
Abstract
Aims:
Mitochondrial homeostasis is essential for maintaining redox balance. Besides canonical autophagy, Rab9-dependent alternative autophagy is a crucial mechanism in metabolic cardiomyopathy. Here, we aim to investigate the role of alternative mitophagy and Beclin 1 haploinsufficiency (Beclin 1 +/− ) in high-fat diet (HFD)–induced metabolic cardiomyopathy.
Results:
Twenty-four-week HFD impaired glucose tolerance and cardiomyocyte contraction in wild-type mice, both of which were rescued in Beclin 1 +/− mice. Beclin 1 haploinsufficiency had little effect on the conventional autophagy mediators (ATG5, LC3 II/LC3 I) but further upregulated Rab9 expression, a marker of alternative autophagy, in response to HFD challenge. Furthermore, either the inhibition of alternative autophagy or Beclin 1 haploinsufficiency abolished palmitic acid (PA)-induced cardiomyocyte contractile anomalies. In vitro, PA overactivated mitophagy, resulting in decreased mitochondrial content in H9C2 cells. These aberrations were alleviated in cells deficient in alternative autophagy but not in cells deficient in conventional autophagy. Mechanistically, HFD promoted reactive oxygen species (ROS) production, activated Rab9-dependent alternative mitophagy, and inhibited mitochondrial biosynthesis. Beclin 1 +/− rescued HFD-induced ROS overflow, mitochondrial biogenesis impairment, and prevented Rab9 translocation from the cytoplasm to the mitochondria, thereby inhibiting Rab9-mediated mitophagy overactivation.
Innovation:
For the first time, this study suggests that prolonged alternative mitophagy exacerbates chronic HFD-induced cardiac dysfunction and supports the protective role of Beclin 1 haploinsufficiency in metabolic cardiomyopathy. This provides additional evidence for a target-based pharmacological intervention.
Conclusion:
Beclin 1 haploinsufficiency protects against HFD-induced cardiac dysfunction by inhibiting Rab9-dependent alternative mitophagy and ROS production, while promoting mitochondrial biogenesis. Modulating Beclin 1 expression holds promise in preventing chronic HFD-related cardiomyopathy.

Introduction
Metabolic cardiomyopathy has emerged as a significant contributor to adverse cardiovascular events in individuals with obesity, insulin resistance, and diabetes. This condition manifests initially as impaired diastolic performance, progresses to compromise systolic function, and may ultimately lead to congestive heart failure or even death (Nakamura and Sadoshima, 2020). The pathogenesis of metabolic cardiomyopathy is complex, involving dysregulated lipid metabolism, oxidative stress, mitochondrial dysfunction, and other factors (Chen et al., 2022).
Innovation
Although timely removal of damaged mitochondria through mitophagy can benefit cardiac function; prolonged mitophagy can be detrimental (Sciarretta et al., 2012). We are the first to report that long-term high-fat diet (HFD) consumption leads to the overactivation of alternative mitophagy, resulting in impaired cardiomyocyte contraction. Furthermore, our study demonstrates that heterozygous deletion of Beclin 1 protects against HFD-induced cardiomyopathy by inhibiting Rab9 translocation from the cytoplasm to mitochondria, thereby reducing the excessive mitophagy. This work enhances our understanding of the molecular mechanisms underlying HFD-induced cardiomyopathy and supports the targeting of overactivated alternative mitophagy as a promising therapy for chronic HFD-related cardiomyopathy.
Oxidative stress plays a pivotal role in the development and progression of metabolic cardiomyopathy (Chen et al., 2022). Mitochondria are the primary cellular sources of reactive oxygen species (ROS) in this context (Henri Honka et al., 2021). In the setting of metabolic disorder, disturbances in cardiac substrate metabolism impair mitochondrial function, leading to an overproduction of ROS (Fukushima and Lopaschuk, 2016; Jia et al., 2018). Excessive mitochondria-derived ROS induce insulin resistance, disrupt calcium cycling, and result in the mechanical dysfunction of cardiomyocytes (Aon et al., 2015; Chen et al., 2022; Tocchetti et al., 2012).
Excessive ROS production also triggers mitochondrial injury, consequently activating mitophagy, a process in which damaged or dysfunctional mitochondria are specifically targeted for degradation and removal. This selective form of macroautophagy is indispensable for maintaining mitochondrial health (Ma et al., 2020). In addition to the well-established conventional autophagy pathway, dependent on autophagy-related genes 5 and 7 (Atg5/7), an alternative pathway emerged in 2009 involving Rab9. This Rab9-dependent alternative pathway orchestrates the fusion of isolation membranes with vesicles derived from the trans-Golgi and late endosomes, providing an additional layer of complexity to the regulation of mitochondrial quality control (Nishida et al., 2009). Both conventional and alternative processes lead to the bulk degradation of cellular proteins, yet they are regulated by distinct autophagic proteins, utilize membranes from different sources, and serve different physiological functions (Nishida et al., 2009).
Mitophagy plays an essential role in maintaining mitochondrial function and redox balance (Aon and Foster, 2015). A prevailing theory proposed mitophagy as a survival mechanism that allows cells cull defective mitochondria to preserve redox environment, compensate energy demand, and maintain cell viability under stress (Hoshino et al., 2013). However, maladaptive mitophagy can also exacerbate pathological conditions by targeting healthy mitochondria rather than injured mitochondria or triggering cell death mechanisms (Wang et al., 2021). Whether mitophagy exerts a protective or injurious role depends on various factors, including cellular homeostasis, stimulus intensity, extent, and duration (Eisenberg-Lerner et al., 2009; Hamacher-Brady et al., 2006; Matsui et al., 2007). Recent study by Saito et al. (2019) demonstrated that mitophagy executed via Rab9-associated autophagosomes plays a protective role in myocardial ischemia. However, the role of Rab9-associated autophagosomes in the mitochondria homeostasis in the context of long-term high-fat diet (HFD)–induced metabolic cardiomyopathy remains largely unexplored.
Beclin 1, a critical protein involved in early autophagosome formation, plays a pivotal role in both conventional and alternative autophagy pathways (Ao et al., 2014; Juenemann and Reits, 2012). Taking advantage of a Beclin 1 haploinsufficiency (Beclin 1 +/−) mouse model, our study aims to elucidate the cellular and molecular mechanisms of alternative mitophagy in the context of metabolic cardiomyopathy induced by long-term HFD. We hypothesized that (i) HFD activates alternative mitophagy and induces an overflow of ROS, consequently reducing the number of functional mitochondria and impairing myocardial function. (ii) Heterozygous deletion of Beclin 1 confers protection against HFD-induced cardiomyopathy likely through alternative mitophagy rather than the conventional form. (iii) Beclin 1 mitigates overactivated alternative mitophagy and ROS production, preserving cardiac mitochondrial function, and exerting a protective role in HFD-induced metabolic cardiomyopathy.
Results
Beclin 1 haploinsufficiency alleviates 24-week HFD-induced insulin resistance and cardiac contractile impairment
To investigate the potential impact of Beclin 1 in HFD-induced cardiomyopathy, we subjected both wild-type (WT) and Beclin 1 +/− mice to either a 24-week HFD or low-fat diet (LFD). As shown in Figure 1, in both strains, mice fed a HFD exhibited reduced daily food intake, and gained more weight over time. Beclin 1 haploinsufficiency itself exerted minimal effect on food intake and mouse body weight (Fig. 1A, B). Furthermore, glucose tolerance test revealed elevated glucose levels and increased area under the curve in response to HFD, the effects of which were mitigated by Beclin 1 haploinsufficiency. This suggests that Beclin 1 +/− offers protection against HFD-induced insulin resistance (Fig. 1C, D).
