to decreased incidence of heart attack and stroke in susceptible patients (Ridker et al., 2017).
In addition, in vitro or ex vivo treatment of macrophages with SPMs including LXA4, RvD1, and RvD2 led to improved efferocytosis (Kasikara et al., 2018). These important atheroprotective roles of the SPMs are supported by a plethora of in vivo studies in which individual SPMs have been delivered to atheroprone rodent models or SPM function was interrupted by deletion of the receptors through which the SPMs signal. These are discussed in greater detail in several recent reviews (Back et al., 2019; Kasikara et al., 2018).
There has been a recent surge of interest in omega-3 fatty acids, the precursors to the SPMs, in the treatment of cardiovascular disease. Clinical trials that treated patients with a combination of EPA and DHA as a means of increasing SPM production yielded mixed results, which has been debated as possibly related to the composition of the omega-3 fatty acid preparations used or the variability of dosing between studies (Sherratt et al., 2023; Sperling and Nelson, 2016).
More recently, however, two large clinical trials known as the Japan EPA Lipid Intervention (JELIS) study and the Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial (REDUCE-IT) demonstrated that treating patients with either EPA or the purified ethyl ester of EPA respectively led to reduced clinical cardiac events (Bhatt et al., 2019; Yokoyama et al., 2007). A subsequent meta-analysis that included these most recent data found that the use of omega-3 fatty acids led to improved cardiovascular outcomes and reduced cardiovascular mortality (Khan et al., 2021).
Interestingly, this meta-analysis suggested that cardiovascular risk reduction was more prominent with EPA alone versus with the EPA+DHA used in most of the earlier trials (Khan et al., 2021). The authors suggest that this may be due to potentially deleterious functions unique to DHA but not EPA, including the ability of DHA to raise LDL levels (Khan et al., 2021). While neither JELIS or REDUCE-IT measured levels of SPMs at the patient level, it is interesting to hypothesize that omega-3 fatty acid supplementation may enhance SPM production in humans as it did in pre-clinical studies (Sobrino et al., 2020).
This is supported by smaller clinical trials in which consumption of omega-3 fatty acid-derived supplements led to increased plasma SPM concentrations along with decreased leukocyte activation and enhanced neutrophil and monocyte phagocytosis (Souza et al., 2020).
Pre-clinical data suggest that administration of exogenous SPMs ameliorates ASCVD; however, their practical use in patients is limited by their short half-lives, particularly for chronic disease. To address this, the development and testing of synthetic analogs of the SPMs is underway (Godson et al., 2023). Acting through the ALX/FPR2 receptor, these agonists limit pro-inflammatory cytokine production and inhibit neutrophil infiltration in peritonitis (De Gaetano et al., 2021; Sun et al., 2009b).
One LXA4 analog, benzo-LXA4, was recently shown to slow the development of atherosclerotic lesions in a mouse model of diabetes-associated atherosclerosis (Brennan et al., 2018). Small-molecule agonists of the ALX/FPR2 receptor are also in development (Galvao et al., 2021; Maciuszek et al., 2021). In vitro, these agonists were shown to decrease neutrophil adhesion (Maciuszek et al., 2021). In an in vivo murine model of arthritis, the ALX/FPR2 agonist AT-01-KG was shown to reduce neutrophil recruitment, lower levels of pro-inflammatory cytokines, and enhance efferocytosis in the articular space and surrounding tissues (Galvao et al., 2021). Future studies will be needed to investigate the effect of these compounds in models of chronic inflammation.
b. Gaseous transmitters
Several gaseous transmitters have been shown to play a role in atherosclerosis. NO is the best studied of these and, as in hypertension, its role in atherosclerotic disease is complex. Loss of eNOS led to accelerated atherosclerosis in atheroprone mice (Kuhlencordt et al., 2001b). Importantly, this effect was found to be independent of the effects of eNOS on hypertension, which is a major risk factor for atherosclerotic disease and could be a potential confounder of these studies (Chen et al., 2001). eNOS-derived NO exerts multiple atheroprotective effects, including the prevention of leukocyte recruitment to the vessel wall and inhibition of LDL oxidation (Forstermann et al., 2017).