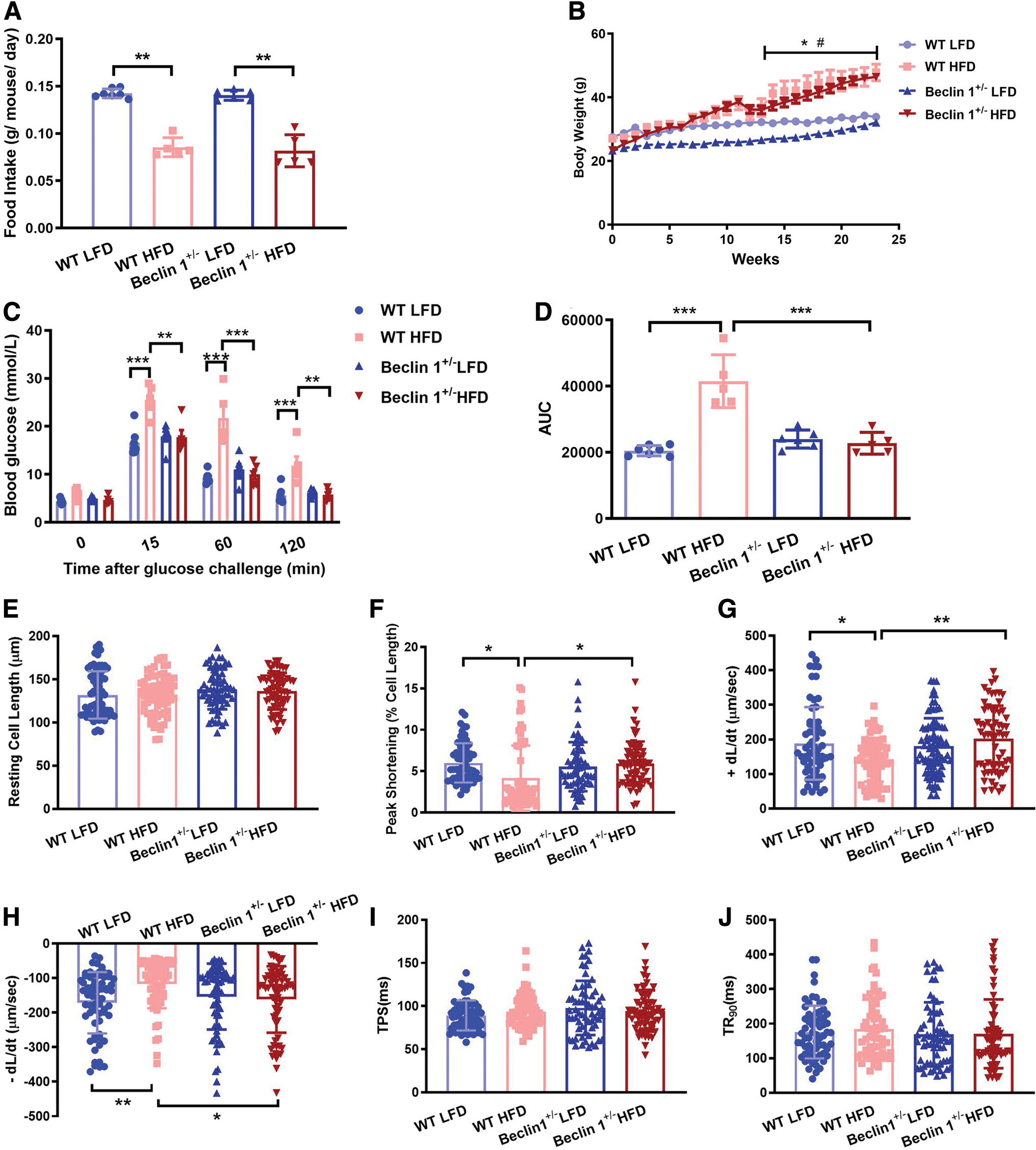
We also assessed the influence of Beclin 1 haploinsufficiency on myocardial contractility. Cardiomyocytes were isolated from both WT and Beclin 1 +/− mice, and their mechanical properties were evaluated. HFD consumption notably decreased peak shortening and maximal velocity of shortening/relengthening while leaving other parameters, such as resting cell length and time to peak shortening, largely unaffected. Beclin 1 haploinsufficiency effectively reversed the mechanical abnormalities induced by HFD without causing any significant effects on its own (Fig. 1E–J). These results indicated that Beclin 1 haploinsufficiency exerted a protective role against HFD-induced insulin resistance and preserves cardiomyocyte contractile properties during metabolic stress.
Influence of Beclin 1 haploinsufficiency on autophagic levels in metabolic stressed heart after 24-week HFD
Dysregulation of autophagy in cardiomyocytes is implicated in various cardiovascular diseases such as myocardial ischemia and heart failure (Dewanjee et al., 2021). Beclin 1, a core component of the autophagy machinery, plays a cardinal role in regulating autophagy and survival of cardiomyocytes (Maejima et al., 2016). To unravel the mechanism behind the cardioprotective responses induced by HFD and Beclin 1 insufficiency, conventional and Rab9-dependent alternative autophagy levels were determined in WT and Beclin 1 +/− mice after 24-week HFD consumption.
As shown in Figure 2, Beclin 1 protein expression was decreased after Beclin 1 heterozygous deletion (Beclin 1 +/−-LFD) and in WT mice subjected to HFD (WT-HFD), compared with WT-LFD mice (Fig. 2B). HFD had a minimal impact on ATG5 and p62 expression and the LC3 II/I ratio but increased Rab9 protein levels in comparison with WT. In addition, we observed similar levels of LC3 II/I and ATG5 between WT and Beclin 1 +/− mice under HFD intake (Fig. 2C–E), suggesting a limited role for LC3 II/I and ATG5-related conventional autophagy in the protection offered by Beclin 1 +/− against HFD-induced insulin resistance and cardiac dysfunction.

Rab9 is a key molecular factor in alternative autophagy responsible for sequestering and degrading damaged organelles without affecting general autophagy (Barbero et al., 2002). Our immunoblot data showed accumulated Rab9 protein in the heart of WT-HFD, and this effect was further intensified by Beclin 1 +/− (Fig. 2F). These findings suggest that Beclin 1 +/− may exert a cardioprotective role, possibly through Rab9-dependent alternative autophagy, as opposed to LC3 II/I and ATG5-associated conventional autophagy.
Alternative autophagic inhibitor alleviates palmitic acid-induced cardiomyocyte contractile impairment
Bafilomycin A1 (BAF) inhibits total autophagy by blocking the fusion of autophagic vacuoles with lysosomes (Mizushima and Yoshimori, 2007). Brefeldin A1 (Bref) is an alternative mitophagy inhibitor that disrupts the Golgi apparatus and autophagosome formation (Nishida et al., 2009). Both BAF and Bref were employed to precisely assess the role of alternative autophagy in WT and Beclin 1 +/− cardiomyocyte contractile properties with palmitic acid (PA) to mimic HFD in vitro.
Our results revealed that PA treatment significantly impaired cardiomyocyte contractile function, evidenced by reduced peak shortening, and maximal velocity of shortening/relengthening compared with WT group. Interestingly, we observed the rescue of PA-induced cardiomyocyte contractile anomalies not only in the autophagy-inhibited group (BAF) but also in the group with alternative autophagy inhibition (Bref) (Fig. 3B–D). These findings emphasize that inhibiting alternative autophagy yields responses similar to total autophagy interruption. Because HFD did not affect conventional autophagy levels (ATG5 and LC3 II/I) but led to an increase in Rab9 protein expression (Fig. 2), the observed cardiac dysfunction induced by PA/HFD might be attributed to the activated alternative autophagy. Furthermore, we are surprised to find that PA-induced cardiomyocytes contractility impairments were abolished in cardiomyocytes from Beclin 1 +/− mice (Fig. 3). Those findings suggest a protective role for alternative autophagy inhibition and Beclin 1 haploinsufficiency in PA-induced cardiomyocyte contraction impairment, with the underlying mechanism remaining unclear.

PA reduces mitochondrial content in a Rab9-dependent manner
To further explore the molecular mechanism of conventional and especially alternative autophagy in HFD-induced metabolic cardiomyopathy, short-hairpin RNA (shRNA) was used to generated cell lines deficient in conventional autophagy (shATG5) and alternative autophagy (shRab9). The knockdown efficiency of ATG5 and Rab9 protein expression is shown in Supplementary Figure S1.
Given the crucial role of mitochondria in ATP production, calcium handling, and redox homeostasis in cardiomyocytes (Aon et al., 2015), transmission electron microscopy was next used to observe mitochondrial morphology and autophagosomes in H9C2, shATG5 or shRab9 cells treated with or without PA. We also utilized BAF to monitor autophagy flux, reflecting the quantity of autophagic vacuoles delivered to and degraded in the lysosome (Mizushima and Yoshimori, 2007).
In general, mitochondria were arranged normally in the cytoplasm, and BAF treatment increased numbers of autophagic vacuoles in the control group. However, mitochondria swelling, vacuolation, or shrinkage were evidenced under PA challenge, and some dysfunctional mitochondria were engulfed and digested in the vacuoles (Fig. 4A). Notably, compared with the BAF-treated group, damaged mitochondria and autolysosomes were significantly increased in the BAF plus PA stimulation group, in both WT, shATG5, and shRab9 cells (Fig. 4B, C).

COX IV is a protein that is an integral part of the mitochondrial electron transport chain, and its presence is indicative of the mitochondria mass (Hancock et al., 2008; Maj et al., 2011). Our results revealed reduced mitochondrial content, as evidenced by lower COX IV expression, in the PA or PA plus BAF groups compared with the control or only BAF groups in WT and shATG5 H9C2 cells; however, a similar amount of COX IV expression was observed after PA stimulation in shRab9 cells (Fig. 4D–F). In summary, PA induces mitochondrial dysfunction, and reduced mitochondrial content in WT and shATG5 H9C2 cells. Intriguingly, while PA also impairs mitochondria in shRab9 cells, the decrease in mitochondrial content observed in response to PA is abolished in shRab9 H9C2 cells.
PA stimulates Rab9-dependent mitophagy
To assess mitophagy levels more directly after PA stimulation, we first transfected cells with a pH-sensitive mt-Keima adenovirus. Red puncta indicate damaged or dysfunctional mitochondria undergoing mitophagy in an acidic environment (lysosome), while green puncta represent healthy mitochondria in a neutral environment, such as the mitochondrial matrix (Katayama et al., 2011). As shown in Figure 5, PA stimulation increased the red mt-Keima fluorescence and reduced the green mt-Keima fluorescence in both H9C2 and shATG5 cell lines, indicating PA activated mitophagy flux and decreased mitochondrial content (CTR vs. PA or BAF vs. PA+BAF) (Fig. 5A, B). However, PA failed to trigger more red fluorescence while preserving green fluorescence in alternative autophagy deficiency cells (shRab9 cell line) (Fig. 5C). These results indicate that inhibiting alternative autophagy could attenuate PA-induced mitophagy activation and maintain mitochondrial content.
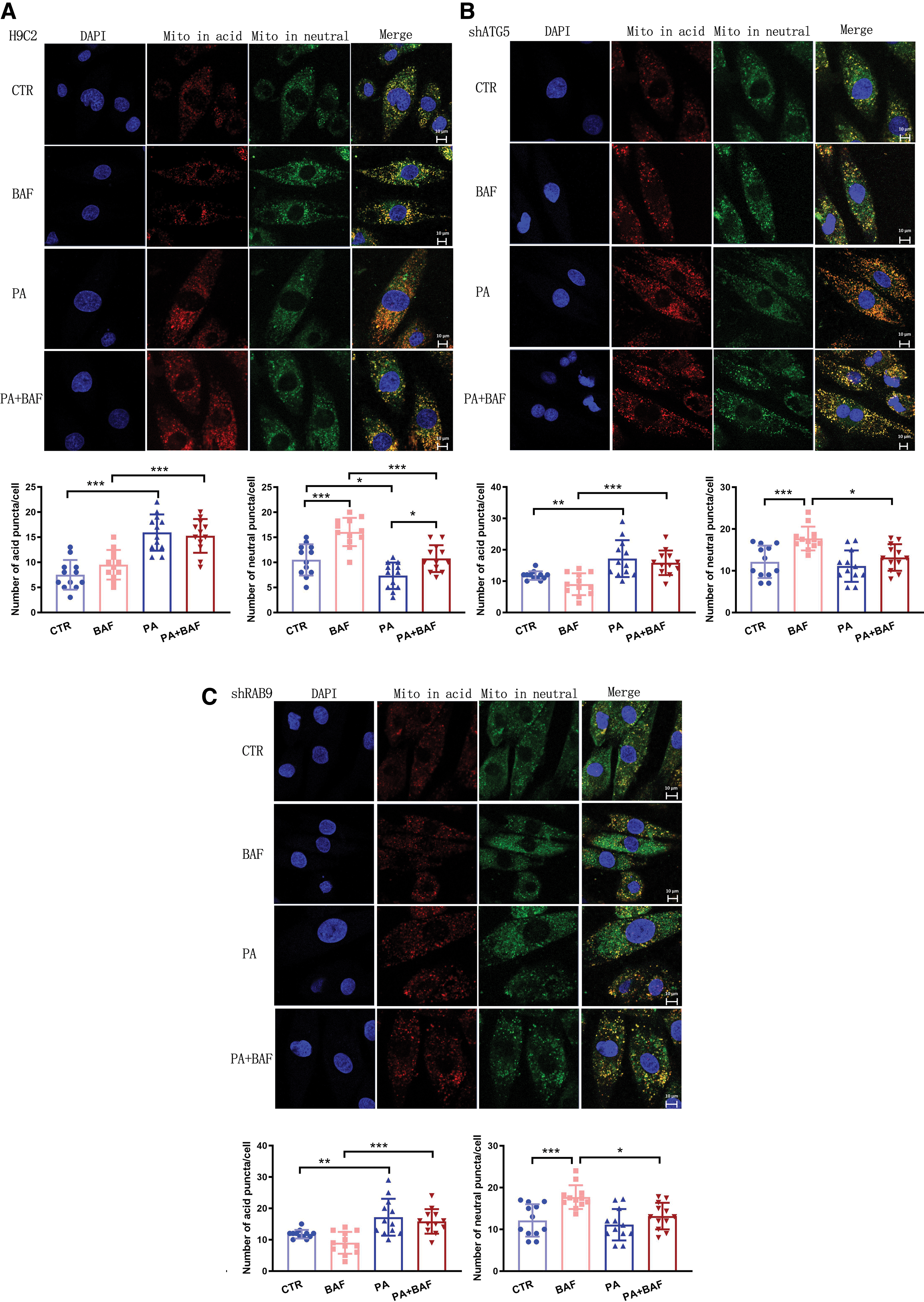
We then isolated mitochondrial and cytoplasmic lysates and total protein from H9C2, shATG5, and shRab9 cells with or without PA challenge. In Figure 6A, the combination of PA plus BAF showed lower levels of mitochondrial Beclin 1, ATG5, and LC3 II/I compared with the BAF-only group, indicating PA suppressed conventional autophagy-related mitochondrial targeting. However, PA stimulation did not increase but instead decreased p62 expression, indicative of enhanced autophagy flux. Typically, p62 accumulates when autophagy is disrupted. Therefore, the reduction in p62 levels might be attributed to Rab9-dependent alternative autophagy, as evidenced by the increased accumulation of mitochondrial Rab9 in the PA plus BAF group compared with the BAF group. This underscores the crucial role of alternative mitophagy in the PA-induced disruption of cardiomyocytes.