In addition, endogenous NO can block IL-1β production by nitrosylating the NLRP3 inflammasome and preventing its assembly in macrophages (Hernandez-Cuellar et al., 2012). Interestingly, genetic deletion of iNOS from atheroprone mice leads to reduced atherosclerotic lesion area, suggesting an inflammatory role that contrasts that of eNOS (Kuhlencordt et al., 2001a). As described for hypertension, this may be due to the high iNOS activity sequestering cofactors necessary for eNOS activity, leading to the uncoupling of eNOS that favors superoxide generation over NO (Forstermann et al., 2017). In addition, higher levels of NO lead to peroxynitrite formation that contributes to oxidative stress and lipid peroxidation in lesions (Forstermann et al., 2017).
Emerging data suggest that there are other pro-resolving gases that are also important in the vasculature. The loss of H2S from atheroprone mice led to increased atherosclerosis, whereas strategies that enhanced the levels of H2S ameliorated plaque burden (Mani et al., 2013; Wang et al., 2009; Zheng et al., 2019). Compared with control mice, atheroprone Apoe−/− mice have lower plasma and aortic levels of H2S, suggesting that H2S production is impaired during atherogenesis (Wang et al., 2009).
The improvements induced by H2S appear to be due, at least in part, to the ability of H2S to enhance efferocytosis and prevent production of IL-1β in an NLRP3-dependent manner (Castelblanco et al., 2018; Zheng et al., 2019). Similarly, CO, which is produced as a byproduct of heme degradation by heme oxygenase-1 (HO-1), also blocks inflammatory cytokine production and inhibits the formation of atherosclerotic lesions (Ishikawa et al., 2001; Siow et al., 1999). Further, CO has been shown to increase the levels of SPMs, linking these two arms of the resolution program (Chiang et al., 2013).
c. MI and ischemic heart failure
Advanced atherosclerotic plaque can impede coronary artery blood flow, due to either obstruction or secondary thrombo-occlusive disease at the site of plaque erosion or rupture, leading to what is clinically recognized as an MI. After MI, the heart undergoes repair and remodeling at both the cellular and molecular levels. Unlike in atherosclerosis, inflammation after heart attack is often acute and resolving. In cases where the extent of infarction is massive, the inflammatory response is excessive, or the process of repair is impaired, there can be maladaptive remodeling leading to heart failure (Prabhu and Frangogiannis, 2016) (Fig. 6). Therefore, proper resolution of inflammation is crucial.
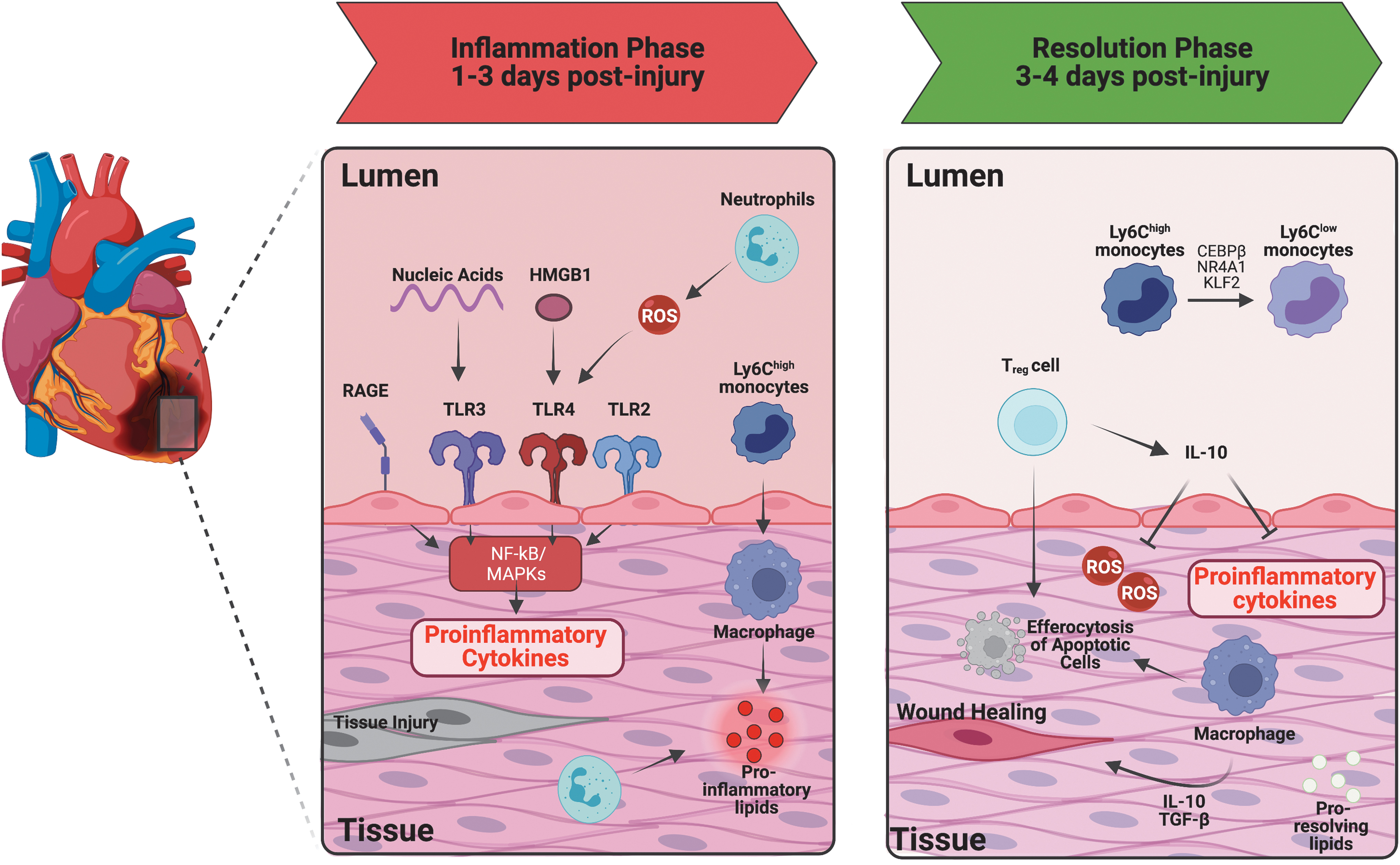
i. Inflammation