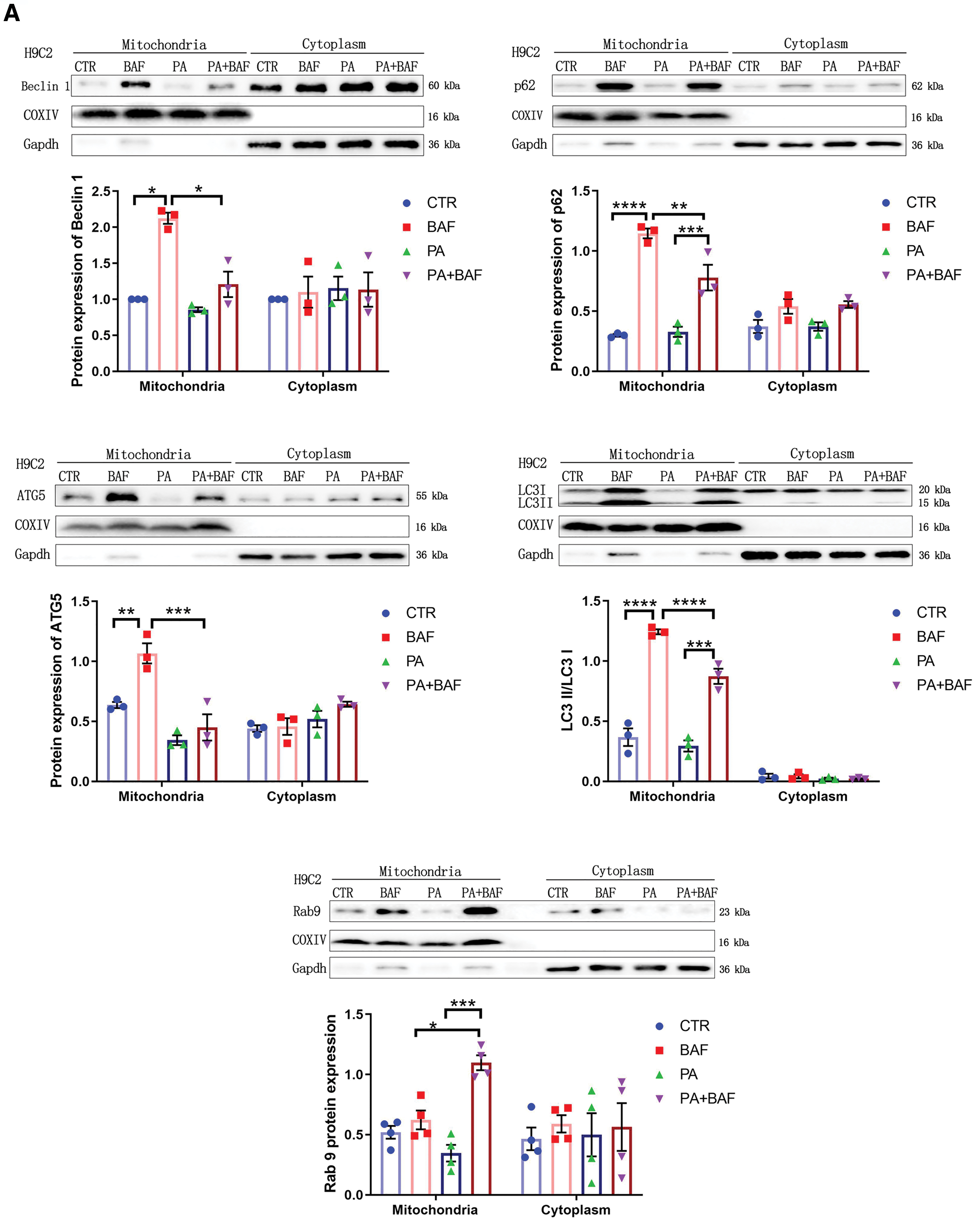


We repeated these immunoblots in shATG5 (Fig. 6B) and shRab9 cells (Fig. 6C). In shATG5 cells, Beclin 1, LC3 II/I, p62, and Rab9 changes mirrored those in H9C2 cells. In shRab9 cells, despite preserved decreased conventional autophagy, p62 increased in the PA group and exhibited a trend to further increase in the BAF plus PA treatment, indicating the abolishment of the increased autophagy flux after PA stimulation upon alternative autophagy suppression. Similar patterns were observed in the total protein fraction between groups (Supplementary Fig. S2). Taken together, these results support the notion that PA-induced changes in autophagy flux occur in Rab9-dependent manner.
Beclin 1 haploinsufficiency attenuates 24-week-HFD intake–induced mitochondrial biogenesis impairment
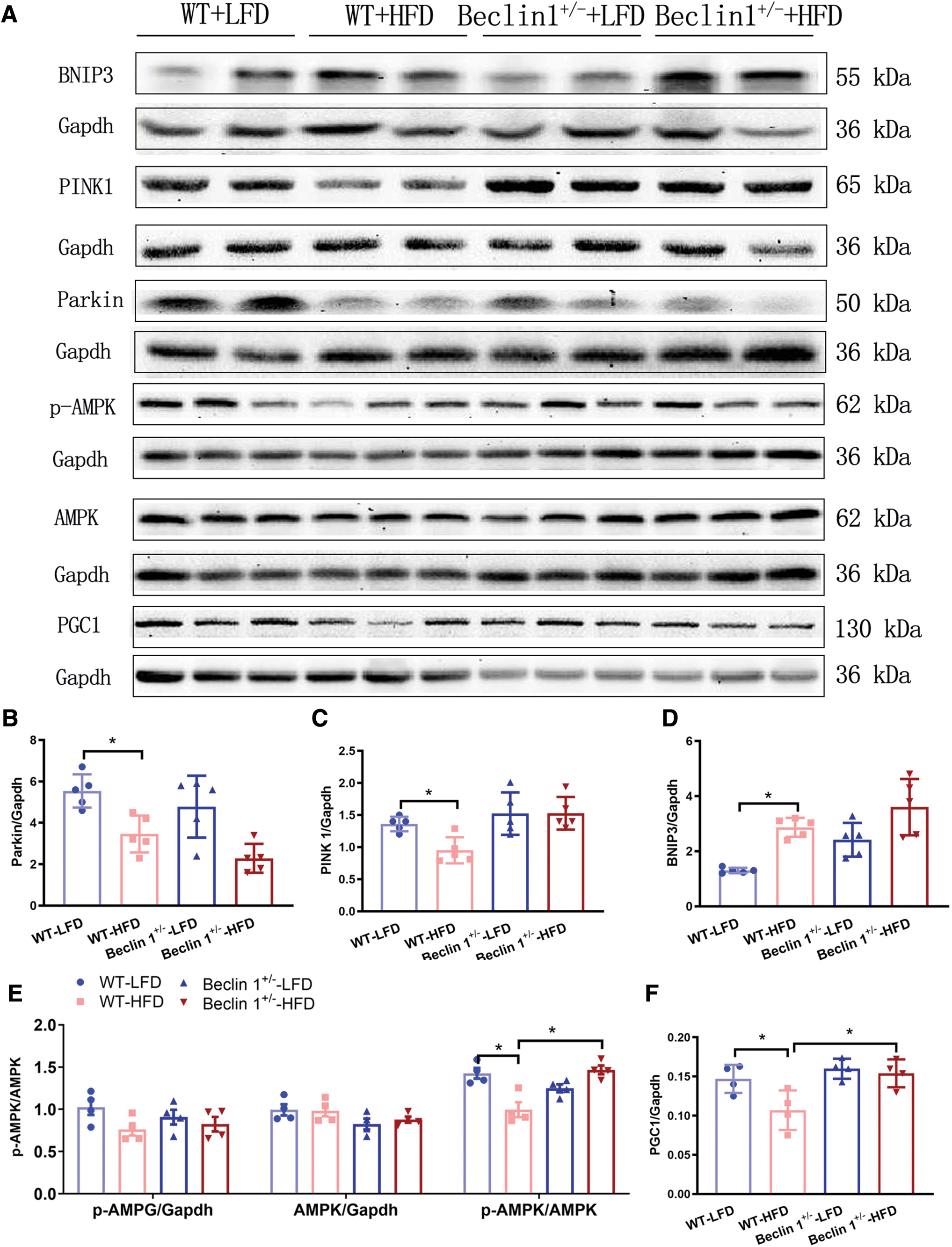
We have previously demonstrated that HFD/PA primarily activates Rab9-dependent alternative mitophagy (Figs. 5 and 6), and the cardioprotective effect of Beclin 1 deficiency is mediated through Rab9-dependent alternative mitophagy (Fig. 2). To further expand our understanding, we investigated the potential involvement of mitophagy mediators such as PINK1/Parkin and BNIP3, alongside markers of mitochondrial biogenesis including PGC1 and the p-AMPK/AMPK ratio.
As shown in Figure 7, the immunoblot analysis revealed that chronic HFD feeding led to a decrease in PINK1/Parkin protein expression while increasing BNIP3 levels when compared with the WT group (Fig. 7B–D). Notably, Beclin 1 haploinsufficiency had minimal impact on the expressions of PINK1, Parkin, and BNIP3 after HFD treatment, suggesting that Beclin 1 insufficiency possibly exerts limited influence on conventional mitophagy. Furthermore, our investigation unveiled a reduction in markers associated with mitochondrial biogenesis, specifically PCG1 and the p-AMPK/AMPK ratio, after a 24-week HFD challenge. Interestingly, this effect was reversed in Beclin 1 +/− hearts (Fig. 7E, F). These results indicate that Beclin 1 insufficiency-induced cardioprotective effects may, in part, be attributed to the attenuation of HFD-induced mitochondrial biogenesis impairment.
Beclin 1 haploinsufficiency inhibited Rab9 translocation from cytoplasm to mitochondria
To unravel the mechanism behind Rab9-dependent mitophagy in Beclin 1 haploinsufficiency-mediated cardioprotection, we isolated cardiomyocytes from both WT and Beclin 1 +/− mice, exposing them to either PA or PA plus Bref. Subsequently, mitochondrial and cytoplasmic lysates were separated for Western blotting. In the WT group, PA treatment activated the Rab9-dependent mitophagy pathway, evident from the decreased COX IV/TOM 20 ratio (indicating increased mitophagy) and an upsurge in Rab9 protein fold within the mitochondrial fraction (Fig. 8A, B, D). These results indicated that Rab9 was primarily located in the mitochondrial fraction in WT mice. However, Beclin 1 haploinsufficiency prevented Rab9 from translocating from the cytoplasm to the mitochondria, as evidenced by the robust expression of Rab9 in the cytosolic fraction. Our results also revealed that PA failed to increase Rab9 expression in the mitochondrial fraction of Beclin 1 haploinsufficient mice, in contrast to what was observed in the WT group (Fig. 8A, C). These intriguing findings suggest that Beclin 1 haploinsufficiency may prevent HFD-induced cardiac dysfunction by inhibiting the translocation of Rab9 from the cytoplasm to the mitochondria to initiate mitophagy. However, the exact mechanism requires further exploration.

Beclin 1 haploinsufficiency mitigates PA-induced mitochondrial ROS production
Basal mitochondrial ROS production is essential for various physiological processes, while excessive ROS levels can lead to oxidative stress, impair mitochondrial function, the initiation of mitophagy, and cardiac dysfunction (Chen et al., 2022). We subsequently investigated the role of mitochondrial ROS in Beclin 1 insufficiency-mediated cardiac protection.
In Figure 9A, small-interfering RNA (siRNA)-Beclin 1–1 (siBCN) exhibited the highest knockdown efficiency after 48 h compared with siRNA-NC (siNC). Thus, we chose Beclin 1 siRNA-1 (siBCN1) to modulate Beclin 1 expression for the following experiment. H9C2 cells transfected with siBCN1 were further subjected to PA treatment or left untreated. The Mito-Sox results revealed a significant increase in mitochondrial ROS production after PA treatment. Notably, Beclin 1 haploinsufficiency attenuated PA-induced mitochondrial ROS overflow, potentially contributing to the alleviation of mitophagy and the improvement of mechanical dysfunction and insulin resistance (Fig. 9B, C). The schematic diagram in the graphical abstract illustrates potential mechanisms involving Beclin 1 +/− in protecting against HFD-induced cardiac anomalies.