Following tissue injury, damaged cardiomyocytes and vascular cells release DAMPs to the extracellular environment, which act via PRRs to initiate a pro-inflammatory cascade of cytokines, chemokines, and cell adhesion molecules (Zhang et al., 2015). In particular, high mobility group box 1 (HMGB1), S100 proteins, heat shock proteins, as well as mitochondrial DNA and RNA released by dying cells trigger the inflammatory response during MI.
The TLRs, NLRs, and the cell-surface receptor for advanced glycation end products (RAGE) are all key PRRs after MI and the loss of these receptors leads to decreased inflammation and reduced infarct size after MI (Andrassy et al., 2008; Shinagawa and Frantz, 2015). The signaling pathways through these receptors have been extensively reviewed elsewhere (Mann, 2011; Prabhu and Frangogiannis, 2016); however, they converge on inflammatory signaling via activation of mitogen-activated protein kinases (MAPKs) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB).
The net effect of this signaling is the upregulation of inflammatory cytokines (IL-1β, IL-6, IL-18, and TNF-α) (Ding et al., 2013), chemokines (CCL2, CCL5, CXCL8) (Kukielka et al., 1995), and pro-inflammatory lipids (PGE2 and LTB4) (Soehnlein and Lindbom, 2010) that promote the recruitment of leukocytes. In cases where perfusion is restored to the myocardium, there can be a second wave of injury resulting from endothelial cell activation, influx of leukocytes, ROS generation, and activation of the complement cascade (Timmers et al., 2012; Vandervelde et al., 2006).
Neutrophils are the first immune cells that infiltrate the infarcted myocardium. They release proteases, generate ROS, and participate in early phagocytosis of dying cardiomyocytes before ultimately becoming apoptotic themselves. Early monocyte populations (days 1–3 in mouse models) are dominated by Ly6chigh monocytes, which originate from the bone marrow and spleen, and give rise to inflammatory macrophages that contribute to the pro-inflammatory signaling cascade (Nahrendorf et al., 2007; Yan et al., 2013). T cells are present in the healthy myocardium at low levels and begin to accumulate in the infarct zone after reperfusion, peaking in later stages as discussed next (Yang et al., 2006).
ii. Resolution
In the later phases (days 3–4 in reperfused MI, days 3–7 in permanent occlusion models), monocyte recruitment slows, and a pro-reparative macrophage phenotype predominates. This shift appears to be due in part not only to the increased recruitment of Ly6clow monocytes, but also to the ability of Ly6chigh monocytes to shift their phenotype through activation of factors such as Interferon-Regulatory Factor 5 (IRF5) and nuclear receptor subfamily 4 group A member 1 (Nr4a1), the latter of which prolongs survival of cardiac phagocytes and is associated with increased MerTK expression by these cells (Courties et al., 2014; Dehn and Thorp, 2018; Hilgendorf et al., 2014).