Discussion
In this study, we demonstrated that prolonged 24-week HFD consumption increased mitochondrial ROS levels, resulting in insulin resistance, cardiac mechanical dysfunction, and activated alternative mitophagy in WT mice. Inhibiting alternative autophagy alleviated HFD-induced cardiomyocyte contraction dysfunction. Mechanistically, PA treatment triggered Rab9-dependent mitophagy, resulting in reduced mitochondrial content and contributing to impaired cardiac function. Beclin 1 haploinsufficiency alleviated HFD-induced alternative mitophagy activation and mitochondrial ROS production, leading to improved cardiac mechanical function. Further analysis revealed that Beclin 1 haploinsufficiency prevented HFD-induced cardiac dysfunction by inhibiting the translocation of Rab9 from the cytoplasm to the mitochondria. This action avoided the overactivation of Rab9-dependent mitochondria depletion in metabolic cardiomyopathy. In addition, Beclin 1 deficiency rescued HFD-induced mitochondrial biogenesis impairment. In summary, our findings enhance our understanding of mitophagy in HFD-induced cardiomyopathy, providing further evidence for potential target-based pharmacological interventions.
Mitochondria play a crucial role in regulating the balance of ROS production and scavenging to maintain signaling-compatible ROS levels (Aon et al., 2015; Li et al., 2015). Beclin 1's protective role in metabolic cardiomyopathy stems from its ability to counter HFD-induced ROS, preventing further mitochondrial damage and cardiac injury (Schönfeld and Wojtczak, 2008). The bursts of mitochondria-derived ROS disrupt cardiac electrocontraction coupling, compromise energetics, and perturb calcium cycling, ultimately resulting in mechanical dysfunction in cardiomyocytes (Tocchetti et al., 2012; Li et al., 2015). The excess of ROS production also activates various serine kinases that phosphorylate insulin receptor substrates proteins, ultimately leading to insulin resistance (Morino et al., 2005). In our study, Beclin 1 haploinsufficiency prevented PA-induced mitochondrial ROS production, thus alleviating ROS-induced mechanical dysfunction and insulin resistance in cardiomyocytes. Given that mitochondria are both sources and target of oxidative stress, Beclin 1 haploinsufficiency mitigated PA-induced ROS production preventing additional ROS-induced mitochondrial damage and averting cardiac injury (Aon et al., 2014).
Excessive mitochondrial ROS production can induce mitochondrial damage and activate mitophagy (Aon et al., 2014). As shown in Figure 5, PA treatment activates Rab9-dependent mitophagy. Notably, Beclin 1 haploinsufficiency, despite not altering BNIP3, PINK1, or Parkin protein expression, effectively counteracts the overactivated Rab9-dependent mitophagy induced by prolonged HFD challenge, thereby preserving mitochondrial numbers (Fig. 8). To restore mitochondrial content, mitochondrial biogenesis is pivotal in generating new and healthy mitochondria. Our results also demonstrate that Beclin 1 haploinsufficiency rescues HFD-induced suppression of p-AMPK and PGC1 during HFD treatment (Fig. 7). Hence, it is the combined reduction in mitophagy and enhancement of mitochondrial biogenesis that underpins the cardioprotective role of Beclin 1 haploinsufficiency under HFD conditions. In response to the prolonged HFD-induced overactivation of Rab9-related mitophagy, leading to a decrease in mitochondrial content, Beclin-1 deficiency upregulates PGC1 to rejuvenate mitochondrial function and promote mitochondrial biogenesis.
The cellular and molecular mechanisms of mitophagy in HFD-induced cardiomyopathy are complex. Conventional mitophagy initiated in mice during the early stages of HFD, while alternative mitophagy was maintained in more advanced disease stages (Jihoon Nah et al., 2022). It remains largely unknown whether these mechanisms are independently controlled and the conditions under which adaptive mitophagy turns maladaptive, contributing to cardiac dysfunction (Rabinovich-Nikitin et al., 2019). A study by Tan et al., 2021 demonstrated that short-term (8 weeks) HFD feeding, but not long-term (16 weeks) feeding, protects against pressure overload-induced heart failure through mitophagy activation. Moreover, Tong et al., 2021 found that conventional mitophagy activation peaks at ∼6 to 8 weeks of HFD consumption and decreases over time. In contrast, a Rab9-dependent alternative mitophagy pathway is activated after 3 weeks of HFD consumption and continues to increase for ∼24 weeks. Their study also showed that transgenic mice with cardiac-specific overexpression of Rab9 are protected from cardiac dysfunction induced by 20 weeks of HFD consumption, as evidenced by reduced left ventricular hypertrophy and improved left ventricular function (Rabinovich-Nikitin et al., 2021; Tong et al., 2021).
In our study, we observed suppressed conventional autophagy and activation of Rab9-dependent alternative mitophagy after 24 week-HFD intake. However, our results demonstrated that PA treatment impaired cardiomyocyte contractile properties in cells isolated from WT mice. This impairment was rescued by an inhibitor of alternative mitophagy, suggesting a detrimental role of Rab9-dependent alternative mitophagy in metabolic cardiomyopathy. The differences in HFD consumption duration may help explain the variations between our findings and those of Tong's work. Chronic mitophagy activation can reduce the number of functional mitochondria, potentially compromising cell function (Kobayashi et al., 2012). During the early stages of HFD consumption, increased alternative mitophagy eliminates damaged and dysfunctional mitochondria, exerting prosurvival effects. However, prolonged alternative mitophagy may trigger the removal of normal mitochondria, leading to a decrease in mitochondria numbers, and reduce energy efficiency, and even cardiac atrophy (He et al., 2013; Mellor et al., 2011; Ouyang et al., 2014; Zou and Xie, 2013). As reported, cardiomyocyte apoptosis typically occurs at the later stages of cardiomyopathy after prolonged hyperglycemia in metabolic patients, resulting in cardiac contractile dysfunction and remodeling due to cell loss (Hu et al., 2017; Joubert et al., 2019). Therefore, the functional role of alternative mitophagy in metabolic cardiomyopathy must be assessed in the specific context, and the mechanisms governing the transition between its beneficial to harmful roles are yet to be clearly understood.
Rab9, primarily localized in late endosomes, facilitates vesicle transportation from endosomes to the trans-Golgi (Barbero et al., 2002). Energy stress triggers the formation of a multiprotein complex, including Ulk1, Rab9, Rip1, and Drp1, which recruits trans-Golgi membranes to damaged mitochondria, initiating alternative mitophagy (Sadoshima, 2022). Previous studies have shown that Rab9 silencing inhibits autophagosome maturation, reducing the number of autophagic vacuoles while increasing the accumulation of isolated membranes in etoposide-treated ATG5 knockout cells. In our study, we observed swelling, vacuolar degeneration, and increased mitochondrial density under PA challenge in shRab9 cells. However, PA-induced mitochondrial content decrease in H9C2 and shATG5 cells was reversed after Rab9 knockdown. It is important to note that Rab9's function is influenced not only by its expression but also by its spatial distribution. In cases of Niemann-Pick disease type C, a neurodegenerative disease, Rab9 is expressed at normal levels in cellular and mouse models. However, large amounts of Rab9 are trapped in a dephosphorylated form on intermediate filament vimentin, rendering it unable to function properly. This leads to the impairment of mannose 6-phosphate receptors' transport out of late endosomes, resulting in vesicle transport disruption and cell death (Ganley and Pfeffer, 2006; Ng et al., 2012).
Our immunoblot data revealed an increase in Rab9 protein levels in the hearts of WT-HFD mice, and this effect was further amplified in Beclin 1 haploinsufficient mice during HFD consumption (Fig. 2F). The heightened Rab9 protein levels in Beclin 1 +/− hearts may be attributed to Beclin 1 haploinsufficiency preventing Rab9 translocation from the cytoplasm to impaired mitochondria, which, in turn, stimulates Rab9 transcription, synthesis, and accumulation in the cytoplasm in an inactive state. Although further investigation is needed to delve into the specific mechanism, our findings provide new insights into the role of alternative mitophagy in metabolic cardiomyopathy and propose an innovative strategy for developing novel therapeutic agents targeting Rab9 translocation in this condition.
Several experimental limitations should be considered in this study. First, we exclusively assessed alternative mitophagy activity after 24 weeks of HFD consumption. Given the distinct roles of mitophagy after long-term and short-term HFD consumption, as shown in our study and Tong's work, further investigations are necessary to elucidate the mechanisms responsible for the transition between the beneficial and deleterious roles of alternative mitophagy in the context of HFD consumption. Second, our study relied solely on Bref to suppress alternative mitophagy in isolated cardiomyocytes from WT and Beclin 1 +/− mice. Additional interventions, such as the use of genetically altered mouse models, are required to further validate our observations. Third, although we observed that Beclin 1 +/− inhibited Rab9 dislocation, preventing overactivated mitophagy and mitigating HFD-induced cardiac dysfunction, further investigations into the molecular mechanisms are needed to identify more specific intervention targets for modulating Rab9-dependent alternative mitophagy.
In summary, this analysis revealed that 24-weeks HFD consumption overactivated Rab9-dependent alternative mitophagy and overwhelmed ROS production, and interrupted mitochondrial biogenesis, leading to impaired insulin sensitivity and cardiac contractile function. Heterozygous deletion of Beclin 1 helps mitigate ROS overflow, suppresses the overactivation of alternative mitophagy, and enhances mitochondrial biogenesis, thereby offering protection against HFD-induced metabolic cardiomyopathy.
Materials and Methods
Animal model
Adult male C57BL/6 mice (WT, 12 weeks old) and heterozygous Beclin 1 deletion mice (C57BL/6J background, Beclin 1 +/−) were used in this study. Beclin 1 +/− mice were kindly provided by Prof. Zhenyu Yue from Mount Sinai School of Medicine with heterozygous deletion of Beclin 1 as described (Liu et al., 2022). Mouse DNA was extracted from tails, and genotyping was performed using primers as follows: forward: 5′-TGG AGG GCA GTC CAT ACC CTG G-3′, reverse: 5′-CGC CTT CTA TCG CCT TCT TGA CGA GTT-3′. WT and Beclin 1 +/− mice were fed HFD (60% kcal diet, HFD) for 24 weeks to induce metabolic cardiomyopathy. A control group of mice matched for age and gender were fed standard chow (10% kcal diet, LFD) for the same period of time. Mice were housed under a standard temperature-controlled environment with a 12 h light/dark cycle, and were given food and water ad libitum. Food intake and body weight were monitored daily until the day of sacrifice. Animal experimental protocols were performed according to the guidelines of “The Guide for the Care and Use of Laboratory Animals” Academy Press (NIH Publication No 85-23, revised 1996), and were approved by the Institutional Animal Care and Use Committee of Central South University.
Intraperitoneal glucose tolerance test
After 24 weeks, mice underwent an intraperitoneal glucose tolerance test, following the procedure described (Palleria et al., 2017). Mice were intraperitoneally injected with 2 g of glucose per kilogram of body weight after a 12-h fast. Blood samples were collected from tail veins immediately before (time 0) and at 15, 60, and 120 min after the injection, using an Accu-Chek III automatic glucometer (Roche Diagnostics, Inc., Indianapolis, IN). The area under the curve was calculated using the trapezoid method (Allison et al., 1995).
PA preparation
To simulate a HFD in vitro, we prepared a PA solution (150 μM, P0500; Sigma Aldrich, St. Louis, MO) using a 10% bovine serum albumin (BSA) solution. In brief, we placed 25.6 mg of PA powder in a tube heater at 70°C. Once melted, PA was dissolved in a 0.1 M NaOH solution to achieve a final concentration of 100 mM at 70°C. Subsequently, the PA solution was further diluted with a 10% fatty acid-free BSA (in ddH2O) to a concentration of 5 mM at 55°C for 10 min. The PA/BSA solution was then cooled to room temperature, filtered through a 0.22 μm pore membrane filter, aliquoted, and stored at −80°C until use.
Cardiomyocyte isolation
Murine ventricular cardiomyocytes were enzymatically isolated as described (Liu et al., 2022; Sun et al., 2014) after 24-weeks HFD/LFD consumption. After ketamine/xylazine sedation via intraperitoneal injection, hearts from WT or Beclin 1 +/− mice were rapidly removed and perfused with the Krebs–Henseleit bicarbonate buffer containing the following components (in mM): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES, and 11.1 glucose. This was followed by collagenase D (Roche Diagnostics, Inc.) digestion for 20 min. The left ventricles were excised and cut into 1-mm pieces. Single cells were obtained through mechanical titration, and the cell suspension was centrifuged at 30 g for 45 s. The cell pellet was then resuspended in prewarmed Medium 199 (11150067; Gibco, Grand Island, NY). Cardiomyocyte yield was ∼75%. Only calcium-tolerant, quiescent, rod-shaped myocytes displaying clear cross striations were used for further experiments. For specific experiments, the isolated cardiomyocytes were challenged with PA in the absence or presence of BAF (50 nM, HY-100558; MedChemExpress, Monmouth Junction, NJ), or Bref (3 μg/mL, 00-4506; eBioscience, San Diego, CA) for 4 h, and cell proteins were collected for Western blot analysis (Hsu et al., 2018).
Measurements of cardiomyocytes contractile properties
Cardiomyocyte contractile properties were assessed at room temperature using the SoftEdgeMyoCam® system (IonOptix Incorporation, Milton, MA) after the isolation of ventricular cardiomyocytes, as outlined in Sun et al. (2014). The isolated cells were positioned in a chamber with a glass coverslip base, mounted on an inverted microscope (IX-70; Olympus, Tokyo, Japan). The chamber was perfused with the Krebs–Henseleit bicarbonate buffer containing 1 mM CaCl2. Myocytes were stimulated using a pair of platinum wires placed on opposite sides of the chamber and connected to an FHC stimulator, delivering suprathreshold voltage at a frequency of 0.5 Hz with a duration of 3 ms. Cell visualization was facilitated through an IonOptixMyoCam camera, and cardiomyocyte mechanical properties, including resting cell length, peak shortening, time to peak shortening, time to 90% relengthening, and maximum velocities of shortening/relengthening, were measured (Aberle et al., 2004). The data obtained from 8–16 consecutive contractions were averaged for analysis.
H9C2 cell culture
Rat cardiomyocyte-derived cell line H9C2, purchased from Cell Bank of Chinese Scientific Academy (Shanghai, China), was cultured in high-glucose Dulbecco's modified Eagle's medium (11965092; Gibco) supplemented with 10% fetal bovine serum (FBS; SFBS; Bovogen, Melbourne, Australia), and 1% Penicillin/Streptomycin (P4333; Sigma Aldrich) in humidified environment at 37°C with 5% CO2. When reaching 70%–80% confluence, cultured cells were left untreated or treated with 400 μM PA for 24 h. Fifty nanomolars BAF was added for 1 h before the addition of 400 μM PA, and the cells were subjected to incubation for 24 h to regulate autophagy (Hsu et al., 2018). Cells were harvested for analysis following specified treatment.