Overall, although Ly6clow monocytes are recruited in this phase, their numbers are far fewer than those of the Ly6chigh monocytes recruited in the inflammatory phase. During this phase, the surviving cardiac fibroblast population expands, and neighboring viable fibroblasts are recruited to the site of infarction, where they are believed to transdifferentiate into myofibroblasts in response to TGF-β (Serini et al., 1998). Macrophages and fibroblasts secrete extracellular matrix proteins that can interact with cells to modulate their behavior and signaling (Prabhu and Frangogiannis, 2016).
Over time, the repaired area matures into scar as extracellular matrix cross-links, reparative cells are deactivated, and myofibroblasts become quiescent (Prabhu and Frangogiannis, 2016). When there is persistent inflammation or a failure of the repair process, the myocardium undergoes adverse remodeling characterized by extensive fibrosis, cardiomyocyte loss, thinning of myocardial tissue, and dilation of the ventricular cavity, all of which lead to loss of cardiac function (Leoni and Soehnlein, 2018).
Clinically, outcomes after MI are dependent on the extent of the initial infarction and the development of an adequate post-MI reparative process. Although timely coronary reperfusion can limit the initial ischemic insult, there are a few therapies that address how to promote efficient resolution. Therefore, a more thorough understanding of the reparative processes post-MI is crucial.
Like monocytes and macrophages, neutrophils also appear to play a dual role in MI. In studies that depleted neutrophils early in the course of myocardial ischemia/reperfusion, both dogs and mice were found to have smaller overall infarct areas (Horckmans et al., 2017; Romson et al., 1983). Strategies that targeted neutralization or deletion of myeloperoxidase at the time of ischemia, a major enzyme that drives ROS production and tissue damage in MI, did not change the overall infarct size but did improve long-term cardiac function and remodeling after MI (Puhl and Steffens, 2019).
More recently, a protective effect has been attributed to neutrophils. In a mouse model of permanent coronary artery ligation, deletion of neutrophils did not have an effect on infarct size 24 hours after infarction; however, it did lead to more ventricular dilation and reduced systolic function after 7–14 days (Horckmans et al., 2017). These hearts were observed to have persistent inflammatory cytokine expression within the myocardium and excessive fibrosis.
Interestingly, the authors discovered that loss of neutrophils led to lower expression of MerTK by macrophages despite an overall upregulation of other pro-reparative macrophage markers, suggesting that neutrophils may promote resolution. They went on to show that neutrophils produce neutrophil-derived gelatinase-associated lipocalin (NGAL), which promotes MerTK expression and efferocytosis35. However, it is not yet known whether there are distinct neutrophil subsets present during or after MI that might explain this functional difference (Puhl and Steffens, 2019). This would appear to highlight the importance of the early inflammatory phase in programming the reparative phase.
Both CD4+ and CD8+ T cells appear to play dual roles in MI and cardiac remodeling. Rag1−/− mice, which lack mature B and T lymphocytes, were found to have significantly smaller infarcts than control mice. Interestingly, adoptive transfer of CD4+ but not CD8+ T cells into Rag1−/− mice reversed this effect, which appeared at least partly due to adenosine receptor signaling and IFN-γ production (Yang et al., 2006). While IFN-γ is one of the cytokines that drives inflammation post MI, the role of IFN-γ in the infarcted myocardium remains largely unknown.
Excess overexpression of IFN-γ is known to elicit systemic inflammation, yet a recent study argues that inhibiting IFN-γ in adult mice leads to a less trafficking of myeloid cells within the infarcted tissue, suggesting a protective function of IFN-γ in MI (Finger et al., 2019). Whether IFN-γ modulates resolution or efferocytosis in a beneficial manner has yet to be explored.