Lentivirus transfection
For RNA interference, recombinant lentivirus vectors containing shRNA sequences targeting ATG5 (shATG5, shRAN 5′-AGAAGATGTTAGTGAGATT-3′) and RAB9 (shRab9, 5′CCGAAGAGTTCTCTTATGAACAGATTCAAGAGATCT GTTCATAAGAGAACTCTTTTTTT-3′) were obtained from Wanlei Technology (Shengyang, China). A scrambled shRNA was used as a control. H9C2 cells (1 × 106/well) were seeded into a 12-well plate with Dulbecco's modified Eagle's medium containing 10% FBS and grown to 50%–60% confluence. Cell lines with deficiencies in conventional autophagy and alternative autophagy were created by transfecting H9C2 cells with lentiviral vectors carrying ATG5 or Rab9 shRNA, following the manufacturer's instructions with a multiplicity of infection of 100. After 72 h of transfection, the cells were harvested to assess the expression of specific genes or for subsequent experiments.
siRNA transfection
Beclin 1 siRNAs were obtained from Ribo Biological Company (Guangzhou, China). H9C2 cells were seeded into six-well plates. When the cells reached ∼50% confluence, transfection was performed using a transfection kit purchased from Ribo (C10511-05, Guangzhou, China), following the manufacturer's instructions. The oligonucleotide sequences of siRNAs were as follows: siNC 5′-TTCTCCGAACGTGTCACGTdTdT-3′; siRNA-Beclin 1–1 5′-CAATAAGATGGGTCTGAAA-3′; siRNA-Beclin 1–2 5′CAGCGAGAATATAGTGAAT-3′; siRNA-Beclin 1–3 5′-GCTCAGTACCAGCGAGAAT-3′. In brief, siBCN or siNC was thoroughly mixed with transfection reagents to achieve a final concentration of 50 nM. H9C2 cells were then incubated with this mixture for 48 h.
MitoSOX Red fluorescence measurement
After 48 h of transfection, experimental cells were exposed to PA for an additional 24 h. Subsequently, the cells were incubated with 5 μM MitoSOX Red (HY-D1055; MCE) for 10 min at 37°C, followed by two washes with phosphate-buffered saline. The fluorescence intensity of MitoSOX Red was captured at 510/580 nm using a Nikon fluorescence microscope (N2-DMi8).
Subcellular fractionation
We conducted subcellular fractionation (mitochondria and cytoplasm) following the procedure described by Hsu et al. (2014). Cells from 10 cm plates were lysed in a 1 mL buffer containing 250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 × proteinase and phosphatase inhibitor cocktails (P1045; Beyotime Biotechnology, Shanghai, China). The lysate was then centrifuged at 720 g for 5 min. The supernatant was collected and centrifuged at 10,000 g for 20 min to obtain the mitochondrial fraction. Subsequently, the supernatant was centrifuged at 100,000 g for 60 min to obtain the membrane and cytosol fractions. We confirmed the purity of the enriched subcellular fractions using specific antibodies for cytosolic (Gapdh) and mitochondrial (COX-IV) proteins.
Western blot
Heart tissue or cardiomyocyte protein extraction involved homogenization in RIPA buffer (P0013B; Beyotime Biotechnology) with 1 × proteinase and phosphatase inhibitor cocktail. Protein concentration was determined using a BCA protein assay kit according to the manufacturer's instructions (P0012; Beyotime Biotechnology). Equal protein amounts were denatured at 100°C for 10 min, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) on a 12.5% gel (PG113 EpiZyme, Shanghai, China), and transferred onto a polyvinylidene difluoride membrane (IPVH00010; Millipore, Billerica, MA). The membrane was blocked for 1 h at room temperature in TBST with 5% nonfat dry milk, followed by overnight incubation at 4°C with primary antibodies against p62 (18420-1-AP; Proteintech, Rosemont, IL), LC3 (14600-1-AP; Proteintech), COX IV (4850S; Cell Signaling Technology, Danvers, MA), Rab9 (5133; CST), Beclin-1 (3495S; CST), Gapdh (10494-1-AP; Proteintech), ATG 5 (12994S; CST), TOM20 (42406S; CST), PGC1 (2178S; CST), p-AMPK (2535S; CST), AMPK (2532; CST), PINK1 (23274-1-AP; Proteintech), Parkin (2132; CST), BNIP3 (3796; CST). Immunoreactive bands were detected using alkaline phosphatase-conjugated antimouse (SA00001-1; Proteintech) or rabbit antibodies (SA00001-2; Proteintech) and an enhanced chemiluminescent substrate (34096; Thermo Fisher Scientific, Waltham, MA). Densitometry was performed with ImageJ (version 1.34S).
Transmission electron microscopy
Cell samples were fixed with 2.5% glutaraldehyde and 1% osmium tetroxide. After dehydration in graded ethanol, samples were impregnated with epoxy-dendritic ester, followed by sectioning and double staining with uranium acetate and lead citrate. Autophagosomes and mitochondria were observed using a transmission electron microscope (JEM1400, Tokyo, Japan) in random fields of view.
mt-Keima adenovirus transfection
mt-Keima, a mitochondrial-localized pH indicator protein, shifts its excitation spectrum from 440 to 586 nm when mitochondria are delivered to acidic lysosomes, changing from green to red (Katayama et al., 2011). For observation of mitophagy in cultured cells, adenovirus harboring mt-Keima was conducted (Hanbio Technology Co., Ltd., Shanghai, China), and transfected according to the manufacturer's instructions. Cells were cultured on 24-well plates with cell slides and transfected with mt-Keima adenovirus (Multiplicity of Infection = 50) for 18 h at 37°C. Afterward, the medium was replaced with fresh medium containing PA or BAF for an additional 24 h. Subsequently, the cells were washed, fixed with 4% paraformaldehyde at room temperature for 15 min, and mounted using Vectashield mounting medium (H-1200; Vector Labs, Burlingame, CA). Statistical analysis was performed by quantifying the number of red and green puncta in each cell using a confocal microscope (LSM 900 with Airyscan 2; ZEISS, Germany).
Statistical analysis
All values in graphs are expressed as mean ± standard error. All analysis was performed using GraphPad Prism 7 software (GraphPad Prism Software, La Jolla, CA). Statistical analysis for multiple groups analysis was conducted using one-way ANOVA followed by Tukey's test (data with normal distribution) or the Kruskal–Wallis test followed by Dunn's test (data with non-normal distribution). For quantification with more than two groups and the groups with subgroups, two-way ANOVA followed by Tukey's test was used. A value of p < 0.05 was considered significant.
Footnotes
Data Availability
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Authors' Contributions
X.L., J.R., and L.W. contributed to study concept and design; X.Z., Z.Y., J.S., F.L., and L.P. assisted with development of methodology and writing, review and revision of the article; C.Z., X.J., A.R., and L.Z. performed acquisition, analysis and interpretation of data, and statistical analysis; all authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (81700279, 82100071, 81873416, 82070055); the Key Research and development program of Hunan Province (2020SK2065); the Research project of Hunan Provincial Health Commission (20220315026); the Youth Fund of Natural Science Foundation of Hunan Province (2020JJ5945); the Natural Science Foundation of Hunan Province (2022JJ40794); VUMC Faculty Research Scholars grant and R01HL095797-08.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.