More recently, it was shown that although the loss of CD8+ T cells in mice did not change infarct size, it did improve overall survival of these mice in the first week after coronary occlusion, suggesting that CD8+ T do play some detrimental role in post-MI (Ilatovskaya et al., 2019). Interestingly, both CD4+ and CD8+ T cells also play an important role in myocardial healing. Mice deficient in functional CD8+ T cells (CD8a tm1mak ) had elevated circulating pro-inflammatory cytokines/chemokines, increased levels of sol-Mer suggesting dysfunctional MerTK, and more accumulated necrotic cells within the infarct.
Further, these mice had increased fibrosis, and higher rates of death from cardiac rupture after MI than control mice (Ilatovskaya et al., 2019). Loss of CD4+ T cells was found to delay the transition from Ly6chigh to Ly6clow monocyte accumulation in the infarcted area (Hofmann et al., 2012).
CD4+ Foxp3+ Tregs also accumulate within the heart during the reparative phase of MI and peak on day 7 post-MI in murine models (Xia et al., 2015; Zhuang et al., 2022). Rodent models that augment Treg numbers through adoptive transfer or via an agonistic antibody treatment that promotes Treg expansion both led to improved post-MI outcomes, whereas deletion of Tregs led to increased inflammation and negatively impacted myocardial remodeling (Weirather et al., 2014; Zhuang et al., 2022).
Therapeutic Treg expansion led to increased expression of IL-10 and TGF-β within the scar and skewing of myeloid cells within the myocardium toward a pro-reparative phenotype with higher expression of Arginase 1, CD206, and osteopontin (Weirather et al., 2014). In a study of patients with atherosclerotic disease, Treg numbers were lower in those with MI than in patients with stable coronary artery disease. Further, a low Treg:CD4+ T cell ratio in circulation predicted a higher rate of cardiovascular events in a prospective cohort (George et al., 2012; Mor et al., 2006; Wigren et al., 2012).
1. Efferocytosis
Efferocytosis of apoptotic cells is a crucial step in the resolution of myocardial inflammation and subsequent repair. Macrophages that perform efferocytosis release pro-resolving cytokines such as IL-10 and TGF-β as well as specialized pro-resolving mediators LXA4 and RvD1 that promote tissue repair. Therefore, impairment of efferocytosis would be expected to impair overall healing post-MI (Fadok et al., 1998; Huynh et al., 2002).
Consistent with this, mice lacking the efferocytosis receptor MerTK develop more severe reperfusion injury in response to coronary artery ligation. As in atherosclerosis, the function of MerTK can be compromised by cleavage of the receptor, resulting in the generation of sol-Mer that may competitively inhibit the interaction of MerTK with its ligands in the post-MI period. Elevated levels of sol-Mer were detected in human plasma after MI as well as in mouse serum after cardiac ischemia/reperfusion (Deberge et al., 2017; Wan et al., 2013).
Mice expressing a cleavage-resistant version of MerTK had smaller infarct areas with improved overall systolic function (Deberge et al., 2017). Recently, it was also observed that extracellular vesicles secreted by cardiosphere-derived cells, which are a type of cardiac progenitor cell, are able to polarize macrophages toward a reparative phenotype and enhance efferocytic ability in rodent models of coronary ligation/reperfusion.
The authors discovered that this occurred via transfer of miR-26a between extracellular vesicles and macrophages, which led to suppression of ADAM17 and upregulation of MerTK, again confirming the key role of MerTK and efferocytosis in resolution post-MI (De Couto et al., 2019). Another protein important for efferocytosis, MFG-E8, acts as a bridging molecule between apoptotic cells and the efferocytosis receptor integrin αvβ5.
Mice genetically deficient in MFG-E8 have increased cardiac inflammation, more necrosis, and worse cardiac dysfunction after coronary artery ligation, and this can be reversed by intracardiac injection of MFG-E8 (Nakaya et al., 2017). Post-MI, MFG-E8 is partially generated by a subpopulation of cardiac myofibroblasts that appear after infarction. These cells are able to act as efficient phagocytes and their ingestion of apoptotic cells leads to decreased expression of pro-inflammatory cytokines and upregulation of pro-resolving cytokines, including TGF-β, which promotes tissue repair (Nakaya et al., 2017).
Cells that become apoptotic in the setting of cardiac ischemia inappropriately retain expression of the “don't-eat-me” signal, CD47. When treated with an anti-CD47 antibody immediately after MI, mice had had smaller infarcts and less cardiac inflammation (Zhang et al., 2017). Together, these studies highlight the importance of efferocytosis in the post-MI repair process. Despite this, not all efferocytosis receptors play the same role in MI.
Although MerTK clearly has a protective role in MI and cardiac repair, it was recently discovered that another member of the TAM receptor family, Axl, plays a maladaptive role. Deletion of Axl from myeloid cells led to reduced infarct size, improved cardiac function, and less myocardial infiltration by neutrophils and Ly6chigh monocytes. The authors found that, in contrast to the cardioprotective role played by macrophage MerTK, Axl augments TLR4-priming of the inflammasome, leading to increased IL-1β production. In addition, Axl signaling promotes a switch to glycolytic metabolism that promotes pro-inflammatory responses in macrophages (Deberge et al., 2021).
ii. Pro-resolving mediators
a. Lipid mediators
Altered production of specialized pro-resolving mediators is also thought to play a role in MI. Patients suffering from an acute MI were found to have an increase in plasma levels of SPMs compared with those with chronic atherosclerotic disease (Fosshaug et al., 2019). After permanent coronary occlusion in mice, the expression of the biosynthetic enzymes that generate the SPMs, 5-LO and 12/15-LO begins to rise within the myocardium. Accordingly, between days 1 and 5 after MI, significant levels of D-series Rv, E-series Rv, protectins, maresins, and LXs also begin to accumulate (Halade et al., 2018).
Intriguingly, leukocytes recruited from the spleen appeared to be the major source of these resolving mediators, lending further strength to the concept that blocking early inflammation may also impair resolution in later phases. It was also observed that patients with congestive heart failure had lower levels of RvD1 as well as its receptor, GPR32, than healthy patients. Whereas mononuclear cells derived from healthy control patients, when treated with RvD1, could suppress the pro-inflammatory cytokine production of CD4+ and CD8+ T cells, those from heart failure patients were unable to modulate T cell responsiveness (Chiurchiu et al., 2019).
Treatment with RvD1 after MI led to reduced accumulation of neutrophils and less fibrosis in a coronary artery ligation model of murine MI (Kain et al., 2015). Similarly, treatment with RvE1 was found to reduce infarct size and post-MI leukocyte infiltration in a rat coronary artery ischemia/reperfusion model (Keyes et al., 2010). Interestingly, the effect of RvE1 was phase-dependent since treatment in the early post-MI period (days 1–7) led to improved cardiac function whereas late treatment (days 7–14) actually led to inhibited cardiac recovery after ischemia.
The authors of this study determined that in the early phase, RvE1 blocked the recruitment of Ly6chigh monocytes, leading to less inflammation and less apoptosis. In the late phase, RvE1 blocked monocyte recruitment and macrophage migration, which led to lower expression of angiogenic factors and limited neovascularization in the peri-infarct region (Liu et al., 2018). LXA4 and the LXA4 derivative, 15-epimer-LXA4 (15-epi-LXA4), were also found to be lower in patients with severe heart failure versus those with stable symptoms (Reina-Couto et al., 2014).
In a mouse model of permanent coronary ligation, the injection of 15-epi-LXA4 led to enhanced neutrophil clearance and reduced ventricular dysfunction than controls when given on either day 1 or day 5 after MI (Kain et al., 2017). Two recent studies have demonstrated that small molecular agonists of the FPR receptors also lead to reduced infarct area and less ventricular dysfunction after MI (Garcia et al., 2019; Qin et al., 2019). It will be important to further explore whether production of SPMs is altered in MI as it is in atherosclerosis, and whether these changes are causal. The pro-resolving proteins, Annexin A1 and Annexin A5 have also been shown to be protective after MI.
After cardiac ischemia/reperfusion, rats treated with Annexin A1 or a peptide derived from the N-terminus of Annexin A1 were found to have less necrosis, less overall inflammation, and improved cardiac function (D'Amico et al., 2000; La et al., 2001; Qin et al., 2019). Similarly, Annexin A5 was also found to limit the post-MI inflammatory response, prevent adverse ventricular remodeling, and improve cardiac function after MI (De Jong et al., 2018).
b. Gaseous transmitters
As in other cardiac diseases, there is a role for the pro-resolving gases in MI. Administration of NO or small-molecule agonists that enhance NO release reduces infarct size in ischemia/reperfusion models of MI (Schulz et al., 2004). However, the role of endogenous NO is more complicated. In an ischemia-reperfusion model of MI, mice lacking eNOS (Nos3−−- ) had similar infarct sizes and cardiac function as control mice (Yang et al., 1999). This may be because eNOS activity peaks in early ischemia but declines with prolonged ischemia as pro-inflammatory cytokines and tissue acidosis drive down eNOS expression (Schulz et al., 2004).
In early ischemia, eNOS-derived NO contributes to decreased cytokine expression leukocyte infiltration to the myocardium, increased vasodilation, and improved contractile function. This same pro-inflammatory milieu that impairs eNOS activity leads to upregulation of iNOS in leukocytes (Schulz et al., 2004). The excessive NO produced by iNOS in ischemia contributes to oxidative stress that promotes apoptosis and necrosis of myocytes, and impairs contractile function (Schulz et al., 2004) and inhibition of iNOS leads to a reduction in infarct size (Wang et al., 1999). Most research on the role of NO has focused on myocyte function and survival, and further studies are needed to determine the impact on cardiac inflammation post-MI.
Treatment of rodents with inhaled CO before induction of ischemia led to reduced infarct area and lower TNF-α expression in tissue (Fujimoto et al., 2004). Use of CO donors before ischemia in rats also led to increased recruitment of c-kit+ progenitor cells to the border of the infarcted area, suggesting that CO promotes repair (Lakkisto et al., 2010). Similarly, administration of H2S or treatment with H2S donors before MI led to a reduction in infarct size and preserved ventricular function (Calvert et al., 2009).
This was attributed to reduced oxidative stress, enhanced resistance of cardiomyocytes to apoptosis, and reduced leukocyte-influx to the region of infarct (Calvert et al., 2009; Snijder et al., 2013). Treatment with H2S donors also led to increased expression of HO-1, suggesting that H2S may also promote the protective effects of CO in this model (Calvert et al., 2009).
3. Future Directions
Given the importance of inflammation in the development of cardiovascular diseases, targeting the inflammatory cascade is desirable. Indeed, in recent years, there have been several clinical trials demonstrating efficacy in treated cardiovascular disease. For example, in patients with known ASCVD, treatment with either an anti-IL-1β antibody or the anti-inflammatory agent colchicine led to a reduction in recurrent clinical cardiac events (Nidorf et al., 2020; Ridker et al., 2017; Tardif et al., 2019). In patients suffering from an acute MI, treatment with anti-IL-1β or anti-IL-6 antibodies led to less inflammation and higher rates of short-term myocardial recovery (Abbate et al., 2020; Broch et al., 2021; Kleveland et al., 2016).
However, these studies have also raised the concern that anti-inflammatory strategies that target host defenses may unwantedly increase the possibility of infectious complications. The MI studies were short term, whereas the studies of ASCVD demonstrated higher rates of pneumonia in patients receiving colchicine and an increased risk of fatal infection in patients receiving IL-1β therapy, respectively (Ridker et al., 2017; Tardif et al., 2019).
Although there were initially concerns about whether augmenting resolution would lead to incomplete clearance of pathogens/injurious stimuli, we now know that specialized pro-resolving mediators not only promote resolution but also augment functions such as phagocytosis and efferocytosis (Chiang et al., 2012). Thus, unlike anti-inflammatory strategies, pro-resolving strategies do not appear to compromise host defense. Therefore, future studies should focus on methods to selectively enhance inflammation resolution over anti-inflammatory strategies alone as a novel therapeutic strategy for the treatment of cardiovascular diseases.
Footnotes
Acknowledgment
The authors wish to thank
Authors' Contributions
A.L.G.: writing—original draft (equal), writing—review and editing (equal); M.M.D.: writing—review and editing (supporting); C.D.S.: writing—review and editing (supporting); M.S.M.: writing—review and editing (supporting); A.C.D.: conceptualization (lead), writing—original draft (equal), writing—review and editing (equal).
Author Disclosure Statement
The authors declare they have nothing to disclose.
Funding Information
This work was supported by an American Heart Association Fellow to Faculty Award 17FTF33660643 (A.C.D.), an American Heart Association Established Investigator Award EIA34480023 (M.S.M.), and NIH grants R01HL159487 (A.C.D.), DP2HL137166 (M.S.M.), R01HL161212 (M.S.M.), F30HL151069 (C.D.S.), T32HL144446 (M.M.D.), and T32AI138932 (A.L.G.).